
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dioxygen controls the nitrosylation reactions of a protein-bound [4Fe4S] cluster†
Daniel B.
Grabarczyk‡
 ,
Philip A.
Ash§
,
William K.
Myers
,
Erin L.
Dodd
and
Kylie A.
Vincent
*
,
Philip A.
Ash§
,
William K.
Myers
,
Erin L.
Dodd
and
Kylie A.
Vincent
*
Department of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: kylie.vincent@chem.ox.ac.uk
First published on 9th September 2019
Abstract
Iron–sulfur clusters are exceptionally tuneable protein cofactors, and as one of their many roles they are involved in biological responses to nitrosative stress. Both iron–sulfur proteins and synthetic model clusters are extremely sensitive to nitrosylation, tending towards rapid multi-step reaction and cluster degradation. Reaction of protein-bound iron–sulfur clusters with nitric oxide can be stopped at partial nitrosylation in vivo, and repair of protein-bound nitrosylated clusters is possible in the cellular environment. We have used a combination of infrared, EPR, and UV-visible spectroscopies to show that a model [4Fe4S] cluster-containing protein, A. ferroxidans high potential iron–sulfur protein (HiPIP), reacts with NO to give a product mixture dominated by Roussin's Black Salt (RBS) and Roussin's Red Ester (RRE) species. We have shown that O2 plays a critical role in controlling the major product of nitrosylation, with RBS-like products favoured under strictly anaerobic conditions and RRE favoured in the presence of trace O2. Moreover, addition of trace O2 to anaerobically nitrosylated samples induces conversion of RBS-like products to RRE. These findings may have implications for mechanisms of iron–sulfur cluster repair following nitrosative stress, suggest a crucial role for trace O2, and provide an important link between nitrosylation chemistry of iron–sulfur proteins and the well-established reactivity of synthetic iron–sulfur clusters.
Introduction
Bacteria are exposed to the free-radical gas nitric oxide (NO) in a wide variety of situations, for example during the mammalian immune response1 and as an intermediate in denitrification.2 Nitric oxide has been implicated in bacterial group behaviour3 and its disruption.4 It is a very reactive species, cytotoxic at high levels.5 The [4Fe4S] cluster motif is highly reactive to NO and bacteria exploit this sensitivity to evade the harmful effects of NO using [4Fe4S] cluster-containing proteins to sense NO and thereby modulate the activity of proteins in order to regulate the nitrosative stress response of the organism.6,7M. tuberculosis WhiB1,8S. coelicolor WhiD,9,10 and E. coli FNR,10–13 as well as the dedicated NO-sensing protein NsrR found in a wide range of microorganisms,14–20 are [4Fe4S]-containing transcription regulator proteins known to have a role in bacterial NO sensing. Understanding bacterial NO responses has broad implications, from soil science to disease treatment.The nitrosylation products of protein-bound iron–sulfur clusters have been identified by comparison with the spectroscopic signatures of small-molecule iron nitrosyl complexes. Synthetic model studies of FeS clusters have provided simplified systems to model the NO reactivity of the protein systems.21–23 A range of nitrosylation products (Scheme 1) have been directly characterized and the combination of infrared (IR) spectroscopy and crystallographic data has provided detailed structural information. The known complexes exhibit well-defined IR features in the NO spectral region that are largely solvent- and counterion-independent24 and function as a valuable spectroscopic fingerprint that can be used to characterize nitrosylation products in proteins.
 | ||
| Scheme 1 Iron–Sulfur clusters and nitrosyl iron–sulfur complexes. | ||
The mononuclear dinitrosyl iron complex (DNIC, Scheme 1), easily detected by a characteristic EPR signal at g = 2.03, is the most well-known product of the nitrosylation of synthetic and protein-bound iron–sulfur clusters.25 When quantified, the yield of this product arising from nitrosylation of protein-bound [4Fe4S] clusters is usually low.10,26–28 Other known products of iron–sulfur cluster protein nitrosylation are EPR-silent and in some cases such species make up the majority of products.7,23 UV-visible spectroscopy has implicated the dinuclear Roussin's Red Ester (RRE, Scheme 1) as a major product, visible by its absorption at 367 nm.29 This RRE product is EPR silent, but can be reduced by S2O42− and quantified indirectly by the g = 2.01 EPR signal of the reduced RRE (rRRE, Scheme 1).30 Nuclear resonance vibrational spectroscopy (NRVS) and IR spectroscopy have been used to distinguish directly the final products of reactions of NO with protein-bound iron–sulfur clusters. Using NRVS, it was shown that a [2Fe2S] Rieske protein formed RRE as the major nitrosylation product,30 whilst a [4Fe4S] ferredoxin formed Roussin's Black Salt (RBS, Scheme 1) as the major product.31 These are consistent with products observed from small molecule nitrosylation.32 Non-denaturing mass spectrometry and NRVS have been used productively to identify further products of the nitrosylation reactions of [4Fe4S] proteins.9,10,14–16,33–35
We previously reported IR spectroscopy alongside other spectroscopic methods to show that [2Fe2S] Spinach ferredoxin (Fd) reacts with NO in a manner dependent on the redox environment of the reaction. Nitrosylation of this protein under anaerobic conditions yields a mixture of DNIC and RRE, whereas, in the presence of trace O2 or the thiolate sequestering reagent, iodoacetamide, RRE is formed as the major product.36 During the mammalian immune response, the iron–sulfur proteins within a pathogenic bacterium would be exposed to NO in the presence of various oxidants and thiolate-reactive agents. Recent studies of the nitrosylation of the [4Fe4S] cluster-containing Endonuclease III have utilized a combination of protein film voltammetry, mass spectroscopy, and hyperfine sublevel correlation (HYSCORE) pulse EPR spectroscopy to show loss of iron and formation of stoichiometrically equal amounts of DNIC and RRE as the final nitrosylation products.28
In this work, we employ IR spectroscopy, in addition to EPR and UV-visible spectroscopy, to explore the reactivity of a model [4Fe4S] cluster protein, Acidithiobacillus ferroxidans high potential iron–sulfur protein (HiPIP), with NO. This HiPIP offers advantages of well-defined redox chemistry, and is O2-stable, allowing us to explore reactions with NO in the presence of O2.37,38 A variant of the protein with a poly-His tag for affinity purification can be prepared in good yields, in air, via an over-expression system in Escherichia coli. For ease of communication, we will use the formula type [FenSm]0/a+/b− to describe specific cluster species throughout this paper, while keeping to the traditional form [4Fe4S] in general cases. Here we identify products of reaction of HiPIP with NO, and demonstrate that trace O2 can alter the reactivity of this protein-bound cluster with NO.
Experimental methods
Cloning, expression and purification of recombinant Acidithiobacillus ferroxidans HiPIP
A strain for expression of recombinant Acidithiobacillus ferroxidans HiPIP was generously donated by Prof. Ben Berks, University of Oxford. HiPIP was in a pQE60 expression plasmid with its native Tat signal peptide and a C-terminal His-tag, expressed in the cytoplasm of an E. coli BL21 strain with a deleted Tat system. The strain was grown aerobically at 37 °C in lysogeny broth media containing 100 mg L−1 ampicillin and kanamycin. When the culture had grown to an A600 of 0.6, cells were induced with 1 mM isopropyl β-D-1-thiogalacto-pyranoside and incubated for a further four hours. Bacteria were harvested by centrifugation and resuspended in 50 mM Tris-HCl pH 8, 500 mM NaCl, 10 mM imidazole with EDTA-free protease inhibitors (Roche) and a few crystals of lysozyme and DNase I (both Sigma-Aldrich). Cells were then lysed by two passages through a French Press (Thermo Scientific) at 65-100 MPa or by sonication (Fisherbrand Q500, Fisher Scientific). Cell debris was removed by centrifugation at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for one hour at 4 °C. The supernatant was loaded on a 5 mL histrap HP (GE Healthcare) column using an ÅKTA FPLC (GE Healthcare) system and washed with ten column volumes of 50 mM Tris-HCl buffer containing 500 mM NaCl and 25 mM imidazole, and then eluted with a gradient from 25 to 210 mM imidazole over ten column volumes. Protein-containing fractions were pooled and then concentrated by ultrafiltration to 500 μL, before final purification by size exclusion chromatography using a Superdex 75 10/300 (GE Healthcare) equilibrated in 25 mM HEPES-NaOH pH 7.5 160 mM NaCl. The concentration of HiPIP was determined by optical absorption using an ε388 of 18000 M−1 cm−1.
000g for one hour at 4 °C. The supernatant was loaded on a 5 mL histrap HP (GE Healthcare) column using an ÅKTA FPLC (GE Healthcare) system and washed with ten column volumes of 50 mM Tris-HCl buffer containing 500 mM NaCl and 25 mM imidazole, and then eluted with a gradient from 25 to 210 mM imidazole over ten column volumes. Protein-containing fractions were pooled and then concentrated by ultrafiltration to 500 μL, before final purification by size exclusion chromatography using a Superdex 75 10/300 (GE Healthcare) equilibrated in 25 mM HEPES-NaOH pH 7.5 160 mM NaCl. The concentration of HiPIP was determined by optical absorption using an ε388 of 18000 M−1 cm−1.
Gas exposure
10% NO gas in N2 (BOC) was first passed through a stainless steel column packed with Ascarite II (20–30 mesh, Sigma Aldrich) to remove higher oxide impurities. The flow rate of each gas was set at 20 mL min−1 using mass flow controllers (Brooks Instruments). All following steps were performed in a N2-filled anaerobic glovebox (<1 ppm O2, Glove Box Technology Ltd). Gases were bubbled through 5 M NaOH to moisture-saturate and remove NOx impurities.The solubility of NO in water is 1.94 mM at 1 bar pressure, dropping by about 10% in a buffered solution containing 150 mM NaCl.2 The solubility of O2 in water is 1.2 mM at 1 bar pressure.3
Infrared spectroscopy
Attenuated Total Reflectance (ATR)-IR measurements were recorded using the external beam of a Varian 680-IR spectrometer coupled to a customised ATR accessory (GladiATR, PIKE Technologies) with a trapezoidal silicon internal reflection element (IRE, Crystal GMBH) and a mercury cadmium telluride detector (InfraRed Associates) cooled to 77 K in a N2-filled anaerobic, dry glove box (<1 ppm O2, <−85 °C dew point, Glove Box Technology Ltd) as described previously.4 Spectra were recorded at 4 cm−1 resolution with an acquisition time of 325 s. The Si Internal Reflection Element (IRE) was washed between the loading of every sample by low power sonication (35 W, Ultrawave U100) in sulfuric acid and then nitric acid, and rinsed in ultra pure (18 MΩ cm) water (MilliQ, Millipore). Samples (10 μL) were loaded onto the prism using a gas-tight syringe (Hamilton) and then immediately sealed in a gas-tight cell to prevent evaporation during acquisition of spectra.UV-visible spectroscopy
A 5 μL aliquot of exposed or native HiPIP (0.8 mM) was transferred from the exposure vial using a gas-tight syringe (Hamilton) and diluted to 30 μM in buffer (25 mM HEPES 160 mM NaCl pH 7.5), centrifuged to remove any precipitate, and transferred to a quartz cuvette (Hellma) with a 10 mm pathlength. Spectra were recorded at a scan rate of 300 nm min−1versus a buffer blank using a Cary 60 UV-visible spectrophotometer (Agilent) housed entirely in the same glove box as the gas exposure system.EPR spectroscopy
CW-EPR spectra were collected in the Centre for Advanced Electron Spin Resonance (CAESR) in the Department of Chemistry at the University of Oxford. X-band measurements were performed under non-saturating conditions with a Bruker-Biospin EMXmicro spectrometer equipped with a PremiumX microwave bridge, a cylindrical TE011 mode resonator (SHQE-W), an ESR-900 cryostat operating with liquid helium, and an ITC-503s temperature controller (Oxford Instruments). Spin quantitation was obtained by double integration and comparison to a 1 mM Cu(II)EDTA standard. For each EPR sample, protein (120 μM) was prepared in buffer (25 mM HEPES, 160 mM NaCl, 25% glycerol, pH 7.5). For experiments involving oxidation/reduction of protein or nitrosylation products, samples of protein (120 μM) were prepared in buffer (25 mM HEPES, 160 mM NaCl, 25% glycerol, pH 7.5) and excess sodium hexachloroiridate (Na2IrCl6) or excess sodium dithionite (Na2S2O4) was added directly.Results and discussion
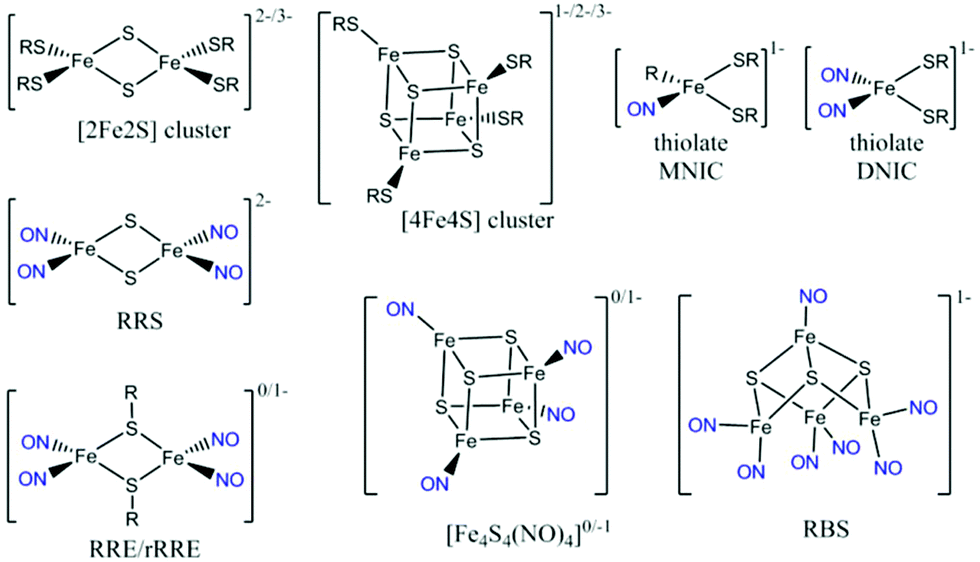
A. ferroxidans HiPIP is isolated in the [Fe4S4]2+ state. The [4Fe4S] cluster is bound to the protein via four cysteine ligands and the protein also contains an internal disulfide bond involving two other cysteine ligands, one of which is directly adjacent to a cluster-binding cysteine (sequence shown in ESI†).37 We first performed UV-visible and EPR spectroscopy to verify the identity of the isolated HiPIP. The UV-visible spectrum shows the characteristic [Fe4S4]2+ maximum at 388 nm (Fig. 1B).38 As-purified, the sample displayed no EPR signal, as anticipated for a [Fe4S4]2+ cluster (Fig. S1†). Oxidation of the sample to the [Fe4S4]3+ state with sodium hexachloroiridate (Na2IrCl6) resulted in a typical [Fe4S4]3+ axial signal as anticipated for the oxidised HiPIP (Fig. 1C(ii)).38 The [Fe4S4]+ state of the cluster is inaccessible,39 and an EPR spectrum of HiPIP treated with sodium dithionite (Na2S2O4) showed no signal (Fig. 1C(iii)). The identity of the protein was further verified by cyclic voltammetry, which showed a reversible redox process centred at +0.52 V vs. SHE (Fig. S2†) consistent with the potential reported by Bruscella et al.38 for untagged A. ferroxidans HiPIP.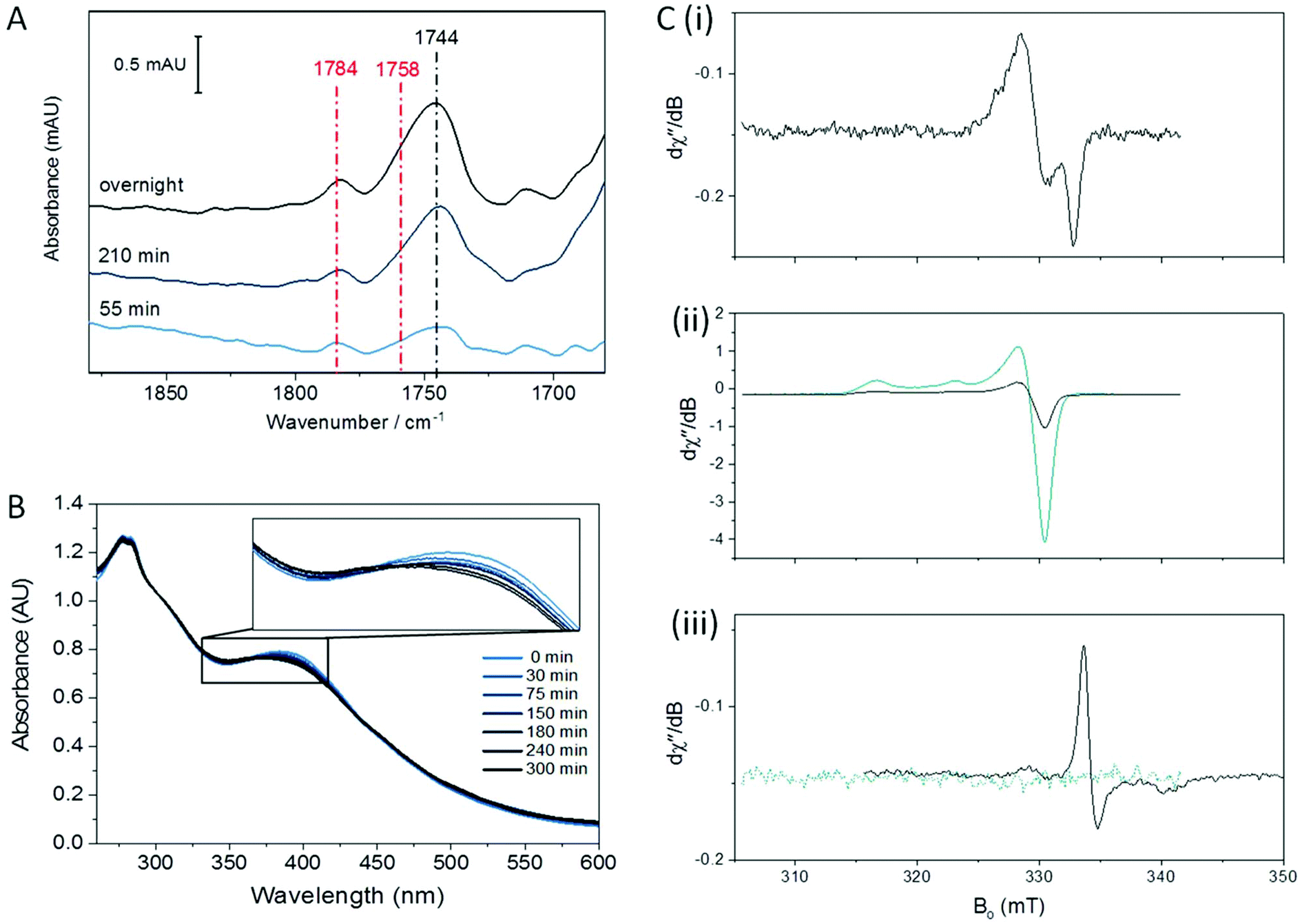 | ||
| Fig. 1 Anaerobic reaction of HiPIP with NO (A) IR spectroscopy: 0.8 mM HiPIP was exposed to a flow of 10% NO above the sample on the ATR prism. Spectra were recorded at the exposure times indicated and processed against a background of the initial unexposed HiPIP sample. (B) UV-visible spectra of 30 μM HiPIP following timed exposure to 10% NO gas. Spectra are normalized at 300 nm to account for small differences in protein concentrations which obscure the spectral changes. (C) EPR spectra at 15 K of 120 μM HiPIP: (i) exposed to 10% NO overnight; (ii) unreacted HiPIP (blue) and HiPIP exposed to 10% NO overnight (black), followed by oxidation by IrCl62−; (iii) unreacted HiPIP (blue) and HiPIP exposed to 10% NO overnight (black), followed by reduction by S2O42−. Spectra were recorded at 15 K. EPR measurement conditions of C, D, & E were all 5 μW microwave power, 0.2 mT field modulation, and microwave frequencies of a 9.379 ± 0.002 GHz range. | ||
Anaerobic nitrosylation of [Fe4S4]2+ HiPIP
The nitrosylation of a [4Fe4S] HiPIP from Allochromatium vinosum was previously studied by UV-visible and EPR spectroscopy.40 The authors observed significant DNIC formation by EPR spectroscopy. Both the UV-visible spectrum of the nitrosylated protein and the EPR spectrum of the nitrosylated protein following reduction were consistent with concomitant formation of RRE products. To investigate the nitrosylation chemistry of A. ferrooxidans HiPIP we first exposed the protein to an atmosphere of 10% NO gas (in N2) under anaerobic conditions (Fig. 1). After 210 min of exposure an IR spectrum showed a range of peaks in the νNO region (Fig. 1A). Features below 1710 cm−1 are likely to be caused by losses of protein secondary structure on a background of small changes in total water and amide absorption: these features are similar in all spectra of NO-exposed HiPIP samples and will not be interpreted further. The most intense peak in the nitrosyl region of the spectrum is at 1744 cm−1, with a shoulder at 1758 cm−1 and another small peak at 1783 cm−1. After overnight anaerobic exposure of HiPIP to 10% NO all peaks had increased in intensity.We assign the 1744 cm−1 peak to formation of an RBS-like species. The formation of RBS has been observed as a major product of the nitrosylation of synthetic [4Fe4S] clusters.23,32 RBS is accepted as the major product of anaerobic nitrosylation of small molecule [4Fe4S] clusters.23,32 The formation of a RBS-like species is also consistent with previous reports of the products of nitrosylation of protein-bound [4Fe4S] clusters based on non-denaturing mass spectrometry and NRVS.7,9,12,16,31,33 The observation by IR spectroscopy of RBS as a significant product of a [4Fe4S] protein nitrosylation is particularly important as this species cannot be readily assigned using EPR spectroscopy.
Salts of the synthetic RBS anion are characterised by a strong absorbance at 1745 cm−1 accompanied by a much weaker band centred at 1796 cm−1.23,24 A weak feature may be present in our data at 1798 cm−1, though it is not significantly distinguishable from noise (vide infra). We hesitate to posit that the species is chemically identical to synthetic RBS, as the data presented later in this paper suggest that the cluster remains bound to the protein. This would not be possible for the true RBS anion, which has only sulfide and nitrosyl ligands to iron. This species has no EPR signal, consistent with the S = 0 of RBS, and shows an identical frequency to RBS for the most intense νNO band in the IR. We have chosen to use the term ‘RBS-like’ species to account for the likely possibility that the product remains bound to the protein and is a derivative of RBS.
The 1758 cm−1 and 1783 cm−1 peaks of near-equal intensity are consistent with the pair of peaks observed for small molecule RRE species.36,41,42 They are also in complete agreement with nitrosyl peaks observed for protein-bound RRE which is formed in low yield following nitrosylation of Spinach Fd under anaerobic conditions.36 The ratio of RBS-like and RRE products observed was reproducible over multiple experiments. UV-visible spectroscopy of the sample exposed to 10% NO as a function of exposure time (Fig. 1B) showed a reaction profile similar to those seen in the nitrosylation of the [4Fe4S] cluster proteins FNR, NsrR, and WhiX.9,12,16,33 For these proteins a decrease in the intensity of the [Fe4S4]2+ peak at approximately 400 nm was observed, accompanied by a very slight increase in intensity at 360 nm, followed shortly by a decrease in intensity for all peaks above 300 nm. The spectra obtained for the anaerobic nitrosylation of HiPIP indicated that the nitrosylation reaction with HiPIP did not proceed to complete loss of cluster signal at 388 nm under these conditions (Fig. 1B). The UV-visible spectrum of RBS formed in small-molecule systems is characterized by peaks at 360, 420 and 550 nm.24 These would not be seen clearly here due to overlap with absorbance from the protein and unreacted cluster, and possibly other nitrosylation products (Fig. 1B).
Fig. 1C shows an EPR spectrum of HiPIP following overnight exposure to 10% NO under anaerobic conditions. EPR Spectra recorded at 15 K or 100 K showed only a characteristic DNIC signal (Fig. 1C(i), Fig. S3†) with a temperature-independent spin concentration of 3.2 μM at 15 K. An EPR spectrum of unreacted HiPIP of equal total protein concentration, oxidised to the EPR-active [Fe4S4]3+ state using IrCl62− (Fig. 1C(ii)), gave a spin concentration of 95 μM. Therefore the DNIC product accounts for only 3% of the original cluster, less than 1% of the total iron. The DNIC EPR signal from nitrosylation of HiPIP also had a noticeable shoulder, which may be due to a very minor DNIC species with cysteine persulfide ligands10 or the presence of a very small amount of a tetranitrosylated cubane cluster,23 or other minor cluster derivative. At these concentrations, any DNIC signal in the IR νNO region would be likely to be obscured by other bands. Lin et al.43 report a pair of νNO bands of near-equal intensity for cysteine-bound DNICs ranging from 1772 cm−1 and 1727 cm−1 for [(Cys)2Fe(NO)2]− to 1767 cm−1 and 1722 cm−1 for a de-novo peptide-bound DNIC. IR data are available for a range of model compounds featuring the DNIC Fe(NO)2 motif, with the νNO bands varying with co-ligand and solvent.44–54 No bands in the spectra for nitrosylated HiPIP resemble a DNIC species.
As both RBS and neutral RRE are EPR-silent, we tested the nitrosylation products both before and after oxidation with IrCl62− (Fig. 1C(ii)) or reduction with S2O42− (Fig. 1C(iii)). An EPR spectrum of the nitrosylated sample following addition of IrCl62− showed only a signal identical to that of HIPIP in the [Fe4S4]3+ state (Fig. 1C(ii)) which we assigned to unreacted cluster; the spin concentration of 20 μM showed that this accounted for 20% of the original cluster. The EPR spectrum of the sample reduced by S2O42− showed a reduced RRE (rRRE) signal (Fig. 1C(iii)) which accounted for a small portion of the original cluster with a spin concentration of 0.9 μM, or 1% of total cluster; it is possible that S2O42− fails to fully reduce the protein bound RRE as the [4Fe4S] cluster is known to be quite buried in HiPIP.37 The presence of rRRE in the sample reduced by S2O42− supports the assignment of the peaks at 1783 cm−1 and 1758 cm−1 in the IR spectrum to RRE.
We cannot rule out loss of some iron-containing material in the sample preparation stage but the result suggests that there may be nitrosylation products of HiPIP which are EPR silent and which cannot be easily oxidised or reduced to an EPR-active product. The major product observed by IR, which we assigned as the RBS-like species with a signal at 1744 cm−1, is unaccounted for in the identified EPR active species. The monoanionic synthetic RBS is an S = 0 complex undetectable by EPR. Our RBS-like complex is also EPR-silent, and we are therefore unable to quantify it using a spin concentration.
Taking these data together we conclude that the major product of anaerobic nitrosylation of [Fe4S4]2+ HiPIP is an RBS-like species, with concomitant formation of a small amount of RRE and a smaller proportion of DNIC.
Synthetic studies suggest that, in situations with high thiolate availability, an overall reducing environment leads to DNIC products predominating from the nitrosylation of a [4Fe4S] cluster, while the presence of trace O255 or nitrosylation by NO+ as opposed to the neutral free radical NO˙ favours RRE products.21 High thiolate availability is here understood to be at least eightfold excess thiolate per [4Fe4S] cluster, or two thiolates per iron centre. In the absence of excess thiolate, RBS has been found to predominate.32 We note that thiolate availability itself would be very much affected by the pH and the redox environment, with thiolate being oxidized to disulfide in oxidising environments (free Cys E0′(CysSH/CysSSCys) = −220 mV at pH 7.0 (ref. 56)).
The observations we have made of the anaerobic nitrosylation of A. ferroxidans HiPIP fit this assessment. The [4Fe4S] cluster of this protein is surrounded by four available cysteine residues in a hydrophobic pocket within the protein, as well as two unavailable, oxidised cysteine residues in the form of a disulfide bridge.37,38 Thus the RBS-like species we observed would be expected in the HiPIP.
Effect of trace O2 on the nitrosylation of HiPIP
Redox environment is emerging as a critical factor in the determination of which nitrosylation products will predominate following reaction of synthetic complexes with NO.22,23,32,42 We have previously shown that DNIC species are the major product when Spinach Fd is exposed to NO in an anaerobic environment, whereas RRE is formed rapidly as the major product in the presence of trace O2.36 We therefore investigated A. ferroxidans HiPIP nitrosylation in the presence of trace O2.Fig. 2 shows IR, UV-visible, and EPR spectra recorded following exposure of HiPIP to a gas mixture containing 10% NO and 0.05% O2 (with N2 as the remaining carrier gas). After 30 minutes, two strong peaks in the IR spectrum at 1783 and 1758 cm−1 could be seen (Fig. 2A), attributed to RRE.41 A feature at 1744 cm−1 was also present, again assigned to an RBS-like species. These species remained with similar relative intensities in IR spectra recorded after 60 and 125 minutes. However, the relative intensity of bands attributed to RRE and RBS-like species is significantly different to that observed following anaerobic nitrosylation (Fig. 1A), with RRE peaks now being much more prominent. Thus there is a balance in the formation of RRE and RBS-like products that is shifted in the direction of RRE when nitrosylation occurs in the presence of a small amount of O2. UV-visible spectra (Fig. 2B) of the sample exposed to 10% NO in the presence of trace O2 for 125 minutes showed loss of characteristic [Fe4S4]2+ features, and an increase in a peak at 365 nm, consistent with extensive RRE formation.36
 | ||
| Fig. 2 Reactions of HiPIP with NO and trace O2 (A) IR spectra of 0.8 mM HiPIP exposed to a 10% NO/0.05% O2 gas mixture for the indicated time. Spectra are presented processed against a background of unexposed HiPIP. (B) UV-visible spectra of the 125 min sample from panel A shown with the unreacted HIPIP sample for comparison. Spectra were recorded of exposed or unexposed HiPIP which had been diluted to 33 uM in buffer. Spectra were normalized at 300 nm to account for small differences in concentration for clarity. (C) EPR spectra at 100 K of 120 μM HiPIP: (i) exposed to 10% NO overnight (black) and a 10% NO/0.05% O2 gas mixture for 150 minutes (red), and (ii) exposed to 10% NO overnight followed by reduction with S2O42− (black) and a 10% NO/0.05% O2 gas mixture for 150 minutes followed by reduction with S2O42− (red). EPR measurement conditions of C & D were all 200 μW microwave power, 0.4 mT field modulation, and microwave frequencies of 9.3813 and 9.3792 GHz, respectively. | ||
EPR spectroscopy was performed on HiPIP exposed to 10% NO and trace O2 for 150 minutes (Fig. 2C). In the absence of additional oxidising or reducing agents, a clear DNIC signal was observed with a spin concentration of 9.9 μM, or 10% of total cluster (Fig. 2C(i)). Reduction of the exposed HiPIP with S2O42− gave rise to a rRRE signal with a spin concentration of 29.9 μM, or 30% of total cluster (Fig. 2C(ii)), compared to only about 1% RRE from the anaerobic nitrosylation of HiPIP (Fig. 1C(iii)). The [Fe4S4]3+ EPR signal of sample exposed to 10% NO and trace O2 for 150 minutes and treated with IrCl62− had a spin concentration of 8.3 μM (9% unreacted cluster). A comparison of the amounts of residual unreacted cluster following aerobic and anaerobic exposure to NO shows that less unreacted cluster remained following reaction with NO in the presence of trace O2 after 150 min than following overnight anaerobic exposure (Fig. S4†).
The preference for the formation of RRE from the reaction of iron–sulfur clusters with NO and trace O2 appears to be a general phenomenon. For formation of RRE from Spinach Fd, we suggested that the role of O2 was to sequester thiolates during the reaction and showed that iodoacetamide, a specific thiolate sequestering reagent and general scavenger of strong nucleophiles could substitute for O2 in this role. However, in HiPIP, thiolate sequestration is an unlikely mechanism due to the buried nature of the cluster site.37 Supporting this, addition of iodoacetamide had no effect on the product distribution for anaerobic reaction of HiPIP with NO (Fig. S5†).
Nitric oxide itself is susceptible to changes in the redox environment, with both the reduced state, nitroxyl (NO−), and the oxidised state, nitrosonium (NO+), being reactive species in their own right. Nitric oxide is well understood to react with oxygen to yield a wide range of reactive NOx products, with the speciation heavily dependent on the reaction conditions.57–59 It is possible that a specific step in the RRE formation pathway is favoured over RBS formation by the presence of the products resulting from the combination of NO and trace O2.
Harrop et al. found that, while the reaction of synthetic [4Fe4S] cluster with NO yielded RBS products in the absence of other reagents, the reaction would instead yield DNIC products when performed in the presence of excess thiolate with the free thiolates participating in the elimination of the bridging sulfides,32 suggesting that the NO reactivity of [4Fe4S] clusters is dependent on reaction conditions. While the number of available thiols is unchanged between the anaerobic and aerobic nitrosylation of A. ferroxidans HiPIP, the O2 could potentially facilitate elimination of the bridging sulfides. Oxidation of the bridging sulfides has been observed in the nitrosylation of other protein-bound [4Fe4S] clusters with the sulfides remaining bound to the protein as persulfide cysteine,15,35 and indeed Crack et al. observed formation of RRE products in the nitrosylation of several [4Fe4S] proteins.7
We know that thiolate DNIC species can be converted to RRE under oxidising conditions with the thiolate sulfides as targets of oxidation and disulfide as a byproduct.42,55,60 Likewise, direct reaction of iron thiolates with nitrosonium (NO+) is known to result in RRE formation.21 In particular, Fitzpatrick et al. found that reaction of thiolate DNIC species with O2 resulted in the formation of RRE species and disulfides. Thus it is not unreasonable that the predominance of RRE following reaction of A. ferridoxans HiPIP with NO in the presence of O2 could arise from the same mechanism proposed for synthetic models, namely oxidation and abstraction of bridging sulfides of the [4Fe4S] cluster, or by analogous reaction with a more oxidised form of NOx.
Effect of HiPIP oxidation state: anaerobic nitrosylation of [Fe4S4]3+ HiPIP
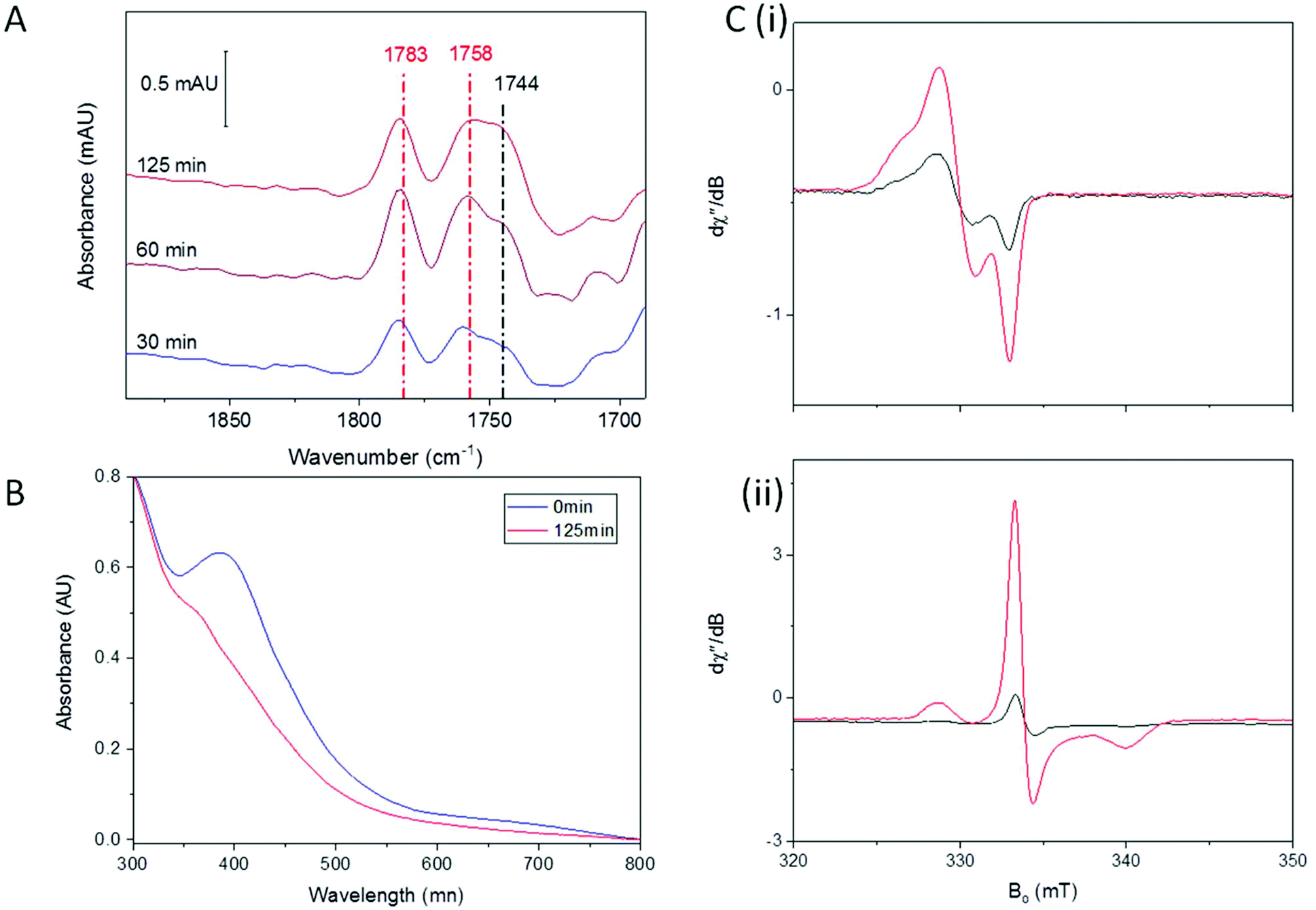
A previous report studied the nitrosylation of Allochromatium vinosum HiPIP and showed that the concentration of DNIC products was dependent on the starting redox state of the cluster, though other products were not investigated in that study.40 Having observed the effect of an oxidative environment in increasing the amount of RRE, we were interested to see if the initial oxidation state of the cluster had a similar effect. We therefore oxidised A. ferroxidans HiPIP using IrCl62−, and removed the IrCl62−/3− by buffer exchange. We then exposed [Fe4S4]3+ HiPIP to 10% NO under anaerobic conditions. The IR spectrum of [Fe4S4]3+ HiPIP exposed to 10% NO overnight (Fig. 3A) showed a similar pattern of peaks as when [Fe4S4]2+ HiPIP was exposed to 10% NO overnight (Fig. 1A). The UV-visible spectrum of [Fe4S4]3+ HiPIP has increased absorption between 400 and 600 nm compared to HiPIP at its resting oxidation state (Fig. 3B) consistent with previous observations.38 A UV-visible spectrum of the NO-exposed sample (Fig. 3B) showed that the increased absorbance between 400–600 nm observed in the spectrum of the oxidised cluster was lost by the reaction with NO. The band at 388 nm lost some intensity as compared to features at 300 nm. | ||
| Fig. 3 Anaerobic reaction of [Fe4S4]3+ HiPIP with NO (A) IR spectrum of 0.8 mM [Fe4S4]3+ exposed to 10% NO overnight processed against a background of unexposed HiPIP. (B) UV-visible spectra of 33 μM HiPIP. Purified HiPIP (green) was oxidised with IrCl62− and desalted (purple). The sample was then exposed to 10% NO overnight (black). Spectra are normalized at 300 nm to account for small differences in protein concentrations which obscure the spectral changes. | ||
From these data we must conclude that similar NO-reactivity was observed for both the reduced [Fe4S4]2+ and oxidised [Fe4S4]3+ forms of the HiPIP cluster. It is possible that NO first reduced [Fe4S4]3+ HiPIP before further reaction to produce nitrosylated products, as previously shown for A. vinosum [Fe4S4]3+ HiPIP using freeze-quench EPR.40
Overall the initial redox state of the cluster has been shown here to cause no significant difference in the relative amounts of RRE and RBS in the final products observed. This is not entirely unexpected, since reported studies of the reaction of FeS clusters with nitrosonium found that it was actually the thiolates that were abstracted and oxidised during the reaction that ultimately yielded RRE products,55 and therefore a one-electron oxidation from [Fe4S4]2+ to [Fe4S4]3+ HiPIP might not provide sufficient change in the overall number of electrons to alter nitrosylation reactivity of the [4Fe4S] cluster.
O2-Induced conversion of HiPIP nitrosylation products
We next investigated whether the species we are assigning as RBS with an intense IR band at 1744 cm−1, generated as a significant product during anaerobic exposure to 10% NO gas, was stable following subsequent exposure to a 10% NO/0.05% O2 gas mixture. HiPIP was exposed to 10% NO anaerobically overnight (Fig. 4, black). Trace O2 (0.05%) was then introduced to the 10% NO atmosphere above that same sample and a spectrum 100 min later showed conversion of a significant proportion of the 1744 cm−1 peak into RRE peaks (Fig. 4, red).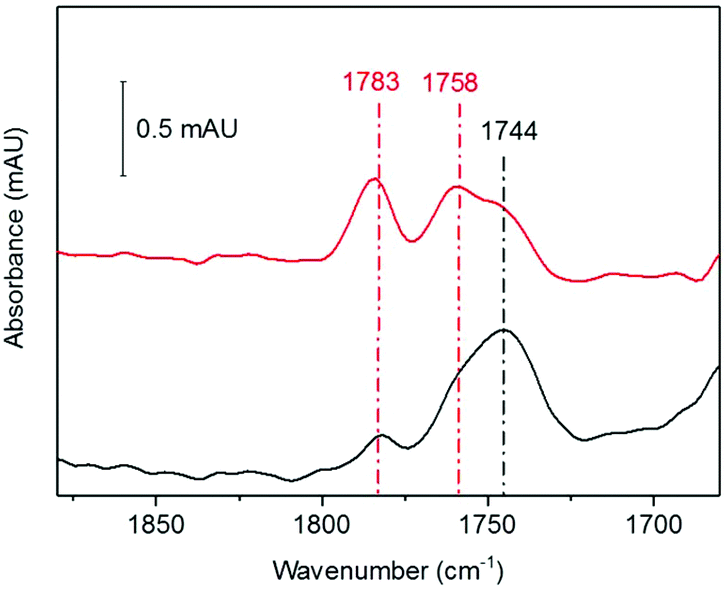 | ||
| Fig. 4 RBS to RRE conversion: step 1: HiPIP exposed to 10% NO overnight (black). Step 2: The same sample of anaerobically nitrosylated HiPIP, exposed further to 10% NO and 0.05% O2 for 100 min (red) was recorded. Spectra were processed against a background of unreacted HiPIP. | ||
Analysis of the protein backbone region of the IR spectra as compared to that of native HiPIP (Fig. S6†) shows no significant change in secondary structure. This suggests that the protein itself remains in largely the same configuration throughout these reaction steps. Comparison of the amide I band intensities before and after O2 addition suggest no significant change in protein concentration (Fig. S7A†), and an IR difference spectrum confirms conversion of RBS to RRE, rather than simply additional nitrosylation of unreacted protein (Fig. S7B†). Furthermore the difference spectrum confirms loss of a peak at 1798 cm−1, supporting our assignment of a RBS-like species. The fact that we see conversion from an RBS-like species to an RRE species suggests that both these products are protein-bound.
In synthetic systems, RBS can undergo conversion to another sulfide-bridged species ‘Roussin's red salt’ (RRS, Scheme 1) under alkaline aqueous conditions.61–63 RRS is diamagnetic, and thus EPR silent, and the sodium salt has two IR nitrosyl bands at νNO = 1717 and 1677 cm−1. No similar bands were observed in our IR spectra. However, this species has been shown to react with alkyl halides62,64 or excess thiolate to form RRE.65 The direct conversion of synthetic RBS to RRE structures is known to require more severe reaction conditions, including reaction with organohalides62,65 or via nucleophilic attack of the S2− on highly electrophilic diazonium (RN2+) and onium (R3O+) ions leading to removal of the bridging sulfides,66 and such a direct conversion is unlikely in our case.
Overall, two common features in the synthetic conversions of (μ-S)-bridged FeS cluster complexes to RRE structures seem to be thiolate availability and redox environment (Scheme 2).32,42,55,60 Excess thiolate will drive the reactions of synthetic [2Fe2S] and [4Fe4S] clusters in the direction of DNIC product.32 DNIC is known to exist in equilibrium with RRE, with the favoured direction of the equilibrium being sensitive to thiolate availability42 and the presence of an oxidising or reducing environment.
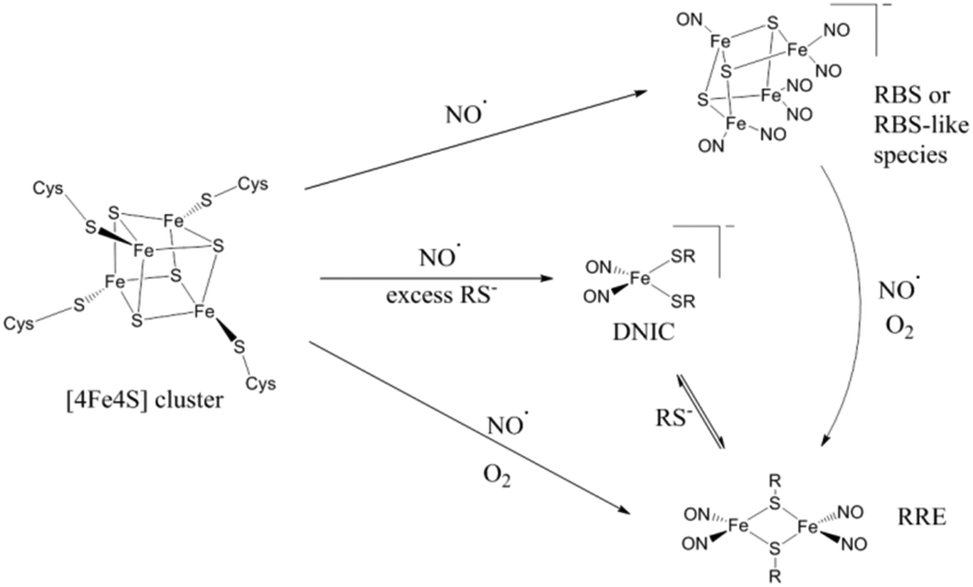 | ||
| Scheme 2 | ||
The nitrosylation of synthetic [4Fe4S] clusters is known to result in either RBS or DNIC products, dependent on the availability of sufficient excess thiolate.32 RBS can be formed directly from DNIC species in synthetic small molecule studies32,67,68 but the reverse reaction is not currently known. We have already speculated as to how the observed RRE product could be produced via oxidative elimination of bridging sulfides upon nitrosylation of A. ferroxidans HiPIP [4Fe4S] cluster in the presence of a small amount of O2.
The trend of converting (μ-S)-bridged FeS clusters to thiolate-bound RRE under oxidative conditions may potentially be extended to a (μ-S)-bridged RBS-like species when that species is held in the cluster binding site of the protein in close proximity to cysteine residues. For the protein-associated RBS-like structure formed from the anaerobic nitrosylation of HiPIP, these thiolates would be poised to interact with the cluster to form the bridging ligands of the RRE species upon further nitrosylation in the presence of O2.
A direct conversion of an RBS-like complex to RRE would necessitate elimination of the three bridging sulfides and concomitant reaction with thiolates to form the new thiolate-bridged RRE structure. This would be difficult to control in a synthetic reaction system. However, the cluster binding site within the protein may provide the ideal environment for such a conversion.
A conversion of RBS to RRE within the protein is fairly strong evidence that the RBS-like species we observe is bound to the protein in the original cluster-binding site. The RBS would need to be held within the original cluster binding site in order to position the nitrosylated cluster to exchange its bridging sulfide ligands for the cysteine thiolates of the HiPIP. Analysis of the amide backbone in the IR confirms that there is no significant change in secondary structure to accompany the formation of either cluster complex, and in fact the protein folding seems to stay fairly unchanged throughout. Thus it is likely that the binding site itself remains as a buried pocket within the protein, and that the cluster products themselves remain within that pocket.
The structure of the protein-bound RBS-like product we have observed is unknown. A survey of the literature to date in which the structures of substituted RBS-like species have been reported suggests that such species, while rare, are indeed known, with nitrosyl substitution reported both at the Fe and bridging sulfide centres.
Holm and co-workers reported a series of species with the formula [Fe4S3(NO)4(PR3)3], in which the axial and apical nitrosyls remain in the same configuration as RBS but the basal ligands are phosphine ligands.69,70 The two bands of the nitrosyl IR band signature for these species were found within the ranges of 1757–1746 cm−1 and 1730–1700 cm−1 with the more basic phosphine ligands pushing the nitrosyl bands to lower wavenumbers. Liaw et al. later proposed SPh-ligated RBS-like species based on IR data of intermediates observed during the [Fe(SPh)4]2− mediated conversion of RBS to [Fe4S4(NO)4]2− towards synthetic repair of a [4Fe4S] cluster.68
No thiolate-ligated analogue of the phosphine-substituted RBS has been isolated to date, and the IR pattern observed in our work does not deviate from the known spectrum of RBS, thus an Fe-substituted species is less likely in our case.
Recent results using NRVS have found a putative ‘Roussin's black ester’ formed in the nitrosylation of the NO-sensing protein NsrR in which an RBS-like structure is bound to protein via bridging thiolato or perthiolato ligands rather than the usual sulfides.34 The cage-like nature of a [4Fe4S] cluster binding site would be the ideal environment to observe such a complex, as it would be able to both limit the access of the site to incoming NO molecules and hold the FeS cluster complex close to the cysteine thiolates. A. ferridoxans HiPIP has a buried cluster site and at least four cysteine residues poised in position to interact with any FeS cluster complex held in that site. The conversion of RBS-like to RRE product observed in the nitrosylation of this HiPIP suggests such a protein-bound RBS-like species may indeed be formed in the anaerobic nitrosylation of this protein.
Overall the nitrosylation of A. ferroxidans HiPIP has been established to lead to multiple products. The identity of the nitrosylation end products seems to be closely linked to the mechanistic pathway by which they are formed, and the formation of both RBS-like and RRE-like products is indicative that several mechanistic pathways are available. The formation of the RBS-type product from the initial [4Fe4S] cluster may proceed via a different nitrosylation mechanism than that which leads to RRE-type product. The type of products formed upon [4Fe4S] cluster nitrosylation appears to be dependent on the redox environment, as the RRE pathway becomes preferred in the presence of trace O2. This phenomenon may be applicable to other [4Fe4S] proteins, including those with biological roles in NO sensing.
Conclusions
We have shown through a combination of IR, EPR, and UV-visible spectroscopies that under anaerobic nitrosylation conditions, the [4Fe4S] cluster of HiPIP from Acidithiobacillus ferroxidans reacts slowly to form a mixture of dinitrosyl iron complexes (DNICs), Roussin's black salt (RBS) or RBS-like species, and Roussin's red ester (RRE) species, with RBS/RBS-like species as the dominant product. When the reaction conditions are altered to include trace O2, the product mixture favours RRE, and the nitrosylation proceeds more quickly.Compellingly, we have shown directly that the anaerobic reaction product RBS can be converted to RRE by NO and trace O2. The conversion of RBS to RRE has been observed previously in synthetic model systems, but this is the first suggestion of this interconversion in a protein. That this was possible in proteins has long been assumed, and the implications for nitrosylation control have driven a strong interest in the literature on synthetic models of such conversions, as well as the identification of mechanisms and the proteins that mediate it.71 This impressive body of work of course strongly overlaps with any exploration of what nitrosylation products would form, as this would influence the starting point for downstream repair. The predominance of larger clusters, as opposed to mononuclear DNIC structures, in the nitrosylation products of a [4Fe4S] cluster is likely to influence the repair process. It will be instructive to see if similar interconversions are observed in [4Fe4S] proteins relevant to NO sensing in biology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Engineering and Physical Sciences Research Council (EPSRC) Small Equipment Award EP/K031503/1 (K. A. V.) and the Biotechnology and Biological Sciences Research Council (BBSRC) BB/P009697/1 (K. A. V. and E. L. D). W. K. M. is supported by the EPSRC (EP/L011972/1, grant to CAESR, the Centre for Advanced Spin Resonance). At the time of research D. B. G. was supported by Biotechnology and Biological Sciences Research Council (BBSRC) studentship BB/F01709X/1. We are grateful to Prof. Ben Berks for the HiPIP expression strain and for use of laboratory equipment for HiPIP purification.References
- M. A. Marletta, P. S. Yoon, R. Iyengar, C. D. Leaf and J. S. Wishnok, Biochemistry, 1988, 27, 8706–8711 CrossRef CAS PubMed.
- V. Fülöp, J. W. B. Moir, S. J. Ferguson and J. Hajdu, Cell, 1995, 81, 369–377 CrossRef.
- S. Hossain, I. Heckler and E. M. Boon, ACS Chem. Biol., 2018, 13, 1964–1969 CrossRef CAS PubMed.
- R. Urbano, J. E. Karlinsey, S. J. Libby, P.-T. Doulias, H. Ischiropoulos, H. I. Warheit-Niemi, D. H. Liggitt, A. R. Horswill and F. C. Fang, Cell Host Microbe, 2018, 23, 594–606 CrossRef CAS PubMed.
- J. B. Hibbs, R. R. Taintor, Z. Vavrin and E. M. Rachlin, Biochem. Biophys. Res. Commun., 1988, 157, 87–94 CrossRef CAS PubMed.
- H. K. Miller and V. Auerbuch, Metallomics, 2015, 7, 943–956 RSC.
- J. C. Crack and N. E. Le Brun, Antioxid. Redox Signaling, 2018, 29, 1809–1829 CrossRef CAS PubMed.
- L. J. Smith, M. R. Stapleton, G. J. M. Fullstone, J. C. Crack, A. J. Thomson, N. E. Le Brun, D. M. Hunt, E. Harvey, S. Adinolfi, R. S. Buxton and J. Green, Biochem. J., 2010, 432, 417–427 CrossRef CAS PubMed.
- J. C. Crack, L. J. Smith, M. R. Stapleton, J. Peck, N. J. Watmough, M. J. Buttner, R. S. Buxton, J. Green, V. S. Oganesyan, A. J. Thomson and N. E. Le Brun, J. Am. Chem. Soc., 2011, 133, 1112–1121 CrossRef CAS PubMed.
- J. C. Crack, M. R. Stapleton, J. Green, A. J. Thomson and N. E. Le Brun, J. Biol. Chem., 2013, 288, 11492–11502 CrossRef CAS PubMed.
- H. Corker and R. K. Poole, J. Biol. Chem., 2003, 278, 31584–31592 CrossRef CAS PubMed.
- J. C. Crack, N. E. Le Brun, A. J. Thomson, J. Green and A. J. Jervis, in Methods Enzymol., ed. R. K. Poole, Academic Press, 2008, vol. 437, pp. 191–209 Search PubMed.
- A. Volbeda, C. Darnault, O. Renoux, Y. Nicolet and J. C. Fontecilla-Camps, Sci. Adv., 2015, 1, e1501086 CrossRef PubMed.
- J. C. Crack, D. A. Svistunenko, J. Munnoch, A. J. Thomson, M. I. Hutchings and N. E. Le Brun, J. Biol. Chem., 2016, 291, 8663–8672 CrossRef CAS PubMed.
- J. C. Crack, C. J. Hamilton and N. E. Le Brun, Chem. Commun., 2018, 54, 5992–5995 RSC.
- J. C. Crack, J. Munnoch, E. L. Dodd, F. Knowles, M. M. Al Bassam, S. Kamali, A. A. Holland, S. P. Cramer, C. J. Hamilton, M. K. Johnson, A. J. Thomson, M. I. Hutchings and N. E. Le Brun, J. Biol. Chem., 2015, 290, 12689–12704 CrossRef CAS PubMed.
- K. Heurlier, M. J. Thomson, N. Aziz and J. W. B. Moir, J. Bacteriol., 2008, 190, 2488–2495 CrossRef CAS PubMed.
- N. P. Tucker, N. E. Le Brun, R. Dixon and M. I. Hutchings, Trends Microbiol., 2010, 18, 149–156 CrossRef CAS PubMed.
- E. T. Yukl, M. A. Elbaz, M. M. Nakano and P. Moënne-Loccoz, Biochemistry, 2008, 47, 13084–13092 CrossRef CAS PubMed.
- A. Volbeda, E. L. Dodd, C. Darnault, J. C. Crack, O. Renoux, M. I. Hutchings, N. E. Le Brun and J. C. Fontecilla-Camps, Nat. Commun., 2017, 8, 15052 CrossRef CAS PubMed.
- T. C. Harrop, D. Song and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 3528–3529 CrossRef CAS PubMed.
- T.-T. Lu, S.-J. Chiou, C.-Y. Chen and W.-F. Liaw, Inorg. Chem., 2006, 45, 8799–8806 CrossRef CAS PubMed.
- E. Victor and S. J. Lippard, Inorg. Chem., 2014, 53, 5311–5320 CrossRef CAS PubMed.
- J. Bourassa, B. Lee, S. Bernard, J. Schoonover and P. C. Ford, Inorg. Chem., 1999, 38, 2947–2952 CrossRef CAS PubMed.
- S. J. Lippard and J. M. Berg, Principles of bioinorganic chemistry, University Science Books, Mill Valley, Calif., 1994 Search PubMed.
- X. Duan, J. Yang, B. Ren, G. Tan and H. Ding, Biochem. J., 2009, 417, 783–789 CrossRef CAS PubMed.
- H. Cruz-Ramos, J. Crack, G. Wu, M. N. Hughes, C. Scott, A. J. Thomson, J. Green and R. K. Poole, EMBO J., 2002, 21, 3235–3244 CrossRef CAS PubMed.
- L. A. Ekanger, P. H. Oyala, A. Moradian, M. J. Sweredoski and J. K. Barton, J. Am. Chem. Soc., 2018, 140, 11800–11810 CrossRef CAS PubMed.
- H. Cruz-Ramos, J. Crack, G. Wu, M. N. Hughes, C. Scott, A. J. Thomson, J. Green and R. K. Poole, EMBO J., 2002, 21, 3235–3244 CrossRef CAS PubMed.
- C. E. Tinberg, Z. J. Tonzetich, H. Wang, L. H. Do, Y. Yoda, S. P. Cramer and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 18168–18176 CrossRef CAS PubMed.
- Z. J. Tonzetich, H. Wang, D. Mitra, C. E. Tinberg, L. H. Do, F. E. Jenney, M. W. W. Adams, S. P. Cramer and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 6914–6916 CrossRef CAS PubMed.
- T. C. Harrop, Z. J. Tonzetich, E. Reisner and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 15602–15610 CrossRef CAS PubMed.
- J. C. Crack, J. Green, A. J. Thomson and N. E. L. Brun, Acc. Chem. Res., 2014, 47, 3196–3205 CrossRef CAS PubMed.
- P. N. Serrano, H. Wang, J. C. Crack, C. Prior, M. I. Hutchings, A. J. Thomson, S. Kamali, Y. Yoda, J. Zhao, M. Y. Hu, E. E. Alp, V. S. Oganesyan, N. E. Le Brun and S. P. Cramer, Angew. Chem., Int. Ed., 2016, 55, 14575–14579 CrossRef CAS PubMed.
- J. C. Crack and N. E. Le Brun, Chem. – Eur. J., 2019, 25, 3675–3684 CrossRef CAS PubMed.
- D. B. Grabarczyk, P. A. Ash and K. A. Vincent, J. Am. Chem. Soc., 2014, 136, 11236–11239 CrossRef CAS PubMed.
- M. Nouailler, P. Bruscella, E. Lojou, R. Lebrun, V. Bonnefoy and F. Guerlesquin, Extremophiles, 2006, 10, 191–198 CrossRef CAS PubMed.
- P. Bruscella, L. Cassagnaud, J. Ratouchniak, G. Brasseur, E. Lojou, R. Amils and V. Bonnefoy, Microbiology, 2005, 151, 1421–1431 CrossRef CAS PubMed.
- R. G. Bartsch, in Methods Enzymol, ed. S. Fleischer and L. Packer, Academic Press, 1978, vol. 53, pp. 329–340 Search PubMed.
- M. W. Foster and J. A. Cowan, J. Am. Chem. Soc., 1999, 121, 4093–4100 CrossRef CAS.
- R. Wang, M. A. Camacho-Fernandez, W. Xu, J. Zhang and L. Li, Dalton Trans., 2009, 777–786 RSC.
- T. C. Harrop, D. Song and S. J. Lippard, J. Inorg. Biochem., 2007, 101, 1730–1738 CrossRef CAS PubMed.
- Z.-S. Lin, F.-C. Lo, C.-H. Li, C.-H. Chen, W.-N. Huang, I. J. Hsu, J.-F. Lee, J.-C. Horng and W.-F. Liaw, Inorg. Chem., 2011, 50, 10417–10431 CrossRef CAS PubMed.
- F.-T. Tsai, S.-J. Chiou, M.-C. Tsai, M.-L. Tsai, H.-W. Huang, M.-H. Chiang and W.-F. Liaw, Inorg. Chem., 2005, 44, 5872–5881 CrossRef CAS PubMed.
- W.-F. Liaw, C.-Y. Chiang, G.-H. Lee, S.-M. Peng, C.-H. Lai and M. Y. Darensbourg, Inorg. Chem., 2000, 39, 480–484 CrossRef CAS PubMed.
- C.-H. Hsieh, R. B. Chupik, T. A. Pinder and M. Y. Darensbourg, Polyhedron, 2013, 58, 151–155 CrossRef CAS.
- C.-H. Hsieh, Ö. F. Erdem, S. D. Harman, M. L. Singleton, E. Reijerse, W. Lubitz, C. V. Popescu, J. H. Reibenspies, S. M. Brothers, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2012, 134, 13089–13102 CrossRef CAS PubMed.
- P. Ghosh, M. Quiroz, R. Pulukkody, N. Bhuvanesh and M. Y. Darensbourg, Dalton Trans., 2018, 47, 11812–11819 RSC.
- V. G. Albano, A. Araneo, P. L. Bellon, G. Ciani and M. Manassero, J. Organomet. Chem., 1974, 67, 413–422 CrossRef CAS.
- C. Chung-Nin, A. Wojcicki, M. Calligaris and G. Nardin, Inorg. Chim. Acta, 1990, 168, 105–113 CrossRef.
- C.-H. Hsieh, S. M. Brothers, J. H. Reibenspies, M. B. Hall, C. V. Popescu and M. Y. Darensbourg, Inorg. Chem., 2013, 52, 2119–2124 CrossRef CAS PubMed.
- M.-L. Tsai, C.-H. Hsieh and W.-F. Liaw, Inorg. Chem., 2007, 46, 5110–5117 CrossRef CAS PubMed.
- J. L. Hess, C.-H. Hsieh, S. M. Brothers, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2011, 133, 20426–20434 CrossRef CAS PubMed.
- J. L. Hess, M. D. Young, C. A. Murillo and M. Y. Darensbourg, J. Mol. Struct., 2008, 890, 70–74 CrossRef CAS.
- J. Fitzpatrick, H. Kalyvas, J. Shearer and E. Kim, Chem. Commun., 2013, 49, 5550–5552 RSC.
- P. C. Jocelyn, Eur. J. Biochem., 1967, 2, 327–331 CrossRef CAS PubMed.
- R. S. Lewis and W. M. Deen, Chem. Res. Toxicol., 1994, 7, 568–574 Search PubMed.
- S. Goldstein and G. Czapski, J. Am. Chem. Soc., 1996, 118, 3419–3425 CrossRef CAS.
- D. A. Wink, R. W. Nims, J. F. Darbyshire, D. Christodoulou, I. Hanbauer, G. W. Cox, F. Laval, J. Laval and J. A. Cook, Chem. Res. Toxicol., 1994, 7, 519–525 Search PubMed.
- J. Fitzpatrick and E. Kim, Inorg. Chem., 2015, 54, 10559–10567 CrossRef CAS PubMed.
- J. Bourassa, W. DeGraff, S. Kudo, D. A. Wink, J. B. Mitchell and P. C. Ford, J. Am. Chem. Soc., 1997, 119, 2853–2860 CrossRef CAS.
- D. Seyferth, M. K. Gallagher and M. Cowie, Organometallics, 1986, 5, 539–548 CrossRef CAS.
- F. Seel, Handbook of Preparative Inorganic Chemistry V2 2nd ed., Academic Press, New York, 1965 Search PubMed.
- T. B. Rauchfuss and T. D. Weatherill, Inorg. Chem., 1982, 21, 827–830 CrossRef.
- S. S. Sung, C. Glidewell, A. R. Butler and R. Hoffmann, Inorg. Chem., 1985, 24, 3856–3859 CrossRef CAS.
- C. Glidewell, R. J. Lambert, M. E. Harman and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1990, 2685–2690 RSC.
- Z. J. Tonzetich, L. H. Do and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 7964–7965 CrossRef CAS PubMed.
- C.-C. Tsou, Z.-S. Lin, T.-T. Lu and W.-F. Liaw, J. Am. Chem. Soc., 2008, 130, 17154–17160 CrossRef CAS PubMed.
- M. J. Scott and R. H. Holm, Angew. Chem., Int. Ed. Engl., 1993, 32, 564–566 CrossRef.
- C. Goh and R. H. Holm, Inorg. Chim. Acta, 1998, 270, 46–54 CrossRef CAS.
- J. Fitzpatrick and E. Kim, Acc. Chem. Res., 2015, 48, 2453–2461 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt00924h |
| ‡ Current address: Department for Structural Biology, Rudolf Virchow Center for Experimental Biomedicine, University of Würzburg, 97080 Würzburg, Germany. |
| § Current address: School of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK. |
| This journal is © The Royal Society of Chemistry 2019 |