
Asymmetric total synthesis of (+)-ovafolinins A and B†
Xianhe
Fang
 ,
Lei
Shen
and
Xiangdong
Hu
*
,
Lei
Shen
and
Xiangdong
Hu
*
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry & Materials Science, Northwest University, Xi’an 710127, China. E-mail: Xiangdonghu@nwu.edu.cn
First published on 16th June 2018
Abstract
(+)-Ovafolinins A and B are two homologous lignans containing unique polycyclic skeletons. Benefiting from a highly diastereoselective alkylation of (S)-Taniguchi lactone, a double Friedel–Crafts reaction, a global debenzylation and a Cu(OAc)2-enabled benzylic oxidative cyclization, we present herein an efficient synthetic approach to (+)-ovafolinins A and B.
Lignans are a large family of natural products widely existing in plants and our food sources, such as wheat, soybeans, broccoli and strawberry.1 Many important biological properties including anticancer,2 antiviral,3 and antioxidant activities,4 alleviating menopausal symptoms, and reducing the risk of cardiovascular disease5 have been disclosed from biological evaluations of this family. In 2010, ovafolinin A, ovafolinin B and other three lignans were discovered during Yun and coworkers’ explorations on Lyonia ovalifolia var. elliptica, a deciduous tree growing in China and Japan.6 Ovafolinin B was also found in Sinocalamus affinis (Rendle) McClure (Poaceae),7 a widely cultivated traditional Chinese medicine named “Ci Zhu Li” and applied in treatments for diseases including cough and phlegm in China.8 Structurally, ovafolinin A has a particular polycyclic skeleton containing an aryl tetralin unit with a tetrahydrofuran motif and a seven-membered benzoxepin bridged-ring. Ovafolinin B possesses a very similar framework except for the opening of the tetrahydrofuran ring. The first asymmetric synthesis of (+)-ovafolinins A and B was achieved by Barker and co-workers9 employing an acyl-Claisen rearrangement developed in their laboratory.10 The unique polycyclic skeleton was achieved through an interesting cascade cyclization enabled by a bulky protecting group. As a pioneering work, Barker and coworkers’ synthesis demonstrated an expedient pathway to the unique skeleton of (+)-ovafolinins A and B. Furthermore, based on optical rotation comparisons between the synthetic compounds (+154.8 (c = 0.16, MeOH) for (+)-ovafolinin A, +150.0 (c = 0.26, MeOH) for (+)-ovafolinin B)9 and the natural samples (−37.3 (c = 0.36, MeOH) for ovafolinin A, +52.0 (c = 0.26, MeOH)6 and +43.3 (c = 0.12, MeOH)7 for ovafolinin B), the exploration convincingly suggested that natural ovafolinins A and B were both isolated in scalemic mixtures. Attracted by their architectural complexity, we started our synthesis with the purpose of devising a new, efficient, and asymmetric route to these lignans.
Based on our retrosynthetic analysis (Fig. 1), (+)-ovafolinin A (1) and (+)-ovafolinin B (2) could be constructed from three building blocks: phenol 5, bromide 8 and (S)-Taniguchi lactone 9. Diastereoselective alkylation between 9 and 8 will be a feasible strategy to set up initially two stereogenic centers of 1 and 2. For introduction of the top-right aromatic ring and formation of the central six-membered ring, a double Friedel–Crafts reaction process between 5 and 6 was originally proposed. Intramolecular Friedel–Crafts hydroxyalkylation of 6 could furnish the central six-membered ring first. Subsequently, intermediate 4 could be formed from a diastereoselective intermolecular Friedel–Crafts alkylation with 5. As a related precedent, Takayama and coworkers reported an expedient construction of complex bridged ring frames through a double Friedel–Crafts reaction between acetal and two different aromatic rings.11 Regarding the construction of the seven-membered benzoxepin bridged-ring unit, we imagined that dehydration cyclization in 4 could be a reasonable solution. Three benzyl protecting groups were designed in 3 for the convenience of synthesis. In light of the close structural relationship of 1 and 2 and their simultaneous generation in the synthesis by Barker and coworkers, we envisaged that 1 could be obtained through benzylic oxidative cyclization of 2.
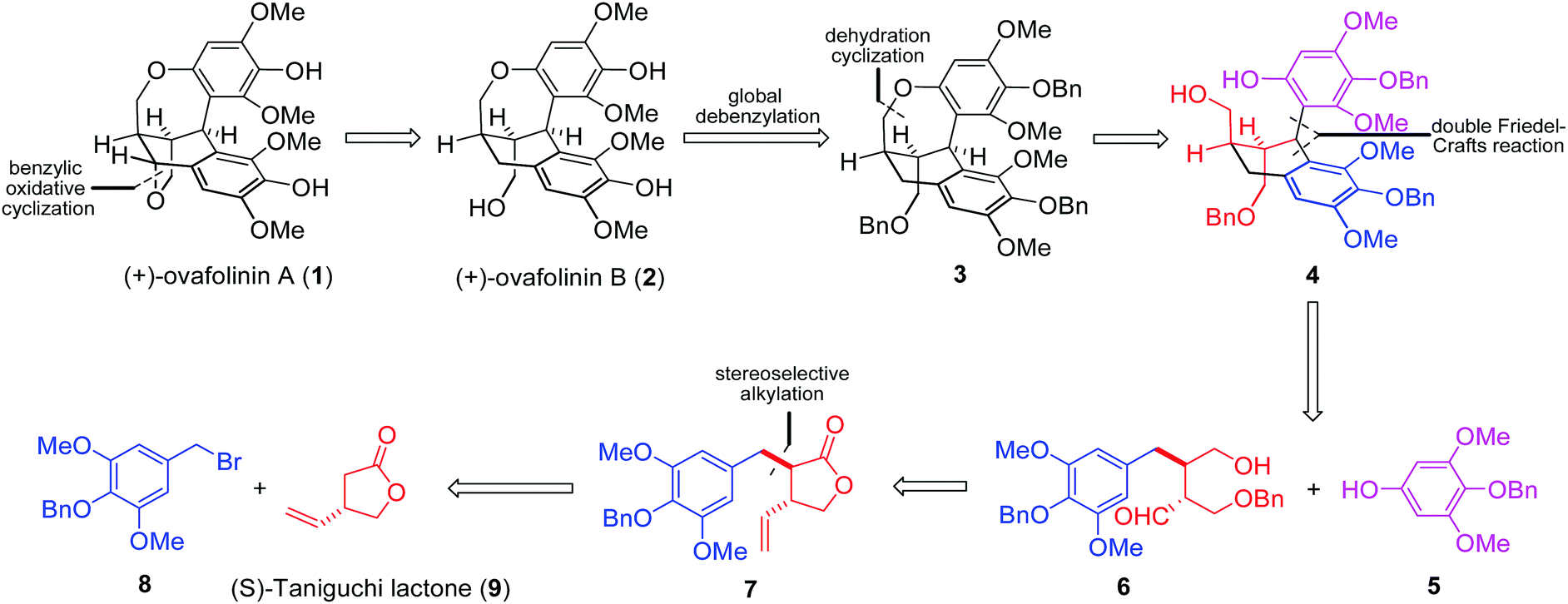 | ||
| Fig. 1 Our original retrosynthetic analysis of (+)-ovafolinins A and B. | ||
Our synthesis started with the preparation of bromide 8 (Scheme 1). The starting material was the commercially available syringaldehyde (10). After benzyl protection, reduction and bromination, 8 was obtained in 66% overall yield. The diastereoselective alkylation of (S)-Taniguchi lactone (9) is a reliable strategy to introduce two adjacent stereogenic centers with defined absolute and relative configurations in the synthesis of natural products.12 According to Kieseritzky's approach,139 was prepared in enantiomerically pure form over three steps. The alkylation process between 8 and 9 successfully afforded 7 in excellent stereoselectivity. The treatment of 7 with an excess amount of benzyl bromide under basic conditions opened the lactone unit smoothly,14 generating ester 12 in 78% yield. After subsequent reduction, product 13 was subjected to vinyl oxidation. The product was hemiacetal 14 generated from the addition of hydroxy to the aldehyde group. The originally proposed double Friedel–Crafts reaction between 515 and 14 was then examined with various Lewis acids. However, no consumption of 5 was observed in all cases.16 As a result, intermolecular Friedel–Crafts reaction seems not a feasible method to couple fragment 5 with 14.
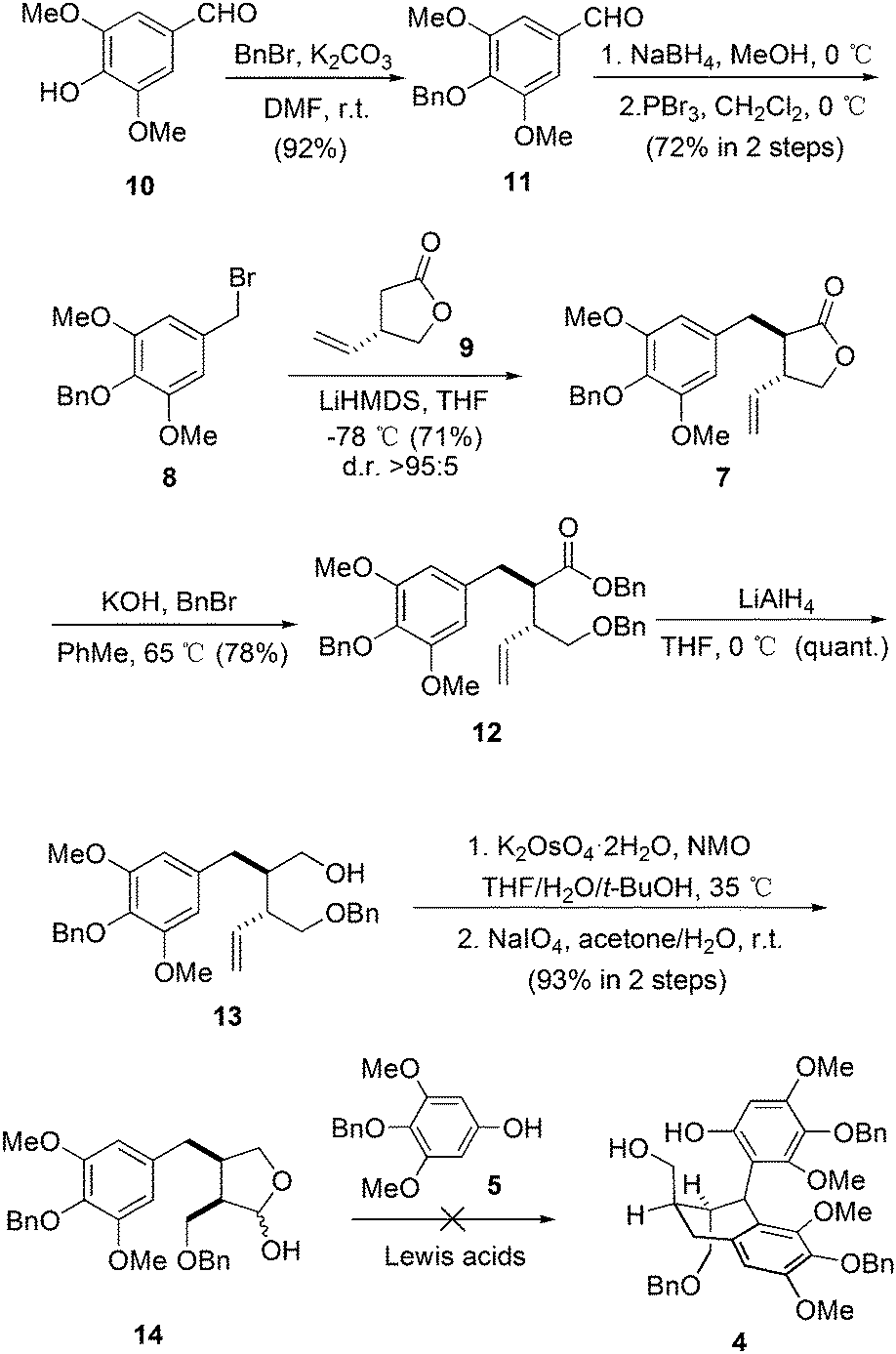 | ||
| Scheme 1 The attempt on the synthesis of 4. | ||
Therefore, we moved our attention to introduce motif 5 into the molecule before the construction of the carbon skeleton. Starting again from 13, motif 5 was readily connected with 13 through a Mitsunobu transformation (Scheme 2). Subsequent vinyl oxidation treatments established the aldehyde group in 16. Notably, during the construction of the unique polycyclic skeletons of 1 and 2, Barker and coworkers explored the cascade cyclization of compounds similar to 16. The bulky tert-butyldiphenylsilyl protecting group on the bottom-left hydroxy was found to be pivotal to enable the expected cyclization. However, methoxymethyl protection will lead to decomposition products.9 In our case, the protecting groups of the three hydroxyl groups in 16 are all benzyl groups. To our delight, treatment of 16 with trifluoroacetic acid established successfully the expected polycyclic skeleton through a double Friedel–Crafts reaction process, affording 3 in 87% yield. The subsequent hydrogenation removed all three benzyl protections and gave (+)-ovafolinin B (2) in quantitative yield. Noteworthily, the final de-protection process in Barker's synthesis led to the formation of not only 2 but also 1, both in poor yields. In our synthesis, there was no formation of 1 observed during the debenzylation process of 2.
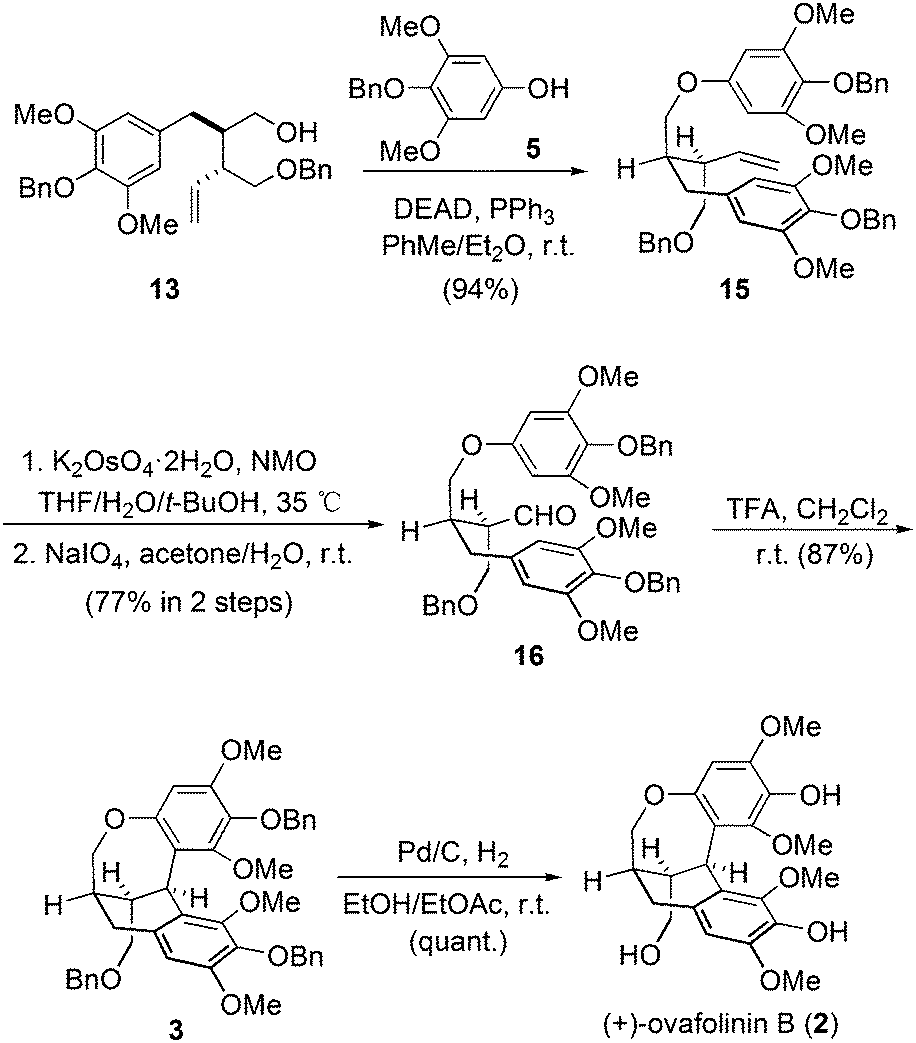 | ||
| Scheme 2 Total synthesis of (+)-ovafolinin B (2). | ||
With the successful development of an asymmetric route to 2, we focused on the synthesis of 1. We envisaged that the benzylic oxidation cyclization of 2 could lead to the formation of p-benzoquinone methide intermediate 17. And the subsequent conjugated addition from the vicinal hydroxy group will furnish 1 in the end. Therefore, 2 was subjected to various conditions reported for the formation of benzoquinone methide intermediates (Scheme 3). The employment of PhI(OAc)217 resulted in the generation of 1 but in poor yield. Oxidation with Ag2O18 and DDQ19 could significantly improve the formation of 1, respectively. The best result was obtained from the treatment with Cu(OAc)2,20 affording 1 in 91% yield. Barker's synthesis conditions were also investigated, which led to the formation of 1 in moderate yield after complete consumption of 2. Out of curiosity, we carried out the aerial oxidation of 2 under neat conditions. Only trace amounts of 1 were formed after three days.
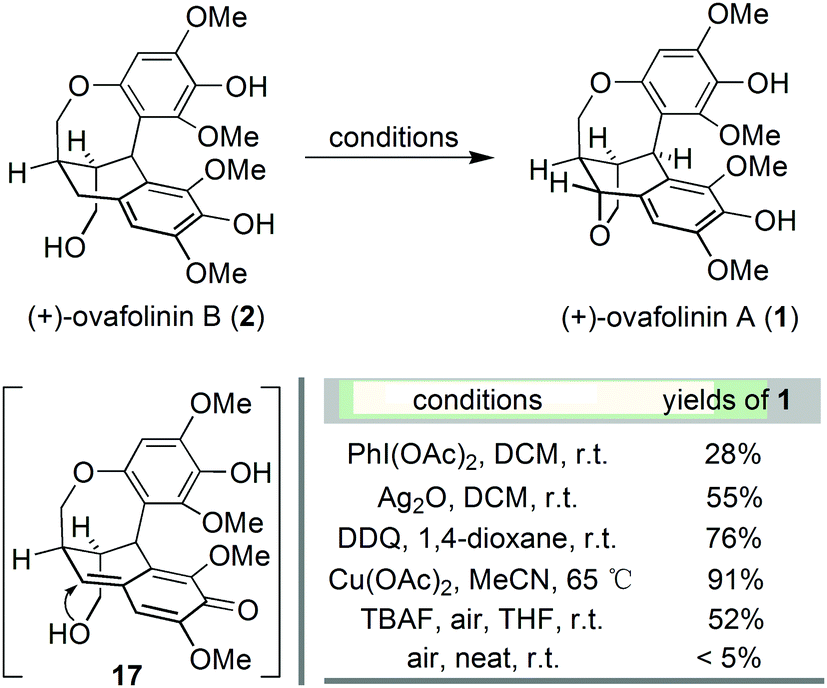 | ||
| Scheme 3 Synthesis of (+)-ovafolinin A. | ||
After the synthesis of 1 and 2 was complete, the optical rotation properties of our synthetic (+)-ovafolinins A and B were investigated. The data (+159.5, (c = 0.36, MeOH) for 1 and +166.0 (c = 0.16, MeOH) for 2) obtained are close to those observed by Baker and coworkers, which supports Barker's conclusion that natural ovafolinins A and B were both isolated in scalemic mixtures.9
In summary, an asymmetric synthetic approach to (+)-ovafolinins A and B has been developed. The entire synthetic route features a highly stereoselective alkylation of (S)-Taniguchi lactone, a double Friedel–Crafts reaction process, a global debenzylation and a Cu(OAc)2-enabled benzylic oxidative cyclization. As a result, the synthesis of (+)-ovafolinin B has been completed in 11 linear steps and 23% total yield. And the synthesis of (+)-ovafolinin A has been achieved in 12 linear steps and 21% total yield.
We are grateful for financial support from the National Natural Science Foundation of China (21772153, 21642006 and 21372184), the Key Science and Technology Innovation Team of Shaanxi Province (2017KCT-37) and the China Postdoctoral Science Foundation (334100041).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Chang, J. Reiner and J. Xie, Chem. Rev., 2005, 105, 4581–4609 CrossRef PubMed; (b) R. B. Teponno, S. Kusari and M. Spiteller, Nat. Prod. Rep., 2016, 33, 1044–1092 RSC.
- T. F. Imbert, Biochimie, 1998, 80, 207–222 CrossRef PubMed.
- J. L. Charlton, J. Nat. Prod., 1998, 61, 1447–1451 CrossRef PubMed.
- M. Fauré, E. Lissi, R. Torres and L. A. Videla, Phytochemistry, 1990, 29, 3773–3775 CrossRef.
- P. C. Li, D. H. F. Mark, M. K. T. Poon, S. P. Ip and K. M. Ko, Phytomedicine, 1996, 3, 217–221 CrossRef PubMed.
- K. Kashima, K. Sano, Y. S. Yun, H. Ina, A. Kunugi and H. Inoue, Chem. Pharm. Bull., 2010, 58, 191–194 CrossRef PubMed.
- L. Xiong, C. Zhu, Y. Li, Y. Tian, S. Lin, S. Yuan, J. Hu, Q. Hou, N. Chen, Y. Yang and J. Shi, J. Nat. Prod., 2011, 74, 1188–1200 CrossRef PubMed.
- H. H. Jia and X. D. Wu, Zhongyaocai, 1992, 15, 35–36 Search PubMed.
- S. J. Davidson and D. Barker, Angew. Chem., Int. Ed., 2017, 56, 9483–9486 CrossRef PubMed.
- (a) C. E. Rye and D. Barker, J. Org. Chem., 2011, 76, 6636–6648 CrossRef PubMed; (b) S. J. Davidson and D. Barker, Tetrahedron Lett., 2015, 56, 4549–4553 CrossRef; (c) C. E. Rye and D. Barker, Synlett, 2009, 3315–3319 Search PubMed.
- T. Suzuki, M. Takamoto, T. Okamoto and H. Takayama, Chem. Pharm. Bull., 1986, 34, 1888–1900 CrossRef.
- (a) F. Ishibashi and E. Taniguchi, Chem. Lett., 1986, 1771–1774 CrossRef; (b) F. Ishibashi and E. Taniguchi, Bull. Chem. Soc. Jpn., 1988, 61, 4361–4366 CrossRef; (c) F. Ishibashi and E. Taniguchi, Phytochemistry, 1998, 49, 613–622 CrossRef; (d) G. Stork, D. Niu, A. Fujimoto, E. R. Koft, J. M. Balkovec, J. R. Tata and G. R. J. Dake, J. Am. Chem. Soc., 2001, 123, 3239–3242 CrossRef PubMed; (e) H. Miyaoka, Y. Hara, I. Shinohara, T. Kurokawa and Y. Yamada, Tetrahedron Lett., 2005, 46, 7945–7949 CrossRef; (f) D. Stadler and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 7557–7559 CrossRef PubMed; (g) K. Ota, N. Sugata, Y. Ohshiro, E. Kawashima and H. Miyaoka, Chem. – Eur. J., 2012, 18, 13531–13537 CrossRef PubMed.
- F. Kieseritzky, Y. Wang and M. Axelson, Org. Process Res. Dev., 2014, 18, 643–645 CrossRef.
- (a) J. Rath, S. Kinast and M. E. Maier, Org. Lett., 2005, 7, 3089–3092 CrossRef PubMed; (b) J. Quancard, A. Labonne, Y. Jacquot, G. Chassaing, S. Lavielle and P. Karoyan, J. Org. Chem., 2004, 69, 7940–7948 CrossRef PubMed.
- Compound 5 was prepared in three steps from syringaldehyde. Please see the ESI† file for experimental details.
- For attempts on the originally proposed double Friedel–Crafts reaction between 5 and 14, please see the ESI† for detail.
- B. Fang, X. Xie, C. Zhao, P. Jing, H. Li, Z. Wang, J. Gu and X. She, J. Org. Chem., 2013, 78, 6338–6343 CrossRef PubMed.
- (a) F. D. Ramdayal, D. J. Kiemle and R. T. LaLonde, J. Org. Chem., 1999, 64, 4607–4609 CrossRef PubMed; (b) K. C. Rice, W. C. Ripka, J. Reden and A. Brossi, J. Org. Chem., 1980, 45, 601–607 CrossRef.
- T. Sengoku, S. Xu, K. Ogura, Y. Emori, K. Kitada, D. Uemura and H. Arimoto, Angew. Chem., Int. Ed., 2014, 53, 4213–4216 CrossRef PubMed.
- J.-A. Jiang, C. Chen, J.-G. Huang, H.-W. Liu, S. Cao and Y.-F. Ji, Green Chem., 2014, 16, 1248–1254 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc03456g |
| This journal is © The Royal Society of Chemistry 2018 |