
Recent advances in thromboresistant and antimicrobial polymers for biomedical applications: just say yes to nitric oxide (NO)
Yaqi
Wo
a,
Elizabeth J.
Brisbois
b,
Robert H.
Bartlett
b and
Mark E.
Meyerhoff
*a
aDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: mmeyerho@umich.edu; Tel: +1 (734) 763-5916
bDepartment of Surgery, University of Michigan Medical Center, Ann Arbor, MI 48109, USA
First published on 26th May 2016
Abstract
Biomedical devices are essential for patient diagnosis and treatment; however, when blood comes in contact with foreign surfaces or homeostasis is disrupted, complications including thrombus formation and bacterial infections can interrupt device functionality, causing false readings and/or shorten device lifetime. Here, we review some of the current approaches for developing antithrombotic and antibacterial materials for biomedical applications. Special emphasis is given to materials that release or generate low levels of nitric oxide (NO). Nitric oxide is an endogenous gas molecule that can inhibit platelet activation as well as bacterial proliferation and adhesion. Various NO delivery vehicles have been developed to improve NO's therapeutic potential. In this review, we provide a summary of the NO releasing and NO generating polymeric materials developed to date, with a focus on the chemistry of different NO donors, the polymer preparation processes, and in vitro and in vivo applications of the two most promising types of NO donors studied thus far, N-diazeniumdiolates (NONOates) and S-nitrosothiols (RSNOs).
 Yaqi Wo | Yaqi Wo is currently pursuing her Ph.D. in Chemistry under Dr Mark E. Meyerhoff at University of Michigan. She received her B.S. degree in Chemistry from Xiamen University, China. Her primary area of research is to develop and characterize novel nitric oxide (NO) releasing polymeric materials. She is also conducting in vitro and in vivo testing to evaluate the hemocompatibility and/or antibacterial properties of these new materials/devices (e.g., intravascular catheters, urinary catheters and wound healing patches). |
 Elizabeth J. Brisbois | Elizabeth J. Brisbois earned her Ph.D. degree in Chemistry at the University of Michigan in 2014. Her dissertation work focused on the design and characterization of novel nitric oxide releasing polymers, based on S-nitrosothiols and diazeniumdiolates, for improved hemocompatibility applications. She is currently a National Institutes of Health Postdoctoral Fellow at the University of Michigan Medical School and has also been awarded with a MICHR Postdoctoral Translational Scholars Program career development award. She is now studying electrochemical methods to generate nitric oxide for potential biomedical applications. |
 Robert H. Bartlett | Robert H. Bartlett earned his M.D. with honors in 1963 from the University of Michigan Medical School and is currently Professor Emeritus in Department of Surgery at the University of Michigan. He is best known for his clinical innovations and development of ECMO, extracorporeal membrane oxygenation, that has saved thousands lives since 1975. His contributions have been recognized by numerous awards, including the American College of Surgeon's Sheen Award for Research, Medal of Special Recognition from the National Academy of Surgery of France, and Medallion of Scientific Achievement from the American Surgical Association. He is also a member of the Institute of Medicine of the National Academy of Science. |
 Mark E. Meyerhoff | Mark E. Meyerhoff earned his Ph.D. degree in Chemistry at the State University of New York at Buffalo, in 1979. He joined the Department of Chemistry at the University of Michigan in 1979 as assistant professor and is currently the Philip J. Elving Collegiate Professor of Chemistry. His active research is in the areas of novel electrochemical and optical chemical sensors for anions and gas detection as well as developing new nitric oxide (NO) release polymers to improve the biocompatibility of implantable biomedical devices. He has been recognized with many awards, including the Ralph N. Adams Award in Bioanalytical Chemistry, the Charles N. Reilley Award in Electroanalytical Chemistry and the ACS Analytical Chemistry Award in Electrochemistry. |
Blood-contacting biomaterials are an integral part of various biomedical devices, ranging from simple catheters to intravascular grafts to extracorporeal circuits and membrane oxygenators, that offer lifesaving treatments to thousands of patients every day. The incorporation of these blood-contacting materials is usually complicated by foreign body response that is initiated by the coagulation cascade1 (see Fig. 1).
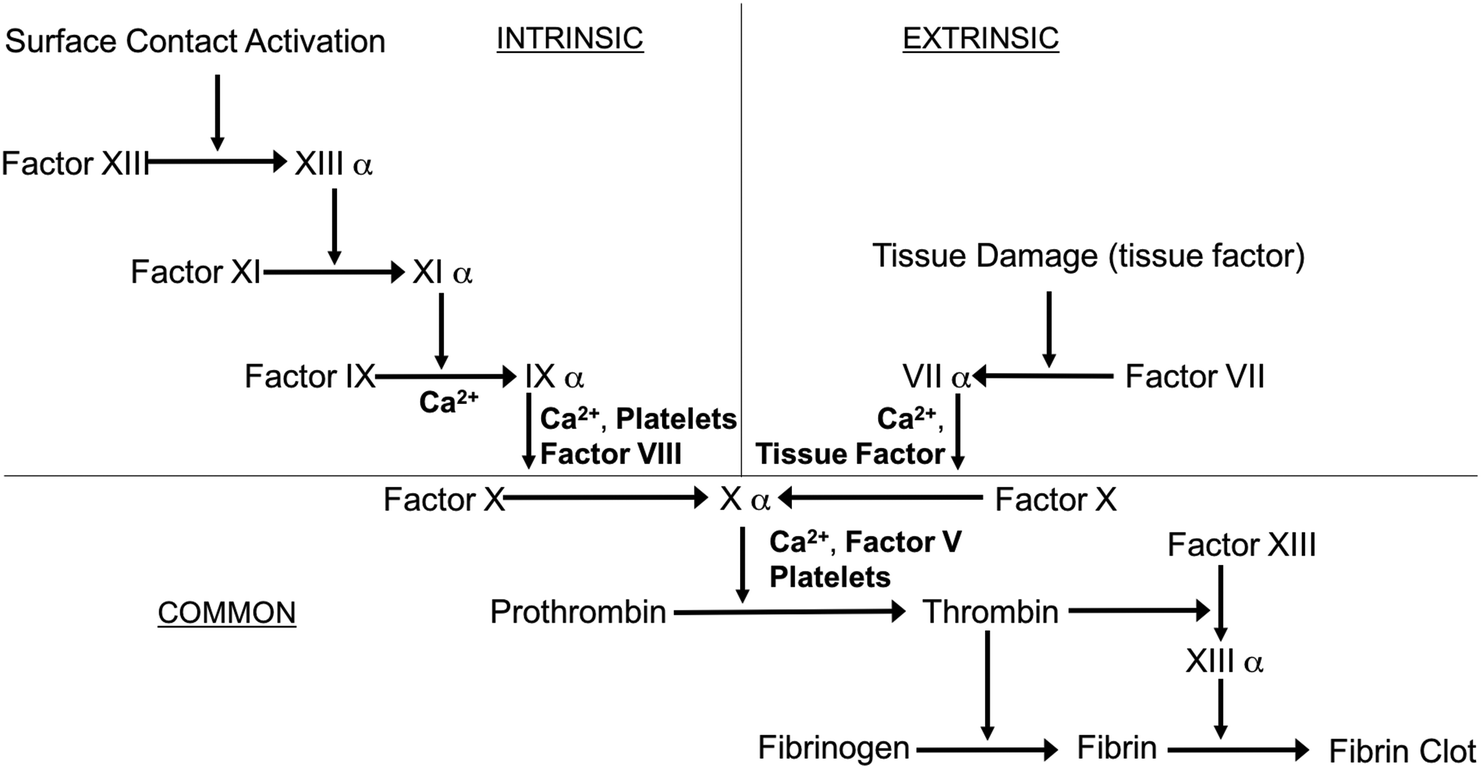 | ||
| Fig. 1 Simplified schematic of the blood-coagulation cascade, where both intrinsic (surface contact) and extrinsic (tissue factors) pathways converge and ultimately form thrombus (modified from review by Sefton et al.2). | ||
Exposure of blood to foreign materials will cause plasma proteins, such as Factor XII and Factor XI, to be activated and adhere to the polymer surface. The activated protein will interact with platelet surface membrane receptor GPIIb/IIIa, that can bind with fibrinogen, von Willebrand Factors (vWF), fibronectin, and vitronectin, which ultimately leads to platelet conformational changes, subsequent release of intracellular agents (e.g., Factor V, VIII and Ca2+, etc.), and initiates platelet activation and aggregation (intrinsic pathway).2,3 Further, surgical procedures that utilize these devices have the potential to injure the vessel wall, disrupt blood homeostasis and release tissue factor, also causing platelet activation and concomitant conformational changes as well (extrinsic pathway).1–6 Both pathways converge and trigger the coagulation cascade as the activated platelets bind with fibrinogen and other clotting factors. Fibrinogen forms insoluble fibrin, which traps red blood cells and ultimately forms a thrombus within a matter of hours7,8 (see Fig. 2).
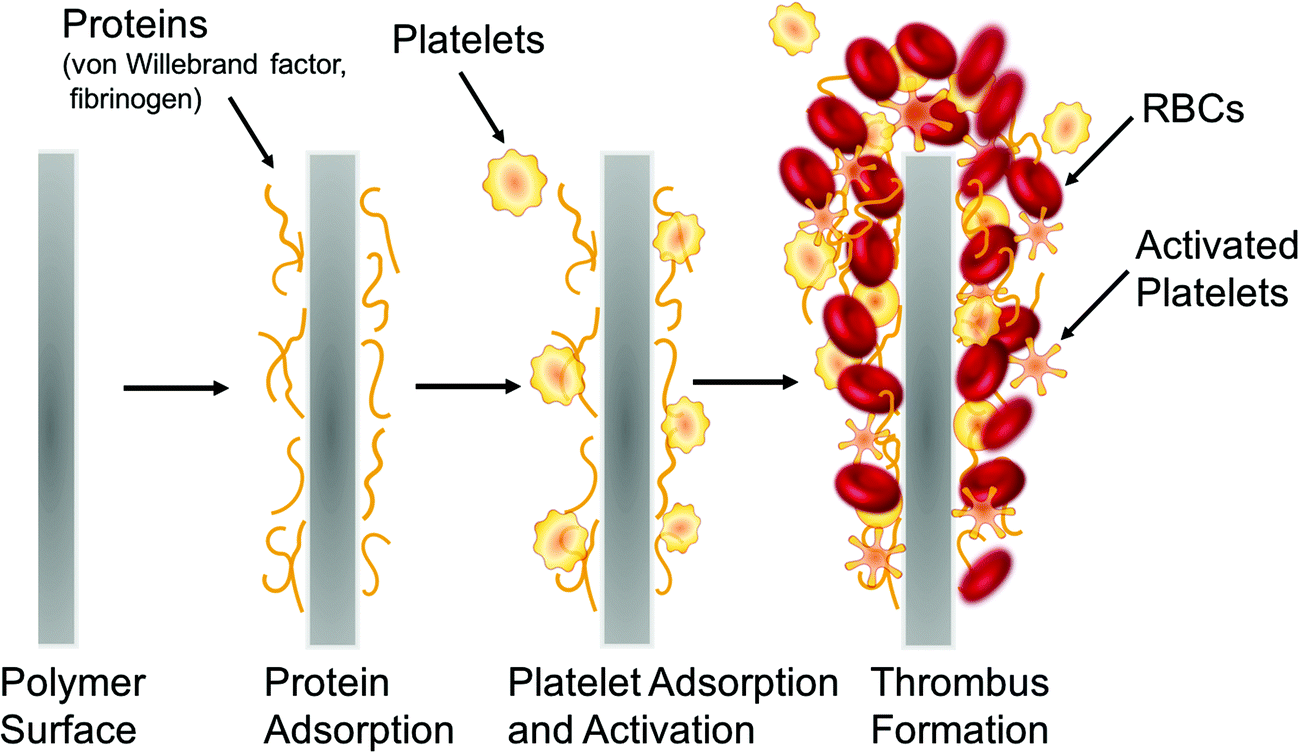 | ||
| Fig. 2 Simplified representation of the processes that lead to thrombus formation on the surfaces of blood-contacting biomedical devices. | ||
In addition to thrombus formation, bacterial infection is also often associated with the use of many biomedical devices. The rise of hospital-acquired infections, also known as nosocomial infections, is a growing concern in healthcare industry. It was reported that an estimated 1.7 million patients suffered from healthcare-associated infections (HAI) in the U.S. hospitals in 2002 and the number of HAI deaths was 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 987 patients, including 30665 resulting from bloodstream infections.9–15 Device-related infections are the result of bacteria adhesion to the biomaterial surface. After planktonic bacteria initially colonize onto the surfaces of polymeric devices, cells start to grow into colonies, and then hydrated matrices of extracellular polymeric substances (EPS) are formed, also known as biofilms,16–18 (see Fig. 3).
987 patients, including 30665 resulting from bloodstream infections.9–15 Device-related infections are the result of bacteria adhesion to the biomaterial surface. After planktonic bacteria initially colonize onto the surfaces of polymeric devices, cells start to grow into colonies, and then hydrated matrices of extracellular polymeric substances (EPS) are formed, also known as biofilms,16–18 (see Fig. 3).
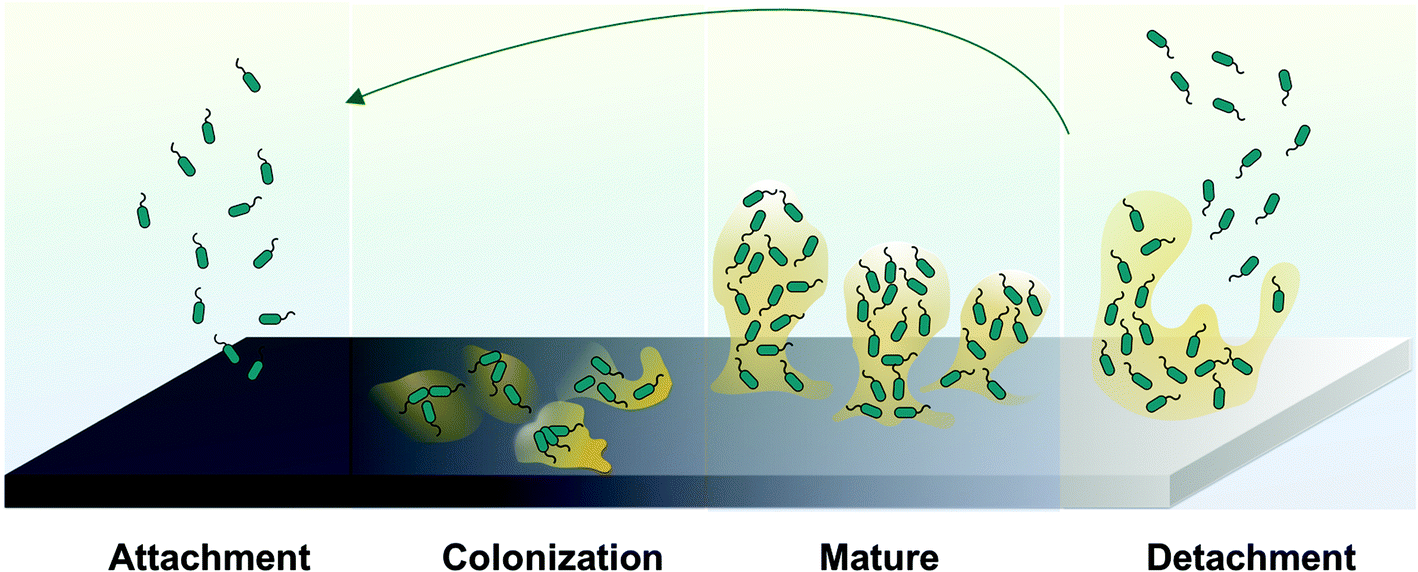 | ||
| Fig. 3 Representative processes involved in biofilm formation and bacteria dispersion. | ||
The EPS holds the bacterial cells together in a mass and firmly attaches the cells to the underlying surface. Ultimately, a mature biofilm will periodically release bacteria cells from the biofilm colony into the surrounding medium. Of note, the EPS is both a physical and a chemical barrier to antibiotics and significantly retards their rate of penetration that makes conventional antibiotic treatment for such infections ineffective. Further, the EPS can possibly foster antibiotic-resistant bacterial gene mutations.19 Ideally, all biomedical devices should be able to prevent bacterial colonization on their surfaces, especially within the first 6 h of blood exposure, which is identified to be the most susceptible and most “decisive period” for the success of a long-term implant.20 Thrombus and bacteria biofilm formation cause device failure, which in many cases can only be solved by device removal and replacement. Besides patients suffering, the increased healthcare costs associated with these infections has also created a significant economic burden.20
The clinical problems of thrombus and infection described above have triggered substantial interest among scientists to develop new and more effective ways to create antithrombotic and antibacterial polymeric surfaces for biomedical devices, especially those in direct contact with blood (e.g., intravascular catheters, vascular grafts, etc.).
Current strategies for prevention of thrombosis
Actively releasing anticoagulants to block the innate coagulation cascade
In a clinical setting, in order to prevent surface-induced thrombosis during cardiopulmonary bypass, hemodialysis, and angioplasty, anticoagulants are routinely administered, the most commonly used one being heparin.2,21 However, systemic heparin administration can lead to hemorrhage and thrombocytopenia. Localized release of heparin with concentrations that are not tolerable at the systemic level may be applied with minimal side-effects.22,23 To date, several approaches have been pursued to achieve localized heparin activity at the surface of implanted devices including the creation of heparin-releasing polymer surfaces via ionic bonding,22,24–26 physical dispersion27–30 and solution swelling,22,31,32 as well as heparin-immobilized polymer surfaces.33–36 For example, Gutowska et al.22 described a novel thermosensitive heparin-releasing poly(N-isopropylacrylamide) (poly(NiPAAm)) coating for prevention of surface-induced thrombosis on polyurethane catheters. Poly(NiPAAm) has a lower critical solution temperature (LCST) in aqueous solution, around 32 °C, which enables it to swell when immersed in a heparin solution at low temperature (e.g., room temperature, RT) and uptake this polyanionic drug. When the temperature is higher than LCST, the swollen coating will collapse dramatically and release heparin. Both the heparin impregnated and control PU catheters were inserted into saphenous veins in a canine model for 1.5 h, and the heparin-releasing PU catheter surfaces demonstrated a significant reduction of thrombus formation after contact with the venous blood. This was demonstrated by SEM images of the explanted catheters when compared to the appropriate controls. Heparin typically has low solubility in organic solvents, but this swelling method introduced the possibility of solution absorption in water and loading of a relatively large amount of the drug into the swollen polymer chains. This method also offers flexibility for the amount and type of anticoagulant drugs that can be loaded into various thermosensitive coatings.22 However, the heparin release kinetics for this system are relatively fast, with the release rates of 1 μg per cm2 per h for up to 6 h. Therefore, the potential application of this material is limited to preventing thrombosis formation in short-term applications, such as for angiography catheters, etc.Heparin has also been covalently linked to the surface of vascular grafts via end-point immobilization and this approach has been commercialized for expanded polytetrafluoroethylene (ePTFE) and Dacron grafts.37–39 The key to the success of heparin-immobilized polymers depends on the covalently bound heparin remaining flexible enough to bind antithrombin III in order to prevent fibrin formation and the ultimate blood clot.3 The literature on immobilization of heparin is vast and has been reviewed extensively elsewhere.37,40 However, due to their very short half-life,41 heparinized polymer surfaces still suffer a great challenge when it comes to long-term in vivo applications. Other anticoagulants such as thrombomodulin,36 direct thrombin inhibitors (e.g., hirudin42 and argatroban43), or antiplatelet drugs (e.g.: prostacyclin (PGI2))36,41,42 have also been immobilized onto polymer surfaces to increase their hemocompatibility, with varying degrees of success.
Chemical modification of the polymer surface to reduce protein adsorption
It is widely known that the first step that initiates activation of the coagulation cascade is protein adsorption (especially fibrinogen and von Willebrand factor; see Fig. 2) to the surface of a blood-contacting medical device. This knowledge has led to many years of research in developing approaches to modify polymer surfaces that focus on reducing such non-specific protein adsorption. Some examples include immobilizing a layer of blood-compatible hydrogels on the device surfaces, such as poly(vinyl alcohol) (PVA), polyacrylamide (PAAm), poly(N-vinyl pyrrolidone) (PNVP), poly(hydroxyethyl methacrylate) (PHEMA), poly(ethylene oxide) (PEO), poly(ethylene glycol) (PEG), poly(ethylene glycol) monomethyl ether (PEGME), and cellulose.3,22,44 Hydrogels are water-swollen polymeric networks containing chemical or physical cross-links and were first used initially as soft contact lenses in the late 1950s.45 Due to their hydrophilicity and excellent water retention properties, they have the natural tendency to prevent cell and protein adhesion and are considered to be very biocompatible and desirable in biomedical applications. For example, immobilization of PEG (–CH2CH2O–) is a widely used method to modify traditional polymer surfaces employed in the medical field, such as plasticized poly(vinyl chloride) (PVC) and polyethylene (PE), etc.46–49 PEG is a non-toxic water-soluble polymer approved by the U.S. Food and Drug Administration (FDA) for internal consumption.50 Lakshmi et al.46 were among the first to graft PEG 4000 onto medical grade PVC sheets and conducted thrombogenicity studies to evaluate platelet adhesion using platelet rich plasma (PRP) and whole blood clotting time with fresh rabbit blood. The scanning electron microscope (SEM) images clearly indicated more platelet adhesion to the bare PVC sheet than the PEG-grafted ones. The PEG grafting also extended the whole blood clotting time from 20 min to more than 70 min as determined by the hemolysis assay. Balakrishnan et al.47 later demonstrated success using bulk modification of PVC resin with PEG 600 that yielded greatly reduced solid/water interfacial free energy and platelet adhesion in in vitro PRP studies. This study expanded the anti-fouling chemistry from surface modification on a finished product to bulk synthesis. However, studies conducted by Sefton and coworkers, who used PEG immobilized PVA hydrogel coated PE tubing in an ex vivo canine arteriovenous (AV) shunt, reported that neither the PEG grafted or control PVA tubing could maintain circulating platelet levels after 4 d.49 Heparin and hydrogel modified surfaces are the most commonly studied approaches for achieving thromboresistant surfaces. For further breadth, readers are guided to reviews that highlight additional approaches developed thus far, such as endothelial cell coated-surfaces, albumin-coated surfaces, pyrolytic carbon-coated surface, phosphorylcholine surfaces, elastin-inspired surfaces, etc.3,42,51–53Current strategies for creating antibacterial surfaces
Surfaces that resist bacteria and reduce initial attachment
Bacteria attachment to device surfaces is the first step required for biofilm formation. Therefore, using biomaterial surfaces that resist bacteria attachment is an intuitive solution to this problem.54 Super-hydrophobic polymer surfaces, such as very smooth silicone, polyurethane, polytetrafluoroethylene (PTFE), poly(vinyl chloride) (PVC), and polyethylene (PE), that have water contact angles larger than 150°, low surface energy,55,56 as well as unique water repellent57 and self-cleaning abilities,56 are known to have resistance to microbial cell adhesion during short-term applications.58 Hydrophilic surface modifications, such as the PEG modified surfaces discussed above, have also been widely characterized in the literature and have demonstrated excellent anti-adhesive properties for bacteria cells and proteins.44 However, the susceptibility of PEG to oxidative damage, especially in the presence of O2, transition metal ions or certain enzymes in vivo, has limited its long-term application in complex media.59,60 Some studies have demonstrated that polymer surfaces with segmented block co-polymers (hard and soft domains) that result in phase-separated structures also exhibit less bacterial adhesion and cell attachment.61–63 Overall, however, passive surfaces without functional active agents may not be ideal for long-term applications because these surfaces may eventually become contaminated due to defects during preparation or deterioration of the coating when in contact with physiological fluids.3,4,64Surfaces that disperse or detach biofilms
Biofilm dispersal is a promising area of research that focuses on how to inhibit biofilm formation using dispersal agents, which induce biofilm bacteria to detach and return to their planktonic form. Several studies have found that bacteria naturally produce biofilm dispersal agents when the community senses a quorum, signaling the detachment process.54 These agents include D-amino acids,65cis-2-decenoic acid (C2DA),16,18,54 peptides and various enzymes (e.g., dispersin B), etc.54,66 However, most mechanisms of action of these compounds are still unclear, which hinders further development and application in this field. Many researchers have studied the effects of these dispersal agents in vitro, by adding the agents directly to pre-formed biofilm in petri dishes or bioreactors.67 Jennings et al. attempted to load C2DA into chitosan sponges for localized delivery by initially immersing the sponges into 1 mL of 100 mg per mL C2DA in 10% enthanol.68 The release of C2DA was determined by HPLC and its release lasted 5 d (200–1000 μg mL−1) with a burst of C2DA release on day 1. The anti-biofilm efficiency of the C2DA loaded chitosan sponge was tested against clinical isolates of methicillin-resistant Staphylococcus aureus (MRSA) and the results showed that C2DA at a concentration (in the sponge) at or above 500 μg mL−1 can inhibit bacterial growth. Current research in this field is aimed at developing vehicles for the controlled and sustained release of such biofilm dispersal agents.54Surfaces that have bactericidal functionalities
The most common mechanism of creating functional antibacterial surfaces is through a bactericidal effect, which includes employing a vast collection of approaches involving quaternary ammonium compounds (QACs)64,69–71 and other polycations (e.g., organometallic dendrimers72 and chlorhexidine73), metal ions (e.g., silver,74,75 copper,76 titanium,77etc.), locally released antibiotics (e.g., gentamicin66,78,79) or bactericidal agents (e.g., bacteriophages,54,80,81 protein synthesis inhibitors,66 antibacterial enzymes such as lysozyme,64,82 antibacterial peptides,83–85 natural biomolecules such as chitosan86–88 and herbal extracts (e.g., flavanones and chalcones,89etc.) and inducing oxidative stress.72,90 In general, the mechanisms of bactericidal surfaces are: (1) contact-based bactericidal activity (e.g., QAC, etc.) which affects the ion-exchange processes and cause general perturbations that destabilize the cytoplasmic membranes of bacteria, resulting in leakage of the intracellular fluid; and (2) release-based bactericidal (e.g., metal ions, antibiotics, etc.), which damage the bacterial cell membrane as well as disrupt the function of bacterial enzymes, DNA, proteins and cell membranes.64 A problem, however, with many bactericidal surfaces is the attachment of dead microorganisms remaining on the antibacterial coatings, which can trigger immune response and inflammation, as well as block a given coating's active functional group.In recent years, in order to achieve improved antibacterial and anti-fouling efficacy, many researchers have developed polymeric surfaces that combine more than one of the antibacterial functionalities mentioned above; for example, creating a surface that not only can attach and kill bacteria but also be able to release any adhered dead bacteria debris. Toward this goal, many pH,59,82,91 thermo,92 or electrical voltage93-responsive polymers, that can be controllably extended or collapsed or altered in their charge carrying properties have been developed. They are often termed “stimuli-responsive smart antibacterial surfaces”. Jiang and coworkers developed a cationic poly(N,N-dimethyl-N-(ethoxycarbonylmethyl)-N-[2′-(methacryloyloxy)ethyl]-ammonium bromide) that has a quaternary ammonium group and was grafted onto a gold surface by surface-initiated atom transfer radical polymerization.59 The cationic surface can kill 99.9% of E. coli bacteria when exposed to a suspension of 1010 cells per mL for 1 h, and then it hydrolyzes into a nonfouling zwitterionic surface and releases 98% of the bacterial residue after 8 d at 37 °C and pH 10.0. This is only a one-time transition between cationic and zwitterionic surface; therefore, in order to achieve a fully reversible “kill and release” functional surface, a surface with morpholinone derivatives that can be switched repeatedly between two equilibrium states was developed by the same authors.91 In neutral or basic aqueous phase, the surface will release dead bacteria and at the same time resist bacteria adhesion. However, under acidic conditions (e.g., in acetic acid for 20 h), the surface will regenerate bacteria-killing function by reforming the quaternary ammonium functional group.
Of note, there are some limitations of this method of using pH change to alter antimicrobial properties of a surface. These include: (1) this surface can kill bacteria attached on the surface while it is dry, since a wet version of this surface is generally resistant to bacterial attachment, suggesting this method mainly works for applications that prevent airborne bacteria; and (2) the requirement of changing environmental pH to achieve surface functionality may be difficult in situ with certain biomedical applications (e.g., blood-contacting devices). Most recently, Chen and coworker developed an on-demand switchable and repeatable antibacterial surface. They used a silicone nanowire grafted pH-responsive poly(methacrylic acid) (SiN-PMAA) for loading a natural antibacterial agent (i.e., lysozyme enzyme), enabling killing of bacteria and releasing dead bacteria debris when alternating the environmental pH between 4, 7 or 10, respectively. The SiN-PMAA surface exhibits a high capacity for binding lysozyme at acidic pH, but can release the majority of the adsorbed lysozyme that serves as biocide to kill bacteria attached to the surface or suspended in solution at neutral pH, and then release the dead bacteria to provide a self-cleaning process when the environment pH is increased to a basic value. This dynamic reservoir concept may serve as the foundation for engineering multifunctional surfaces that could find many practical applications (such as biocatalysis and biosensing) in both biomedical and biotechnology fields. However, achieving the needed changes in pH within blood, which is a relatively strong buffer, will be more challenging.
Lastly, bacterial interference is a different concept that uses active bacteria (either probiotic bacteria54,94,95 or bacteria with less virulence96–98) to inhibit the targeted bacteria by competing for common resources in the same environment. This approach can be leveraged to prevent infections from exogenous sources. Trautner et al. reported a prospective clinical trial of using nonpathogenic bacteria colonized on urinary catheters in patients who require indwelling catheter drainage, to examine their effect in preventing bacteriuria commonly present in patients with indwelling urinary tract catheters.98 Commercial urinary catheters were incubated in broth of Escherichia coli (E. coli) HU2117, a genetic strain that can cause persistent colonization without symptomatic infection,99 for 48 h before insertion into patients for 28 d. The patients’ urine samples were collected at various time points until E. coli HU2117 was no longer detected. Ten (83%) out of 12 subjects were successfully colonized with E. coli HU2117 for 14 d or more (range, 15–165 d) after inoculation by insertion of the catheter. One patient had urinary tract infection (UTI) symptoms caused by Pseudomonas but none of the patients experienced UTI attributable to the colonization of E. coli HU2117. The overall rate of symptomatic UTI for this study was 0.15 cases per 100 patient-days of colonization, compared to the reported rate of 2.72 cases per 100 patient-days, which suggested that this organism may have a protective function in patients who used the E. coli colonized catheters.100 However, a larger study group is needed to test the safety and efficacy of E. coli HU2117 coated catheters in highly problematic populations.
Creating dual-functionality hemocompatible surfaces
The ultimate truly hemocompatible polymer for blood-contacting biomedical devices should have both antithrombotic and antibacterial functionalities. Many dual-functional materials have been investigated, including zwitterion-based surfaces, submicron-patterned surfaces, and surfaces with multiple functional moieties, etc.Zwitterionic polymers are polymers with equimolar number of homogeneously distributed anionic and cationic groups on their polymer chains, that form a hydration layer on the surface of the material through electrostatic interaction and therefore resists plasma protein and bacterial cell adhesion.64,101,102 An alternative strategy towards creating hemocompatible surfaces is to change the polymer's surface topography, thereby mediating subsequent biological responses. Creating a submicron-textured surface can dramatically reduce the accessible contact area for platelets or bacteria to interact with the surface, thereby minimizing the opportunities for bacteria and platelet adsorption.103–105 Polymers that combine multiple functional groups on one surface have also been tested, such as combining a synthetic heparin-mimetic polymer or hydrophilic polymer brushes (e.g., PEG) with antibacterial quaternary compounds (QAC) or Ag nanoparticles.106–108
Despite some successes in the research laboratory with the methods reported above, newer approaches to reduce the possibility of thrombus and/or infection on indwelling device surfaces are still in great demand within the medical community. Therefore, the main focus of the remaining content of this review is the utilization of polymer-based NO delivery to prepare dual functioning antithrombotic and antibacterial surfaces for biomedical applications.
Nitric oxide (NO) to the rescue
Nitric oxide (NO), a diatomic free radical, was identified as the endothelium-derived relaxation factor (EDRF) in the Nobel Prize-winning discovery by Ignarro, Furchgott, and Murad in 1987.20,109–114 Many researchers have later unveiled NO's various physiological functions in the human body, including preventing platelet activation and adhesion, inhibiting bacterial proliferation and biofilm formation, enhancing endothelization, signaling in the immune system's response, and promoting angiogenesis and the wound healing process.113,115–118The human body synthesizes a large quantity of bio-regulatory NO from the substrate L-arginine via three distinct isoforms of nitric oxide synthase (NOS), including endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS).119–121 The NO produced by eNOS contributes to the thromboresistant properties of the endothelial lining of blood vessels by inhibiting platelet activation. The activation of platelets is mediated by NO through the soluble guanylate cyclase (sGC) pathway. NO binds to the heme iron moiety of sGC and subsequently increases intracellular concentrations of cyclic guanosine monophosphate (cGMP).122,123 In addition, the cGMP increases cyclic adenosine monophosphate (cAMP) levels indirectly through phosphodiesterase III, which will also decrease the intracellular calcium concentration. The NO activated sGC ultimately results in reduced intracellular calcium, inhibition of platelet phosphoinositide 3-kinase (PI3), and reduced affinity as well as number of surface membrane fibrinogen binding sites on platelets (GPIIb/IIIa).118,122–127 It has also been suggested that many NO-related species are essential for NO's potent antimicrobial effects. First, NO can react with superoxide (O2−)128 and form peroxynitrite (OONO−), which is a lethal oxidizing agent and can induce oxidative stress, nitrosate amino acids and thereby alter protein functionality, oxidize and break DNA strands, and cause cell membrane damage to the bacteria it comes in contact with via lipid peroxidation.20,128–130 A second possible route reported for NO mediated cytotoxicity relies upon the formation of S-nitrosothiols (RSNO) in which after oxidation of NO to N2O3 the N2O3 can react with sulfhydryl groups on cysteine residues of membrane proteins to create RSNO structures that alter protein functionality, leading to cell stasis or cell death.109 Indeed, NO released or generated from polymer matrices has been shown to have similar antiplatelet and antimicrobial effects, as further described in detail below.
Some surgery interventions or trauma that destroys the endothelial lining, as well as certain vascular diseases, will lead to an increased level of superoxide anions and that react with NO and reduce its bioavailablity.120,131 Reduced NO levels are associated with many health complications and/or diseases, such as high blood pressure, deep vein thrombosis, intimal hypertension, restenosis, endothelial dysfunction, and prolonged wound healing times in diabetic patients, etc.1,132,133 There are three main strategies for increasing NO bioavailability: (1) by participating in physical exercise and controlling certain dietary components (e.g., nitrate-rich diet);133,134 (2) by administrating drugs that alter the enzymatic production of NO through nitric oxide synthases (NOSs) and increase the biosynthesis of NO endogenously;118 and (3) by using modified polymeric materials that can actively deliver NO exogenously to the sites of interest.3,110,111 Various NO-related drugs have been used in the clinical settings for many years: nitroglycerin (converted to NO by enzymes) for chest pain, sodium nitroprusside for controlling blood pressure, and molsidomine for pulmonary hypertension.132 This has triggered substantial interest in developing polymers that can be functionalized as an artificial endothelium-like surface to therapeutically deliver NO locally at the polymer/blood interface.
Since NO is highly active in vivo (with a short-half life on the order of seconds due to its rapid reaction with oxyhemoglobin, oxygen, and thiols, etc.), many NO donors have been synthesized and used for achieving the goal of prolonged and controlled NO delivery. Some examples include organic nitrates or nitrate esters (e.g., nitroglycerin or glyceryl trinitrate (GTN) and pentaerythrityl tetranitrate (PETN)), metal-NO complexes (e.g., sodium nitroprusside), nitrite, N-diazeniumdiolates (NONOate), and S-nitrosothiols (RSNO),135–141 (see Fig. 4). Nitroglycerin, the most commonly used organic nitrate for clinically treating hypertension and angina pain, is known to release 1 mole equivalent of NO upon bioactivation by mitochondrial aldehyde dehydrogenase (mtALDH).112 However, patients often develop nitrate tolerance (tachyphylaxis) after prolonged use because the reactive oxygen species generated by nitroglycerin can oxidize the thiol group of the mtALDH and result in enzyme dysfunction.112,142–144 This low bioavailability limits nitroglycerin's use as an efficient NO donor and complicates clinical treatments.
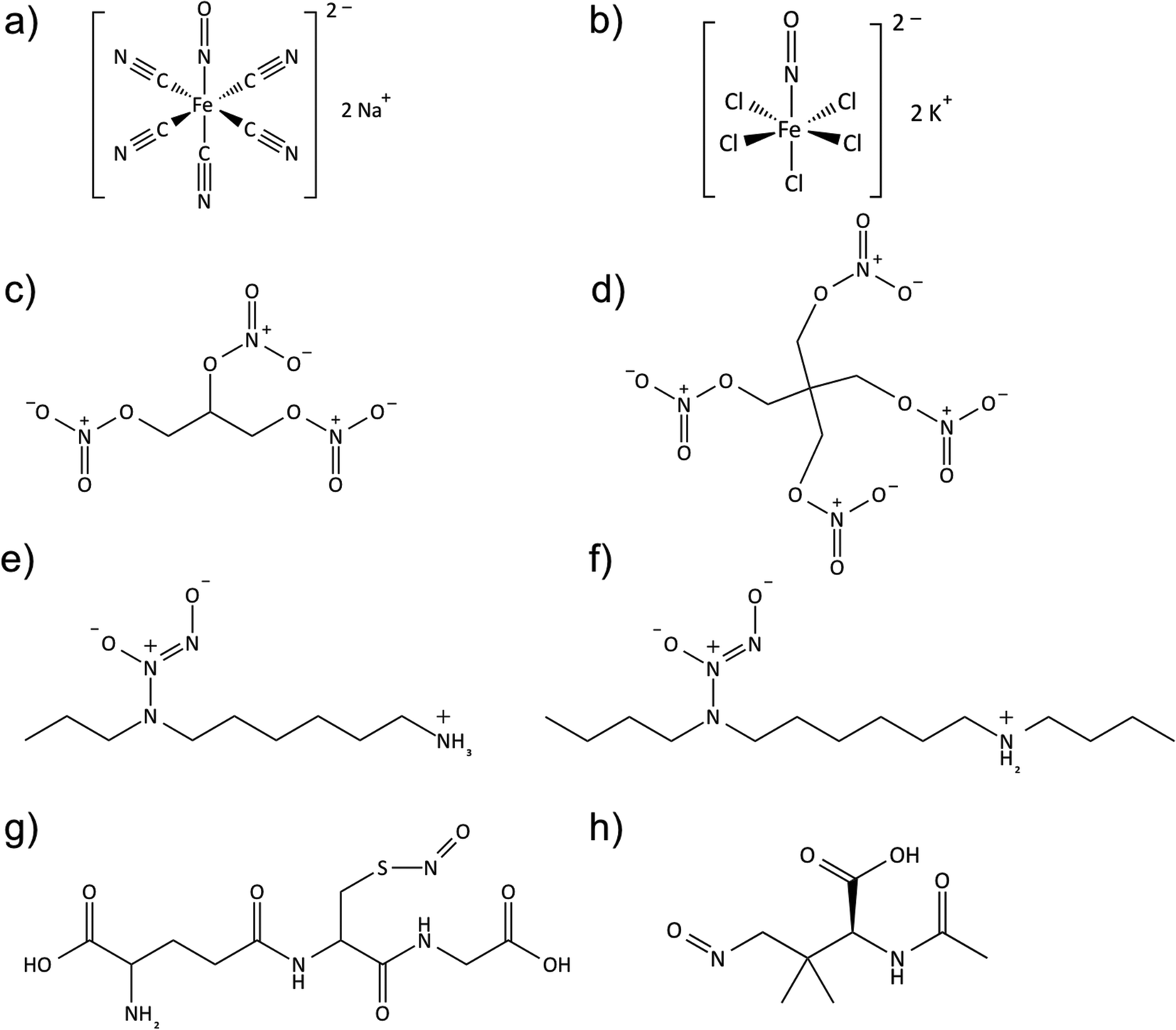 | ||
| Fig. 4 Structures of commonly studied NO donors in biomedical applications, (a) sodium nitroprusside; (b) potassium nitrosylpentachlororuthenate; (c) nitroglycerin; (d) pentaerythrityl tetranitrate; (e) diazeniumdiolated N-(6-aminohexyl)aminopropane; (f) diazeniumdiolated N,N′-dibutylhexamethylenediamine; (g) S-nitrosoglutathione; (h) S-nitroso-N-acetylpenicillamine. | ||
Metal NO complexes (metal nitrosyls) represent another class of NO donors used in biological testing. Many different metal NO complexes have been reported, including manganese,109,137 iron145 and ruthenium–NO109,112,137 complexes, with sodium nitroprusside (Na2[Fe(CN)5NO], SNP) being the most common. SNP is often used as a potent vasodilator in hypertensive emergencies,145 and releases NO in the presence of reducing agents (e.g., thiol-containing compounds such as cysteine or glutathione) or by illumination with near-infrared or visible light.112,121,145 However, cellular toxicity concerns due to the release of cyanide and cytotoxic peroxynitrite as byproducts have made metal nitrosyl complexes less attractive as medicinally used NO donors.112,141
Reduction of nitrite can generate NO via both enzymatic (nitrite reductase, xanthine oxidoreductase, etc.) and non-enzymatic (gastrointestinal acid, ascorbate, myoglobin, etc.) pathways.133 Recently, it was demonstrated that nitrite can also be used to generate NO via electrochemical reduction at an electrode surface with either Cu(I) ion generated from oxidation of Cu0 (ref. 146 and 147) or via the use of Cu(II)–ligand complexes that mimic the active site of nitrite reductase (e.g., Cu(II)-tri(2-pyridylmethyl)amine, Cu(II)TPMA).148
Many researchers over the past two decades have focused on using N-diazeniumdiolates (NONOates) and S-nitrosothiols (RSNOs) as leading candidates for controlled NO delivery due to their relatively high stability, their ability to spontaneously release NO under physiological conditions (e.g., no enzyme required) in a predictable manner, and their higher bioavailability in vivo.3,121,136 Furthermore, the tissue and metabolite independent release also avoids the build-up of tolerance over time, which makes these agents more suitable for biomedical applications.
N-Diazeniumdiolates (1-amino-substituted diazen-1-ium-1,2-diolate) are a class of NO donor molecules formed by reactions between primary or secondary amines with NO under high pressure (e.g., 5 atm) in the presence of base (either an unreacted amine substrate or an added metal alkoxide base) at low temperatures.1,137 These species are able to generate two mole equivalents of NO per mole of donor, via a proton-driven reaction (hydrolysis), in the physiological environment such as when the compound is exposed to blood or tissue fluids (see Fig. 5). NONOates can also release NO via thermal, photochemical or enzymatically (e.g., esterase) reactions.137,149
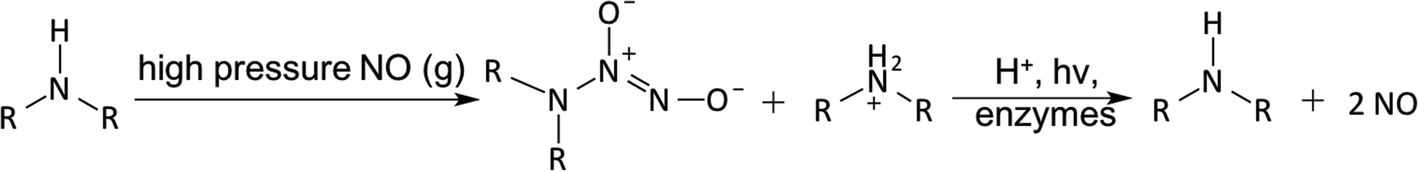 | ||
| Fig. 5 N-Diazeniumdiolates (NONOates) formation and decomposition. | ||
The half-lives of the synthesized NONOates strongly depend on the structure of the amine precursors and hydrogen bonding stabilization from additional amines within the molecule. It has also been reported that hydroxyl groups within the donor molecule, or the matrix it is within, can also contribute to the hydrogen bonding with the diazeniumdiolates.150,151 For example, half-lives can range from the shortest reported t1/2 of 1.8 s for the diazeniumdiolated amino acid proline (PROLI/NO) to t1/2 of 20 h for diethylenetriamine (DETA/NO).136,137 One drawback of the diazeniumdiolate compounds is that they may potentially form carcinogenic nitrosamines.152,153 Batchelor et al. synthesized more lipophilic NONOates for use in reducing the NO donor leaching into blood from polymers that had been doped with such NO donors.154 Since the NO release process produces a lipophilic amine byproduct that increases the pH of organic phase, many additive compounds (such as tetraphenylborate or other borate derivatives154) have been included within the organic polymeric phase to serve as counter ions for organic ammonium cations (when amines form after NO release and proton is extracted into the polymer to create the ammonium species) and thereby partly prevent a pH increase within the organic polymer phase. This can greatly prolong the NO release lifetime from such NO donors when incorporated into biomedical polymers. Unfortunately, the borate additives are not ideal because of their inability to extend the NO release to more than a few days and their cytotoxicity toward endothelial cells.155,156 Therefore, Handa et al.155,156 as well as Cai et al.157 studied various poly(lactic-co-glycolic acid) (PLGA) additives with different hydrolysis rates as a means to slowly produce protons within the organic polymer phase that can continue to drive NO release reaction from NONOates. PLGA is a biocompatible and biodegradable polymer, with tunable mechanical properties and wide range of erosion times. Further, it is already approved for use by the FDA for the development of devices for controlled delivery of small molecule drugs, proteins and other macromolecules.158 These studies demonstrated that the presence of PLGAs in the base polymer containing diazeniumdiolate species can extend NO release under physiological conditions for up to at least 2 weeks.
S-Nitrosothiols (RSNO) represent an endogenous class of NO donors and natural transporters of NO within tissues and blood.121,159 Such molecules include S-nitrosoalbumin (SNO-Alb), S-nitrosohemoglobin (SNO-Hb), S-nitrosocysteine (SNO-Cys) and S-nitrosoglutathione (GSNO).160 RSNOs can be synthesized via the thiol nitrosation reaction (such as nitrous acid and alkyl nitrite, etc. in an acidic environment).141 Owing to the natural occurrence of RSNOs in vivo, these molecules pose a relatively low risk of toxicity to cells/tissues in comparison to NONOates. The transnitrosation reaction transfers the NO+ functional group from an RSNO species to another existing free thiol, thus achieving a circulating unlimited supply of NO in vivo.161
RSNOs have characteristic UV-Vis spectra. In general, they appear green for tertiary RSNOs (such as S-nitroso-N-acetylpenicillamine, SNAP) or pink for primary and secondary RSNOs (such as SNO-Cys, S-nitroso-N-acetylcysteine (SNAC) and GSNO). There are two primary bands in the UV-Vis spectra. The strong band in the UV region is between 330 and 350 nm (ε ∼ 103 M−1 cm−1), which is attributed by the n0 → π* transition.141,162–164 A weak band in the visible region is between 550 to 600 nm (ε ∼ 20 M−1 cm−1), which is attributed to the nN → π* transition.141 It is known that RSNOs can release NO via multiple pathways.164–169 Thermal or photo-initiated decomposition will lead to homolytic cleavage of the S–N bond, and form thiyl and NO radicals, where thiyl radical (RS˙) will react with another RSNO and generate disulfide (RSSR) and another NO radical. Metal ions (such as copper or ferrous ions)170,171 or organoselenium compounds can catalyze RSNO decomposition.115,172–174 It is reported that Cu+ (generated from reduction of Cu2+ by trace amounts of thiol) can react with RSNO and form the corresponding thiolate and Cu2+, which then react to regenerate Cu+ and a disulfide to continue the catalytic cascade. RSNOs can also react with ascorbate to generate NO via two pathways. In one, ascorbate (at low concentration) acts as a reducing agent to generate Cu+ from trace copper ion impurities in solution, or at high concentration ascorbate can act as a nucleophile, attacking the nitroso group to generate NO, a thiolate and dehydroascorbate.8,175,176 Further, certain enzymes such as copper-containing superoxide dismutase (CuZn-SOD)141,177 or the selenium-containing enzyme glutathione peroxidase (GPx)141,172,174 can also convert RSNO to NO in the presence of a reducing agent, such as glutathione (GSH) (see Fig. 6).
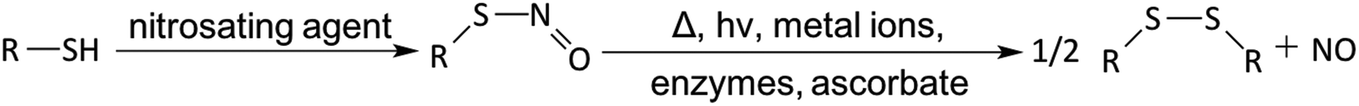 | ||
| Fig. 6 S-Nitrosothiol (RSNO) formation and decomposition. | ||
GSNO and SNAP are two commonly used RSNOs that have been intensively studied in a variety of biomedical applications. GSNO is present in blood endogenously, which makes it innately more biocompatible and attractive for many applications, such as promoting wound healing process in mice or rats.178–181 The precursor for SNAP synthesis is N-acetylpenicillamine (NAP), whose ultimate hydrolysis product, penicillamine, is already a FDA approved chelator for treating heavy metal poisoning, such as Wilson's disease.182–185 SNAP is reportedly one of the most stable NO donors available, due to its intermolecular hydrogen bonding,163,186 and has already been shown to be a very promising candidate for fabricating long-term NO releasing polymeric materials.162,163,187 Lipophilic analogs of SNAP (such as N-substituted derivatives of SNAPs) have also been developed by addition of bulky side-chains to N-acetylpenicillamine and preventing Cu+ catalysis through steric hindrance.188,189 Other lipophilic RSNOs (e.g., S-nitroso-tert-dodecyl mercaptan (SNTDM), logP = 5.3)190 (note: P = partition coefficient of a molecule between octanol and water) with a higher logP value than SNAP (logP = 0.4)163 have been synthesized for reducing the NO donor leaching from the hydrophobic polymer phase.
Polymer-based strategies for NO delivery
For the past two decades, many research groups have focused on developing techniques to deliver NO locally, continuously and efficiently from polymeric devices where blood clotting and/or bacteria infection are major complications. To improve the NO payload and stability, achieve targeted NO delivery through multi-functionalization and elongate NO releasing lifetime, many different scaffolds have been used as NO-releasing or NO-generating vehicles. These include micelles,139,191,192 microbubbles,193 proteins,194–198 liposomes,127,199 inorganic nanoparticles (such as silica,71,200–204 gold205,206), microparticles,199,207 zeolites,208,209 metal–organic frameworks (MOFs),210–213 dendrimers,72,214,215 xerogels,216–218 electrospun fibers,89,101,219 natural polymers (chitosan,86–88,220–222 gelatin,223etc.), and other organic polymers (polymethacrylate,224 polyester,225,226 polydimethylsiloxane (PDMS),227 polysaccharides,228,229 hydrogel,179,180,230,231 PVA,49,232,233 polyurethanes,162,163,234,235 and PVC155). The approaches and benefits of different NO-delivery materials have been highlighted in numerous reviews.1,112,121,132,136–139,236 Here, an overview of the most promising polymer-based NO delivery strategies are discussed, including covalently bound NO donors within/on a polymer matrix, physically dispersed NO donors within a polymer matrix, and catalytically generated NO from various RSNO reservoirs in the body (see Fig. 7), and their representative examples in antithrombotic and antibacterial applications are described.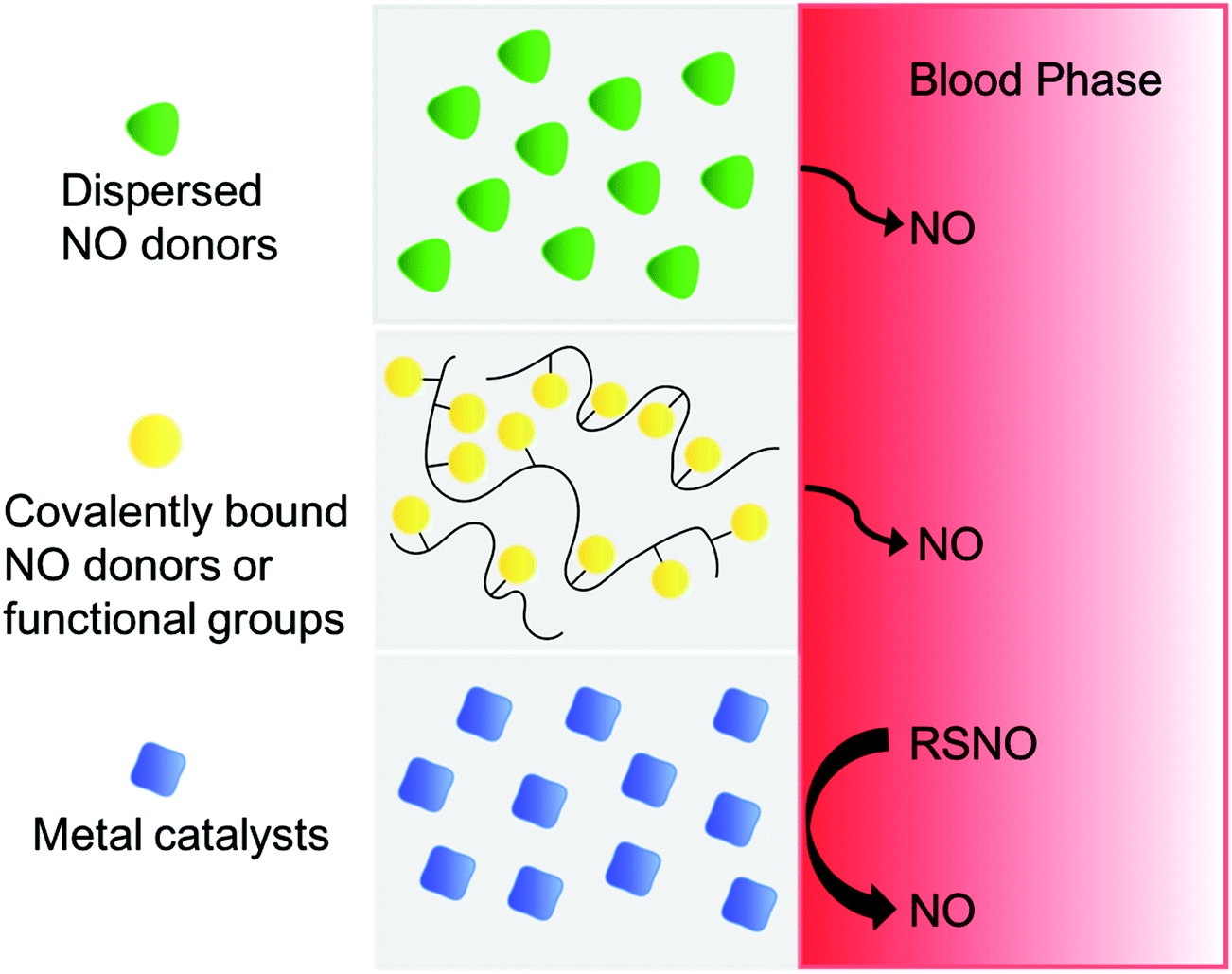 | ||
| Fig. 7 Three different strategies to fabricate of polymer surfaces that release or generate NO, including physical dispersion of NO donor into polymeric matrix, covalently bound NO donor functionalities onto polymer backbone, or NO generation from endogenous RSNO species in blood by metal catalysts embedded in or covalently bound to the surface of the polymer. | ||
Covalently bound NO releasing polymers
N-Diazeniumdiolates are one of the most studied NO donors, and many polymers with secondary amine groups have been chemically modified to create the NONOate moiety.110,111,137 Meyerhoff and coworkers were among the first to link these NO donors to particle fillers (such as fumed silica particles237) or to the polymer backbone of polymers such as PVC,34,238 polyurethane (PU),34,111,234 and silicone rubber (SR).239 Zhang et al. synthesized NO-releasing fumed silica (FS) particles (0.2–0.3 μm) by tethering alkylamines onto the surface of the FS particles using amine-containing silylation reagents (coupling efficiency 50–70%), and then converted them to corresponding NONOate groups with final NO loading of ca. 0.6 μmol per mg of particle.237 The half-lives of the particles are significantly longer than the solution phase NO donor analogs, owing to the fact that a large quantity of amines present at the particle surface can increase the local pH, which then reduces the NO release rate. Of note, when local pH decreases significantly, it results in partial release of the stored NO due to the proton-driven NO release mechanism. Therefore, some methods utilize additional acid-generating compounds to facilitate continual release of the entire NO payload.Keefer and coworkers were one of the earliest groups to covalently attach a diazeniumdiolate species to the backbone of poly(ethylenimine) (PEI) to form PEI/NONOate, which later was used to provide a flexible NO release coating for PTFE vascular grafts.240 The NO release from the sample was at a relatively constant rate (0.5 pmol min−1 mg−1) for 5 weeks in PBS buffer at 37 °C. In subsequent studies, Saavedra et al. prepared methoxymethyl-protected diazeniumdiolated piperazine PVC;238 however, the NO release rate was slow due to the fact that the NO could only be released from the NONOate group after the hydrolysis of the methoxymethyl protecting group.34 In addition, this PVC film can concurrently release toxic methanol and formaldehyde as byproducts of the methoxymethyl-protecting group, which is undesirable for use as materials in biomedical applications. Zhang et al. later incorporated diazeniumdiolated functionalities into more biocompatible silicone rubber.239 Briefly, a diaminoalkyltrimethoxysilane crosslinking agent (DACA) was reacted with the terminal hydroxyl groups of the PDMS, and then the aminated PDMS macromolecules were cross-linked to each other (condensation-curing) in the presence of ambient moisture before forming the NONOates species at the amine sites (via reaction of the polymer with NO(g) at high pressure). Likewise, N-diazeniumdiolated NO donors have been covalently linked to polyester,241 polymethacrylate,224 polyurethane (PU),35,111,234,235 PVA233 and other hydrogels242 as well. The synthetic methods are somewhat similar to that described above, with the exception of one type of PU containing a diamine chain extender, where sodium salts (NaX, where X is an anion) are incorporated into the reaction mixture during the NO addition process to facilitate the formation of anionic diazeniumdiolates.111
Dendrimers are another class of popular NO delivery vehicles due to their ability to store a large reservoir of NO, have tunable sizes, and to be multi-functionalized for targeted delivery.215,243 Stasko et al. were the first to modify generation 3 and 5 polypropylenimine dendrimers at their exterior with different amine functionalities that were then converted to diazeniumdiolated dendrimers via reaction with NO(g) at high pressure. The secondary amine/NONOates yield the highest storage capacity for NO, with up to 5.6 μmol NO per mg of polymer, and a NO release duration of >16 h under physiological conditions.243
Frost et al. was the first to modify FS particles (7–10 nm in diameter) with an RSNO functionality,200,201 such as S-nitrosocysteine-FS (SNO-Cys-FS), S-nitroso-N-acetylcysteine-FS (SNAC-FS) and S-nitroso-N-acetylpenicillamine-FS (SNAP-FS). This was accomplished, by tethering the thiol containing amino acids or N-acetylpenicillamine thiolactone onto aminopropyl-FS. In these efforts, a total NO loading of up to 1.38 ± 0.29 × 10−7 mol NO per mg particles was achieved. The SNAC-FS and SNAP-FS particles can be blended into polyurethane (PU) or trilayer silicone rubber (SR) matrices to create films that release NO at different rates by tuning the chemical identity, water uptake, or thickness of the PU polymer.201 However, the SR films containing SNAC-FS or SNAP-FS in the middle layer do not release NO upon exposure to copper ions or ascorbate in PBS solution because the very high hydrophobicity of the SR blocks the contact between the buffer and the RSNOs.200 However, such films are able to release NO at levels proportional to the intensity of visible light that is allowed to shine on the polymer, providing the first hydrophobic materials that have an external on/off trigger that precisely controls the rate of NO release via a photodecomposition reaction. The authors determined that 590 nm light is primarily responsible for the release of NO from the SNAP-FS-SR films. Evidence suggested that 67% of the photoinitiated NO was accounted for by the longer wavelength (590 nm) and 33% by shorter wavelengths (centered around 340 nm). SNAP has also been covalently attached to PDMS,227 gelatin223 and a macrocycle (e.g., cyclam).244 The ability to generate programmable sequences of NO flux from these light-sensitive materials can offer precise spatial and temporal control of the NO release and potentially provides a platform to systematically study, at a fundamental level, the in vivo physiological response to implanted devices.227 Of note, SNAP-cyclam is capable of releasing physiological relevant levels of NO for up to 3 months in vitro when blended into poly(L-lactic acid) thin films and irradiated by a 385 nm LED, and this represents the longest NO release from SNAP-based polymer films reported to date.244
RSNOs have also been used to form functionalized dendrimers214 or hyperbranched polymers (e.g., hyperbranched polyamidoamine (HPAMAM) or hyperbranched polyethers241) to achieve a high NO payload. Stasko et al. functionalized generation 4 polyamidoamine (PAMAM) dendrimers with N-acetylpenicillamine or N-acetylcysteine and both macromolecular scaffolds have an NO storage of ca. 2 μmol NO per mg polymer after the required nitrosation raction.214
Another category of NO delivering materials with large NO storage capabilities are xerogel films. Riccio et al. designed thiol-modified xerogels derived from 3-mercaptopropyltrimethoxysilane (MPTMS) and methyltrimethoxysilane (MTMS).217 The subsequent thiol nitrosation generates xerogels with NO loadings up to 1.31 ± 0.07 μmol NO mg−1, and films of these materials are capable of releasing NO for up to 2 weeks under physiological conditions. Similar to N-diazeniumdiolates, many other polymer modification studies have been conducted using S-nitrosothiols (RSNOs) as well, including those utilizing PDMS,227 PVA hydrogel,180 polyester and poly(methylmethacrylate),225 as the base polymer materials.
Lastly, several biodegradable polymers, such as citrate-based226 or saccharide-based polymers (e.g., dextran228,245 and chitosan87,88,221) provide very attractive scaffolds for NO delivery because of their natural occurrence, biodegradability, tolerability to mammalian cells and accessibility for NO donor functionalization reactions.221,245,246 Reynolds and coworkers synthesized two S-nitrosated dextran thiomers (dextran-cystamine and dextran-cysteine), with up to 0.205 ± 0.003 μmol per mg NO loading and the capability to release NO for 6 h in PBS buffer at 37 °C. It has been further demonstrated that these dextran-based materials are susceptible to enzymatic degradation by dextranase, and the degradation of dextran derivatives is slower and partial when compared to the unmodified dextran because of the chemical modification of the dextran backbone.245
Physical encapsulation-based NO releasing polymers
Physical encapsulation methods for preparing NO releasing polymers are based on embedding the NO donor within a polymer matrix physically without chemical bonding. This provides a much less complicated method to prepare NO release polymers than covalent attaching NO donors to particle fillers or the polymer backbones. This approach is a more attractive strategy for future commercialization if stable long-term NO release can be achieved by dispersing the NO donor within a polymer matrix. Toward this goal, polymer193 or phospholipid247 shelled microbubbles have been developed in recent years to create a hydrophobic microenvironment for NO transportation and therapeutic delivery. Paradossi and co-workers prepared NO-loaded PVA-shelled microbubbles (4.6 μm ± 0.4 μm diameter and 0.4 μm shell thickness) for treating acute vascular disease, such as thrombosis.193 The microbubbles were loaded with NO by exposing to gas phase NO at 1.5 bar for 2 h. The NO encapsulation is about 1% of the NO present in the reaction vessel during the loading procedure, with the final content being 3.6 μmol per mg microbubbles. The NO release measured by the Griess assay showed that ca. 90% of the NO is released during a 50 min period in phosphate buffer saline solution. Ideally, the NO delivery should be controlled and selective bursts at a specific time and place, but the release can also be spontaneous and nonspecific.193 Unfortunately, the in vitro stability of such NO-loaded microbubbles is relatively low.247Gas storage in porous materials, such as zeolites, is also another attractive method for medicinal applications. Wheatley et al. have used cobalt-exchanged zeolite-A to absorb and store NO (1.2–1.3 μmol NO per mg zeolite). The NO release was triggered by contacting with water at physiological temperature and pH, replacing the gaseous NO with cobalt–water interactions. These NO-zeolite samples were also blended with powdered PTFE or PDMS to increase their mechanical stability and then pressed as disks for in vitro testing. The t1/2 values of NO release from zeolite polymer disks was significantly increased (from 340 s for zeolite powder to 509 s for PTFE disk and 3076 s for PDMS disk).208 The results obtained in a platelet aggregometry assay also suggested that the NO-zeolites greatly inhibits platelet aggregation in vitro.
Liposomes are also a popular choice to protect and deliver gaseous NO. McPherson and coworkers prepared liposomes encapsulated with NO and argon gas (NO/Ar-ELIP) by a pressured-freeze and thawing method. They used these capsules for attenuating intimal hyperplasia and reducing arterial wall thickness.199 Other researchers have conducted further studies to encapsulate NO donors within pH127 or thermo-sensitive248 liposomes to achieve controlled release. In short, when N-diazeniumdiolates are encapsulated within liposomes, pH or temperature fluctuations change the proton influx through the biolayer membrane and subsequently induce a significant NO release. Koehler et al. have also encapsulated SNAP within liposome vesicles and then doped the resulting vesicles within xerogels for photosensitive NO release.249
Biodegradable poly(ethylene oxide-co-lactic acid) (PELA)250 and poly(lactic-co-glycolic acid) (PLGA)207,220,250 have also been shown to be suitable vehicles for encapsulating NO donors to prolong the NO release. Yoo and coworkers developed PLGA (5050DLG5E) nanoparticles (NO/PPNPs) with diazeniumdiolated polyethylenimine (PEI) for long term NO release for antimicrobial applications.220 The average size of the NO/PPNPs was 162 ± 19 nm as confirmed by scanning electron microscopy (SEM), with a NO release capacity of 1.4 μmol NO per mg polymer. The incorporation of PEI/NONOate into hydrophobic PLGA nanoparticle matrix successfully restricts the spontaneous degradation of the NONOate group and extends the NO release lifetime from 12 h to over 6 d.220 More recently, Lautner et al. demonstrated the encapsulation of SNAP into PLGA (RG 503H and RG 504) microspheres (20–125 μm in diameter) using a solid-oil-in water (S/O/W) method251 and achieved up to 4 weeks of controlled release of SNAP from PLGA RG 504, which then generates localized NO in the presence of copper(II) and ascorbate.207
Mowery et al.34 prepared diazeniumdiolated N,N-dimethyl-1,6-hexanediamine (DMHD) or linear PEI and dispersed them into plasticized PVC or PU. Unfortunately, NO donor leaching was observed by HPLC and this raises potential concerns regarding toxicity if nitrosamine formation occurs, and such nitrosamines are also leached into the surrounding aqueous environment. Bachelor et al.154 synthesized more lipophilic dialkylhexamethylenediamine-based NONOates with different alkyl groups (from methyl to didodecyl) which can enhance the retention of potential byproducts within the hydrophobic PVC films and thereby reduce their toxicity threat to biological systems.
Seabra et al. incorporated GSNO and SNAC into Synperonic F-127 gel using a ‘cold method’252 and formed a hydrogel that contained 0.3 mol of GSNO or 0.6 mol of SNAC per gram of hydrogel.252 Kinetic studies showed that SNAC releases NO thermally 3.6 times faster than GSNO (11 ± 0.4 min−1vs. 3.1 ± 0.8 min−1); however, approximately 50% of both NO donors had decomposed after only 3 h.232 Kim et al. developed NO-releasing films by dispersing GSNO into biopolymer chitosan, where 75% GSNO remains after storing at room temperature for 4 weeks. The 20 wt% GSNO/chitosan film released NO over 48 h in PBS buffer at 37 °C.222 Recently, Brisbois et al. discovered that SNAP is exceptionally stable when doped within several low water uptake biomedical grade polymers, such as Elast-eon E2As and CarboSil, and can release NO for up to 3 weeks at physiological conditions.156,162 Wo et al. later demonstrated that the SNAP in CarboSil mainly exists in crystalline form when the SNAP concentration exceeds its solubility in the polymer. Ultimately, a slow crystal dissolution process leads to its long-term NO release under physiological conditions.163 Additionally, this crystal-polymer composite is very stable during storage for at least 8 months, not only because of the hydrophobicity of the CarboSil polymer that limits water diffusion primarily to only the polymer surface, but also because the crystalline SNAP is stabilized by intermolecular hydrogen bonds.163,253
In order to apply NO release to any pre-made or off-the-shelf biomedical devices, Colletta et al.19 developed a simple solvent impregnation method to load SNAP into commercially available silicone Foley urinary tract catheters at room temperature. This impregnation process takes place under very mild conditions, which is beneficial because, like many NO donors, SNAP or its analogs are sensitive to high temperatures used during industrial catheter extrusion processes. In this approach, SNAP or a SNAP analog is dissolved in an organic solvent that can swell the polymer to a great extent without dissolving it, and as the polymer uptakes the solvent, the NO donors are loaded into the polymers. After drying to remove the solvent, the resulting polymer contains a stabilized form of the SNAP or analog of SNAP. As an initial example, commercial silicone Foley catheters (i.d. of 0.30 cm and o.d. of 0.59 cm) were swelled in a SNAP solution prepared in tetrahydrofuran (THF) (125 mg mL−1) for 24 h, resulting in SNAP impregnation of 5.43 ± 0.15 wt% SNAP in the final dried catheter. This level of SNAP loading enabled the catheters to achieve stable NO release above physiological levels for >4 weeks.19
Catalysis-based NO generating polymers
Instead of incorporating NO donors or NO functionalities into polymers, many efforts have been devoted to generate NO from endogenous RNSOs or infusion of RSNO solutions through thiol transnitrosations,161,254 and subsequent catalytic reactions using immobilized copper or selenium-based species (e.g., copper nanoparticles,255 copper(II)-cyclen,171,256 copper-complexes,257,258 copper-containing metal–organic frameworks (MOFs),210–213 and organoselenium species115,172–174). Meyerhoff and coworkers were the first to covalently attach copper(II)-cyclen onto commercial biomedical grade polyurethane, which can generate physiological levels of NO (1–2 × 10−10 mol cm−2 min−1) when in contact with endogenous RSNO and RSH species, such as 10 μM GSNO/GSH in PBS buffer containing 3 μM EDTA.171 This polymer has also been used as an outer coating at the distal end of an amperometric NO sensor that can generate electrochemical response toward the RSNO species in whole blood.171 However, it was reported that 50% of copper(II) ion leached out after soaking in GSNO/GSH containing PBS buffer at RT for 7 d, which may limit the use of such polymers for long-term applications and generate cytotoxicity concerns.Reynolds and coworkers are developing many MOF-NO generating catalysts with accessible catalytic sites and that have resistance to degradation, including Cu3(BTC)2 (BTC: 1,3,5-benzenetricarboxylate), Cu(II)1,3,5-benzene-tris-triazole (CuBTTri) and Cu(II)1,3,5-tricarboxylate (CuBTC).211–213 It has been reported that CuBTC can be extruded within Tecoflex SG80A polyurethane into single lumen tubing while maintaining the catalyst structure and functionality. The in vitro NO release generated from endogenous RSNO species ranges from 1 h to 16 h with tunable dosage, depending on the specific RSNO levels present in the soaking solution.211
As previously mentioned, Meyerhoff and coworkers have also demonstrated an electrochemical NO generation via a Cu+ ion mediated reduction reaction of inorganic nitrite. The Cu+ ion is generated from oxidation of a copper wire146,147 or the reduction of copper(II)-tri(2-pyridylmethyl)amine (Cu(II)TPMA) complex.148 The electrochemically generated NO can be modulated by changing the potentials applied to the electrodes and can achieve fluxes between 0.5 and 3.5 × 10−10 mol cm−2 min−1 (for catheter applications), which is in the range of physiological relevant levels of NO released by the endothelial cells. This approach has been utilized to develop a new generation of multi-lumen catheters in which one lumen is dedicated to generating NO electrochemically to reduce thrombus and microbial biofilm formation on the surface of the catheters.148,259
Applications of NO releasing/generating polymers for preparing antithrombotic and antibacterial biomedical devices
Inhibition of thrombosis formation
In a healthy vasculature, endothelial NOS (eNOS) in the endothelial cells that line the inner wall of blood vessels produce NO with a surface flux of 0.5–4.0 × 10−10 mol cm−2 min−1 to prevent platelet activation and thereby control the balance between thrombosis and hemorrhage.120 However, many procedures (such as placing stents, grafts, catheters, or other biomedical devices) disrupt the endothelial cells lining and destroy the delicate balance of vascular homeostasis.133 The tissue factor release and protein absorption trigger the coagulation cascade and the lack of NO production leads to platelet activation, aggregation, and ultimately thrombus formation. Therefore, NO and its unique antiplatelet/antithrombotic activity represents a very promising approach to prevent thrombus associated complications in many biomedical applications.Mowery et al. reported that PVC- or PU-based polymer films, prepared with NO releasing diazeniumdiolate functionality via either dispersion or covalent bonding of NO donors, can exhibit significant improvements in biocompatibility during in vitro platelet adhesion tests using platelet-rich plasma (PRP).34 Wu et al. prepared PVC films (with borate additives and di(2-ethylhexyl) ester (DOS)) mixed with 0.5, 1.0, 2.0, 4.0 wt% of diazeniumdiolated N,N′-dibutyl-1,6-hexanediamine (DBHD/N2O2) to determine the effect on platelet adhesion at the surface of polymeric films with various NO flux levels.260 A lactate dehydrogenase (LDH) assay was used to determine the amount of platelets adhered onto the polymer surface. LDH, which is normally stored within intact platelet, is released into the bulk solution when the platelets are lysed by surfactant and is a very useful for indication of the amount of cells adhered.260 Fewer platelets adhered on the polymer surfaces with higher NO flux levels, and NO successfully reduced the amount of platelets adhered on the polymer surfaces from 14.0 ± 2.1 × 105 cells per cm−2 on the controls to 2.96 ± 0.18 × 105 cells per cm−2 on the surfaces with highest NO release of 7.05 × 10−10 mol cm−2 min−1.
Thrombosis is also an important risk factor in any blood exposure to synthetic materials, such as grafts, stents, intravascular sensors, catheters, extracorporeal circuits (ECC) or hemodialysis membranes.120,121,261,262 Efforts to use NO release polymers for these applications are summarized below based on the type of biomedical device.
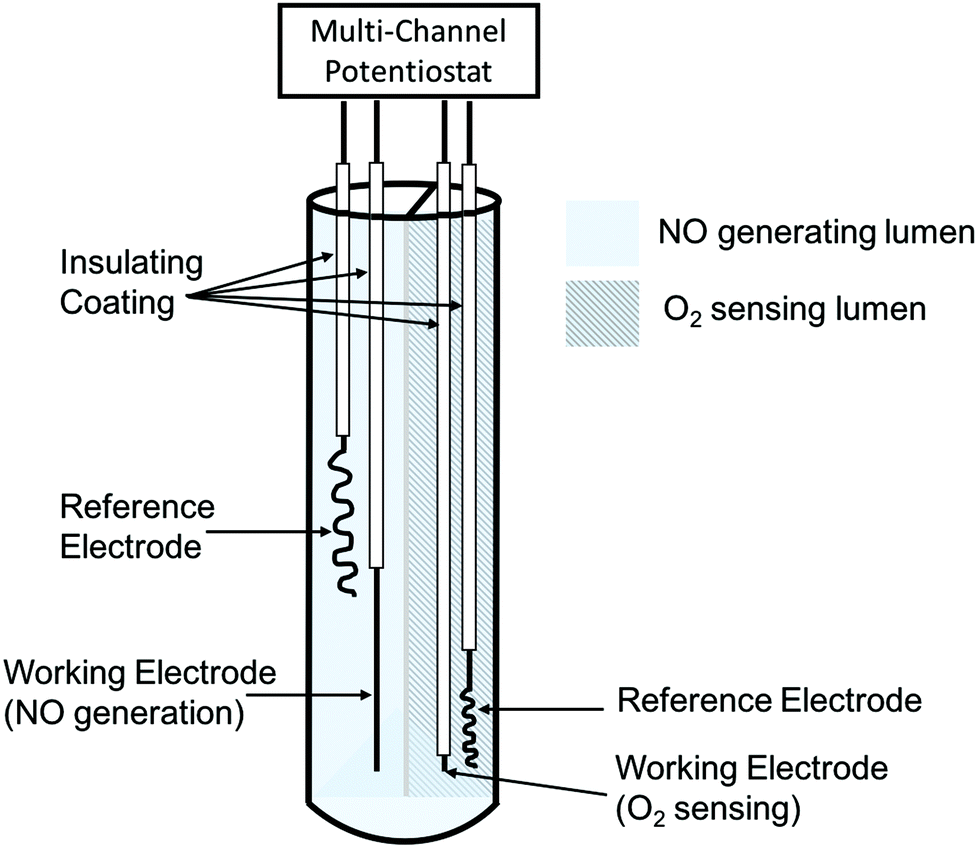 | ||
| Fig. 8 Simplified schematic of dual-lumen catheter-type oxygen sensor with electrochemical NO generation from nitrite solution via Cu(II)TPMA (figure not drawn to scale). | ||
Applying different potentials on the electrodes can modulate the rate of NO generation, which offers a steady, controllable, and physiologically relevant flux of NO, compared to the chemically-based NO generation. The performance of the sensor was evaluated in rabbit veins and pig arteries for 7 and 21 h, respectively. The sensors in the arteries were challenged with a wider range of oxygen levels by changing the pig's fraction of inspired oxygen between 100% and 21%.259 In both cases, the sensors with electrochemical NO generation provided very accurate oxygen responses, while control sensors deviated from the real values by 30–40% after 5 h of implantation because the local oxygen was consumed by the activated platelets and other cells trapped in the thrombus formed on the surface of the control sensors. Similar to oxygen sensors, NO releasing intravascular glucose,272–276 pH,276,277 and CO2276,278 sensors using diazeniumdiolated NO donors have also been tested, demonstrating much improved electrochemical responses over the corresponding control sensors.
| Summary | ECC coating material | NO donor, additives, etc. | NO level (×10−10 mol min−1 cm2) | Platelet count after 4 h (% of initial) | Ref | |
|---|---|---|---|---|---|---|
| NO release | Covalent attachment | Tygon tubings with Carbonthane 3573A coating | N-Diazeniumdiolated FS fillers | 4.1 | ca. 85 ± 15% (NO) vs. 58 ± 5% (PU) | 237 |
| Silicone rubber tubings | N-Diazeniumdiolated PDMS | >4.0 at 23 °C; >10.0 at 37 °C | 86 ± 24% (NO) vs. 65 ± 10% (SR) | 239 | ||
| Physical dispersion | Tygon tubings with PVC/DOS coating | DBHD/NONOate, borate | 12.5 | 79 ± 7% (NO) vs. 58 ± 7% (PVC/DOS) | 279 | |
| Tygon tubings with PVC/DOS coating | DBHD/NONOate, PLGA | >20 | 79 ± 11% (NO) vs. 54 ± 6% (PVC/DOS) | 155 | ||
| Tygon tubings with Elasteon-E2As coating | DBHD/NONOate, PLGA | 6 | 97 ± 10% (NO) vs. 58 ± 3% (E2As) | 156 | ||
| Tygon tubings with Elasteon-E2As coating | DBHD/NONOate, PLGA, argatroban | 6.5 | ca. 90% (NO/argatroban) vs. 58 ± 3% (E2As) | 43 | ||
| Tygon tubings with Elasteon-E2As coating | SNAP | 2 | 100 ± 7% (NO) vs. 60 ± 6% (E2As) | 162 | ||
| NO generation | Tygon tubings with Tecophilic SP-60D-60 coating containing 10 wt% Cu0 nanoparticle | Endogenous RSNO and/or infused SNAP | >10 (in presence of 1 μM GSNO, 30 μM GSH, 5 μM EDTA) | ca. 90% (10 wt% Cu0 and SNAP) vs. 75% (10 wt% Cu0 only) vs. 50% (SNAP only) | 255 | |
| Inherent polymer properties | Tygon tubings with Tecoflex SG 80A, Tecophilic SP-60D-60, PVC/DOS coating | N/A | N/A | 44 ± 4% (SG 80A) vs. 41 ± 5% (SP-60D-60) vs. 46 ± 3% (PVC/DOS) | 156 | |
Zhang et al. prepared PVC tubing with PU coating containing 20 wt% diazeniumdiolated FS particle fillers237 and SR tubing with covalently attached to DACA/N2O2239 as NO releasing polymer tubing circuits for 4 h ECC experiments in a rabbit model. Both types of NO release tubing exhibited less overall platelet adhesion and thrombus surface coverage compared to the controls. However, it still remains a challenge to achieve long-term NO release at physiologically relevant conditions, stable storage capability at room temperatures and high NO donor loading. Handa et al. recently prepared ECC tubing composed of 25 wt% DBHD/N2O2 and 10 wt% PLGA (5050DLG7E) additives in 2:1 PVC/DOS polymer matrix and achieved up to 14 d of NO release between 7–18 × 10−10 mol cm−2 min−1 at 37 °C.155 This circuit tubing successfully preserved the platelet count during 4 h of experiments, with 79 ± 11% vs. 54 ± 6% for the NO release circuits compared to PVC/DOS controls. In a subsequent study by the same authors, four different biomedical grade polymers were evaluated for their inherent hemocompatibility in 4 h ECC experiments in rabbits.156 The type of polymer material can ultimately influence their efficiency as NO releasing coatings. E2As polymer was found to be the most biocompatible material amongst the four tested. E2As coated ECC tubing can preserve 56% of baseline platelet after 4 h versus 48, 40 and 47% for PVC/DOS, Tecophilic SP-60D-60 and Tecoflex SG 80A, respectively. Major et al. later used E2As polyurethane with DBHD/N2O2 and a direct thrombin inhibitor argatroban (AG) as an ECC coating to better mimic the vascular endothelium. The results showed that the combined AG/DBHD polymer coatings can better prevent thrombus formation after 4 h of blood exposure compared to control ECCs or ECCs with coatings containing only DBHD/N2O2 or AG alone.43
In addition to diazeniumdiolates, RSNOs has also been used to create NO releasing coatings for ECC applications. Brisbois et al.162 were the first to discover that SNAP-doped E2As polymer films exhibit unprecedented shelf-life stability, with 82% of the initial SNAP remaining after 2 months storage at 37 °C. The 10 wt% SNAP/E2As films can release NO for up to 20 d at levels above the physiological NO flux range. In the ECC experiments with rabbits, the inner walls of PVC ECC circuits were coated with the SNAP/E2As polymer. Such coated tubing successfully preserved the platelet count during the 4 h of experiments (at 100 ± 7% vs. 60 ± 6% for controls), with 33% less thrombus formation on the tubings’ inner surfaces.
Another related approach to increase the biocompatibility of ECC tubing is to generate NO catalytically using infused RSNOs.255 Hydrophilic SP-60D-60 polyurethane polymer containing 10 wt% Cu0 nanoparticles (NP) (80 nm) coated on the inner walls of ECC circuit tubing were tested in the rabbit thrombogenicity model. Experiments were conducted with and without intravenous SNAP infusion (0.1182 μmol kg−1 min−1) over a 4 h period. The Cu0 NP embedded ECC circuit with SNAP infusion yields considerably less thrombus (0.4 ± 0.2 pixels per cm2) on the surface of the chamber after 4 h, as compared to the Cu0 NP circuit without SNAP infusion (3.2 ± 0.2 pixels per cm2) or the control circuit without Cu0 NP (4.9 ± 0.5 pixels per cm2). Of note, since the endogenous RSNO levels are low (e.g., 1–10 μmol L−1 for SNO-albumin),281 this approach requires consistent supply of infused RSNOs to provide continuous catalytic generation of NO at the flowing blood/polymer tubing interface.255
Prevention bacterial infection or biofilm formation
Nitric oxide (NO) has antimicrobial activity against a growing list of microorganisms, including bacteria, fungi, parasites, viruses, and yeast.20,112,121,128,178,282,283 However, for medical device applications, the focus has thus far been on its antibacterial/antibiofilm properties, especially for the bacteria that most commonly cause CRBSIs (catheter related bloodstream infections), such as Staphylococcus epidermidis (S. epidermidis), Staphylococcus aureus (S. aureus), and Pseudomonas aeruginosa (P. aeruginosa).66,104 It has been clearly demonstrated that NO releasing/generating polymers can have strong bactericidal effects,19,147,148,163,202,228,284,285 even for bacteria that are able to metabolize and deactivate NO, such as P. aeruginosa, that possesses NO reductase enzyme that converts NO to nitrous oxide (N2O) and ultimately nitrogen (N2).87,202,220 Of note, NO possesses broad-spectrum antibacterial activity against both Gram-positive and Gram-negative bacteria, including methicillin-resistant Staphylococcus aureus (MRSA).220,282 Moreover, the dose of NO required to kill bacteria (e.g., 200 ppm of gaseous NO)282,283,286 does not show any cytotoxic effects in human dermal fibroblasts when exposed for 48 h.217,221 In addition to its bactericidal activity at high dosage, low levels of NO (picomolar to nanomolar range in solution phase)287 also serve as a key mediator that minimize planktonic bacteria adhesion and colonization, as well as disperse mature biofilm and release the bacteria trapped in the EPS film back to their planktonic state.287–289 It has been proposed that exposure to low doses of NO restore the sensitivity of biofilm and dispersed bacteria towards several types of antibiotics, greatly increasing their efficacy.287 Overcoming bacteria colonization on the surfaces of biomedical devices through continuous NO release represents an innovative and highly desirable approach to reduce risk of infections, which can ultimately increase device functionality and success rates, reduce morbidity and mortality, and improve patient outcomes.Frost et al. successfully develop a S-nitroso-N-acetyl-D-penicillamine functionalized polymer matrix (e.g., PDMS,227 cyclam244 and gelatin223) that can release NO via light-initiated SNAP decomposition. SNAP modified purified gelatin can release NO up to 1 × 10−8 μmol mg−1 s−1 under a 527 nm wavelength light-emitting diode (LED,) and it also shows continuous and light intensity-responsive NO release over 24 h, with a total payload of 0.06 μmol NO mg−1.223 The antibacterial activity of SNAP/gelatin was tested against S. aureus (3 × 105 CFU mL−1) and it demonstrated the ability to create a zone of inhibition of 1.2 ± 0.7 mm and 0.75 ± 0.3 mm, when exposed to the 4.5 V and 3 V LED light, respectively, over 24 h. Schoenfisch and coworkers have evaluated the antibacterial characteristics of NO releasing sol–gel coatings290 and xerogel films217 which are capable of releasing NO for 5 and 14 d, respectively, against a 108 colony forming units (CFU) per mL saline suspension of P. aeruginosa for up to 4 h. NO successfully reduced bacterial cell coverage on these surfaces by up to 40%. Recently, Reynolds and coworkers developed a water-soluble NO-releasing polysaccharide derivative that can release 100% of its NO storage capacity (49.5 ± 5.0 μmol g−1) over 24 h.228 The bactericidal activity of this species was evaluated against E. coli, Acinetobacter baumannii (A. baumannii) and S. aureus and the reported data demonstrated an 8-log unit reduction in viable bacteria cell counts for all three types of bacteria after 24 h of incubation. The absence of CFU counts after 72 h corroborated that there was no bacterial growth recovery after exposure to NO. This is the first time a water-soluble antibacterial agent has reached an industrially relevant level of antimicrobial activity.228
Further, it is also well known that bacteria in mature biofilms are much harder to eradicate than when they are in planktonic stage. Meyerhoff and coworkers have conducted many experiments where the antibacterial properties of NO releasing polymer were evaluated against mature biofilm, developed in CDC bioreactors prior to contacting with NO.146,148 Electrochemical NO generating catheters using inner copper wire working electrode and inorganic nitrite salt solution can generate NO through the walls of the catheters with a flux >1.0 × 10−10 mol cm−2 min−1 for more than 60 h.147 First, the catheters were mounted in a drip-flow bioreactor, which is a standardized model that allows growth of high biomass-biofilm of E. coli at the air liquid interface when incubated with in LB (Luria Bertani) broth for 2 d and 4 d without the NO turned on. After allowing biofilm to form on catheter surfaces, the electrochemical NO release was “turned on” for only 3 h at the end of the experiments. The short period of NO release was effective at dispersing and killing the E. coli biofilm, and could lower the number of live bacteria adhered on the catheter surfaces by >99.9%. In a separate study, Backlund et al.215 synthesized diazeniumdiolated PAMAM dendrimers with different alkyl chain modifications and examined their antibacterial properties against 24 h old Streptococcus mutans biofilm (S. mutans) at pH 7.4 and 6.4. The bactericidal action of the NO releasing dendrimers was reported to increase with alkyl chain length of the dendrimer and lower pH. In another experiment conducted by Schoenfisch and coworkers, a Center for Disease Control (CDC) bioreactor, that mimics bacterial growth under different sheer force conditions, was used to grow P. aeruginosa biofilms over a 48 h period. The biofilms were then exposed to a water-soluble NO releasing chitosan oligosaccharide (0.17–0.46 μmol NO mL−1) for 24 h. The presence of the NO release elicited a 5-log unit reduction in viable bacterial counts with a minimal bactericidal concentration (MBC) of the dendrimer preparation as low as 400 μg mL−1.221
Continuous NO release during bacterial proliferation has also been proven to prevent biofilm formation. Cai et al. fabricated NO releasing silicone rubber films containing DBHD/N2O2 along with PLGA (RG 502H) additives that can release NO for 10 d at physiological conditions. pH sensitive dyes were incorporated in the films to help visualize the pH change within the polymeric matrix due to slow hydrolysis of the PLGA that promotes the extended NO release.157 Both the NO release and control films were incubated in a CDC bioreactor for 7 d at 37 °C and the number of live bacteria on the surfaces of the silicone rubber was assessed. The NO release films exhibited substantial antibiofilm properties against both Gram-positive S. aureus (98.4% reduction) and Gram-negative E. coli (99.9% reduction) when compared to controls.157 In subsequent studies, Colletta et al.19 and Ketchum et al.190 used the solvent impregnation method previously described above (see section Physical Encapsulation-based NO Releasing Polymers) to load silicone-based tubing with NO donors (SNAP and lipophilic SNAP derivatives, respectively) and demonstrated that they have antibacterial effects toward several microorganisms (e.g., Staphylococcus epidermis, Proteus mirabilis or S. aureus). These results offer a new strategy to reduce bacterial infections associated with not only intravascular catheters but also urinary catheters.
In addition to in vitro experiments, NO releasing polymers (e.g., catheters) have also exhibited both antithrombotic and antibacterial properties in vivo. Brisbois et al. conducted experiments where both SNAP-doped E2As catheter and control E2As catheters were implanted in sheep jugular veins for 7 d.187 At the end of the 7 d period, the catheters were carefully explanted and both the clot area as well as the number of live bacteria adhered on the catheter surfaces were assessed. The 10 wt% SNAP/E2As catheters exhibited enhanced hemocompatibility (70% less thrombus formation) and bactericidal activity (90% reduction of bacteria adhesion) when compared to the controls.
Nitric oxide releasing polymers have also provided a potential strategy for use as active wound healing dressings, capable of reducing bacterial infections at wound sites and accelerating the wound healing process for incisional and excisional wounds,180,220,222 burn wounds,291 and diabetic wounds.233 While normal fibroblasts do not synthesize NO, it has been demonstrated that fibroblasts isolated from the regular wound can release NO.292 The iNOS is synthesized in the early phase of wound healing by inflammatory cells, such as macrophages, monocytes, and other immune lineage cells.120,121,293 The NO released through iNOS enhances angiogenesis, regulates collagen formation, cell proliferation and wound contractions.112,293 Studies using induced NOS (iNOS) knock out mice showed diminished collagen deposition, with the restoration of normal collagen deposits upon exposure to SNAP, indicating the important relationship between NO production and wound healing.294 Therefore, elevating the local NO concentration at a wound site by an exogenous NO supply represents a promising strategy to facilitate the wound healing process.
Yoo and coworkers developed PEI/NONOate nanoparticles (NPs) capable of prolonged NO release, which were then used to treat MRSA-infected excisional wounds (5 mm diameter, full thickness) in mice.220 MRSA was inoculated in the wound for 1 d to develop infection and then the wounds were treated with the NO releasing NPs or control NPs over 6 d. The photographs of the wounds taken at day 0, 1, 4 and 7 clearly showed an accelerated wound healing process for mice treated with NPs, with wound area reduced to 25% at the end of 7 d. The untreated wounds showed a thicker scab with no reduction in the wound area. In a subsequent study, GSNO-doped chitosan films, which exert strong antimicrobial activity against a broad spectrum of bacteria, were evaluated as a wound healing dressing on full thickness excisional wounds (1.5 cm × 1.5 cm) in a rat model for 15 d.222 Comparing the NO-treated and untreated wounds, those treated with the GSNO/Chitosan dressing were almost completely healed at the end of the 15 d, with less bacterial burden and decreased inflammatory cell infiltration, but enhanced re-epithelialization.121,222 Brisbois et al. tested the antibacterial efficacy of DBHD/N2O2 and PLGA (5050DLG1A) doped biomedical grade PU films as a dressing for burn wounds (10 cm2 and partial thickness) in a mouse model.291 Burn wounds are quite common, but difficult to treat and often lead to infections.112 The burn wounds in mice were inoculated by A. baumannii for 24 h prior to application of the NO and control wound dressings. After 24 h of treatment, the wound area skin tissue was harvested and assessed for number of viable bacteria adhered at the wound sites. A 4-log unit bacterial reduction was observed for the NO treated wounds when compared to the control would sites.
Conclusions
As detailed above, there are many approaches that have been examined to improve the biocompatibility and antimicrobial activity of biomedical polymers/devices. Most of the conventional approaches, (e.g., creating heparin-releasing surfaces, bacteria-resistant or biocide-releasing surfaces, etc.), can only improve either the hemocompatibility or the antibacterial properties of the devices. However, NO release or generating polymers have demonstrated very positive effects in terms of preventing both platelet activation and thrombus formation as well as exhibiting significant antimicrobial activity toward infection/biofilm formation without bacterial resistance. Although this review mainly focused on the use of NO release polymers for antithrombotic and antibacterial applications, these polymers also have many other potential applications for use on skin, bones, connective tissue, gastroenterology, or studying NO-induced cellular responses.112,178,295,296Recently, considerable effort has been devoted to develop combination therapy and hybrid drugs that combine NO release functionality with other active agents for increased efficacies, including anticoagulants,35,127 direct thrombin inhibitors,36,43 antimicrobial metal ions,216 quaternary ammonium compounds,71 PEG,116 antibiotics,297 and non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen.141 The remaining challenge for NO delivery polymers in order to be used in clinical settings lies in the development of more stable NO donors and polymer scaffolds and delivering the desired amount NO in a controlled spatial and temporal manner. In many cases, long-term NO release is desired, especially for using NO-releasing, blood compatible polymeric materials for preparing intravascular grafts, stents, catheters, and other vascular access devices. Further research should focus on the dose-dependent biological responses to different NO concentrations and aim towards developing clinically relevant NO delivery materials for that purpose, that can ultimately expedite the translation of these materials from the laboratory to clinical use. Overall, both in vitro and in vivo (animals) studies of some of the NO releasing/generating polymers developed to date have clearly demonstrated the immense potential of this approach to improve the hemocompatibility/antimicrobial activity of a variety of biomedical devices that are required to improve patient care. Hopefully, the first commercial devices that employ this strategy will soon become available for routine use within hospitals.
Acknowledgements
We thank the National Institutes of Health (grants HL-128337 and HL-119403) for funding research in our laboratory on the development of novel NO release biomedical polymers/devices.References
- N. Naghavi, A. De Mel, O. S. Alavijeh, B. G. Cousins and A. M. Seifalian, Small, 2013, 9, 22–35 CrossRef CAS PubMed
.
- M. B. Gorbet and M. V. Sefton, Biomaterials, 2004, 25, 5681–5703 CrossRef CAS PubMed
-
E. J. Brisbois, H. Handa and M. E. Meyerhoff, in Advanced Polymers in Medicine, ed. F. Puoci, Springer International Publishing, Cham, 2015, pp. 481–511 Search PubMed
- M. V. Sefton, C. H. Gemmell and M. B. Gorbet, J. Biomater. Sci., Polym. Ed., 2000, 11, 1165–1182 CrossRef CAS PubMed
-
M. B. Gorbet and M. V. Sefton, in The Biomaterials: Silver Jubilee Compendium, Elsevier, 2004, vol. 25, pp. 219–241 Search PubMed
- T. A. Horbett, Cardiovasc. Pathol., 1993, 2, 137–148 CrossRef
-
J. D. Andrade and V. Hlady, in Advances in Polymer Science, 1986, vol. 79, pp. 1–63 Search PubMed
- Y. Wu and M. E. Meyerhoff, Talanta, 2008, 75, 642–650 CrossRef CAS PubMed
- R. M. Klevens, J. R. Edwards, C. L. Richards, T. C. Horan, R. P. Gaynes, D. A. Pollock and D. M. Cardo, Public Health Rep., 2007, 122, 160–166 Search PubMed
- C. Abad and N. Safdar, Infect. Dis. Spec. Ed., 2011 Search PubMed
- H. Shah, W. Bosch, K. M. Thompson and W. C. Hellinger, Neurohospitalist, 2013, 3, 144–151 CrossRef PubMed
- L. A. Mermel, M. Allon, E. Bouza, D. E. Craven, P. Flynn, N. P. O'Grady, I. I. Raad, B. J. Rijnders, R. J. Sherertz and D. K. Warren, Clin. Infect. Dis., 2009, 49, 1–45 CrossRef CAS PubMed
- A. Sitges-Serra and M. Girvent, World J. Surg., 1999, 23, 589–595 CrossRef CAS PubMed
- M. R. Goede and C. M. Coopersmith, Surg. Clin. North Am., 2009, 89, 463–474 CrossRef PubMed
- R. Gahlot, C. Nigam, V. Kumar, G. Yadav and S. Anupurba, Int. J. Crit. Illn. Inj. Sci., 2014, 4, 162–167 Search PubMed
- M. Chen, Q. Yu and H. Sun, Int. J. Mol. Sci., 2013, 14, 18488–18501 CrossRef PubMed
- R. M. Donlan, Clin. Infect. Dis., 2001, 33, 1387–1392 CrossRef CAS PubMed
- J. B. Kaplan, J. Dent. Res., 2010, 89, 205–218 CrossRef CAS PubMed
- A. Colletta, J. Wu, Y. Wo, M. Kappler, H. Chen, C. Xi and M. E. Meyerhoff, ACS Biomater. Sci. Eng., 2015, 1, 416–424 CrossRef CAS PubMed
- E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780–789 RSC
- J. Hirsh, N. Engl. J. Med., 1991, 324, 1565–1574 CrossRef CAS PubMed
- A. Gutowska, Y. H. Bae, H. Jacobs, F. Mohammad, D. Mix, J. Feijen and S. W. Kim, J. Biomed. Mater. Res., 1995, 29, 811–821 CrossRef CAS PubMed
- T. E. Warkentin, C. P. Hayward, C. A. Smith, P. M. Kelly and J. G. Kelton, J. Lab. Clin. Med., 1992, 120, 371–379 CAS
- H. Tanzawa, Y. Mori, N. Harumiya, H. Miyama, M. Hori, N. Ohshima and Y. Idezuki, ASAIO J., 1973, 19, 188–194 CrossRef CAS
- V. L. Gott, J. D. Whiffen and R. C. Dutton, Science, 1963, 142, 1297–1298 CrossRef CAS PubMed
- R. Shibuta, M. Tanaka, M. Sisido and Y. Imanishi, J. Biomed. Mater. Res., 1986, 20, 971–987 CrossRef CAS PubMed
- C. A. Hufnagel, P. W. Conrad, J. F. Gillespie, R. Pifarre, A. Llano and T. Yokoyama, Ann. N. Y. Acad. Sci., 1968, 146, 262–270 CrossRef CAS PubMed
- J. Y. Lin, C. Nojiri, T. Okano and S. W. Kim, ASAIO Trans., 1987, 33, 602–605 CAS
- P. W. Heyman, C. S. Cho, J. C. McRea, D. B. Olsen and S. W. Kim, J. Biomed. Mater. Res., 1985, 19, 419–436 CrossRef CAS PubMed
-
C. Ebert, J. McRea and S. W. Kim, in Controlled Release of Bioactive Materials, Elsevier, 1980, pp. 107–122 Search PubMed
- A. Gutowska, Y. H. Bae, J. Feijen and S. W. Kim, J. Controlled Release, 1992, 22, 95–105 CrossRef CAS
- S. Wan Kim and H. Jacobs, Blood Purif., 1996, 14, 357–372 CrossRef
- N. Ayres, D. J. Holt, C. F. Jones, L. E. Corum and D. W. Grainger, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7713–7724 CrossRef CAS PubMed
- K. A. Mowery, M. H. Schoenfisch, J. E. Saavedra, L. K. Keefer and M. E. Meyerhoff, Polymer, 2000, 21, 9–21 CAS
- Z. Zhou and M. E. Meyerhoff, Biomaterials, 2005, 26, 6506–6517 CrossRef CAS PubMed
- B. Wu, B. Gerlitz, B. W. Grinnell and M. E. Meyerhoff, Biomaterials, 2007, 28, 4047–4055 CrossRef CAS PubMed
- S. E. Sakiyama-Elbert, Acta Biomater., 2014, 10, 1581–1587 CrossRef CAS PubMed
- P. C. Begovac, R. C. Thomson, J. L. Fisher, A. Hughson and A. Gallhagen, Eur. J. Vasc. Endovasc. Surg., 2003, 25, 432–437 CrossRef CAS PubMed
- C. Devine and C. McCollum, J. Vasc. Surg., 2004, 40, 924–931 CrossRef PubMed
- P. Olsson, J. Sanchez, T. E. Mollnes and J. Riesenfeld, J. Biomater. Sci., Polym. Ed., 2000, 11, 1261–1273 CrossRef CAS PubMed
- A. G. Kidane, H. Salacinski, A. Tiwari, K. R. Bruckdorfer and A. M. Seifalian, Biomacromolecules, 2004, 5, 798–813 CrossRef CAS PubMed
- S. W. Jordan and E. L. Chaikof, J. Vasc. Surg., 2007, 45, 104–115 CrossRef PubMed
- T. C. Major, E. J. Brisbois, A. M. Jones, M. E. Zanetti, G. M. Annich, R. H. Bartlett and H. Handa, Biomaterials, 2014, 35, 7271–7285 CrossRef CAS PubMed
- N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward and J. Zhang, Annu. Rev. Biomed. Eng., 2000, 2, 9–29 CrossRef CAS PubMed
- O. Wichterle and D. Lím, Nature, 1960, 185, 117–118 CrossRef
- S. Lakshmi and A. Jayakrishnan, Artif. Organs, 1998, 22, 222–229 CrossRef CAS PubMed
- B. Balakrishnan, D. S. Kumar, Y. Yoshida and A. Jayakrishnan, Biomaterials, 2005, 26, 3495–3502 CrossRef CAS PubMed
- C. R. Blass, C. Jones and J. M. Courtney, Int. J. Artif. Organs, 1992, 15, 200–203 CAS
- G. R. Llanos and M. V. Sefton, J. Biomed. Mater. Res., 1993, 27, 1383–1391 CrossRef CAS PubMed
-
J. M. Harris, Poly(Ethylene Glycol) Chemistry, Springer US, Boston, MA, Netherlands, 1992, vol. 11 Search PubMed
- M. C. Tanzi, Expert Rev. Med. Devices, 2005, 2, 473–492 CrossRef CAS PubMed
- S. K. Williams, D. G. Rose and B. E. Jarrell, J. Biomed. Mater. Res., 1994, 28, 203–212 CrossRef CAS PubMed
- P. Zilla, M. Deutsch, J. Meinhart, R. Puschmann, T. Eberl, E. Minar, R. Dudczak, H. Lugmaier, P. Schmidt and I. Noszian, J. Vasc. Surg., 1994, 19, 540–548 CrossRef CAS PubMed
- S. R. Shah, A. M. Tatara, R. N. D'Souza, A. G. Mikos and F. K. Kasper, Mater. Today, 2013, 16, 177–182 CrossRef CAS
- S.-H. Park, S. Lee, D. Moreira, P. R. Bandaru, I. Han and D.-J. Yun, Sci. Rep., 2015, 5, 15430 CrossRef PubMed
- W. Ye, Q. Shi, J. Hou, J. Jin, Q. Fan, S.-C. Wong, X. Xu and J. Yin, J. Mater. Chem. B, 2014, 2, 7186–7191 RSC
- X. Gao and L. Jiang, Nature, 2004, 432, 36 CrossRef CAS PubMed
- U. Nydegger, R. Rieben and B. Lämmle, Transfus. Sci., 1996, 17, 481–488 CrossRef CAS PubMed
- G. Cheng, H. Xue, Z. Zhang, S. Chen and S. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8831–8834 CrossRef CAS PubMed
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS
- S.-H. Hsu, Y.-C. Kao and Z.-C. Lin, Macromol. Biosci., 2004, 4, 464–470 CrossRef CAS PubMed
- S.-H. Hsu and Y.-C. Kao, Macromol. Biosci., 2005, 5, 246–253 CrossRef CAS PubMed
- L.-C. Xu, P. Soman, J. Runt and C. A. Siedlecki, J. Biomater. Sci., Polym. Ed., 2007, 18, 353–368 CrossRef CAS PubMed
- Q. Yu, Z. Wu and H. Chen, Acta Biomater., 2015, 16, 1–13 CrossRef CAS PubMed
- I. Kolodkin-Gal, D. Romero, S. Cao, J. Clardy, R. Kolter and R. Losick, Science, 2010, 328, 627–629 CrossRef CAS PubMed
- M. R. Kiedrowski and A. R. Horswill, Ann. N. Y. Acad. Sci., 2011, 1241, 104–121 CrossRef CAS PubMed
- S. Sepehr, A. Rahmani-Badi, H. Babaie-Naiej and M. R. Soudi, PLoS One, 2014, 9 Search PubMed
- J. A. Jennings, H. S. Courtney and W. O. Haggard, Clin. Orthop. Relat. Res., 2012, 470, 2663–2670 CrossRef PubMed
- M. C. Jennings, L. E. Ator, T. J. Paniak, K. P. C. Minbiole and W. M. Wuest, ChemBioChem, 2014, 15, 2211–2215 CrossRef CAS PubMed
- M. C. Jennings, K. P. C. Minbiole and W. M. Wuest, ACS Infect. Dis., 2015, 288–303 CrossRef CAS
- A. W. Carpenter, B. V. Worley, D. L. Slomberg and M. H. Schoenfisch, Biomacromolecules, 2012, 13, 3334–3342 CrossRef CAS PubMed
- A. S. Abd-El-Aziz, C. Agatemor, N. Etkin, D. P. Overy, M. Lanteigne, K. McQuillan and R. G. Kerr, Biomacromolecules, 2015, 16, 3694–3703 CrossRef CAS PubMed
- N. D. Allan, K. Giare-Patel and M. E. Olson, J. Biomed. Biotechnol., 2012, 2012 Search PubMed
- K. Wu, Y. Yang, Y. Zhang, J. Deng and C. Lin, Int. J. Nanomed., 2015, 10, 7241–7252 Search PubMed
- L. Mei, Z. Lu, X. Zhang, C. Li and Y. Jia, ACS Appl. Mater. Interfaces, 2014, 6, 15813–15821 CAS
- S. K. Sehmi, S. Noimark, J. Weiner, E. Allan, A. J. MacRobert and I. P. Parkin, ACS Appl. Mater. Interfaces, 2015, 7, 22807–22813 CAS
- L. Zhang, C. Ning, T. Zhou, X. Liu, K. W. K. Yeung, T. Zhang, Z. Xu, X. Wang, S. Wu and P. K. Chu, ACS Appl. Mater. Interfaces, 2014, 6, 17323–17345 CAS
- S. Rossi, A. O. Azghani and A. Omri, J. Antimicrob. Chemother., 2004, 54, 1013–1018 CrossRef CAS PubMed
- L. Zhao, P. K. Chu, Y. Zhang and Z. Wu, J. Biomed. Mater. Res., Part B, 2009, 91, 470–480 CrossRef PubMed
- R. Cademartiri, H. Anany, I. Gross, R. Bhayani, M. Griffiths and M. A. Brook, Biomaterials, 2010, 31, 1904–1910 CrossRef CAS PubMed
- U. Puapermpoonsiri, J. Spencer and C. F. van der Walle, Eur. J. Pharm. Biopharm., 2009, 72, 26–33 CrossRef CAS PubMed
- T. Wei, Q. Yu, W. Zhan and H. Chen, Adv. Healthcare Mater., 2016, 5, 449–456 CrossRef CAS PubMed
- G. Cado, R. Aslam, L. Séon, T. Garnier, R. Fabre, A. Parat, A. Chassepot, J.-C. Voegel, B. Senger, F. Schneider, Y. Frère, L. Jierry, P. Schaaf, H. Kerdjoudj, M.-H. Metz-Boutigue and F. Boulmedais, Adv. Funct. Mater., 2013, 23, 4801–4809 CAS
- K. V. Holmberg, M. Abdolhosseini, Y. Li, X. Chen, S.-U. Gorr and C. Aparicio, Acta Biomater., 2013, 9, 8224–8231 CrossRef CAS PubMed
- J. Li, Z. Chen, M. Zhou, J. Jing, W. Li, Y. Wang, L. Wu, L. Wang, Y. Wang and M. Lee, Angew. Chem., Int. Ed., 2016, 55, 2592–2595 CrossRef CAS PubMed
- W. J. Yang, T. Cai, K. G. Neoh, E. T. Kang, G. H. Dickinson, S. L. M. Teo and D. Rittschof, Langmuir, 2011, 27, 7065–7076 CrossRef CAS PubMed
- K. P. Reighard and M. H. Schoenfisch, Antimicrob. Agents Chemother., 2015, 59, 6506–6513 CrossRef CAS PubMed
- K. P. Reighard, D. B. Hill, G. A. Dixon, B. V. Worley and M. H. Schoenfisch, Biofouling, 2015, 31, 775–787 CrossRef CAS PubMed
- J. Choi, B. J. Yang, G. N. Bae and J. H. Jung, ACS Appl. Mater. Interfaces, 2015, 7, 25313–25320 CAS
- J. Dolanský, P. Henke, P. Kubát, A. Fraix, S. Sortino and J. Mosinger, ACS Appl. Mater. Interfaces, 2015, 7, 22980–22989 Search PubMed
- Z. Cao, L. Mi, J. Mendiola, J. R. Ella-Menye, L. Zhang, H. Xue and S. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2602–2605 CrossRef CAS PubMed
- Q. Yu, J. Cho, P. Shivapooja, L. K. Ista and G. P. Lopez, ACS Appl. Mater. Interfaces, 2013, 5, 9295–9304 CAS
- M. Dargahi, Z. Hosseinidoust, N. Tufenkji and S. Omanovic, Colloids Surf., B, 2014, 117, 152–157 CrossRef CAS PubMed
- S. Tamura, H. Yonezawa, M. Motegi, R. Nakao, S. Yoneda, H. Watanabe, T. Yamazaki and H. Senpuku, Oral Microbiol. Immunol., 2009, 24, 152–161 CrossRef CAS PubMed
- S. A. Ishijima, K. Hayama, J. P. Burton, G. Reid, M. Okada, Y. Matsushita and S. Abe, Appl. Environ. Microbiol., 2012, 78, 2190–2199 CrossRef CAS PubMed
- B. W. Trautner, R. A. Hull and R. O. Darouiche, Urology, 2003, 61, 1059–1062 CrossRef PubMed
- B. W. Trautner, R. A. Hull and R. O. Darouiche, Curr. Opin. Infect. Dis., 2005, 18, 37–41 CrossRef PubMed
- B. W. Trautner, R. A. Hull, J. I. Thornby and R. O. Darouiche, Infect. Control Hosp. Epidemiol., 2007, 28, 92–94 CrossRef PubMed
- P. Andersson, I. Engberg, G. Lidin-Janson, K. Lincoln, R. Hull, S. Hull and C. Svanborg, Infect. Immun., 1991, 59, 2915–2921 CAS
- A. Esclarin De Ruz, E. Garcia Leoni and R. Herruzo Cabrera, J. Urol., 2000, 164, 1285–1289 CrossRef CAS PubMed
- A. R. Unnithan, A. Ghavami Nejad, A. R. K. Sasikala, R. G. Thomas, Y. Y. Jeong, P. Murugesan, S. Nasseri, D. Wu, C. H. Park and C. S. Kim, Chem. Eng. J., 2016, 287, 640–648 CrossRef CAS
- A. Vaterrodt, B. Thallinger, K. Daumann, D. Koch, G. M. Guebitz and M. Ulbricht, Langmuir, 2016, 32, 1347–1359 CrossRef CAS PubMed
- L.-C. Xu and C. A. Siedlecki, Biomed. Mater., 2014, 9, 035003 CrossRef PubMed
- L. C. Xu and C. A. Siedlecki, Acta Biomater., 2012, 8, 72–81 CrossRef CAS PubMed
- L. Xu, J. W. Bauer and C. A. Siedlecki, Colloids Surf., B, 2014, 124, 49–68 CrossRef CAS PubMed
- L. Wang, H. Li, S. Chen, C. Nie, C. Cheng and C. Zhao, ACS Biomater. Sci. Eng., 2015, 1, 1183–1193 CrossRef CAS
- Z.-Q. Shi, H.-F. Ji, H.-C. Yu, X.-L. Huang, W.-F. Zhao, S.-D. Sun and C.-S. Zhao, Colloid Polym. Sci., 2016, 294, 441–453 CAS
- J. Deng, L. Ma, X. Liu, C. Cheng, C. Nie and C. Zhao, Adv. Mater. Interfaces, 2016, 3, 1500473 Search PubMed
- A. B. Seabra and N. Durán, J. Mater. Chem., 2010, 20, 1624 RSC
- M. C. Frost, M. M. Reynolds and M. E. Meyerhoff, Biomaterials, 2005, 26, 1685–1693 CrossRef CAS PubMed
- M. M. Reynolds, M. C. Frost and M. E. Meyerhoff, Free Radicals Biol. Med., 2004, 37, 926–936 CrossRef CAS PubMed
- S. P. Nichols, W. L. Storm, A. Koh and M. H. Schoenfisch, Adv. Drug Delivery Rev., 2012, 64, 1177–1188 CrossRef CAS PubMed
- E. M. Hetrick and M. H. Schoenfisch, Annu. Rev. Anal. Chem., 2009, 2, 409–433 CrossRef CAS PubMed
- M. M. Reynolds and G. M. Annich, Organogenesis, 2011, 7, 42–49 CrossRef PubMed
- Y. Wang, S. Chen, Y. Pan, J. Gao, D. Tang, D. Kong and S. Wang, J. Mater. Chem. B, 2015, 3, 9212–9222 RSC
- L. J. Taite, P. Yang, H.-W. Jun and J. L. West, J. Biomed. Mater. Res., Part B, 2007, 84, 108–116 Search PubMed
- I. L. A. Geenen, D. G. M. Molin, N. M. S. van den Akker, F. Jeukens, H. M. Spronk, G. W. H. Schurink and M. J. Post, J. Tissue Eng. Regen. Med., 2015, 9, 564–576 CrossRef CAS PubMed
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC
- M. Vaughn, L. Kuo and J. Liao, Am. J. Physiol., 1998, 274, 2163–2176 Search PubMed
- K. M. Vural and M. Bayazit, Eur. J. Vasc. Endovasc. Surg., 2001, 22, 285–293 CrossRef CAS PubMed
- M. C. Jen, M. C. Serrano, R. van Lith and G. A. Ameer, Adv. Funct. Mater., 2012, 22, 239–260 CrossRef CAS PubMed
- J. Loscalzo, Circ. Res., 2001, 88, 756–762 CrossRef CAS PubMed
- G. Walford and J. Loscalzo, J. Thromb. Haemostasis, 2003, 1, 2112–2118 CrossRef CAS
- J. S. Isenberg, M. J. Romeo, C. Yu, C. K. Yu, K. Nghiem, J. Monsale, M. E. Rick, D. A. Wink, W. A. Frazier and D. D. Roberts, Blood, 2008, 111, 613–623 CrossRef CAS PubMed
- A. Gries, C. Bode, K. Peter, A. Herr, H. Böhrer, J. Motsch and E. Martin, Circ., 1998, 97, 1481–1487 CrossRef CAS
- M. W. Radomski, R. M. Palmer and S. Moncada, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5193–5197 CrossRef CAS
- D. J. Suchyta, H. Handa and M. E. Meyerhoff, Mol. Pharm., 2014, 11, 645–650 CrossRef CAS PubMed
- D. O. Schairer, J. S. Chouake, J. D. Nosanchuk and A. J. Friedman, Virulence, 2012, 3, 271–279 CrossRef PubMed
- H. Rubbo, R. Radi, M. Trujillo, R. Telleri, B. Kalyanaraman, S. Barnes, M. Kirk and B. A. Freeman, J. Biol. Chem., 1994, 269, 26066–26075 CAS
- F. C. Fang, Nat. Rev. Microbiol., 2004, 2, 820–832 CrossRef CAS PubMed
- L. Ignarro and G. Buga, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 9265–9269 CrossRef CAS
- S. R. Hanson, T. C. Hutsell, L. K. Keefer, D. L. Mooradian and D. J. Smith, Adv. Pharmacol., 1995, 34, 383–398 CAS
- J. O. Lundberg, M. T. Gladwin and E. Weitzberg, Nat. Rev. Drug Discovery, 2015, 14, 623–641 CrossRef CAS PubMed
- J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Crit. Care Med., 2008, 7, 156–167 CAS
- R. Scatena, P. Bottoni, G. Martorana and B. Giardina, Expert Opin. Invest. Drugs, 2005, 14, 835–846 CrossRef CAS PubMed
- D. A. Riccio and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731–3741 RSC
- J. A. Hrabie and L. K. Keefer, Chem. Rev., 2002, 102, 1135–1154 CrossRef CAS PubMed
- V. N. Varu, N. D. Tsihlis and M. R. Kibbe, Vasc. Endovascular Surg., 2009, 43, 121–131 CrossRef PubMed
- J. Saraiva, S. S. Marotta-Oliveira, S. A. Cicillini, J. D. O. Eloy and J. M. Marchetti, J. Drug Delivery, 2011, 2011 Search PubMed
- S. Sortino, Chem. Soc. Rev., 2010, 39, 2903–2913 RSC
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed
- T. Münzel, A. Daiber and A. Mülsch, Circ. Res., 2005, 97, 618–628 CrossRef PubMed
- K. Sydow, A. Daiber, M. Oelze, Z. Chen, M. August, M. Wendt, V. Ullrich, A. Mülsch, E. Schulz, J. F. Keaney, J. S. Stamler and T. Münzel, J. Clin. Invest., 2004, 113, 482–489 CrossRef CAS PubMed
- A. Daiber, M. Oelze, M. Coldewey, M. Bachschmid, P. Wenzel, K. Sydow, M. Wendt, A. L. Kleschyov, D. Stalleicken, V. Ullrich, A. Mulsch and T. Munzel, Mol. Pharmacol., 2004, 66, 1372–1382 CrossRef CAS PubMed
- L. Grossi and S. D'Angelo, J. Med. Chem., 2005, 48, 2622–2626 CrossRef CAS PubMed
- L. Höfler, D. Koley, J. Wu, C. Xi and M. E. Meyerhoff, RSC Adv., 2012, 2, 6765–6767 RSC
- H. Ren, A. Colletta, D. Koley, J. Wu, C. Xi, T. C. Major, R. H. Bartlett and M. E. Meyerhoff, Bioelectrochemistry, 2015, 104, 10–16 CrossRef CAS PubMed
- H. Ren, J. Wu, C. Xi, N. Lehnert, T. Major, R. H. Bartlett and M. E. Meyerhoff, ACS Appl. Mater. Interfaces, 2014, 6, 3779–3783 CAS
- J. R. Hwu, C. S. Yau, S.-C. Tsay and T.-I. Ho, Tetrahedron Lett., 1997, 38, 9001–9004 CrossRef CAS
- A. Wan, Q. Gao and H. Li, J. Mater. Sci. Mater. Med., 2009, 20, 321–327 CrossRef CAS PubMed
- S. Hong, J. Kim, Y. S. Na, J. Park, S. Kim, K. Singha, G. Il Im, D. K. Han, W. J. Kim and H. Lee, Angew. Chem., Int. Ed., 2013, 52, 9187–9191 CrossRef CAS PubMed
- C. M. Maragos, D. Morley, D. A. Wink, T. M. Dunams, J. E. Saavedra, A. Hoffman, A. A. Bove, L. Isaac, J. A. Hrabie and L. K. Keefer, J. Med. Chem., 1991, 34, 3242–3247 CrossRef CAS PubMed
- L. K. Keefer, R. W. Nims, K. M. Davies and D. A. Wink, Methods Enzymol., 1996, 268, 281–293 CAS
- M. M. Batchelor, S. L. Reoma, P. S. Fleser, V. K. Nuthakki, R. E. Callahan, C. J. Shanley, J. K. Politis, J. Elmore, S. I. Merz and M. E. Meyerhoff, J. Med. Chem., 2003, 46, 5153–5161 CrossRef CAS PubMed
- H. Handa, E. J. Brisbois and T. C. Major, J. Mater. Chem. B, 2013, 1, 3578–3587 RSC
- H. Handa, T. C. Major, E. J. Brisbois, K. A. Amoako, M. E. Meyerhoff and R. H. Bartlett, J. Mater. Chem. B, 2014, 2, 1059–1067 RSC
- W. Cai, J. Wu, C. Xi and M. E. Meyerhoff, Biomaterials, 2012, 33, 7933–7944 CrossRef CAS PubMed
- H. K. Makadia and S. J. Siegel, Polymer, 2011, 3, 1377–1397 CAS
- J. S. Stamler, O. Jaraki, J. Osborne, D. I. Simon, J. Keaney, J. Vita, D. Singel, C. R. Valeri and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7674–7677 CrossRef CAS
- D. Giustarini, A. Milzani, R. Colombo, I. Dalle-Donne and R. Rossi, Clin. Chim. Acta, 2003, 330, 85–98 CrossRef CAS
- X. Duan and R. S. Lewis, Biomaterials, 2002, 23, 1197–1203 CrossRef CAS PubMed
- E. J. Brisbois, H. Handa, T. C. Major, R. H. Bartlett and M. E. Meyerhoff, Biomaterials, 2013, 34, 6957–6966 CrossRef CAS PubMed
- Y. Wo, Z. Li, E. J. Brisbois, A. Colletta, J. Wu, T. C. Major, C. Xi, R. H. Bartlett, A. J. Matzger and M. E. Meyerhoff, ACS Appl. Mater. Interfaces, 2015, 7, 22218–22227 CAS
- D. L. H. Williams, Chem. Commun., 1996, 1085–1091 RSC
- D. L. H. Williams, Transition Met. Chem., 1996, 21, 189–191 CrossRef CAS
- D. L. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS
- K. Szacilowski and Z. Stasicka, Prog. React. Kinet. Mech., 2001, 26, 1–58 CrossRef CAS
- R. J. Singh, N. Hogg, J. Joseph and B. Kalyanaraman, J. Biol. Chem., 1996, 271, 18596–18603 CrossRef CAS PubMed
- C. T. Aravindakumar, M. M. Veleeparampil and U. K. Aravind, Adv. Phys. Chem., 2009, 2009 Search PubMed
- J. McAninly, D. L. H. Williams, S. C. Askew, A. R. Butler and C. Russell, J. Chem. Soc., Chem. Commun., 1993, 1758–1759 RSC
- S. Hwang and M. E. Meyerhoff, Biomaterials, 2008, 29, 2443–2452 CrossRef CAS PubMed
- W. Cha and M. E. Meyerhoff, Biomaterials, 2007, 28, 19–27 CrossRef CAS PubMed
- J. Yang, J. L. Welby and M. E. Meyerhoff, Langmuir, 2008, 24, 10265–10272 CrossRef CAS PubMed
- W. Cai, J. Wu, C. Xi, A. J. Ashe and M. E. Meyerhoff, Biomaterials, 2011, 32, 7774–7784 CrossRef CAS PubMed
- A. J. Holmes and D. L. H. Williams, Chem. Commun., 1998, 1711–1712 RSC
- A. J. Holmes and D. L. H. Williams, J. Chem. Soc., Trans. 2, 2000, 1639–1644 CAS
- D. Jourd'heuil, F. S. Laroux, A. M. Miles, D. A. Wink and M. B. Grisham, Arch. Biochem. Biophys., 1999, 361, 323–330 CrossRef PubMed
-
A. B. Seabra, in Nanocosmetics and Nanomedicines, ed. R. Beck, S. Guterres and A. Pohlmann, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 253–268 Search PubMed
- R. Vercelino, T. M. Cunha, E. S. Ferreira, F. Q. Cunha, S. H. Ferreira and M. G. De Oliveira, J. Mater. Sci. Mater. Med., 2013, 24, 2157–2169 CrossRef CAS PubMed
- F. S. Schanuel, K. S. Raggio Santos, A. Monte-Alto-Costa and M. G. De Oliveira, Colloids Surf., B, 2015, 130, 182–191 CrossRef CAS PubMed
- M. G. de Oliveira, Basic Clin. Pharmacol. Toxicol., 2016 Search PubMed
- T. R. Borthwick, G. D. Benson and H. J. Schugar, J. Lab. Clin. Med., 1980, 95, 575–580 CAS
- V. Parameshvara, Br. J. Ind. Med., 1967, 24, 73–76 CAS
- K. A. Graeme and C. V. Pollack, J. Emerg. Med., 1998, 16, 45–56 CrossRef CAS PubMed
- M. M. Jones, A. D. Weaver and W. L. Weller, Res. Commun. Chem. Pathol. Pharmacol., 1978, 22, 581–588 CAS
- N. Arulsamy, D. S. Bohle, J. A. Butt, G. J. Irvine, P. A. Jordan and E. Sagan, J. Am. Chem. Soc., 1999, 121, 7115–7123 CrossRef CAS
- E. J. Brisbois, R. P. Davis, A. M. Jones, T. C. Major, R. H. Bartlett, M. E. Meyerhoff and H. Handa, J. Mater. Chem. B, 2015, 3, 1639–1645 RSC
- I. L. Megson, S. Morton, I. R. Greig, F. A. Mazzei, R. A. Field, A. R. Butler, G. Caron, A. Gasco, R. Fruttero and D. J. Webb, Br. J. Pharmacol., 1999, 126, 639–648 CrossRef CAS PubMed
- I. L. Megson, Drugs Future, 2002, 27, 777 CrossRef CAS
- A. Ketchum, M. Kappler, J. Wu, C. Xi and M. E. Meyerhoff, J. Mater. Chem. B, 2015, 4, 422–430 RSC
- Y. S. Jo, A. J. van der Vlies, J. Gantz, T. N. Thacher, S. Antonijevic, S. Cavadini, D. Demurtas, N. Stergiopulos and J. A. Hubbell, J. Am. Chem. Soc., 2009, 131, 14413–14418 CrossRef CAS PubMed
- N. Kanayama, K. Yamaguchi and Y. Nagasaki, Chem. Lett., 2010, 39, 1008–1009 CrossRef CAS
- F. Cavalieri, I. Finelli, M. Tortora, P. Mozetic, E. Chiessi, F. Polizio, T. B. Brismar and G. Paradossi, Chem. Mater., 2008, 20, 3254–3258 CrossRef CAS
- J. A. Hrabie, J. E. Saavedra, P. P. Roller, G. J. Southan and L. K. Keefer, Bioconjugate Chem., 1999, 10, 838–842 CrossRef CAS PubMed
- D. I. Simon, M. E. Mullins, L. Jia, B. Gaston, D. J. Singel and J. S. Stamler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4736–4741 CrossRef CAS
- J. F. Ewing, D. V. Young, D. R. Janero, D. S. Garvey and T. A. Grinnell, J. Pharmacol. Exp. Ther., 1997, 283, 947–954 CAS
- Y. Ishima, T. Akaike, U. Kragh-Hansen, S. Hiroyama, T. Sawa, A. Suenaga, T. Maruyama, T. Kai and M. Otagiri, J. Biol. Chem., 2008, 283, 34966–34975 CrossRef CAS PubMed
- H. Katsumi, M. Nishikawa, F. Yamashita and M. Hashida, J. Pharmacol. Exp. Ther., 2005, 314, 1117–1124 CrossRef CAS PubMed
- S.-L. Huang, P. H. Kee, H. Kim, M. R. Moody, S. M. Chrzanowski, R. C. Macdonald and D. D. McPherson, J. Am. Coll. Cardiol., 2009, 54, 652–659 CrossRef CAS PubMed
- M. C. Frost and M. E. Meyerhoff, J. Am. Chem. Soc., 2004, 126, 1348–1349 CrossRef CAS PubMed
- M. C. Frost and M. E. Meyerhoff, J. Biomed. Mater. Res., Part A, 2005, 72, 409–419 CrossRef PubMed
- A. W. Carpenter, D. L. Slomberg, K. S. Rao and M. H. Schoenfisch, ACS Nano, 2011, 5, 7235–7244 CrossRef CAS PubMed
- M. H. Kafshgari, A. Cavallaro, B. Delalat, F. J. Harding, S. J. McInnes, E. Mäkilä, J. Salonen, K. Vasilev and N. H. Voelcker, Nanoscale Res. Lett., 2014, 9, 333 CrossRef PubMed
- D. L. Slomberg, Y. Lu, A. D. Broadnax, R. A. Hunter, A. W. Carpenter and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2013, 5, 9322–9329 CAS
- A. R. Rothrock, R. L. Donkers and M. H. Schoenfisch, Society, 2005, 127, 9362–9363 CAS
- M. A. Polizzi, N. A. Stasko and M. H. Schoenfisch, Langmuir, 2007, 23, 4938–4943 CrossRef CAS PubMed
- G. Lautner, M. E. Meyerhoff and S. P. Schwendeman, J. Controlled Release, 2016, 225, 133–139 CrossRef CAS PubMed
- P. S. Wheatley, A. R. Butler, M. S. Crane, S. Fox, B. Xiao, A. G. Rossi, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2006, 128, 502–509 CrossRef CAS PubMed
- M. Neidrauer, U. K. Ercan, A. Bhattacharyya, J. Samuels, J. Sedlak, R. Trikha, K. A. Barbee, M. S. Weingarten and S. G. Joshi, J. Med. Microbiol., 2014, 63, 203–209 CrossRef CAS PubMed
- J. L. Harding and M. M. Reynolds, J. Am. Chem. Soc., 2012, 134, 3330–3333 CrossRef CAS PubMed
- J. L. Harding and M. M. Reynolds, J. Mater. Chem. B, 2014, 2, 2530–2536 RSC
- J. L. Harding, J. M. Metz and M. M. Reynolds, Adv. Funct. Mater., 2014, 24, 7503–7509 CrossRef CAS
- M. J. Neufeld, J. L. Harding and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2015, 7, 26742–26750 CAS
- N. A. Stasko, T. H. Fischer and M. H. Schoenfisch, Biomacromolecules, 2008, 9, 834–841 CrossRef CAS PubMed
- C. J. Backlund, B. V. Worley and M. H. Schoenfisch, Acta Biomater., 2016, 29, 198–205 CrossRef CAS PubMed
- W. L. Storm, J. A. Johnson, B. V. Worley, D. L. Slomberg and M. H. Schoenfisch, J. Biomed. Mater. Res., Part A, 2014, 1–11 Search PubMed
- D. A. Riccio, K. P. Dobmeier, E. M. Hetrick, B. J. Privett, H. S. Paul and M. H. Schoenfisch, Biomaterials, 2009, 30, 4494–4502 CrossRef CAS PubMed
- P. Nacharaju, C. Tuckman-Vernon, K. E. Maier, J. Chouake, A. Friedman, P. Cabrales and J. M. Friedman, Nitric Oxide, 2012, 27, 150–160 CrossRef CAS PubMed
- P. N. Coneski, J. A. Nash and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2011, 3, 426–432 CAS
- J.-W. Yoo, H. Nurhasni, J. Cao, M. Choi, I. Kim, B. Luel Lee and Y. Jung, Int. J. Nanomed., 2015, 10, 3065–3080 CrossRef PubMed
- Y. Lu, D. L. Slomberg and M. H. Schoenfisch, Biomaterials, 2014, 35, 1716–1724 CrossRef CAS PubMed
- J. O. Kim, J. K. Noh, R. K. Thapa, N. Hasan, M. Choi, J. H. Kim, J. H. Lee, S. K. Ku and J. W. Yoo, Int. J. Biol. Macromol., 2015, 79, 217–225 CrossRef CAS PubMed
- C. Vogt, Q. Xing, W. He, B. Li, M. C. Frost and F. Zhao, Biomacromolecules, 2013, 14, 2521–2530 CrossRef CAS PubMed
- P. G. Parzuchowski, M. C. Frost and M. E. Meyerhoff, J. Am. Chem. Soc., 2002, 124, 12182–12191 CrossRef CAS PubMed
- A. B. Seabra, R. Da Silva, G. F. P. De Souza and M. G. De Oliveira, Artif. Organs, 2008, 32, 262–267 CrossRef CAS PubMed
- J. P. Yapor, A. Lutzke, A. Pegalajar-Jurado, B. H. Neufeld, V. B. Damodaran and M. M. Reynolds, J. Mater. Chem. B, 2015, 3, 9233–9241 RSC
- G. E. Gierke, M. Nielsen and M. C. Frost, Sci. Technol. Adv. Mater., 2011, 12, 055007 CrossRef
- A. Pegalajar-Jurado, K. A. Wold, J. M. Joslin, B. H. Neufeld, K. A. Arabea, L. A. Suazo, S. L. McDaniel, R. A. Bowen and M. M. Reynolds, J. Controlled Release, 2015, 220, 617–623 CrossRef CAS PubMed
- A. Wan, Q. Gao and H. Li, J. Mater. Sci. Mater. Med., 2008, 20, 321–327 CrossRef PubMed
- J. Park, J. Kim, K. Singha, D. K. Han, H. Park and W. J. Kim, Biomaterials, 2013, 34, 8766–8775 CrossRef CAS PubMed
- J. Kim, Y. Lee, K. Singha, H. W. Kim, J. H. Shin, S. Jo, D. K. Han and W. J. Kim, Bioconjugate Chem., 2011, 22, 1031–1038 CrossRef CAS PubMed
- A. B. Seabra, A. Fitzpatrick, J. Paul, M. G. De Oliveira and R. Weller, Br. J. Dermatol., 2004, 151, 977–983 CrossRef CAS PubMed
- K. Masters, S. Leibovich, P. Belem, J. L. West and L. Poole-Warren, Wound Repair Regen., 2002, 10, 286–294 CrossRef PubMed
- M. M. Reynolds, J. A. Hrabie, B. K. Oh, J. K. Politis, M. L. Citro, L. K. Keefer and M. E. Meyerhoff, Biomacromolecules, 2006, 7, 987–994 CrossRef CAS PubMed
- M. M. Reynolds, J. E. Saavedra, B. M. Showalter, C. A. Valdez, A. P. Shanklin, B. K. Oh, L. K. Keefer and M. E. Meyerhoff, J. Mater. Chem., 2010, 20, 3107–2114 RSC
- B. Parzuchowska and P. Parzuchowski, Polimery, 2014, 59, 105–194 CrossRef
- H. Zhang, G. M. Annich, J. Miskulin, K. Stankiewicz, K. Osterholzer, S. I. Merz, R. H. Bartlett and M. E. Meyerhoff, J. Am. Chem. Soc., 2003, 125, 5015–5024 CrossRef CAS PubMed
- J. E. Saavedra, M. N. Booth, J. A. Hrabie, K. M. Davies and L. K. Keefer, J. Org. Chem., 1999, 64, 5124–5131 CrossRef CAS
- H. Zhang, G. M. Annich, J. Miskulin, K. Osterholzer, S. I. Merz, R. H. Bartlett and M. E. Meyerhoff, Biomaterials, 2002, 23, 1485–1494 CrossRef CAS PubMed
- D. J. Smith, D. Chakravarthy, S. Pulfer, M. L. Simmons, J. A. Hrabie, M. L. Citro, J. E. Saavedra, K. M. Davies, T. C. Hutsell, D. L. Mooradian, S. R. Hanson and L. K. Keefer, J. Med. Chem., 1996, 39, 1148–1156 CrossRef CAS PubMed
- Y. Kou and A. Wan, Bioorg. Med. Chem. Lett., 2008, 18, 2337–2341 CrossRef CAS PubMed
- Y. Kang, J. Kim, Y. M. Lee, S. Im, H. Park and W. J. Kim, J. Controlled Release, 2015, 220, 624–630 CrossRef CAS PubMed
- N. A. Stasko and M. H. Schoenfisch, J. Am. Chem. Soc., 2006, 128, 8265–8271 CrossRef CAS PubMed
- C. W. McCarthy, J. Goldman and M. C. Frost, ACS Appl. Mater. Interfaces, 2016, 8, 5898–5905 CAS
- V. B. Damodaran, L. W. Place, M. J. Kipper and M. M. Reynolds, J. Mater. Chem., 2012, 22, 23038 RSC
- T. Kean and M. Thanou, Adv. Drug Delivery Rev., 2010, 62, 3–11 CrossRef CAS PubMed
- C. Wang, F. Yang, Z. Xu, D. Shi, D. Chen, J. Dai, N. Gu and Q. Jiang, Thromb. Res., 2013, 131, e31–e38 CrossRef CAS PubMed
- L. A. Tai, Y. C. Wang and C. S. Yang, Nitric Oxide - Biol. Chem., 2010, 23, 60–64 CrossRef CAS PubMed
- J. J. Koehler, J. Zhao, S. S. Jedlicka, D. M. Porterfield and J. L. Rickus, J. Phys. Chem. B, 2008, 112, 15086–15093 CrossRef CAS PubMed
- H. S. Jeh, S. Lu and S. C. George, J. Microencapsulation, 2004, 21, 3–13 CrossRef CAS PubMed
- C. Wischke and S. P. Schwendeman, Int. J. Pharm., 2008, 364, 298–327 CrossRef CAS PubMed
- S. M. Shishido, A. B. Seabra, W. Loh and M. G. de Oliveira, Biomaterials, 2003, 24, 3543–3553 CrossRef CAS PubMed
- M. G. de Oliveira, S. M. Shishido, A. B. Seabra and N. H. Morgon, J. Phys. Chem. A, 2002, 106, 8963–8970 CrossRef CAS
- H. Gappa-Fahlenkamp and R. S. Lewis, Biomaterials, 2005, 26, 3479–3485 CrossRef CAS PubMed
- T. C. Major, D. O. Brant, C. P. Burney, K. A. Amoako, G. M. Annich, M. E. Meyerhoff, H. Handa and R. H. Bartlett, Biomaterials, 2011, 32, 5957–5969 CrossRef CAS PubMed
- S. C. Puiu, Z. Zhou, C. C. White, L. J. Neubauer, Z. Zhang, L. E. Lange, J. A. Mansfield, M. E. Meyerhoff and M. M. Reynolds, J. Biomed. Mater. Res., Part B, 2009, 91, 203–212 CrossRef PubMed
- B. K. Oh and M. E. Meyerhoff, Biomaterials, 2004, 25, 283–293 CrossRef CAS PubMed
- V. Wonoputri, C. Gunawan, S. Liu, N. Barraud, L. H. Yee, M. Lim and R. Amal, ACS Appl. Mater. Interfaces, 2015, 7, 22148–22156 CAS
- H. Ren, M. A. Coughlin, T. C. Major, S. Aiello, A. Rojas Pena, R. H. Bartlett and M. E. Meyerhoff, Anal. Chem., 2015, 8067–8072 CrossRef CAS PubMed
- Y. Wu, Z. Zhou and M. E. Meyerhoff, J. Biomed. Mater. Res., Part A, 2007, 81, 956–963 CrossRef PubMed
- A. M. Skrzypchak, N. G. Lafayette, R. H. Bartlett, Z. Zhou, M. C. Frost, M. E. Meyerhoff, M. M. Reynolds and G. M. Annich, Perfusion, 2007, 22, 193–200 CrossRef PubMed
- C. W. McCarthy, R. J. Guillory, J. Goldman and M. C. Frost, ACS Appl. Mater. Interfaces, 2016, 8, 10128–10135 CAS
- B. M. Bloom, J. Grundlingh, J. P. Bestwick and T. Harris, Eur. J. Emerg. Med., 2014, 21, 81–88 CrossRef PubMed
- D. S. Martin and M. P. W. Grocott, Crit. Care Med., 2013, 41, 423–432 CrossRef CAS PubMed
- The Juvenile Diabetes Research Foundation Continuous Glucose Monitoring Study Group, N. Engl. J. Med., 2008, 359, 1464–1476 CrossRef PubMed
- K. E. A. Burns and A. McLaren, Can. Respir. J., 2009, 16, 163–165 CrossRef PubMed
- S. M. Marxer, M. E. Robbins and M. H. Schoenfisch, Analyst, 2005, 130, 206–212 RSC
- K. A. Mowery, M. H. Schoenfisch, N. Baliga, J. A. Wahr and M. E. Meyerhoff, Electroanalysis, 1999, 11, 681–686 CrossRef CAS
- M. H. Schoenfisch, K. A. Mowery, M. V. Rader, N. Baliga, J. A. Wahr and M. E. Meyerhoff, Anal. Chem., 2000, 72, 1119–1126 CrossRef CAS PubMed
- M. C. Frost, S. M. Rudich, H. Zhang, M. A. Maraschio and M. E. Meyerhoff, Anal. Chem., 2002, 74, 5942–5947 CrossRef CAS PubMed
- Y. Wu, A. P. Rojas, G. W. Griffith, A. M. Skrzypchak, N. Lafayette, R. H. Bartlett and M. E. Meyerhoff, Sens. Actuators, B, 2007, 121, 36–46 CrossRef CAS
- Q. Yan, T. C. Major, R. H. Bartlett and M. E. Meyerhoff, Biosens. Bioelectron., 2011, 26, 4276–4282 CrossRef CAS PubMed
- A. K. Wolf, Y. Qin, T. C. Major and M. E. Meyerhoff, Chin. Chem. Lett., 2015, 26, 464–468 CrossRef CAS
- J. H. Shin, S. M. Marxer and M. H. Schoenfisch, Anal. Chem., 2004, 76, 4543–4549 CrossRef CAS PubMed
-
M. C. Frost, A. K. Wolf and M. E. Meyerhoff, in Detection Challenges in Clinical Diagnostics, 2013, pp. 129–155 Search PubMed
- R. K. Meruva and M. E. Meyerhoff, Biosens. Bioelectron., 1998, 13, 201–212 CrossRef CAS PubMed
- K. P. Dobmeier, G. W. Charville and M. H. Schoenfisch, Anal. Chem., 2006, 78, 7461–7466 CrossRef CAS PubMed
- M. C. Frost, M. M. Batchelor, Y. Lee, H. Zhang, Y. Kang, B. Oh, G. S. Wilson, R. Gifford, S. M. Rudich and M. E. Meyerhoff, Microchem. J., 2003, 74, 277–288 CrossRef CAS
- T. C. Major, D. O. Brant, M. M. Reynolds, R. H. Bartlett, M. E. Meyerhoff, H. Handa and G. M. Annich, Biomaterials, 2010, 31, 2736–2745 CrossRef CAS PubMed
- T. C. Major, H. Handa, G. M. Annich and R. H. Bartlett, J.
Biomater. Appl., 2014, 29, 479–501 CrossRef CAS PubMed
- J. S. Stamler, Circ. Res., 2004, 94, 414–417 CrossRef CAS PubMed
- A. Ghaffari, C. C. Miller, B. McMullin and A. Ghahary, Nitric Oxide, 2006, 14, 21–29 CrossRef CAS PubMed
- D. P. Arora, S. Hossain, Y. Xu and E. M. Boon, Biochemistry, 2015, 54, 3717–3728 CrossRef CAS PubMed
- Y. Lu, D. Slomberg, B. Sun and M. Schoenfisch, Small, 2013, 9, 2189–2198 CrossRef CAS PubMed
- R. Vumma, C. S. Bang, R. Kruse, K. Johansson and K. Persson, J. Antibiot., 2015, 1–4 Search PubMed
- B. B. McMullin, D. R. Chittock, D. L. Roscoe, H. Garcha, L. Wang and C. C. Miller, Respir. Care, 2005, 50, 1451–1456 Search PubMed
- N. Barraud, M. J. Kelso, S. A. Rice and S. Kjelleberg, Curr. Pharm. Des., 2015, 21, 31–42 CrossRef CAS PubMed
- N. Barraud, D. J. Hassett, S.-H. Hwang, S. A. Rice, S. Kjelleberg and J. S. Webb, J. Bacteriol., 2006, 188, 7344–7353 CrossRef CAS PubMed
- N. Barraud, M. V. Storey, Z. P. Moore, J. S. Webb, S. A. Rice and S. Kjelleberg, Microb. Biotechnol., 2009, 2, 370–378 CrossRef CAS PubMed
- B. J. Nablo and M. H. Schoenfisch, J. Biomed. Mater. Res., Part A, 2003, 67, 1276–1283 CrossRef PubMed
- E. J. Brisbois, J. Bayliss, J. Wu, T. C. Major, C. Xi, S. C. Wang, R. H. Bartlett, H. Handa and M. E. Meyerhoff, Acta Biomater., 2014, 4136–4142 CrossRef CAS PubMed
- M. R. Schäffer, P. A. Efron, F. J. Thornton, K. Klingel, S. S. Gross and A. Barbul, J. Immunol., 1997, 158, 2375–2381 Search PubMed
- M. B. Witte and A. Barbul, Am. J. Surg., 2002, 183, 406–412 CrossRef CAS PubMed
- H. P. Shi, D. T. Efron, D. Most and A. Barbul, Surgery, 2001, 130, 225–229 CrossRef CAS PubMed
- G. Richardson and N. Benjamin, Clin. Sci., 2002, 105, 99–105 CrossRef
- G. E. Romanowicz, W. He, M. Nielsen and M. C. Frost, Redox Biol., 2013, 1, 332–339 CrossRef CAS PubMed
- T.-K. Nguyen, R. Selvanayagam, K. K. K. Ho, R. Chen, S. K. Kutty, S. A. Rice, N. Kumar, N. Barraud, H. T. T. Duong and C. Boyer, Chem. Sci., 2016, 7, 1016–1027 RSC
| This journal is © The Royal Society of Chemistry 2016 |