
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination-induced reversible electrical conductivity variation in the MOF-74 analogue Fe2(DSBDC)†
Lei
Sun
 a,
Christopher H.
Hendon
b and
Mircea
Dincă
*a
a,
Christopher H.
Hendon
b and
Mircea
Dincă
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: mdinca@mit.edu
bMaterial Science Institute, Department of Chemistry and Biochemistry, University of Oregon, Eugene, Oregon 97403, USA
First published on 2nd July 2018
Abstract
Inner-sphere changes at the open Fe centers in Fe2(DSBDC) (DSBDC4− = 2,5-disulfidobenzene-1,4-dicarboxylate), as caused by coordination and release of solvent molecules, lead to reversible structural and electrical conductivity changes. Specifically, coordination of N,N-dimethylformamide (DMF) to the open Fe sites improves the room-temperature electrical conductivity by three orders of magnitude. Supported by additional density functional theory calculations, we attribute the electrical conductivity enhancement to partial electron transfer from Fe to DMF, which generates hole carriers and improves the charge carrier density in Fe2(DSBDC).
Introduction
Traditionally raising interest due to their highly porous nature,1 metal–organic frameworks (MOFs) offer an excellent platform for investigating and tuning electrical, magnetic, and optical processes in organic-based materials, some of which have already led to applications in lithium-ion batteries,2–4 thermoelectrics,5,6 and supercapacitors,7 among others. The electronic properties may be modulated either inherently, through changes in chemical composition, or through host–guest interactions. Guest molecules may cause structural variation,8,9 tune electrical conductivity,10,11 induce spin state transitions,12,13 modulate luminescence wavelength,14,15 and even participate in guest-guest interactions.16 These phenomena are of fundamental interest and form the basis of various applications including chemiresistive sensors,11,17,18 fluorescence sensors,19 and cooperative adsorption.13,16Although several strategies are effective for increasing the electrical conductivity in MOFs,20,21 among the most promising, and certainly more tractable synthetically, are those that involve post-synthetic doping. Guest molecules introduced after the formation of a MOF may tune either the charge mobility or the charge density in the skeleton of a given material, allowing in either case continuous enhancement of electrical conductivity over several orders of magnitude. Chemically, the dopant molecules may engage with the MOF either through outer-sphere electron transfer (i.e. redox reactivity)22–25 or through inner-sphere reactivity. The latter involves binding of guest molecules to coordinatively unsaturated metal centers to form charge transport pathways and/or to inject charge carriers.10 The inner sphere mechanism is proposed to be operative, for instance, in TCNQ-infiltrated Cu3(benzene-1,3,5-tricarboxylate)2 (TCNQ = 7,7,8,8-tetracyanoquinodimethane), wherein TCNQ molecules coordinate to Cu sites and generate increasingly efficient charge percolation pathways and million-fold increase in the electrical conductivity of the parent MOF.10
Herein, we show that simple coordination of solvent molecules such as DMF modulates the electrical conductivity of Fe2(DSBDC)26 by three orders of magnitude. Fe2(DSBDC) is a structural analogue of the well-known MOF-74 series M2(2,5-dihydroxybenzene-1,4-dicarboxylate) (M2(DOBDC), M = Mg, Mn, Fe, Co, Ni, Cu, Zn)27–34 where phenoxide groups on the DOBDC ligand have been replaced by thiophenoxide. Each Fe atom in Fe2(DSBDC) is coordinated by two trans thiophenoxide groups, three meridionally coordinated carboxylate groups, and one DMF molecule. The latter can be removed to yield coordinatively unsaturated Fe sites. The Fe and thiophenoxide S atoms form (–Fe–S–)∞ chains, which are bridged by DSBDC4− ligands to form a three-dimensional framework containing one-dimensional hexagonal pores (Fig. 1). Previously, we reported that the regular hexagonal pores of DMF-filled Fe2(DSBDC) (Fe2(DSBDC)(DMF)2·x(DMF)) distort significantly when the unbound guest DMF molecules are replaced by dichloromethane (DCM) and the material is activated 80 °C to give Fe2(DSBDC)(DMF)2.26 The distortion is reversible: soaking Fe2(DSBDC)(DMF)2 in DMF regenerates the regular hexagonal pores and original structure. We extend the structural and electrical properties study of this system here by investigating the fully desolvated material, Fe2(DSBDC), which shows divergent behaviour from its solvated congeners.
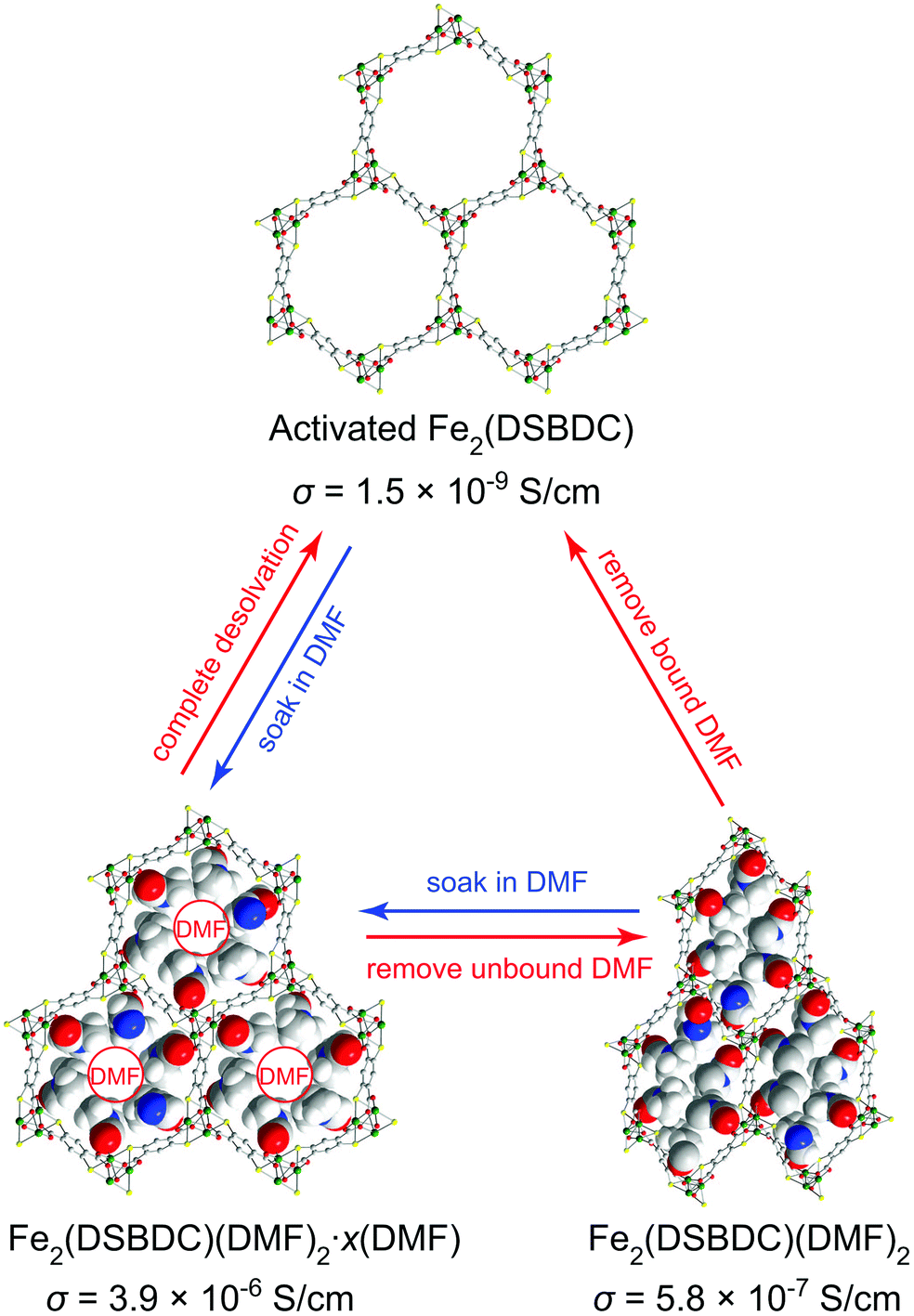 | ||
| Fig. 1 Structural changes induced in Fe2(DSBDC) by coordination and release of DMF, with respective electrical conductivity (σ) values. The structures of Fe2(DSBDC)(DMF)2·xDMF and Fe2(DSBDC)(DMF)2 were determined by single-crystal XRD and supported by DFT simulations, respectively.26 The structure of the activated Fe2(DSBDC) is proposed based on its PXRD pattern. Green, yellow, red, blue, grey, and white spheres represent Fe, S, O, N, C, and H atoms, respectively. Unbound DMF molecules and framework H atoms are omitted for clarity. | ||
Results and discussion
Fe2(DSBDC)(DMF)2·x(DMF) was synthesized according to a previously reported procedure.26 Owing to the sensitivity of this material to both water and air, all subsequent manipulations were performed in a dry, oxygen-free atmosphere. Thermogravimetric analysis (TGA) of this material revealed two consecutive weight loss events at 210–335 °C and 320–560 °C, respectively (Fig. S1†). The first weight loss step can be correlated to the loss of both bound and unbound DMF molecules, while the second likely corresponds to framework decomposition. Indeed, powder X-ray diffraction (PXRD) confirmed that heating Fe2(DSBDC)(DMF)2·x(DMF) at 200 °C under vacuum for 2 days causes amorphization (Fig. S2†). In an attempt to activate the material without decomposition, we exchanged DMF by soaking the as-synthesized MOF in lower-boiling methanol (MeOH), to produce Fe2(DSBDC)(MeOH)2·x(MeOH). Near-complete displacement of DMF by MeOH was confirmed by infrared (IR) spectroscopy, which revealed significantly decreased intensity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1654 cm−1 and appearance of a MeOH C–O stretching band at 1008 cm−1 as well as a broad O–H stretching band at ∼3300 cm−1 (Fig. 2). Although Fe2(DSBDC)(MeOH)2·x(MeOH) remains crystalline when kept in liquid MeOH (Fig. 3), it also decomposes upon desolvation (Fig. S2†), possibly because of a stronger interaction between MeOH and the framework. We therefore sought to replace methanol with a weaker interacting solvent and soaked the methanol-exchanged material in tetrahydrofuran (THF) to produce Fe2(DSBDC)(THF)2·x(THF). IR spectroscopy once again confirmed near-quantitative exchange, with complete disappearance of the bands at 1008 and 3300 cm−1 and rise of characteristic C–H stretching bands for THF at 2969 and 2866 cm−1, which are blue-shifted from the C–H stretching bands for MeOH at 2938 and 2831 cm−1. Note that direct replacement of DMF by THF is not successful, presumably because THF is too poorly coordinating and cannot displace Fe-bound DMF, thus requiring the intermediacy of MeOH.
O stretch at 1654 cm−1 and appearance of a MeOH C–O stretching band at 1008 cm−1 as well as a broad O–H stretching band at ∼3300 cm−1 (Fig. 2). Although Fe2(DSBDC)(MeOH)2·x(MeOH) remains crystalline when kept in liquid MeOH (Fig. 3), it also decomposes upon desolvation (Fig. S2†), possibly because of a stronger interaction between MeOH and the framework. We therefore sought to replace methanol with a weaker interacting solvent and soaked the methanol-exchanged material in tetrahydrofuran (THF) to produce Fe2(DSBDC)(THF)2·x(THF). IR spectroscopy once again confirmed near-quantitative exchange, with complete disappearance of the bands at 1008 and 3300 cm−1 and rise of characteristic C–H stretching bands for THF at 2969 and 2866 cm−1, which are blue-shifted from the C–H stretching bands for MeOH at 2938 and 2831 cm−1. Note that direct replacement of DMF by THF is not successful, presumably because THF is too poorly coordinating and cannot displace Fe-bound DMF, thus requiring the intermediacy of MeOH.
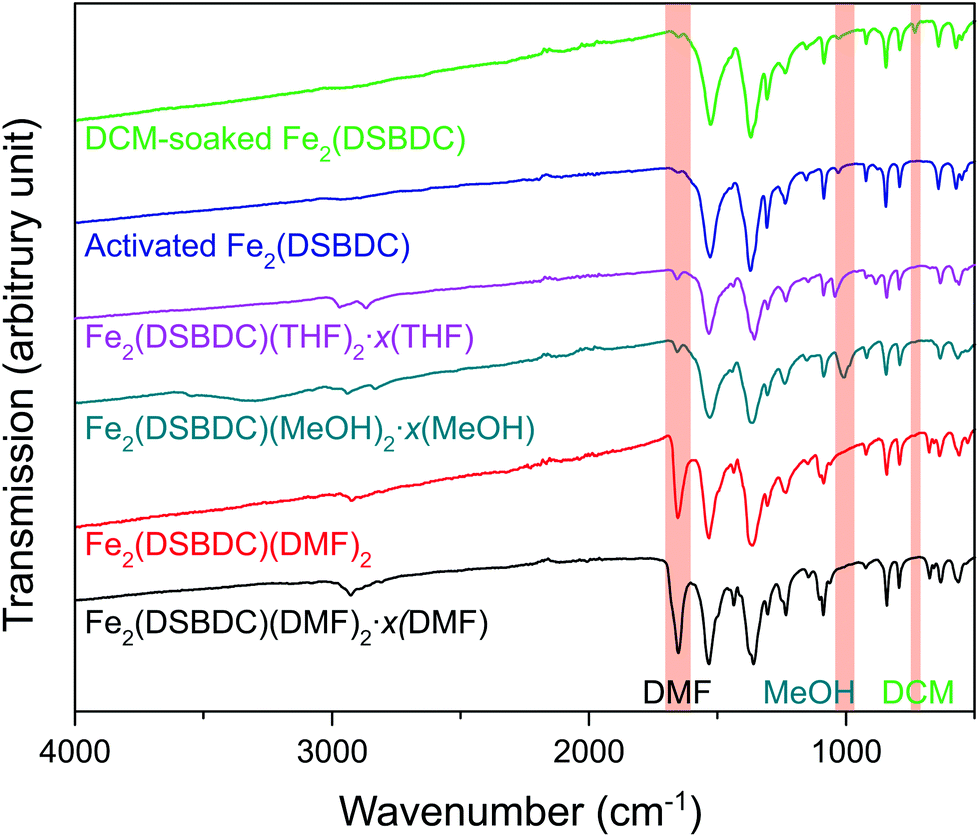 | ||
| Fig. 2 IR spectra of various forms of Fe2(DSBDC). Characteristic peaks of DMF, MeOH, and DCM are highlighted. | ||
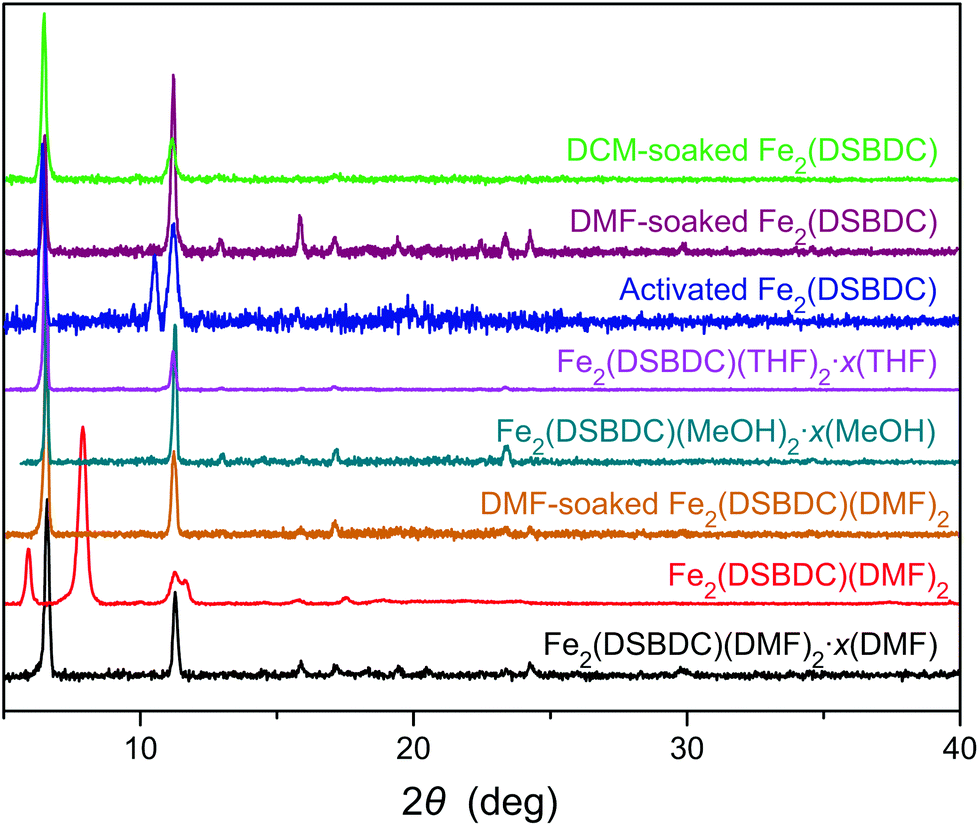 | ||
| Fig. 3 PXRD patterns of various phases of Fe2(DSBDC). | ||
TGA of Fe2(DSBDC)(THF)2·x(THF) confirmed that THF can be removed even below 100 °C, much lower than the decomposition temperature of approximately 270 °C (Fig. S3†). Therefore, to remove all solvent and produce completely activated MOF, Fe2(DSBDC)(THF)2·x(THF) was heated at 170 °C under dynamic vacuum (<3 mTorr) for 36 h. This yielded a dark red powder whose elemental microanalysis revealed a formula that matched that of fully desolvated Fe2(DSBDC), whose IR spectrum also indicates the complete absence of DMF, MeOH, or THF characteristic bands (Fig. 2 and ESI†). Most tellingly, an N2 adsorption of Fe2(DSBDC) exhibits a type I isotherm with a saturation uptake of approximately 160 cm3 of N2 per g at 77 K (Fig. 4) and an apparent Brunauer–Emmett–Teller (BET) surface area of 624 m2 g−1 (211 m2 mmol−1) (Fig. S4†). Although smaller than the value observed for related Mn2(DSBDC) (329 m2 mmol−1)35 and MOF-74 analogues (287–416 m2 mmol−1),30,32,33 possibly due to a further structural distortion (vide infra), the apparent surface area of Fe2(DSBDC) is significantly larger than that of Fe2(DSBDC)(DMF)2 (83 m2 g−1, 40 m2 mmol−1) (Fig. 4),36 as would be expected upon removal of pore-blocking DMF molecules.
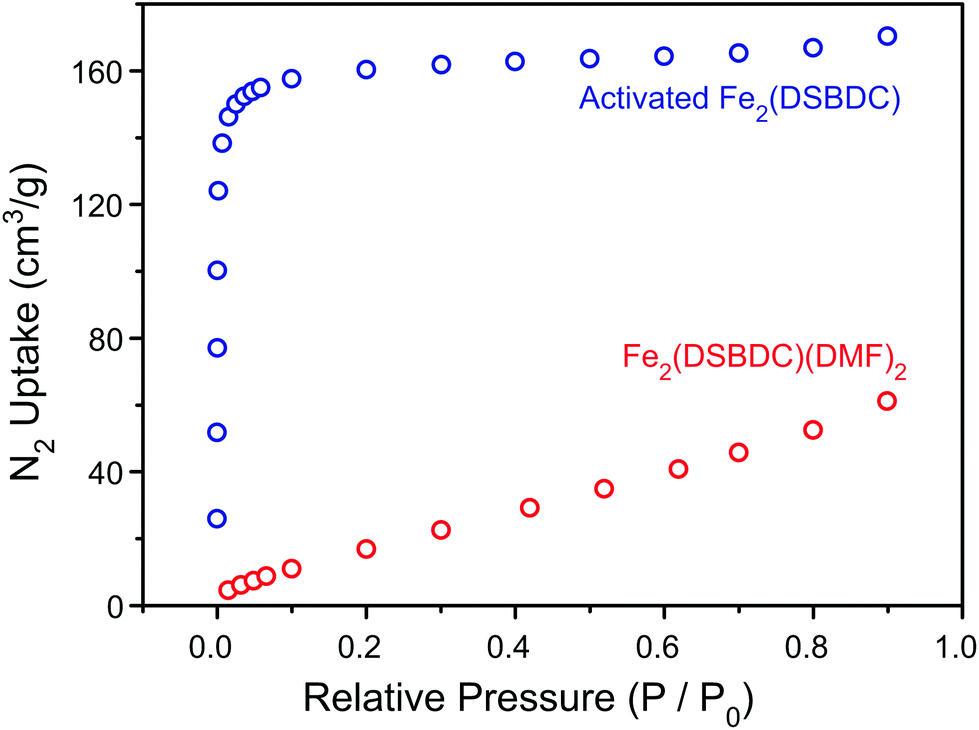 | ||
| Fig. 4 N2 sorption isotherms of Fe2(DSBDC)(DMF)2 and activated Fe2(DSBDC). | ||
Although Fe2(DSBDC) is crystalline, its PXRD pattern diverges from those of both Fe2(DSBDC)(DMF)2·x(DMF) and Fe2(DSBDC)(DMF)2: peaks at 6.6° and 11.3° corresponding to the [2–10] and [300] planes in the original regular hexagonal lattice are intact, but a new peak at 10.5° appears (Fig. 3). Qualitatively, this confirms that the hexagonal symmetry and pore shape is retained in Fe2(DSBDC), but the lattice suffers a distortion along the crystallographic c direction, which runs parallel to the (–Fe–S–)∞ chains. Unfortunately, the relatively low crystallinity of the activated material does not offer sufficient quality for further structural refinement. However, soaking Fe2(DSBDC) in DMF or DCM produces materials whose PXRD patterns are identical to that of original Fe2(DSBDC)(DMF)2·x(DMF) (Fig. 3).‡ This confirms that the distortion associated with solvent exchange and evacuation does not affect the connectivity of the framework. Similarly, any changes in electrical properties stemming from these distortions should also be reversible.
The electrical properties of Fe2(DSBDC) were investigated with two-contact-probe devices in air-free conditions at 297 K using pressed pellets made by in situ press, an apparatus that was introduced previously.37,38 Single-crystal electrical conductivity measurements are unfeasible for all phases of Fe2(DSBDC) because of the small crystallite size and extreme sensitivity to air. As shown in Fig. 5, current–voltage curves of all samples are linear, allowing the use of Ohm's Law for resistance calculations. Notably, these measurements revealed that the electrical conductivity increases with the degree of solvation: it is 3.9 × 10−6 S cm−1 for Fe2(DSBDC)(DMF)2·x(DMF), it drops to 5.8 × 10−7 S cm−1 for Fe2(DSBDC)(DMF)2 upon removal of unbound DMF, and decreases further upon complete removal of DMF to only 1.5 × 10−9 S cm−1 in fully desolvated Fe2(DSBDC). In line with the reversible structural distortions discussed above, the electrical properties are also recovered upon soaking Fe2(DSBDC) or Fe2(DSBDC)(DMF)2 in DMF, with the recovery values of 3.1 × 10−7 S cm−1 and 2.3 × 10−6 S cm−1, respectively, in good agreement with the conductivity of pristine, as-synthesized Fe2(DSBDC)(DMF)2·x(DMF) (Scheme S1†).
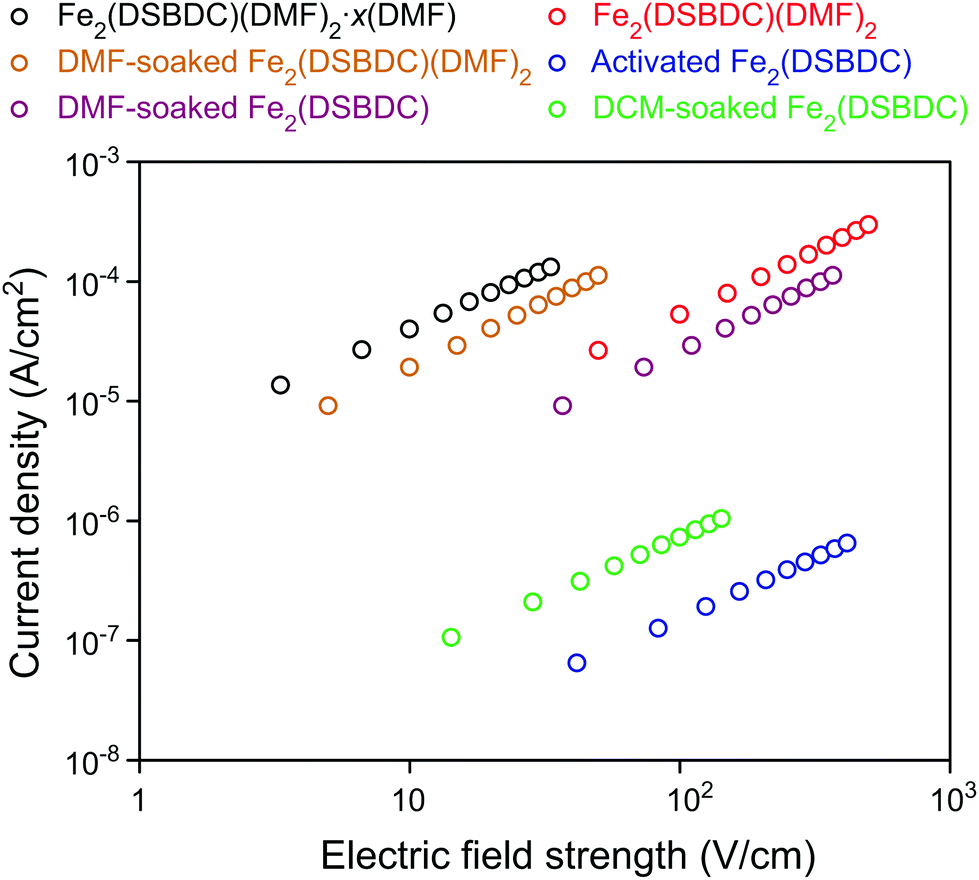 | ||
| Fig. 5 Plots of current density versus electric field strength of various forms of Fe2(DSBDC). | ||
It should be noted, however, that coordination to the Fe center is critical for improved electrical conductivity. Indeed, although soaking fully desolvated Fe2(DSBDC) in DCM recovers the skeleton structure observed in Fe2(DSBDC)(DMF)2·xDMF (Fig. 3), DCM treatment does not recover electrical conductivity, which remains at 7.3 × 10−9 S cm−1 (Fig. 5), similar to that of activated Fe2(DSBDC) itself. In other words, swelling with DCM (or presumably other non-coordinating solvents) recovers the original structure, but does not recover the electrical properties, which are more critically dependent on metal coordination rather than structural deformations.
Density functional theory (DFT) calculations were further used to probe the origin of the coordination-induced electrical conductivity enhancement (Fig. 6). The crystal structure of the activated Fe2(DSBDC) was allowed to reach geometric and electronic equilibrium, resulting in regular hexagonal pores that resemble the structure of the DCM-soaked Fe2(DSBDC). The projected density of states (PDOS) shows that the valance band of activated Fe2(DSBDC) is dominated by Fe and S orbitals, suggesting that perturbation to either Fe or S valence will directly modify the valence band character. We hence conjecture that the major charge transport mechanism in activated Fe2(DSBDC) is hole hopping between Fe and S within the (–Fe–S–)∞ chains, similar to the situation proposed in Fe2(DSBDC)(DMF)2.26,36
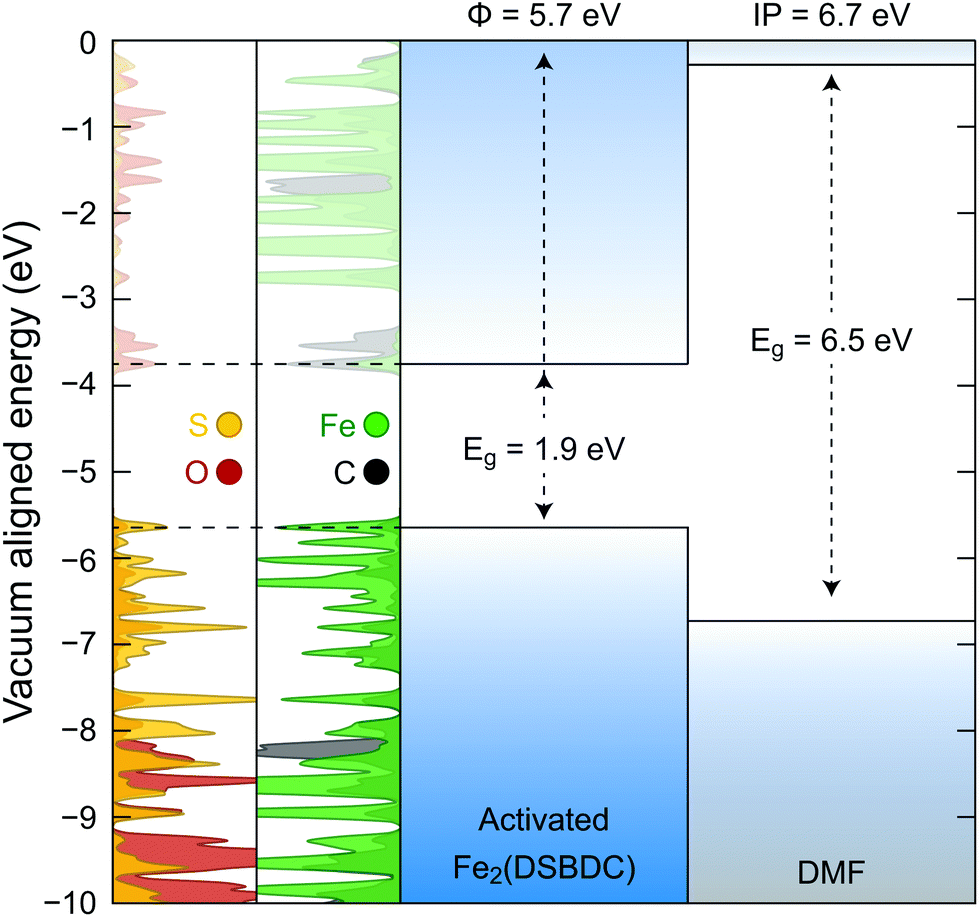 | ||
| Fig. 6 Electronic band structure of activated Fe2(DSBDC) and molecular orbital energy levels of DMF. The left two panels are projected density of states in Fe2(DSBDC). Eg represents the band gap for Fe2(DSBDC) and the HOMO–LUMO gap for DMF. Φ and IP represent work function and ionization potential, respectively. | ||
Because the skeletons of Fe2(DSBDC)(DMF)2 and the DCM-soaked Fe2(DSBDC) are isostructural, DMF likely does not affect charge mobility. Instead, it modulates charge density in the skeleton of the framework. Indeed, both a comparison of solid-state and truncated cluster calculations show that the work function of the activated Fe2(DSBDC) is smaller than the ionization potential of DMF by 1 eV (Fig. 6). As a result, partial electron transfer occurs when DMF binds to Fe centers in activated Fe2(DSBDC), as evidenced by approximately 1% reduction in local electron density of Fe, observed in both the solid-state and cluster calculations.§ Electron transfer thus generates holes as charge carriers, induces effectively mixed valency in (–Fe–S–)∞ chains, and improves charge density and electrical conductivity.36
Conclusions
DMF binding to the coordinatively unsaturated Fe centers in Fe2(DSBDC) leads to thousand-fold increase in the electrical conductivity at 297 K. DFT calculations identify electron transfer from Fe centers to the bound DMF molecules as a possible mechanism for improving charge density in the skeleton of the MOF. These results reinforce the importance of redox-matching between the framework and guest molecules to achieve electrically conductive MOFs, and point out the possibility of tuning electrical conductivity via coordinating guest molecules as well as applying MOFs with open metal sites for chemiresistive sensing of coordinating molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The experimental work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (DE-SC0018235). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. We thank Dr D. Sheberla, Dr B. M. Wiers and Prof. J. R. Long for helpful discussions.Notes and references
- H. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef PubMed.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112 CrossRef PubMed.
- Z. Zhang, H. Yoshikawa and K. Awaga, Chem. Mater., 2016, 28, 1298 CrossRef.
- M. L. Aubrey and J. R. Long, J. Am. Chem. Soc., 2015, 137, 13594 CrossRef PubMed.
- K. J. Erickson, F. Léonard, V. Stavila, M. E. Foster, C. D. Spataru, R. E. Jones, B. M. Foley, P. E. Hopkins, M. D. Allendorf and A. A. Talin, Adv. Mater., 2015, 27, 3453 CrossRef PubMed.
- L. Sun, B. Liao, D. Sheberla, D. Kraemer, J. Zhou, E. A. Stach, D. Zakharov, V. Stavila, A. A. Talin, Y. Ge, M. D. Allendorf, G. Chen, F. Léonard and M. Dincă, Joule, 2017, 1, 168 CrossRef.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220 CrossRef PubMed.
- K. Barthelet, J. Marrot, D. Riou and G. Férey, Angew. Chem., Int. Ed., 2002, 41, 281 CrossRef PubMed.
- C. R. Murdock, B. C. Hughes, Z. Lu and D. M. Jenkins, Coord. Chem. Rev., 2014, 258–259, 119 CrossRef.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66 CrossRef PubMed.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349 CrossRef PubMed.
- X. Bao, H. J. Shepherd, L. Salmon, G. Molnár, M.-L. Tong and A. Bousseksou, Angew. Chem., Int. Ed., 2013, 52, 1198 CrossRef PubMed.
- D. A. Reed, B. K. Keitz, J. Oktawiec, J. A. Mason, T. Runčevski, D. J. Xiao, L. E. Darago, V. Crocellà, S. Bordiga and J. R. Long, Nature, 2017, 550, 96 Search PubMed.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- N. B. Shustova, B. D. McCarthy and M. Dincă, J. Am. Chem. Soc., 2011, 133, 20126 CrossRef PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303 CrossRef PubMed.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, J. Am. Chem. Soc., 2015, 137, 13780 CrossRef PubMed.
- M. Campbell and M. Dincă, Sensors, 2017, 17, 1108 CrossRef PubMed.
- K. Müller-Buschbaum, F. Beuerle and C. Feldmann, Microporous Mesoporous Mater., 2015, 216, 171 CrossRef.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566 CrossRef PubMed.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994 RSC.
- Y. Kobayashi, B. Jacobs, M. D. Allendorf and J. R. Long, Chem. Mater., 2010, 22, 4120 CrossRef.
- L. S. Xie, L. Sun, R. Wan, S. S. Park, J. A. DeGayner, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2018, 140, 7411 CrossRef PubMed.
- M. L. Aubrey, B. M. Wiers, S. C. Andrews, T. Sakurai, S. E. Reyes-Lillo, S. M. Hamed, C.-J. Yu, L. E. Darago, J. A. Mason, J.-O. Baeg, F. Grandjean, G. J. Long, S. Seki, J. B. Neaton, P. Yang and J. R. Long, Nat. Mater., 2018, 17, 625 CrossRef PubMed.
- J. G. Park, M. L. Aubrey, J. Oktawiec, K. Chakarawet, L. E. Darago, F. Grandjean, G. J. Long and J. R. Long, J. Am. Chem. Soc., 2018, 140 DOI:10.1021/jacs.8b03696.
- L. Sun, C. H. Hendon, M. A. Minier, A. Walsh and M. Dincǎ, J. Am. Chem. Soc., 2015, 137, 6164 CrossRef PubMed.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504 CrossRef PubMed.
- P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., Int. Ed., 2005, 44, 6354 CrossRef PubMed.
- P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvåg, Chem. Commun., 2006, 959 RSC.
- W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268 CrossRef PubMed.
- P. D. C. Dietzel, R. Blom and H. Fjellvåg, Eur. J. Inorg. Chem., 2008, 3624 CrossRef.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814 CrossRef PubMed.
- R. Sanz, F. Martínez, G. Orcajo, L. Wojtas and D. Briones, Dalton Trans., 2013, 42, 2392 RSC.
- W. L. Queen, M. R. Hudson, E. D. Bloch, J. A. Mason, M. I. Gonzalez, J. S. Lee, D. Gygi, J. D. Howe, K. Lee, T. A. Darwish, M. James, V. K. Peterson, S. J. Teat, B. Smit, J. B. Neaton, J. R. Long and C. M. Brown, Chem. Sci., 2014, 5, 4569 RSC.
- L. Sun, T. Miyakai, S. Seki and M. Dincǎ, J. Am. Chem. Soc., 2013, 135, 8185 CrossRef PubMed.
- L. Sun, C. H. Hendon, S. S. Park, Y. Tulchinsky, R. Wan, F. Wang, A. Walsh and M. Dincă, Chem. Sci., 2017, 8, 4450 RSC.
- L. Sun, S. S. Park, D. Sheberla and M. Dincă, J. Am. Chem. Soc., 2016, 138, 14772 CrossRef PubMed.
- F. Wudl and M. R. Bryce, J. Chem. Educ., 1990, 67, 717 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, computation details, thermogravimetric analysis profiles, powder X-ray diffraction patterns, BET surface area analysis. See DOI: 10.1039/c8dt02197j |
| ‡ The structural reversibility and crystallinity of activated Fe2(DSBDC) rule out desolvation-induced local destruction because it tends to be irreversible and leads to amorphization. |
| § The calculation results of Fe2(DSBDC)(DMF)2 reported in ref. 26 were used to analyze the electron density on Fe centers in Fe2(DSBDC)(DMF)2 |
| This journal is © The Royal Society of Chemistry 2018 |