
On-surface synthesis approach to preparing one-dimensional organometallic and poly-p-phenylene chains
Xiong
Zhou
a,
Fabian
Bebensee
b,
Qian
Shen
a,
Regine
Bebensee
b,
Fang
Cheng
a,
Yang
He
a,
Hui
Su
c,
Wei
Chen
de,
Guo Qin
Xu
de,
Flemming
Besenbacher
b,
Trolle R.
Linderoth
*b and
Kai
Wu
*ae
aBNLMS, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: kaiwu@pku.edu.cn
bSino-Danish Center for Molecular Nanostructures on Surfaces and Interdisciplinary Nanoscience Center, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: trolle@inano.au.dk
cEngineering Department of Materials and Chemistry, Heilongjiang Institute of Technology, Harbin 150050, China
dDepartment of Chemistry, National University of Singapore, Singapore 117543, Singapore
eSPURc, 1 CREATE Way, #15-01, CREATE Tower, Singapore 138602, Singapore
First published on 7th September 2016
Abstract
On-surface Ullmann coupling reactions of dibromo-biphenyl and diiodo-biphenyl molecules on Ag(111), Cu(111), Au(111) and Au(100) and by deposited extrinsic Ag and Ni atoms were systematically explored. Poly-p-phenylene polymer chains turned out to be the final coupling products. Different organometallic polymer chains, [–Ph2Ag–]n, [–Ph2Cu–]n, and [–Ph2Ni–]n, were synthesized as stable intermediates during the coupling reactions. These intermediates were confirmed and verified by X-ray photoelectronic spectroscopy, scanning tunnelling spectroscopy and tip manipulations. On Au surfaces, coupling products were achieved directly from halogenated precursors without formation of the Au-containing organometallic intermediates. Magnetic organometallic polymer chains were prepared by Ullmann coupling of dibromo-biphenyl activated by extrinsic Ni atoms on Au(100) surface and were confirmed by Kondo resonance measurement. Such an on-surface preparation strategy may provide an efficient approach to preparing unentangled organometallic and pure organic polymer chains and subsequently measuring their properties at surfaces.
Introduction
Recently, on-surface synthesis has become a highly versatile strategy to fabricate new and stable molecular structures.1–7 Since the involved reactants, products and intermediates may be confined in a two-dimensional (2D) space, such a strategy presents some unique properties that traditional homogeneous and heterogeneous catalytic processes in a three-dimensional (3D) space do not possess. Firstly, it brings the possibility to prepare compounds which are insoluble in solution or can only be produced via new reaction pathways created by the 2D confinement effect.8–11 Secondly, a wide range of advanced surface science techniques including scanning tunnelling microscopy (STM), X-ray photoelectron spectroscopy (XPS), etc., can be directly applied to explore reaction processes, providing a detailed understanding of the reaction mechanisms and even precise control of the reaction steps.12–15 Thirdly, it offers the opportunity to build functional devices and subsequent in situ property tests on surface.16–20 Finally, due to the interactions of the polymers produced with the substrates, one-dimensional (1D) straight polymers may be feasibly fabricated on surface without tangled structures, which are frequently encountered in wet chemistry.21–23Organometallic or pure organic polymers with or without metal atoms periodically linked by organic ligands have been exploited in many areas due to their potential electronic, optical and magnetic properties.24–27 These properties of traditional polymers usually originate from the collective behaviour of multiple polymer chains. However, a single 1D polymer chain can behave as a stand-alone molecular device with its own properties and functionalities.17,28 As the on-surface synthesis approaches advance, various coupling reactions have been successfully employed to prepare 1D polymer chains, including aryl–aryl coupling (Ullmann coupling), alkyne coupling (Glaser–Hay coupling),29–32 dehydrogenative coupling,8–11etc. These 1D polymer chain preparation approaches bring new vigour into polymer science and promising applications in molecular electronics.
Ullmann coupling, the most commonly applied on-surface synthesis strategy, was discovered by Fritz Ullmann in 1901.33 The classical Ullmann reaction couples two aryl halides into a biphenyl catalyzed by copper powders, which has been extensively used in homogeneous organic synthesis.34,35 In 1992, Xi and Bent first carried out this reaction on a well-defined Cu(111) surface.36,37 In 2007, Grill et al.38 constructed covalent nano-architectures including chains and networks via Ullmann coupling of brominated porphyrin derivatives. Since then, on-surface Ullmann coupling has become a versatile and extensively used approach for bottom-up fabrication of covalent organic nanostructures such as molecular chains,16,17,21 nanoribbons,18,19,22,39–42 nanographene43–45 and organic networks.38,46–48
Studies on the Ullmann coupling reaction mechanism have also attracted tremendous attention with the aim to better predict and control the reaction processes. Generally, two main steps exist in Ullmann coupling of halogenated aryls on metal substrates: (1) detachment of the halogen atoms (dehalogenation), and (2) combination of the aryl species (C–C coupling).49 During these steps, the metal surface plays an active chemical role to provide substitution to split off the halogens, and an organometallic intermediate10,23,50–56 is widely identified after dehalogenation. In some cases, dehalogenated aryl species are anchored to metal surface atoms to form another type of intermediate, surface-bonded aromatic species.37,57–64 The actual intermediates involved in the coupling reaction are determined by the competition of surface adatoms and lattice atoms to detach halogens.65 Surface-bonded aromatic species usually appear in the cases with high catalytic activities for the halogen detachments, either on active metal substrates such as Co island57 and kinked Au(10,7,7)58 or for active reactants such as aryl iodides,37,59–62 leading to direct halogen detachment. In contrast, more active surface adatoms come into play and replace the halogens to form organometallic intermediates. In terms of the Ullmann coupling reaction to synthesize pure organic polymer chains, organometallic polymer chains are often generated as the intermediates, which provides a useful methodology to fabricate organometallic polymer chains.
Either of the two steps involved in the Ullmann coupling could be rate-limiting, depending on the reaction systems employed. When step (2) is rate-limiting, i.e., the energy barrier in step (2) is a controlling factor, the organometallic polymer chains become stable and hence can be obtained. However, when step (1) becomes rate-limiting, step (2) occurs spontaneously and no organometallic polymer chains can be achieved.66 Therefore, two reaction conditions become kinetic prerequisites to synthesize organometallic polymer chains on a metal surface: a higher energy barrier for C–C coupling, and a not-too-low energy barrier for dehalogenation.
With this general picture in mind, it becomes more feasible to understand the mechanisms of a series of Ullmann coupling reactions described below to synthesize organometallic and pure organic polymer chains. This article reports and summarizes our experimental results, aiming to offer a blueprint for on-surface Ullmann coupling reactions and providing insights into their future potential applications.
Results and discussion
Poly-p-phenylene (PPP), one of the simplest pure organic polymers, was chosen to be our target product, whereby para-position halogenated phenyl or phenyls are needed for Ullmann coupling. Diiodo-benzene,67 dibromo-benzene21 and dibromo-terphenyl23 have been previously employed. Organometallic polymer chains appear as intermediates in cases of dibromo-benzene on Cu(110), dibromo-terphenyl on Cu(111), but not observed during the reaction of diiodo-benzene on Cu(110) due to the feasible detachment of iodine atoms as described above (benzene anchored to surface atoms).In this article, dibromo-biphenyl (BrPh2Br) and diiodo-biphenyl (IPh2I) were coupled on Ag(111), Cu(111), Au(111) and Au(100), aiming to tackle the synthetic issues of PPP and organometallic polymer chains on various substrates.
Ullmann coupling of dibromo-biphenyl on Ag(111)
Ag-containing organometallic intermediates have been reported in many processes of on-surface Ullmann coupling reactions.51,68–70 As anticipated, before the final Ullmann coupling of BrPh2Br on Ag(111), organometallic intermediates are formed as expressed in the chemical formula shown in Fig. 1a. Fig. 1b–f show the real reaction processes in our experiments. When deposited on Ag(111) at room temperature (RT, about 300 K) and rapidly cooled down by liquid nitrogen (LN2) or liquid helium (LHe) to low temperatures (typically 77.4 K or 4.2 K), the BrPh2Br molecules molecularly adsorb and self-assemble at the surface, as shown by the STM image in Fig. 1b. The BrPh2Br molecules are held together by weak intermolecular interactions, i.e., synergetic halogen and hydrogen bonds, to form a rectangular brick-wall-like structure. Upon annealing at RT for a short period of time (about 5 minutes), the BrPh2Br molecules start to detach their Br atoms and convert into the intermediate (Fig. 1c) which is approximately twice (∼2.1 nm) as long as the intact BrPh2Br molecule (∼1.0 nm). Such an intermediate shows a distinct bright protrusion at its center in the STM image. This intermediate is tentatively assigned as BrPh2AgPh2Br and consists of two BrPh2 moieties after Br detachments and one surface Ag atom. The small red dots located near the intermediates are detached Br atoms. When the sample is annealed at RT for a sufficiently long period of time (more than 30 min), all BrPh2Br molecules transform into the necklace-like [–Ph2Ag–]n organometallic polymer chains (Fig. 1d). Again in between the [–Ph2Ag–]n chains, the detached Br atoms could be clearly identified. Further annealing of the sample at 373 K for 5 minutes leads to some [–Ph2Ag–]n chains eliminating the coordinated Ag atoms (Fig. 1e). Finally, heating up to 473 K results in full conversion of all [–Ph2Ag–]n chains into the coupling products, the PPP polymer chains (Fig. 1f). Note that the detached Br atoms do not desorb and stay nearby after completion of the coupling reaction at 473 K. It is also noted that all chains obtained during the reaction lie flat on the surface and do not entangle themselves, unlike the products prepared in solution chemistry. The typical distance between the neighboring chains is about 1.02 nm, therefore the polymer chains produced can be envisioned as an array of single chains. To achieve isolated single polymer chains, a low coverage of the precursors is required. | ||
| Fig. 1 Sequential evolution of the Ullmann coupling of BrPh2Br on Ag(111) via thermal activation. V = 0.5 V, I = 0.1 nA. (a) The catalytic reaction process. (b) STM image of the intact and assembled BrPh2Br molecules with superimposed molecular models on Ag(111) at RT. (c) Some BrPh2Br molecules converted into the BrPh2AgPh2Br intermediate species at RT for 5 min. (d) All BrPh2Br molecules converted into the necklace-like [–Ph2Ag–]n organometallic polymer chains at RT for an extended period of time. In between the [–Ph2Ag–]n chains are detached Br atoms. (e) Some [–Ph2Ag–]n chains convert into the coupling products at 373 K. (f) All [–Ph2Ag–]n chains converted into the coupling products, PPP polymer chains, at 473 K. | ||
The above-mentioned case study shows a typical on-surface Ullmann coupling reaction process. Many techniques have been employed to explore the reaction mechanism, particularly for the identification of the organometallic intermediates. Gutzler et al.68 used STM, Wang et al.23 combined STM, scanning tunneling spectrum (STS) and density functional theory (DFT) calculations, and Giovannantonio et al.21 conducted XPS and near-edge X-ray absorption fine structure (NEXAFS) measurements to verify the existence of the organometallic intermediates.
Here, we make use of XPS, STS and STM manipulation to assign the observed intermediates. Fig. 2 shows corresponding XPS peaks originating from C 1s and Br 3d. When BrPh2Br molecules are molecularly adsorbed at low temperature (110 K), the XPS peak in the C 1s region can be fitted with a majority contribution at a binding energy (BE) of 284.4 eV, attributed to benzyl ring carbon not directly bonded to Br, and a minority contribution at a higher BE of 285.3 eV, ascribed to C–Br. The two contributions show a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 intensity ratio, in good agreement with the stoichiometry of BrPh2Br. The Br 3d BE (3d5/2 = 71.4 eV and 3d3/2 = 70.3 eV) indicates no abstraction of Br from the molecules. After RT annealing, Br 3d XPS feature shows a new dominant doublet peak at lower BE (3d5/2 = 68.9 eV and 3d3/2 = 67.8 eV) characteristic of AgBr,71 confirming that Br is detached from the BrPh2Br molecule and binds to the Ag substrate. In terms of the C 1s XPS, the main peak is shifted significantly toward a lower BE, compared with the situation before annealing, and displays a new shoulder at its low BE side. Qualitatively, these changes demonstrate that the Ph2 moieties created by the Br detachment instead couple to Ag atoms which are considerably less electronegative than Br, and can therefore donate electrons, causing a downshift in BE. The C 1s XPS peak can be fitted with a component at a BE of 283.2 eV (ascribed to C in the phenyl rings without hetero-atom attachments) and a small feature at 282.5 eV (ascribed to C–Ag). The intermediate chains are accordingly attributed to the [–Ph2Ag–]n intermediate chains formed by the reaction between the Ph2 moieties and Ag adatoms. Further annealing of the sample leads to the C 1s peak profoundly narrowing down and losing its shoulder at low BE, indicating a uniform chemical state for the C atoms. The peak position (BE 283.8 eV) is in between the features for BrPh2Br and [–Ph2Ag–]n where the biphenyl groups are connected to electronegative (Br) and electropositive (Ag) species, respectively. We therefore conclude that the Ullmann coupling reaction has completed and attribute the smooth and bright chains observed in Fig. 1f to the final PPP chains.
:1 intensity ratio, in good agreement with the stoichiometry of BrPh2Br. The Br 3d BE (3d5/2 = 71.4 eV and 3d3/2 = 70.3 eV) indicates no abstraction of Br from the molecules. After RT annealing, Br 3d XPS feature shows a new dominant doublet peak at lower BE (3d5/2 = 68.9 eV and 3d3/2 = 67.8 eV) characteristic of AgBr,71 confirming that Br is detached from the BrPh2Br molecule and binds to the Ag substrate. In terms of the C 1s XPS, the main peak is shifted significantly toward a lower BE, compared with the situation before annealing, and displays a new shoulder at its low BE side. Qualitatively, these changes demonstrate that the Ph2 moieties created by the Br detachment instead couple to Ag atoms which are considerably less electronegative than Br, and can therefore donate electrons, causing a downshift in BE. The C 1s XPS peak can be fitted with a component at a BE of 283.2 eV (ascribed to C in the phenyl rings without hetero-atom attachments) and a small feature at 282.5 eV (ascribed to C–Ag). The intermediate chains are accordingly attributed to the [–Ph2Ag–]n intermediate chains formed by the reaction between the Ph2 moieties and Ag adatoms. Further annealing of the sample leads to the C 1s peak profoundly narrowing down and losing its shoulder at low BE, indicating a uniform chemical state for the C atoms. The peak position (BE 283.8 eV) is in between the features for BrPh2Br and [–Ph2Ag–]n where the biphenyl groups are connected to electronegative (Br) and electropositive (Ag) species, respectively. We therefore conclude that the Ullmann coupling reaction has completed and attribute the smooth and bright chains observed in Fig. 1f to the final PPP chains.
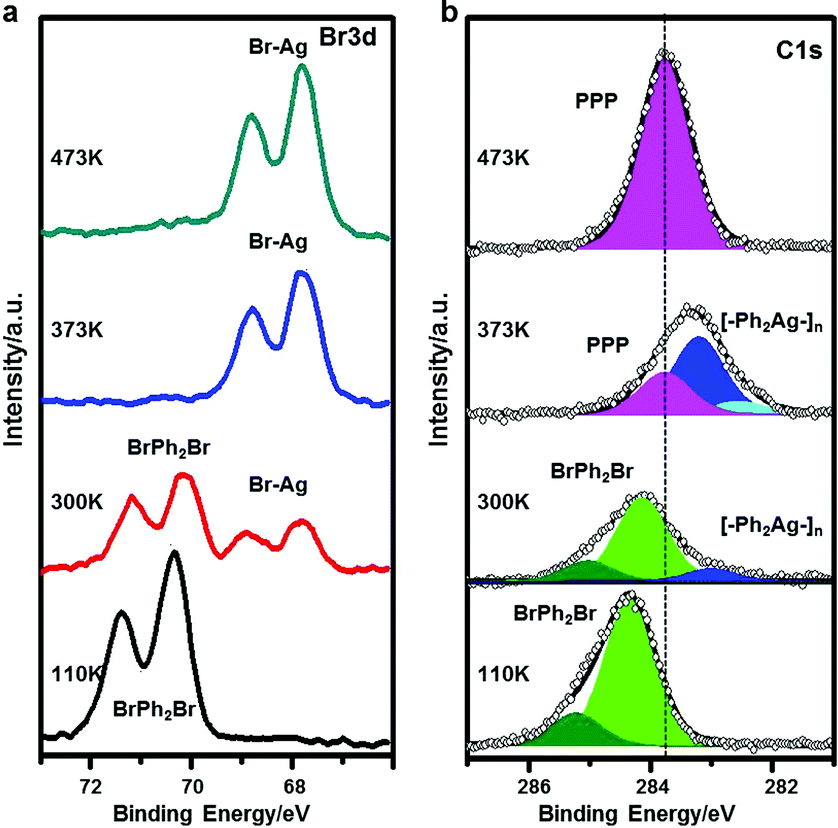 | ||
| Fig. 2 XPS identification of the surface species during the coupling reaction process for BrPh2Br. Corresponding XPS data (a) Br 3d and (b) C 1s features from the BrPh2Br reactant molecularly adsorbed on Ag(111) at 110 K at full coverage (bottom row), the [–Ph2Ag–]n chains formed at RT (2nd row), partially polymerized PhxAgy at 373 K (3rd row) and the final polymer products, PPP chains formed at 473 K (top row). | ||
To further verify the existence of the [–Ph2Ag–]n chains, we have performed STM manipulation experiments at sub-monolayer coverage. An isolated [–Ph2Ag–]n chain could be moved as a whole by the STM tip, indicating that they are discrete chains (Fig. 3a). In contrast, the detached Br atoms adhering to the [–Ph2Ag–]n chain could be moved independently. To prove that the bright central protrusion is indeed an Ag atom, we targeted one intermediate chain (upper panel in Fig. 3b) by the STM tip and applied a sudden voltage pulse. As shown in Fig. 3b, this manipulation knocks off the coordinated Ag atom. The knocked-off Ag atom stays close to the broken chain (lower panel in Fig. 3b). We further carried out local STS measurements at different spots above the [–Ph2Ag–]n chain. The STS spectrum acquired with the tip above the biphenyl group (red curve in Fig. 3c) shows a distinct peak (1.7 eV) for the lowest unoccupied molecular orbital (LUMO) region which is missing in the spectrum acquired with the tip above the central Ag atom (black). This agrees well with the reported spectra23 and STM images obtained at different scanning bias voltages. Taken together, these experimental observations unambiguously show that mild temperature annealing results in the [–Ph2Ag–]n chains.
 | ||
| Fig. 3 Experimental identifications of the [–Ph2Ag–]n organometallic polymer chain. V = 0.5 V, I = 0.1 nA, T = 4.2 K. (a) STM images of an individual [–Ph2Ag–]n chain before (top) and after (bottom) lateral manipulation by the STM tip. (b) STM images of an individual [–Ph2Ag–]n intermediate chain before (top) and after (bottom) a sudden voltage pulse (3 V, 1 s) on a coordinated Ag atom. (c) STS spectra collected with the tip at two different positions of [–Ph2Ag–]n intermediate chain, right above the biphenyl group (red curve) and the coordinated Ag atom (black curve). | ||
Thus, a typical process of on-surface Ullmann coupling is demonstrated and ordered [–Ph2Ag–]n organometallic and PPP polymer chains are fabricated on the Ag(111) surface.
Ullmann coupling of diiodo-biphenyl on Ag(111) and dibromo-biphenyl on Cu(111)
As is well-known, the C–I bond is weaker than C–Br and Cu is more active than Ag, thus halogen atoms are expected to be more easily detached from IPh2I molecules on Ag(111) or BrPh2Br molecules on Cu(111). DFT calculations by Björk et al.72 state that the energy barrier of dehalogenation for iodobenzene is about 0.3 eV lower than that for bromobenzene, and 0.13 eV lower on Cu(111) than on Ag(111). In echoing the DFT calculation results, our experiments show that molecularly adsorbed reactants cannot be obtained when deposited at RT. For both cases of IPh2I on Ag(111) and BrPh2Br on Cu(111), these reactants quickly detach halogen atoms to form the intermediates. However, variation of either the halogen atom or the substrate is not vigorous enough to change the reaction pathway to produce surface-bonded aromatic species. Similar organometallic polymer chains can still be observed during the reaction. As shown in Fig. 4, a similar intact molecular assembly of the reactants appears at about 120 K achieved by LN2 cooling (Fig. 4a and d). By warming the sample up to RT, [–Ph2Ag–]n (Fig. 4b) or [–Ph2Cu–]n (Fig. 4e) chains are generated with the detached halogen atoms dispersed in between the organometallic chains. For the former, subsequent reaction is to eliminate the coordinated Ag atoms in the [–Ph2Ag–]n chains, which is the same as dibromo-biphenyl on Ag(111). Therefore, upon heating the sample at the same temperature 473 K, PPP chains are on-surface synthesized (Fig. 4c). For the latter, the sole difference is that the Ag adatoms in the necklace-like organometallic chains are replaced by Cu adatoms. Eventually, a warm-up of the sample to 450 K leads to PPP chains produced on surface (Fig. 4f).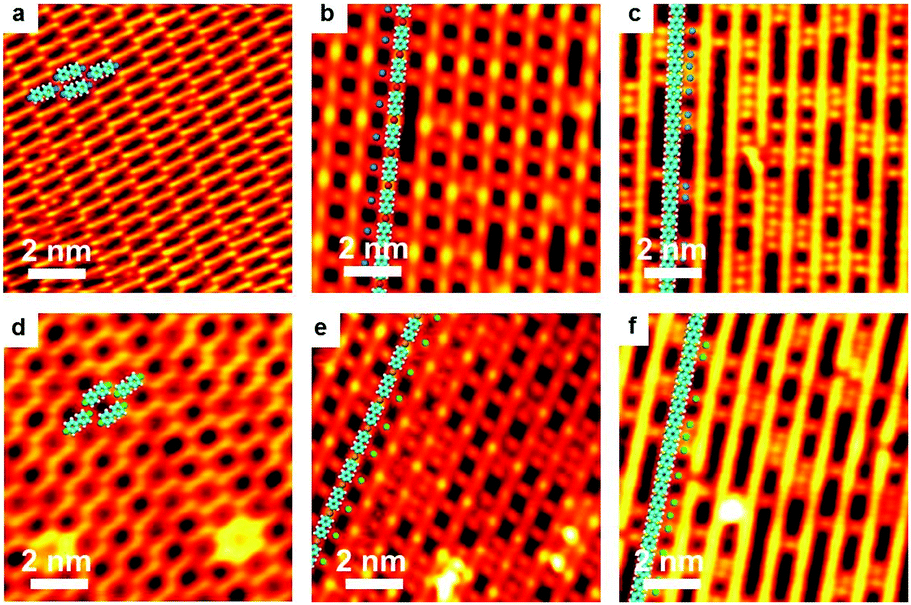 | ||
| Fig. 4 Main reaction processes for Ullmann coupling of IPh2I on Ag(111) and BrPh2Br on Cu(111). V = 0.5 V, I = 0.1 nA. (a) STM image of the intact and assembled IPh2I molecules on Ag(111) at about 120 K. (b) IPh2I molecules converted into the [–Ph2Ag–]n organometallic chains at RT. In between the [–Ph2Ag–]n intermediate chains are detached I atoms. (c) [–Ph2Ag–]n organometallic chains converted into the coupling products, PPP chains, at 473 K. (d) STM image of the intact and assembled BrPh2Br on Cu(111) at about 120 K. (e) BrPh2Br molecules converted into [–Ph2Cu–]n organometallic chains at RT. (f) [–Ph2Ag–]n organometallic chains converted into PPP chains at 450 K. | ||
Ullmann coupling of dibromo-biphenyl on Au(111) and Au(100)
As stated above, the organometallic polymer chains have been experimentally identified and verified as an intermediate state on both Ag and Cu surfaces. However, on Au surface, such a step to form the organometallic chain intermediate vanishes and the coupling products are generated directly from the halogenated aryls,16,22,39,46,73–77 except in some cases where stable Au organometallic intermediates are reported.40,66,78,79 A possible scenario is that the bonding ability for Au is weaker than that for Ag or Cu, so that the energy barrier for halogen detachment on Au is higher than that on Ag or Cu, which means that dehalogenation is activated at RT or a mild temperature on Cu and Ag surfaces, but a much higher temperature is required to trigger the dehalogenation on Au.49,54 Meanwhile, the final C–C coupling of the dehalogenated precursors may require a relatively high temperature annealing on Ag and Cu surfaces, but could commence even at RT on Au surface. Thus, the rate-limiting step changes from step (2) to step (1). Whenever the dehalogenation occurs, C–C coupling follows and finishes immediately. In exceptional cases,40,66,78,79 the designed precursors may not be able to complete the coupling reaction due to their steric hindrance.In our experiments, Ullmann coupling of the BrPh2Br molecules takes place on Au(111) and Au(100) surfaces (Fig. 5). The BrPh2Br molecules are quite stable at RT on the Au surface. Upon RT deposition, different assembly structures by BrPh2Br can be obtained. On Au(111) it is a zig-zag structure (Fig. 5a), while on Au(100), a lamellar structure appears (Fig. 5c). Holding at RT for a long time does not cause noticeable changes of the BrPh2Br molecules. Even at temperatures up to 500 K, no organometallic intermediates containing Au atoms could be observed on the Au surfaces. Instead, the coupling products, oligomers, are directly generated. As can be seen in Fig. 5b and d, oligomers are generated while some unreacted BrPh2Br molecules still exist at the surface. Notably, the oligomers still have bromine ends which can form assembly structures by hydrogen and halogen bonding as Fig. 5b shows. In other words, the anticipated organometallic polymer chains containing Au atoms cannot be identified on the Au surfaces. Moreover, owing to the weak adsorption capability of the Au surfaces, some BrPh2Br molecules obviously desorb from the surfaces during the warming up of the substrates. Although coupling products can be achieved in our experiments, they are typically of short length with a low yield.
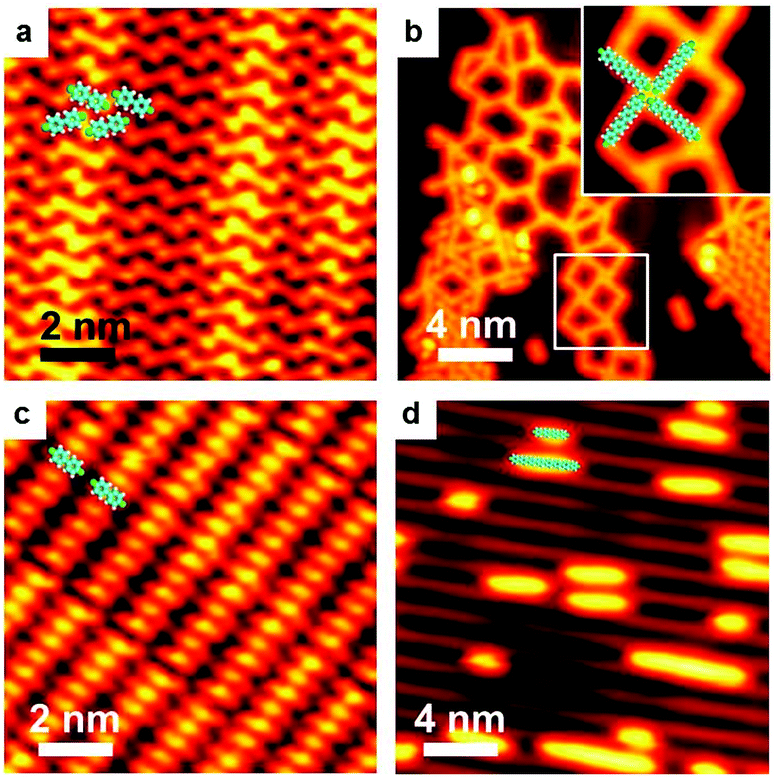 | ||
| Fig. 5 STM images during Ullmann coupling reaction processes of BrPh2Br on Au(111) and Au(100). V = 0.2 V, I = 50 pA. (a) Intact and assembled BrPh2Br at RT and (b) annealed at 500 K, on Au(111). Inset: Enlarged image of the marked area with superimposed molecular models. (c) Intact BrPh2Br molecules at RT, and (d) annealed at 500 K, on Au(100). | ||
Ullmann coupling of dibromo-biphenyl on Au(100) triggered by extrinsic Ag and Ni atoms
In the above discussions, the Ullmann coupling reactions are exclusively triggered by intrinsic surface atoms. As a matter of fact, the Ullmann coupling can also be activated by extrinsic metal atoms prepared via the thermal deposition approach. Dong et al.6 have deposited Pt on Ag(111), and Adisoejoso and co-workers80 have prepared Pd on Au(111) to initiate the Ullmann coupling reactions. Such an approach may provide the possibility to tweak the on-surface Ullmann coupling reactions and widen their potential applications.After RT deposition of the BrPh2Br molecules on Au(100), about 0.1 monolayer Ag was deposited onto the surface at RT. As shown in Fig. 6, the BrPh2Br molecules immediately convert into oligomers which are longer than the BrPh2Br reactant (Fig. 6a). Fig. 6b shows the enlarged STM image of oligomers with terminal bromine atoms. Upon warming the sample up to 500 K, the produced oligomers hardly desorb from the surface, and high-quality PPP chains are accordingly fabricated on surface (Fig. 6c). Such a coupling process is in sharp contrast with those on Ag surfaces where the final coupling products have to go through the organometallic intermediates containing Ag atoms. This shows that the Ullmann coupling process can be mediated by varying the substrates and deposited hetero-atoms.
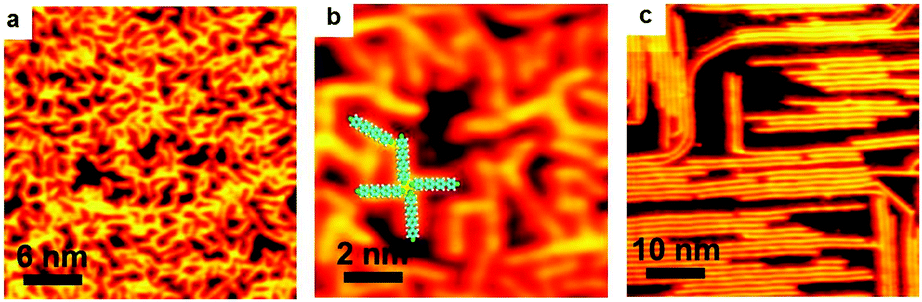 | ||
| Fig. 6 Sequential evolution of the Ullmann coupling of BrPh2Br triggered by Ag deposited on Au(100). (a) BrPh2Br molecules converted into oligomers by Ag deposited at RT. V = 0.1 V, I = 60 pA. (b) Magnified STM image of some oligomers with superimposed molecular models. V = 0.1 V, I = 60 pA. (c) Oligomers converted into the coupling products, PPP chains, at 500 K. V = 1.0 V, I = 50 pA. | ||
The above-described deposition approach can also be used to synthesize special organometallic chains on surface. In Fig. 8a, the adsorbed BrPh2Br molecules are intact on Au(100) at RT. Subsequent deposition of Ni as the extrinsic hetero-atoms can immediately yield the Ni-containing organometallic polymer chains (Fig. 8b).
As a general guidance, the formation temperatures of the involved intermediates and covalently linked products are summarized in Table 1.
| Reactant | Substrate | Extrinsic atoms | Intermediate | Oligomer | PPP chain |
|---|---|---|---|---|---|
| Note: “No” and “—” mean that the species is not observed, and the datum is not available, respectively. | |||||
| BrPh2Br | Ag(111) | RT | — | 473 K | |
| IPh2I | Ag(111) | <RT | — | 473 K | |
| BrPh2Br | Cu(111) | <RT | — | 450 K | |
| BrPh2Br | Au(111) | No | 500 K | No | |
| BrPh2Br | Au(100) | No | 500 K | No | |
| BrPh2Br | Au(100) | Ag | No | RT | 500 K |
| BrPh2Br | Au(100) | Ni | RT | — | — |
Properties of on-surface synthesized single polymer chains
Single polymer wires may behave quite differently from their bulk counterparts in the electric conductance. The electric conductance of an individual polymer wire can seriously deviate from Ohm's law, and show instead an exponential decay as a function of its length. Grill and colleagues16 have measured the electric conductance of a single polyfluorene wire by pulling it up from an Au(111) surface with the STM tip.The above strategy can be applied to measure the electric conductance of various single polymer chains as well. Activated by extrinsic Ag atoms, high-quality PPP chains can be prepared on the Au(100) surface. Due to the weak adsorption capability of the Au surface, the PPP polymer chains can be lifted up, unlike the situations for Ag and Cu surfaces where the prepared polymer chains can easily be broken during the lift-up manipulations with an STM tip. Fig. 7 shows a lifted PPP chain by the STM tip to measure its electric conductance. To do this, the STM tip is located at one end of the PPP chain, and then the feedback of the circuit is cut-off. Subsequently, the STM tip slowly approaches the PPP chain until a sudden jump of the tunnelling current occurs, indicating that the tip has touched the PPP chain. Afterwards, I–z spectroscopy is used to record the circuit current as the tip is moved away. Fig. 7a shows the STM images before and after tip manipulation, showing that the PPP chain is gradually lifted up without any damage. Fig. 7b shows the lnI–z curve during the lift-up process. The curve can be divided into two parts. The left part is the current passing when the STM tip is connected to the PPP chain. When the current suddenly drops down, it means that the STM tip is disconnected from the polymer chain. Then the tunnelling current between the STM tip and the substrate becomes dominant. A linear fitting is carried out for both parts in the lnI–z curve. According to the slope of the blue line, the work function of the Au(100) surface is deduced to be about 4.9 eV, which is close to the reported value, 5.1 eV.81 The well-fitting linear relationship of the left part (red line) indicates the exponential decay of the electric conductance as a function of the lifted PPP chain length, which, based on the fitting, can be expressed as G = 6.1 μSe−0.52z.
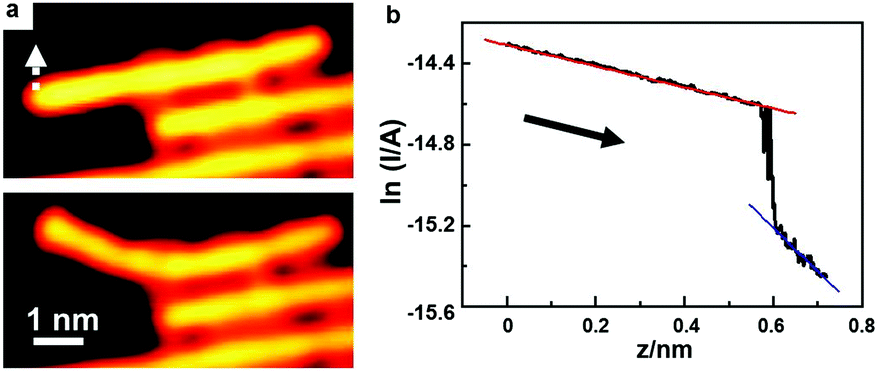 | ||
| Fig. 7 Lift-up of a single PPP chain to measure its electric conductance. T = 4.2 K. (a) STM images before (top) and after (bottom) lifting-up of the PPP chain. V = 0.5 V, I = 0.1 nA. (b) lnI–z curve showing the relationship between the passing circuit current I and the retracting tip height z. V = 0.1 V. | ||
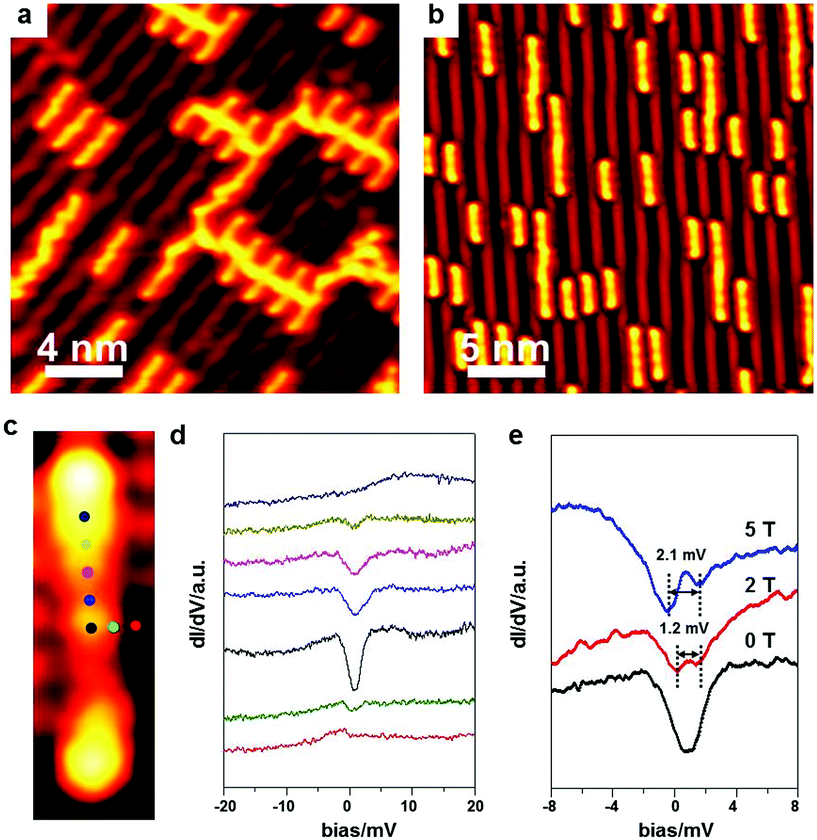 | ||
| Fig. 8 On-surface synthesis of magnetic organometallic chains on Au(100). V = 0.1 V, I = 50 pA, T = 500 mK. (a) STM image of the intact BrPh2Br molecules on Au(100) at RT. (b) BrPh2Br molecules converted into [–Ph2Ni–]n chains triggered by Ni deposited at RT. (c) Enlarged STM image of a BrPh2NiPh2Br intermediate showing STS measurement positions. (d) STS spectra collected at different spots above an individual BrPh2NiPh2Br intermediate in (c). (e) STS spectra collected above the coordinated Ni atom under different magnetic fields. | ||
Besides the electric conductance of the single polymer chains, their magnetism is also of great importance in molecular devices. DiLullo et al.82 have assembled the magnetic molecular chains by covalently linking their molecular building blocks on Au(111), and studied the Kondo effect of the molecular blocks inside the chains where the magnetic centre is held in the precursor. Here, we utilize the extrinsic hetero metal atoms to combine pure organic precursors together, which is more convenient to monitor the Kondo effect. To prove that the Ni-containing organometallic intermediates still possess the magnetic properties, STS measurements are performed right above the intermediate at 500 mK. Fig. 8d shows the STS spectra collected at different spots above an individual BrPh2NiPh2Br intermediate with the measurement positions marked in Fig. 8c. The Kondo signal appears around the coordinated Ni atom, which suggests the existence of unpaired electron spins. A magnetic field of different strength is then applied to generate the Kondo signal splitting. The width of the Kondo feature is about 1.2 meV at 2 T and becomes 2.1 meV at 5 T. The positive correlation between the Kondo feature width and the applied magnetic field further confirms the existence of an unpaired electron spin of the coordinated Ni atom in the intermediate. Such an experiment demonstrates that magnetic organometallic chains can be on-surface synthesized by this strategy.
Conclusions
In summary, on-surface Ullmann coupling reactions of BrPh2Br and IPh2I triggered by intrinsic Ag, Cu and Au substrates, and extrinsic Ag and Ni atoms have been demonstrated and summarized in this study. The reaction intermediates and pathways can be tuned by different precursors and substrates. Different kinds of organometallic polymer chains, [–Ph2Ag–]n, [–Ph2Cu–]n, and [–Ph2Ni–]n, appear as stable intermediates and can be isolated during the reactions. On Au surfaces, the coupling products are directly prepared from the BrPh2Br precursor without going through the Au-containing organometallic intermediate. Magnetic organometallic polymer chains can be successfully fabricated by the Ullmann coupling of BrPh2Br with extrinsic Ni atoms deposited on Au(100). Kondo resonance clearly exists for on-surface prepared Ni-containing organometallic chains, confirming that they are electron spin carriers. Such an on-surface synthesis strategy provides an effective and efficient methodology to prepare functional organometallic and pure organic polymer chains on surface which may find widespread applications, for example, in molecular electronics.Experimental
STM experiments
All STM experiments were performed on a Unisoku UHV LT-STM with a base pressure of 1 × 10−10 Torr. Ag, Cu and Au single crystals were purchased from MaTeck Company and were cleaned by repeated cycles of sputtering and annealing until confirmed by STM images. BrPh2Br and IPh2I were bought from Alfa Aesar (≥98% purity) and dosed onto the substrate from a self-made Ta boat at room temperature without further purification. In order to avoid undesired dosing, the Ta boat was kept in an individually pumped compartment separated from the main chamber by a gate valve. The coverage was controlled by the dosing time. Ag and Ni rods (Aldrich, 99.99% purity) were employed for thermal deposition in an evaporator which was carefully degassed before use. The evaporation temperature was about 1200 K (Ag) and 1700 K (Ni), respectively. The STM measurements were carried out at a base temperature of 77.4 K.XPS measurements
The XPS experiments were performed at the bending magnet beamline SX-700 at the ASTRID storage ring. Clean samples were obtained after repeated cycles of sputtering with Ar+ ions and annealing. Cleaning cycles were repeated until no carbon or bromine contamination could be detected in XPS. BrPh2Br was bought from ACROS (≥99% purity) and dosed onto the Ag(111) crystal from a quartz crucible at RT which was quite similar to the procedure used in the STM chamber. All spectra were collected at normal emission with the X-rays impinging onto the sample at an angle of 45° with respect to the plane of the sample. Each Br 3d spectrum was obtained by adding up 10 sweeps of the electron energy range with a step size of 0.1 eV and an acquisition time of 1 s per step. In order to obtain a signal-to-noise ratio sufficient to capture the slight differences in the C 1s spectra, 120 sweeps were added up per spectrum and the step size was reduced to 0.05 eV per step. The XPS data for molecular adsorption was collected at 110 K, while other XPS data were collected at RT which is either equal to or lower than the annealing temperature, so the molecular chemical states would be retained. Curve fitting was carried out using the software package OriginPro 8. All curves were fitted with Voigt profiles, since the peak shapes are typically determined by a Lorentzian contribution due to the limited lifetime of the core hole state and a Gaussian broadening, mostly due to line shape of the exciting X rays from an X-ray monochromator, electron scattering and detection in the spectrometer. Gaussian contributions may also be related to thermal excitation processes. Chemical, structural and electronic (by dopants) inhomogeneities in the surroundings of the emitting atoms often contribute to the Gaussian broadening.Acknowledgements
This work was jointly supported by NSFC (21133001, 21333001, 91527303, 21261130090), China and NRF CREATE-SPURc project (R-143-001-205-592), Singapore. We acknowledge support from the Danish Council for Independent Research Natural Sciences and the Danish National Research Foundation. F. Bebensee thanks the Alexander von Humboldt-Foundation for a Feodor Lynen-Fellowship. We thank Zheshen Li for technical support in the synchrotron XPS measurements at the SX-700 beamline at ASTRID.Notes and references
- A. Gourdon, Angew. Chem., Int. Ed., 2008, 47, 6950–6953 CrossRef CAS PubMed.
- J. Mendez, M. F. Lopez and J. A. Martin-Gago, Chem. Soc. Rev., 2011, 40, 4578–4590 RSC.
- G. Franc and A. Gourdon, Phys. Chem. Chem. Phys., 2011, 13, 14283–14292 RSC.
- X. Zhang, Q. Zeng and C. Wang, Nanoscale, 2013, 5, 8269–8287 RSC.
- R. Lindner and A. Kühnle, ChemPhysChem, 2015, 16, 1582–1592 CrossRef CAS PubMed.
- L. Dong, P. N. Liu and N. Lin, Acc. Chem. Res., 2015, 48, 2765–2774 CrossRef CAS PubMed.
- J. Björk, J. Phys.: Condens. Matter, 2016, 28, 083002 CrossRef PubMed.
- D. Zhong, J.-H. Franke, S. K. Podiyanachari, T. Blömker, H. Zhang, G. Kehr, G. Erker, H. Fuchs and L. Chi, Science, 2011, 334, 213–216 CrossRef CAS PubMed.
- Q. Sun, C. Zhang, H. Kong, Q. Tan and W. Xu, Chem. Commun., 2014, 50, 11825–11828 RSC.
- C. Zhang, Q. Sun, H. Chen, Q. Tan and W. Xu, Chem. Commun., 2015, 51, 495–498 RSC.
- L. Cai, Q. Sun, C. Zhang, Y. Ding and W. Xu, Chem. – Eur. J., 2016, 22, 1918–1921 CrossRef CAS PubMed.
- S.-W. Hla, L. Bartels, G. Meyer and K.-H. Rieder, Phys. Rev. Lett., 2000, 85, 2777 CrossRef CAS PubMed.
- Y. Okawa and M. Aono, Nature, 2001, 409, 683–684 CrossRef CAS PubMed.
- P. Maksymovych, D. C. Sorescu, K. D. Jordan and J. T. Yates, Science, 2008, 322, 1664–1667 CrossRef CAS PubMed.
- Y. Okawa, M. Akai-Kasaya, Y. Kuwahara, S. K. Mandal and M. Aono, Nanoscale, 2012, 4, 3013–3028 RSC.
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Grill, Science, 2009, 323, 1193–1197 CrossRef CAS PubMed.
- A. Basagni, G. Vasseur, C. A. Pignedoli, M. Vilas-Varela, D. Peña, L. Nicolas, L. Vitali, J. Lobo-Checa, D. G. de Oteyza, F. Sedona, M. Casarin, J. E. Ortega and M. Sambi, ACS Nano, 2016, 10, 2644–2651 CrossRef CAS PubMed.
- P. Ruffieux, J. Cai, N. C. Plumb, L. Patthey, D. Prezzi, A. Ferretti, E. Molinari, X. Feng, K. Müllen, C. A. Pignedoli and R. Fasel, ACS Nano, 2012, 6, 6930–6935 CrossRef CAS PubMed.
- P. Ruffieux, S. Wang, B. Yang, C. Sánchez-Sánchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489–492 CrossRef CAS PubMed.
- G. Vasseur, Y. Fagot-Revurat, M. Sicot, B. Kierren, L. Moreau, D. Malterre, L. Cardenas, G. Galeotti, J. Lipton-Duffin, F. Rosei, M. Di Giovannantonio, G. Contini, P. Le Fevre, F. Bertran, L. Liang, V. Meunier and D. F. Perepichka, Nat. Commun., 2016, 7, 10235 CrossRef CAS PubMed.
- M. angleDi Giovannantonio, M. El Garah, J. Lipton-Duffin, V. Meunier, L. Cardenas, Y. Fagot Revurat, A. Cossaro, A. Verdini, D. F. Perepichka, F. Rosei and G. Contini, ACS Nano, 2013, 7, 8190–8198 CrossRef PubMed.
- A. Basagni, F. Sedona, C. A. Pignedoli, M. Cattelan, L. Nicolas, M. Casarin and M. Sambi, J. Am. Chem. Soc., 2015, 137, 1802–1808 CrossRef CAS PubMed.
- W. Wang, X. Shi, S. Wang, M. A. Van Hove and N. Lin, J. Am. Chem. Soc., 2011, 133, 13264 CrossRef CAS PubMed.
- P. Nguyen, P. Gómez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515–1548 CrossRef CAS PubMed.
- K. A. Williams, A. J. Boydston and C. W. Bielawski, Chem. Soc. Rev., 2007, 36, 729–744 RSC.
- J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef CAS PubMed.
- Y.-Q. Zhang, N. Kepčija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V. Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed.
- H.-Y. Gao, J.-H. Franke, H. Wagner, D. Zhong, P.-A. Held, A. Studer and H. Fuchs, J. Phys. Chem. C, 2013, 117, 18595–18602 CAS.
- J. Liu, Q. Chen, L. Xiao, J. Shang, X. Zhou, Y. Zhang, Y. Wang, X. Shao, J. Li, W. Chen, G. Q. Xu, H. Tang, D. Zhao and K. Wu, ACS Nano, 2015, 9, 6305–6314 CrossRef CAS PubMed.
- F. Klappenberger, Y.-Q. Zhang, J. Björk, S. Klyatskaya, M. Ruben and J. V. Barth, Acc. Chem. Res., 2015, 48, 2140–2150 CrossRef CAS PubMed.
- F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- M. Xi and B. E. Bent, Surf. Sci., 1992, 278, 19–32 CrossRef CAS.
- M. Xi and B. E. Bent, J. Am. Chem. Soc., 1993, 115, 7426 CrossRef CAS.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Mullen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed.
- H. Zhang, H. Lin, K. Sun, L. Chen, Y. Zagranyarski, N. Aghdassi, S. Duhm, Q. Li, D. Zhong, Y. Li, K. Müllen, H. Fuchs and L. Chi, J. Am. Chem. Soc., 2015, 137, 4022–4025 CrossRef CAS PubMed.
- P. Han, K. Akagi, F. Federici Canova, H. Mutoh, S. Shiraki, K. Iwaya, P. S. Weiss, N. Asao and T. Hitosugi, ACS Nano, 2014, 8, 9181–9187 CrossRef CAS PubMed.
- A. Batra, D. Cvetko, G. Kladnik, O. Adak, C. Cardoso, A. Ferretti, D. Prezzi, E. Molinari, A. Morgante and L. Venkataraman, Chem. Sci., 2014, 5, 4419–4423 RSC.
- K. A. Simonov, N. A. Vinogradov, A. S. Vinogradov, A. V. Generalov, E. M. Zagrebina, G. I. Svirskiy, A. A. Cafolla, T. Carpy, J. P. Cunniffe, T. Taketsugu, A. Lyalin, N. Mårtensson and A. B. Preobrajenski, ACS Nano, 2015, 9, 8997–9011 CrossRef CAS PubMed.
- M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2011, 3, 61–67 CrossRef CAS PubMed.
- L. Jiang, T. Niu, X. Lu, H. Dong, W. Chen, Y. Liu, W. Hu and D. Zhu, J. Am. Chem. Soc., 2013, 135, 9050–9054 CrossRef CAS PubMed.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Nat. Chem., 2012, 4, 215–220 CrossRef CAS PubMed.
- C. Sánchez-Sánchez, S. Brüller, H. Sachdev, K. Müllen, M. Krieg, H. F. Bettinger, A. Nicolaï, V. Meunier, L. Talirz, R. Fasel and P. Ruffieux, ACS Nano, 2015, 9, 9228–9235 CrossRef PubMed.
- T. Lin, X. S. Shang, J. Adisoejoso, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2013, 135, 3576–3582 CrossRef CAS PubMed.
- Q. Fan, J. M. Gottfried and J. Zhu, Acc. Chem. Res., 2015, 48, 2484–2494 CrossRef CAS PubMed.
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604 CAS.
- J. Park, K. Y. Kim, K.-H. Chung, J. K. Yoon, H. Kim, S. Han and S.-J. Kahng, J. Phys. Chem. C, 2011, 115, 14834–14838 CAS.
- E. A. Lewis, C. J. Murphy, M. L. Liriano and E. C. H. Sykes, Chem. Commun., 2014, 50, 1006–1008 RSC.
- Q. Fan, C. Wang, Y. Han, J. Zhu, J. Kuttner, G. Hilt and J. M. Gottfried, ACS Nano, 2014, 8, 709–718 CrossRef CAS PubMed.
- Q. Fan, C. Wang, Y. Han, J. Zhu, W. Hieringer, J. Kuttner, G. Hilt and J. M. Gottfried, Angew. Chem., Int. Ed., 2013, 52, 4668–4672 CrossRef CAS PubMed.
- M. Chen, J. Xiao, H.-P. Steinrück, S. Wang, W. Wang, N. Lin, W. Hieringer and J. M. Gottfried, J. Phys. Chem. C, 2014, 118, 6820–6830 CAS.
- I. Píš, L. Ferrighi, T. H. Nguyen, S. Nappini, L. Vaghi, A. Basagni, E. Magnano, A. Papagni, F. Sedona, C. Di Valentin, S. Agnoli and F. Bondino, J. Phys. Chem. C, 2016, 120, 4909–4918 Search PubMed.
- E. A. Lewis, C. J. Murphy, A. Pronschinske, M. L. Liriano and E. C. H. Sykes, Chem. Commun., 2014, 50, 10035–10037 RSC.
- A. Saywell, J. Schwarz, S. Hecht and L. Grill, Angew. Chem., Int. Ed., 2012, 51, 5096–5100 CrossRef CAS PubMed.
- M. Bieri, M.-T. Nguyen, O. Gröning, J. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669 CrossRef CAS PubMed.
- P. S. Weiss, M. M. Kamna, T. M. Graham and S. J. Stranick, Langmuir, 1998, 14, 1284 CrossRef CAS.
- A. Rastgoo Lahrood, J. Bjork, W. M. Heckl and M. Lackinger, Chem. Commun., 2015, 51, 13301–13304 RSC.
- A. F. Carley, M. Coughlin, P. R. Davies, D. J. Morgan and M. Wyn Roberts, Surf. Sci., 2004, 555, L138–L142 CrossRef CAS.
- M. M. Blake, S. U. Nanayakkara, S. A. Claridge, L. C. Fernández-Torres, E. C. H. Sykes and P. S. Weiss, J. Phys. Chem. A, 2009, 113, 13167 CrossRef CAS PubMed.
- Q. Fan, C. Wang, L. Liu, Y. Han, J. Zhao, J. Zhu, J. Kuttner, G. Hilt and J. M. Gottfried, J. Phys. Chem. C, 2014, 118, 13018–13025 CAS.
- N. Lin, D. Payer, A. Dmitriev, T. Strunskus, C. Wöll, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2005, 44, 1488 CrossRef CAS PubMed.
- A. Basagni, L. Ferrighi, M. Cattelan, L. Nicolas, K. Handrup, L. Vaghi, A. Papagni, F. Sedona, C. D. Valentin, S. Agnoli and M. Sambi, Chem. Commun., 2015, 51, 12593–12596 RSC.
- J. A. Lipton-Duffin, O. Ivasenko, D. F. Perepichka and F. Rosei, Small, 2009, 5, 592–597 CrossRef CAS PubMed.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 4456–4458 RSC.
- K.-H. Chung, B.-G. Koo, H. Kim, J. K. Yoon, J.-H. Kim, Y.-K. Kwon and S.-J. Kahng, Phys. Chem. Chem. Phys., 2012, 14, 7304–7308 RSC.
- J. Eichhorn, T. Strunskus, A. Rastgoo-Lahrood, D. Samanta, M. Schmittel and M. Lackinger, Chem. Commun., 2014, 50, 7680–7682 RSC.
- G. Tian, Y. Chen, H.-L. Bao, X. Meng, K. Pan, W. Zhou, C. Tian, J.-Q. Wang and H. Fu, J. Mater. Chem., 2012, 22, 2081–2088 RSC.
- J. Björk, F. Hanke and S. Stafström, J. Am. Chem. Soc., 2013, 135, 5768–5775 CrossRef PubMed.
- S. Schlögl, W. M. Heckl and M. Lackinger, Surf. Sci., 2012, 606, 999–1004 CrossRef.
- G. Eder, E. F. Smith, I. Cebula, W. M. Heckl, P. H. Beton and M. Lackinger, ACS Nano, 2013, 7, 3014–3021 CrossRef CAS PubMed.
- J. Eichhorn, D. Nieckarz, O. Ochs, D. Samanta, M. Schmittel, P. J. Szabelski and M. Lackinger, ACS Nano, 2014, 8, 7880–7889 CrossRef CAS PubMed.
- M. O. Blunt, J. C. Russell, N. R. Champness and P. H. Beton, Chem. Commun., 2010, 46, 7157–7159 RSC.
- J. C. Russell, M. O. Blunt, J. M. Garfitt, D. J. Scurr, M. Alexander, N. R. Champness and P. H. Beton, J. Am. Chem. Soc., 2011, 133, 4220 CrossRef CAS PubMed.
- H. Zhang, J.-H. Franke, D. Zhong, Y. Li, A. Timmer, O. D. Arado, H. Mönig, H. Wang, L. Chi, Z. Wang, K. Müllen and H. Fuchs, Small, 2014, 10, 1361–1368 CrossRef CAS PubMed.
- A. Saywell, W. Greń, G. Franc, A. Gourdon, X. Bouju and L. Grill, J. Phys. Chem. C, 2014, 118, 1719–1728 CAS.
- J. Adisoejoso, T. Lin, X. S. Shang, K. J. Shi, A. Gupta, P. N. Liu and N. Lin, Chem. – Eur. J., 2014, 20, 4111–4116 CrossRef CAS PubMed.
- N. E. Singh-Miller and N. Marzari, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235407 CrossRef.
- A. DiLullo, S.-H. Chang, N. Baadji, K. Clark, J.-P. Klöckner, M.-H. Prosenc, S. Sanvito, R. Wiesendanger, G. Hoffmann and S.-W. Hla, Nano Lett., 2012, 12, 3174–3179 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2017 |