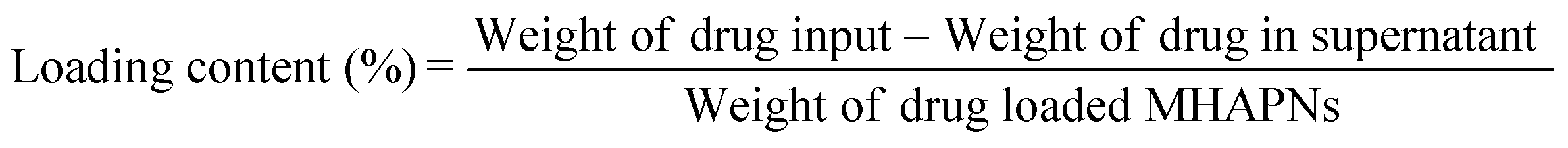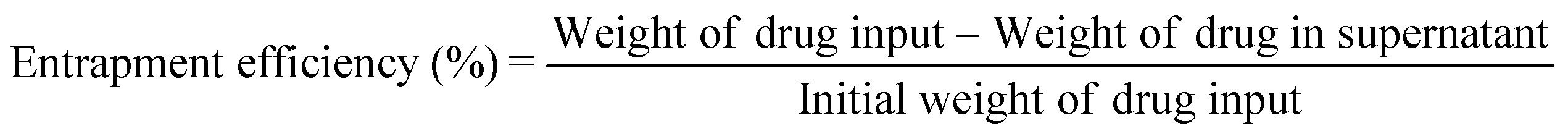DOI:
10.1039/C5BM00228A
(Paper)
Biomater. Sci., 2016,
4, 272-280
Preparation of pH-responsive mesoporous hydroxyapatite nanoparticles for intracellular controlled release of an anticancer drug†
Received
6th July 2015
, Accepted 5th October 2015
First published on 20th October 2015
Abstract
A well-defined core–shell nano-carrier (PAA–MHAPNs) was successfully synthesized based on a graft-onto method by using mesoporous hydroxyapatite nanoparticles (MHAPNs) as the core and polyacrylic acid (PAA) as the shell. Given that MHAPNs are regarded as one of the most promising drug delivery vehicles due to their excellent performance and the nature of their cancer cell anti-proliferative effect, and the grafted PAA, as a pH-responsive switch, could improve the loading amount of the drug doxorubicin hydrochloride (DOX) effectively by electrostatic interactions, all these advantages mean that the designed models show promise for application in pH-responsive drug delivery systems. The loading content and entrapment efficiency of DOX could reach up to 3.3% and 76%, respectively. The drug release levels of the constructed DOX@PAA–MHAPNs were low under normal physiological conditions (pH 7.4), but they could be increased significantly with a decrease of pH. Cytotoxicity assays indicated that the PAA–MHAPNs was biocompatible, and more importantly, the DOX@PAA–MHAPNs demonstrated an obvious ability to induce apoptosis of cancer cells. Overall, the synthesized systems should show great potential as drug nanovehicles with excellent biocompatibility, high drug loading, and pH-responsive features for future intracellular drug delivery.
1 Introduction
In recent years, much attention has been paid to developing new drug-delivery systems with obvious advantages compared to conventional forms of dosage, such as enhanced bioavailability, higher efficiency, lower toxicity, controlled release and so on. Up to now, many materials including polymeric micelles,1 dendrimers,2 liposomes,3 and various inorganic nanoparticles,4–6 have been utilized as drug carriers in drug delivery systems. Among these, mesoporous hydroxyapatite nanoparticles (MHAPNs) are now attracting more and more interest, with particular attention in the drug storage and release field given their large pore volumes, large surface area, and easily modified surface features for site-specific delivery.7–10 Hydroxyapatite [Ca10(PO4)6(OH)2] (HAP), which is a major component of bone and teeth, is a key biomaterial in view of its excellent biocompatibility, bioactivity, nontoxicity and noninflammatory nature.11,12 Moreover, considering that its degradation products could be absorbed by the body, such a material would be an excellent candidate as a drug delivery carrier. Mesoporous hydroxyapatite nanoparticles (MHAPNs) are often prepared by a surfactant templating method.13,14 Due to their unique advantages including biocompatibility, solubility and low toxicity, it is highly desirable to design a drug delivery system based on MHAPNs with controlled drug release behavior in a specific environment in response to external stimuli. To achieve this goal, a variety of systems based on MHAPNs have been reported which show the controlled release of drug molecules in response to external stimuli.15–19 A critical step in constructing the stimuli-responsive controlled drug release system was finding how to seal the mesopores to block the drug molecules inside the pores with “zero release” and “open” functions of the sealants in response to external stimuli. From reports in the literature, the current study found that the release of guest molecules could be achieved in response to external stimuli such as temperature,20,21 pH,22–27 light,28–30 redox reagents,31–33 ultrasound,34,35 enzymes,36–38etc. Among these specific stimulation systems, the pH-responsive system is of special interest for cancer therapy since both the tumor extracellular environment (pH 6.8) and endosomes (pH 5.0) are more acidic than normal tissues (pH 7.4), which enables the carriers to release anticancer drugs in a pH dependent manner.39,40 Up to now, although many ordered mesoporous materials have been developed to build pH-responsive systems,41–43 in most of these systems the drug molecules were physically adsorbed in the channels and the drug-loading capacity was generally low, so to further optimize this system, by the construction of a smart controlled-release system for intracellular drug delivery, in particular with a high drug loading efficiency, should be very interesting and this still requires considerable effort.
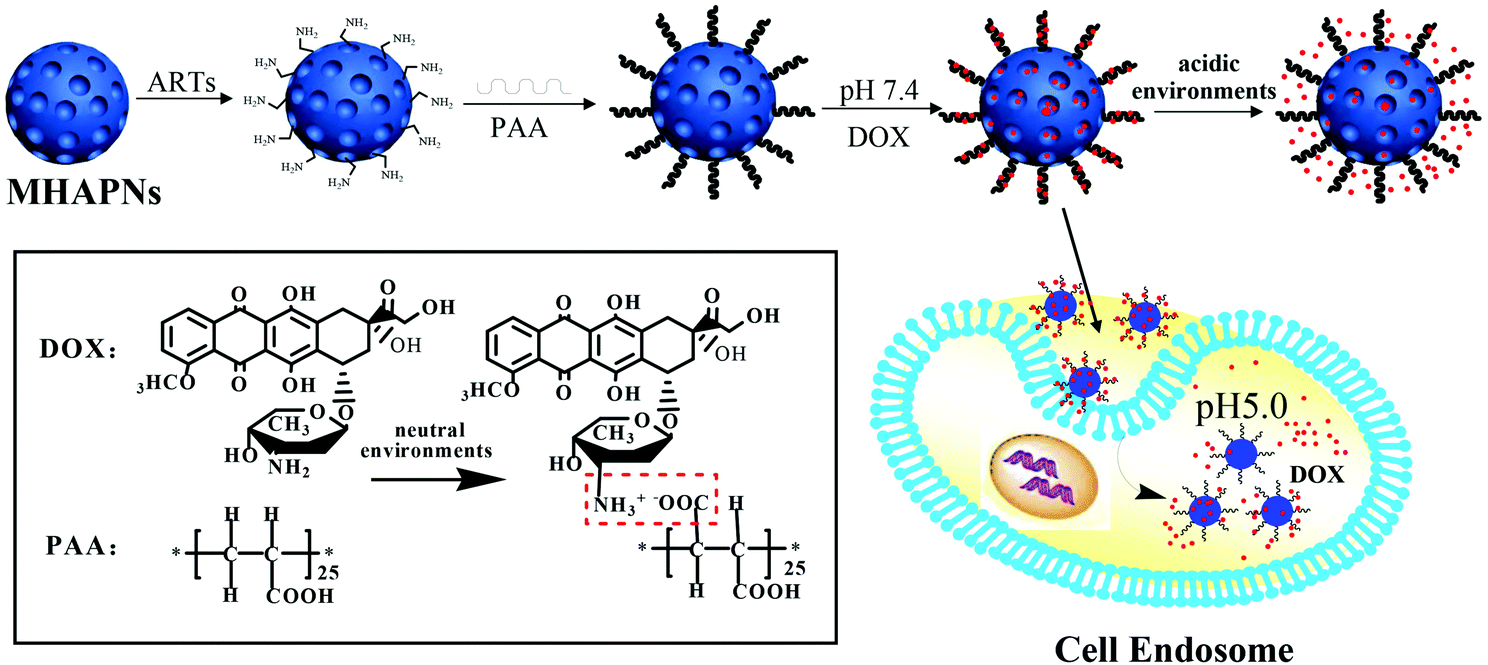
Herein, we designed and constructed a core–shell nanocarrier (PAA–MHAPNs) based on mesoporous HAP as the core which was end-capped with PAA as the shell, for the study of its great potential for pH-responsive controlled drug release behavior. PAA is a superior choice due to its favorable properties, such as biocompatibility, hydrophilicity, easy modification and significant blocking effect.44,45 Moreover, the charge of PAA is pH tunable. The covalently grafted PAA chains acted not only as a switch to modulate the release of the loaded drug, but also as a binding site to improve the loading content of the drug due to electrostatic interactions between the drug and the carboxyl groups in the side chain of the PAA. In our study, doxorubicin hydrochloride (DOX) was chosen as a model drug to assess the drug loading and release behaviors of PAA–MHAPNs. In physiological medium (pH 7.4), DOX was protonized and more of the PAA was deprotonized, thus the positively charged DOX was adsorbed onto the surface of the negatively charged PAA–MHAPNs by strong electrostatic interactions. Meanwhile, due to the fact that the concentration of DOX in the solution was higher than that in the interior channel of the PAA–MHAPNs, the DOX would load constantly into the channels of the PAA–MHAPNs due to the diffusion effect. When PAA–MHAPNs loaded with DOX was in acidic subcellular environments, DOX would be released owing to the dissociation of electrostatic interactions between the DOX and PAA–MHAPNs. Thus, the system could serve as a selective tumor drug delivery system. To the best of our knowledge, this is the first study to employ PAA molecules as switches to construct intracellular pH-responsive controlled release systems based on MHAPNs for drug delivery.
2 Materials and methods
2.1 Materials
PEO99PPO65PEO99 (F127), N-hydroxysuccinimide (NHS), doxorubicin hydrochloride (DOX), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC), and poly(acrylic acid) (PAA) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Calcium pantothenate (C18H32CaN2O10), dipotassium hydrogenphosphate trihydrate (K2HPO4·3H2O), 1,3,5-trimethylbenzene (TMB), 3-aminopropyltriethoxysilane (ARTS), trypan blue and triton X-100 were obtained from J&K Scientific Co. (BeiJing, PR China). Human hepatocellular liver carcinoma cells (HepG2 cells), bovine serum, penicillin and streptomycin were provided by Sanggon biotech Co. (Shanghai, PR China). All the initial chemicals in the work were used without further purification.
2.2 Preparation of MHAPNs
The MHAPNs were synthesized using a templating method according to a previous report.35 The block co-polymer pluronic F127 was selected as the template, and TMB was used as the pore-expanding agent. Firstly, 2.26 g of F127 and 18.43 g of calcium pantothenate (C18H32CaN2O10) were co-dissolved in 100 mL of distilled water and stirred vigorously for 2 h, then 16 mL of 1,3,5-trimethylbenzene (TMB) was added to get a clear micellar solution as the calcium ion source. 5.16 g of dipotassium hydrogenphosphate trihydrate (K2HPO4·3H2O) was dissolved in 50 mL of distilled water to produce the phosphate ion source. Subsequently, the pH of the phosphate solution was adjusted to 12.0 with ammonia (NH3·H2O). Finally, the PO43− solution was added dropwise to the F127/Ca2+ solution. The mixture solution was heated to 100 °C under reflux for 36 h and filtered to collect a precipitate. The white precipitate was subsequently dried in an oven at 100 °C for 48 h, and then calcined at 650 °C for 4 h in a muffle furnace.
2.3 The construction of drug loading systems (DOX@PAA–MHAPNs)
Firstly, 1.46 g of MHAPNs and 2.5 mL of 3-aminopropyltriethoxysilane (ARTS) were dissolved in 100 mL of toluene under stirring conditions. The mixture solution was heated to 80 °C under reflux for 36 h and filtered to collect a precipitate. The white precipitate was subsequently dried at 100 °C for 48 h in a vacuum oven to get NH2-MHAPNs. Then, 1.1 g of NH2-MHAPNs was dispersed in 150 mL of N,N-dimethylformamide (DMF), and 1.8 g of PAA (Mw = 3000) was added to the mixture. Subsequently, the reaction mixture was stirred at 120 °C for 4 h, and 140 °C for 6 h. After the reaction, the mixture was centrifuged to obtain a precipitate. The precipitate was washed with ethanol and water ten times to remove the solvent and unreacted PAA. Finally, the product was dried in a vacuum oven at 100 °C for 24 h to obtain PAA–MHAPNs. Doxorubicin hydrochloride (DOX), a well-known anticancer drug, was utilized as a model drug. Firstly, 30 mg of DOX and 15 mg of PAA–MHAPNs were dissolved in 100 mL of pH 7.4 buffer solution and stirred for 48 h at room temperature. Then the nanoparticles were centrifuged and washed thoroughly with pH 7.4 PBS ten times to remove the unloaded and physically absorbed DOX. The DOX@PAA–MHAPNs was obtained after vacuum drying. The drug loading content and entrapment efficiency were calculated using the following equations:
2.4 Drug loading and release assay
The PAA–MHAPNs was used as a carrier for DOX. The end-capping efficiency and release behavior of DOX@MHAPNs and DOX@PAA–MHAPNs were analysed by UV-vis spectroscopy. 3.0 mg of the DOX@MHAPNs and DOX@PAA–MHAPNs powders was dispersed in 10 mL of phosphate buffer solution (PBS) at pH 7.4, 6.5 or 5.0. The dispersion was transferred into a dialysis bag (molecular weight cut-off = 8000 g mol−1), and the bag was then immersed in 100 mL of PBS solution with the same pH conditions. The volume of the dissolution media was maintained at 100 mL at 37 °C. 1.0 mL of solution was withdrawn at a given time interval, followed by the addition of the same volume of fresh PBS solution. The amount of released drug in the PBS solution was measured using a UV-vis spectrophotometer at 480 nm. In order to investigate the rapid release response, in another assay, 3.0 mg of DOX@PAA–MHAPNs was suspended in 10 mL of PBS (pH 7.4). The suspension was transferred into a dialysis bag (molecular weight cut-off = 8000 g mol−1), and subsequently placed in a beaker containing 100 mL of PBS buffer with the same pH conditions for 5 h. Then the pH of the solution was adjusted to pH 5.0. At a predetermined time, 1 mL of the nanoparticle suspension was withdrawn, followed by the addition of the same volume of fresh PBS solution. The supernatant was taken for UV-vis spectrophotometric analysis at 480 nm to determine the amount of released DOX.
2.5 Cell culture
HepG2 cells were cultured in low-glucose Dulbecco's Modified Eagle's Medium supplemented with 10% bovine serum (FBS; Gibco), 100 U mL−1 of penicillin and 100 μg mL−1 of streptomycin in a 5% CO2 incubator at 37 °C under 95% humidity. The cell culture media was changed every 2 days. When reaching confluence, the cells were detached with 0.25% trypsin in 1 mM tetrasodium EDTA, then centrifuged and resuspended in complete medium for reseeding in new culture flasks.
2.6 Cytotoxicity assay
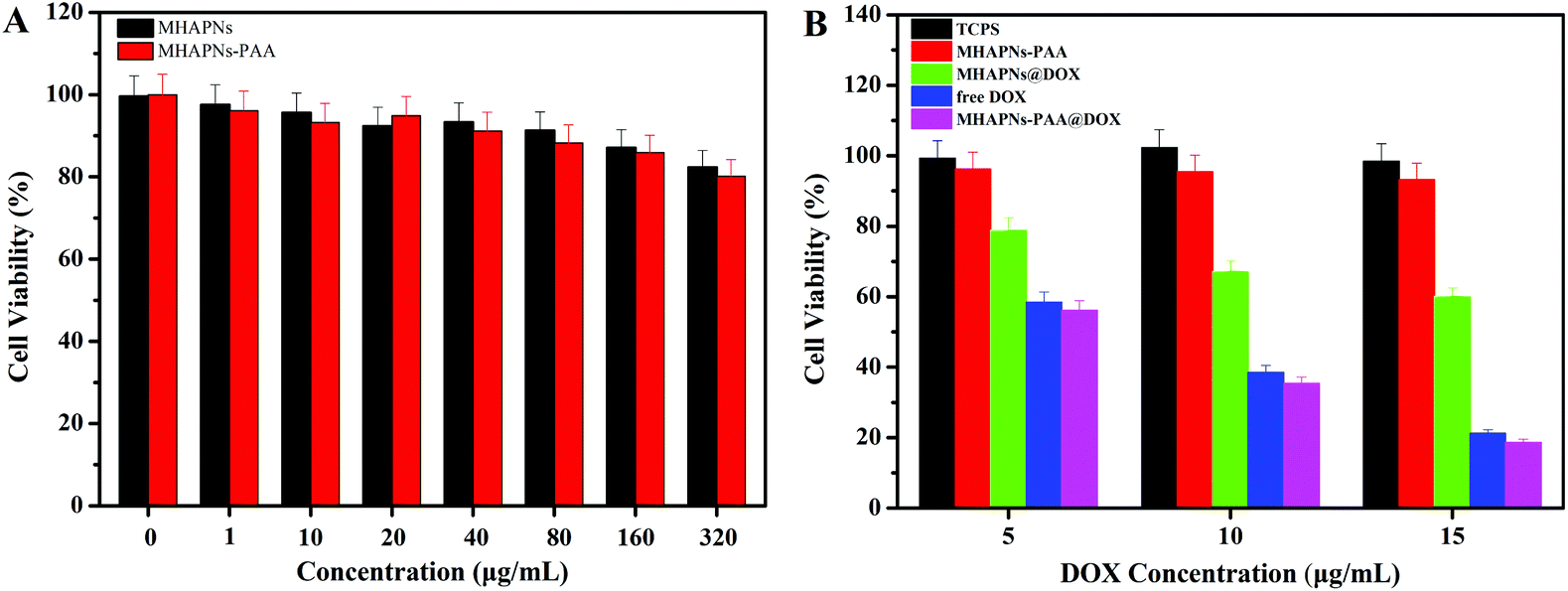
HepG2 cells were used to evaluate the cytotoxicity of MHAPNs, PAA–MHAPNs, DOX@MHAPNs, DOX@PAA–MHAPNs, and free DOX by MTT assay. HepG2 cells were seeded on 24-well plates with an initial seeding density of 2 × 104 cells per cm2. The cells were then rinsed with PBS. Next, MHAPNs, PAA–MHAPNs, DOX@MHAPN, free DOX and DOX@PAA–MHAPNs were added to each well co-culture in 5% CO2 at 37 °C for 24 h. Free DOX was used as a positive control at the same dose as the PAA–MHAPNs loading. After culturing for 24 h, the cells were rinsed with PBS solution and placed in fresh culture medium. About 0.1 mL of MTT (5 mg mL−1) was added to each well, and incubated in the CO2 incubator at 37 °C for another 4 h. Then, the MTT containing medium was removed and 0.5 mL of dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals that had formed. The absorbance values of formazan were determined with a Bio-Rad model-680 microplate reader at 490 nm. Six replicates were done for each treatment group.
2.7 Confocal laser scanning microscopy (CLSM) observation
Suspensions of 20 μg mL−1 of DOX@MHAPNs and DOX@PAA–MHAPNs in PBS were introduced into the culture medium overnight to mimic the blood circulation process prior to cellular uptake. After incubation with the nanoparticles in the medium for 12 h and 24 h, the cells were washed five times with pH 7.4 PBS to remove the residual nanoparticles. Then 200 μg mL−1 trypan blue was added to quench the extracellular fluorescence for 10 min. The cells were fixed with 2% glutaraldehyde at 4 °C for 20 min, then the fixed samples were rinsed with excess PBS buffer and permeabilized with 0.2% Triton X-100 at 4 °C for 10 min. The nuclei of the cells were stained with 10 μg ml−1 Hoechst 33258 at 4 °C for 10 min. Finally, the stained samples were mounted in 90% glycerine. The distribution of DOX in the HepG2 cells was observed with confocal laser scanning microscopy (CLSM).
2.8 Characterization
A series of characterization techniques was used to analyze the structural properties of each product. The morphology and mesoporous aperture of the nanoparticles were observed by transmission electron microscopy (TEM; Philips EM20). The structure of the nanoparticles was analyzed by Fourier transform infrared (FTIR; VECTOR22, BRUKER) spectroscopy within a scanning range of 4000–400 cm−1 using the KBr pellet technique. 1H-NMR analysis was performed with a 1-Bay 500 NMR instrument (500 MHz, Bruker, Germany) with D2O as the solvent. Powder X-ray diffraction (XRD) was recorded with a Bruker D4 X-ray diffractometer with Ni-filtered Cu Kα radiation (40 KV, 40 mA). The size distribution of the nanoparticles was measured by dynamic light scattering (DLS) using a Mastersizer 3000. N2 adsorption–desorption isotherms were measured with an automatic surface area and porosity analyzer (3H-2000PS2, Beishide) at 77 K. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas using adsorption data in a relative pressure range from 0.05 to 0.95. The pore volumes and pore size distributions were derived from the desorption branches of the isotherms using the Barrett–Joyner–Halanda (BJH) method. The zeta potential of the nanoparticles was measured using a zeta potential analyzer (Nanotrac wave, Microtrac) at 25 °C with DI H2O as the solvent. Thermogravimetric analysis (TGA) curves were record using a Perkin Elmer PYRIS 1 DSC at a heating rate of 10 °C min−1 in a nitrogen flow, from 0 to 800 °C. The pH-responsive release of the drug was detected by a UV-vis spectrophotometer (LS50B, PerkinElmer) at 480 nm. Confocal laser scanning microscopy (CLSM, LSM 510Metanlo, Zeiss) was used to detect the distribution of the DOX within the cells.
3 Results and discussion
The general procedure used to construct the pH-responsive nanocarrier DOX@PAA–MHAPNs is shown in Fig. 1. The MHAPNs were synthesized using the templating method. PAA was grafted onto the surface of the MHAPNs by an amidation reaction. DOX was loaded into the MHAPNs by both electrostatic interactions and the diffusion effect. The loading content and the entrapment efficiency of DOX could be as high as 3.3% and 76%, respectively, at a weight ratio of DOX and PAA–MHAPNs of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. However, the loading content and the entrapment efficiency of DOX in DOX@MHAPNs were only 0.26% and 43%, respectively.
:1. However, the loading content and the entrapment efficiency of DOX in DOX@MHAPNs were only 0.26% and 43%, respectively.
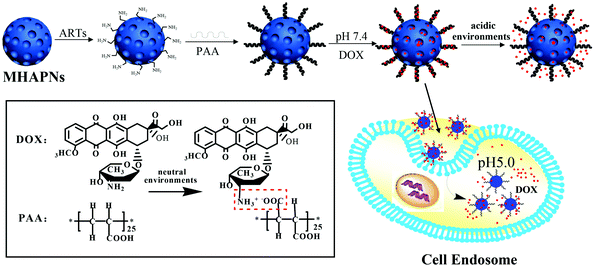 |
| | Fig. 1 Schematic illustration of the preparation of DOX@PAA–MHAPNs and the intracellular pH-responsive drug delivery system. | |
3.1 Characterization of the DOX@PAA–MHAPNs system
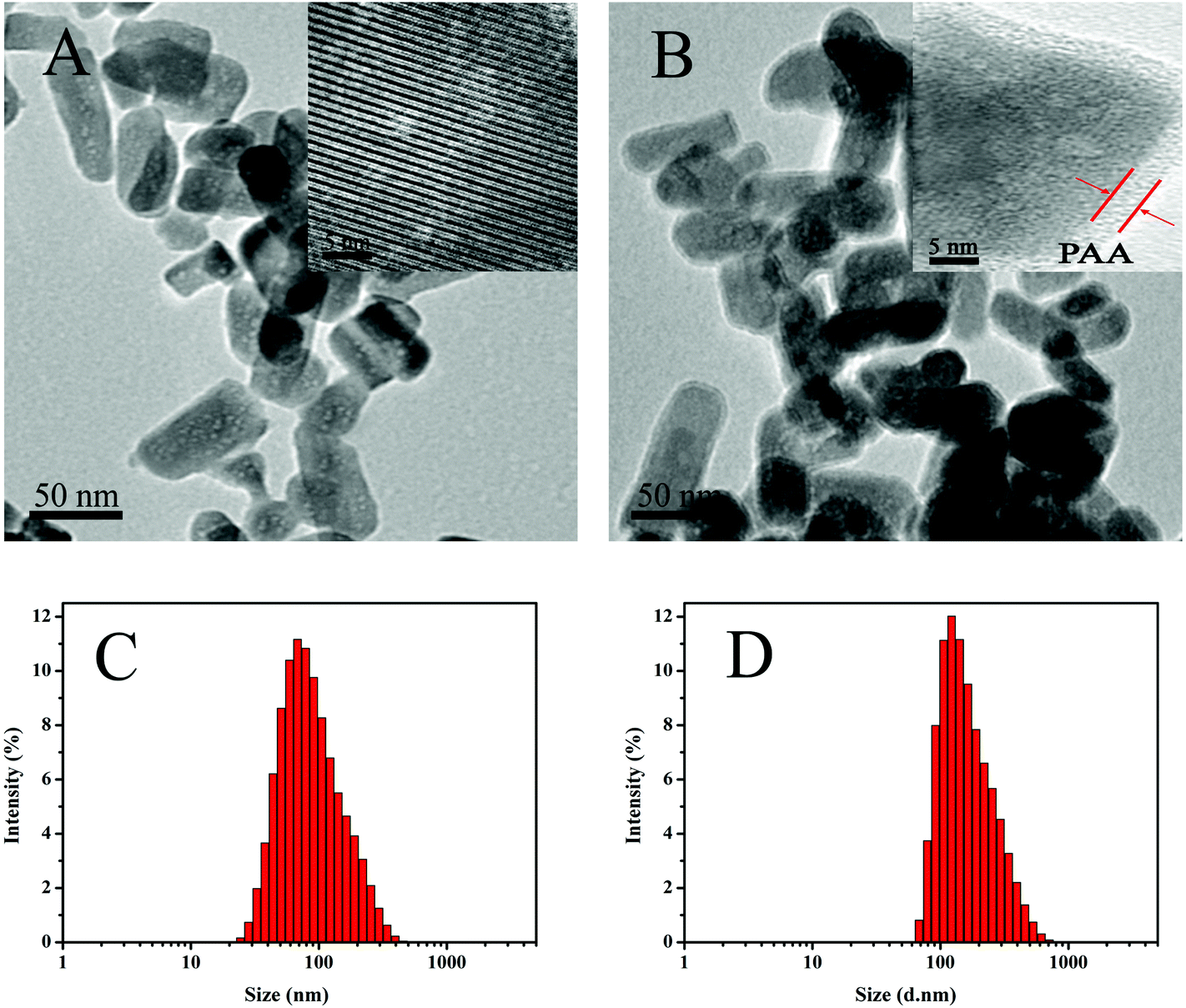
Fig. 2A and B show the TEM images of the MHAPNs and PAA–MHAPNs. It can be seen that the prepared MHAPNs were uniform rod-like nanoparticles with a mean width of 40 nm and length of 80 nm (Fig. S1A and 1B†). In the subsequently magnified TEM image (Fig. 2A, inset), a highly ordered mesoporous network was clearly observed, which was characteristic of the MHAPNs. In Fig. 2B, the rod-like nanoparticles also suggested that the anchored PAA increased the width and length of the MHAPNs (Fig. S1C and 1D†). From the highly magnified image (Fig. 2B, inset), the blurry pore structure and the border around the MHAPNs indicated the existence of a PAA shell around the particle with a thickness of about 3.8 nm despite the low contrast of PAA in TEM imaging. In addition, the diameter and size distribution of the MHAPNs and PAA–MHAPNs were measured by DLS. As depicted in Fig. 2C and D, the diameter of the MHAPNs was about 100 nm with a polydispersity index (PDI) of 0.351, which was larger than that observed from TEM because of the reunion of some of the MHAPNs in deionized water. The diameter of PAA–MHAPNs was about 128 nm and the PDI was about 0.266, indicating that the PAA–MHAPNs system showed a better dispersibility in water, which should be one prerequisite for PAA–MHAPNs serving as an excellent drug carrier.
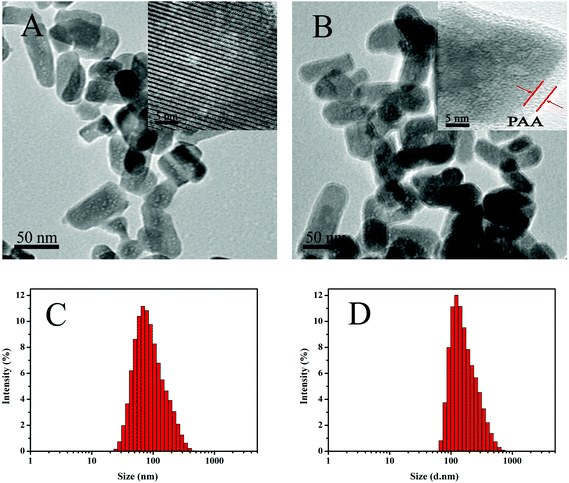 |
| | Fig. 2 TEM images of MHAPNs (A) and PAA–MHAPNs (B), and the size distribution of MHAPNs (C) and PAA–MHAPNs (D). | |
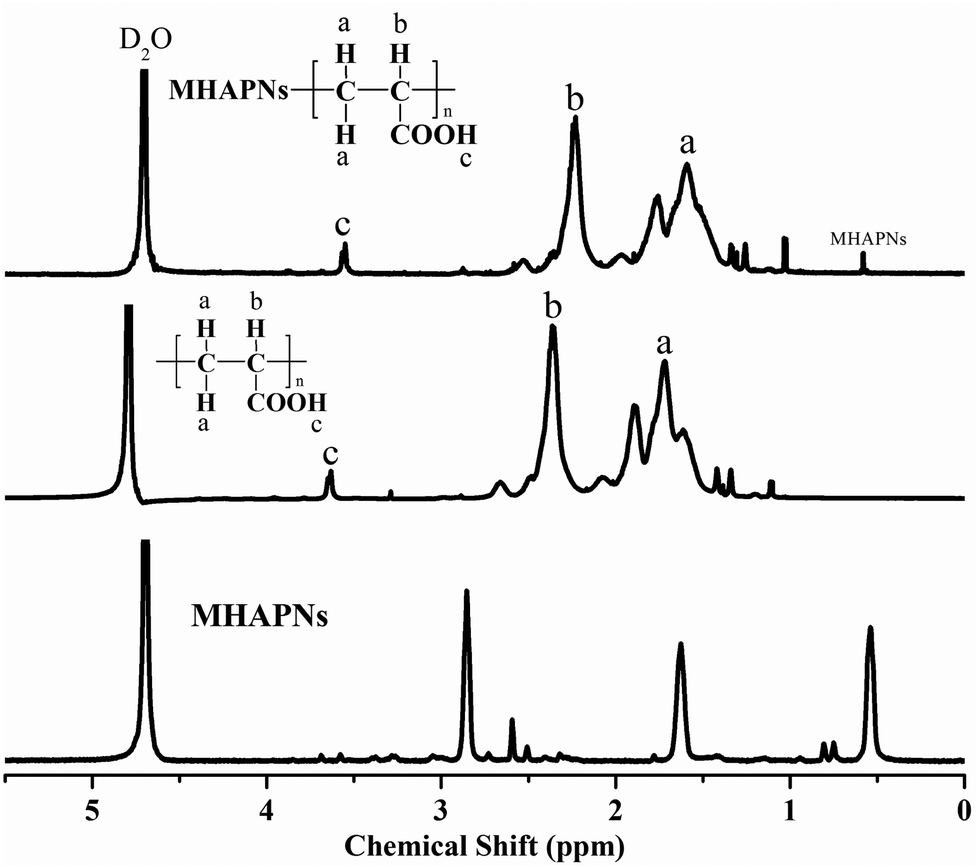
The successful preparation and surface modification of the MHAPNs were further confirmed by a 1H-NMR study. The 1H-NMR spectra of the MHAPNs, PAA and PAA–MHAPNs are shown in Fig. 3. Compared to the 1H-NMR of the MHAPNs, there were some new peaks located at 1.5, 2.2, and 3.6 ppm in the PAA and PAA–MHAPNs, which were ascribed to CH2 protons (a), CH protons (b), and carboxyl protons (c) of the PAA segments, respectively. At the same time, due to the hydroxyl of the MHAPNs being aminated, the 1H-NMR of the hydroxyl was weakened or even disappeared in the PAA–MHAPNs system. Therefore, the 1H-NMR results showed that PAA as a comb chain was successfully grafted onto the MHAPNs.
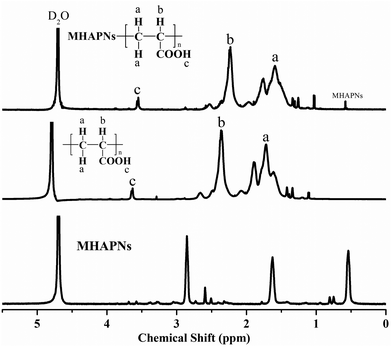 |
| | Fig. 3
1H-NMR spectra of the MHAPNs, PAA and PAA–MHAPNs. | |
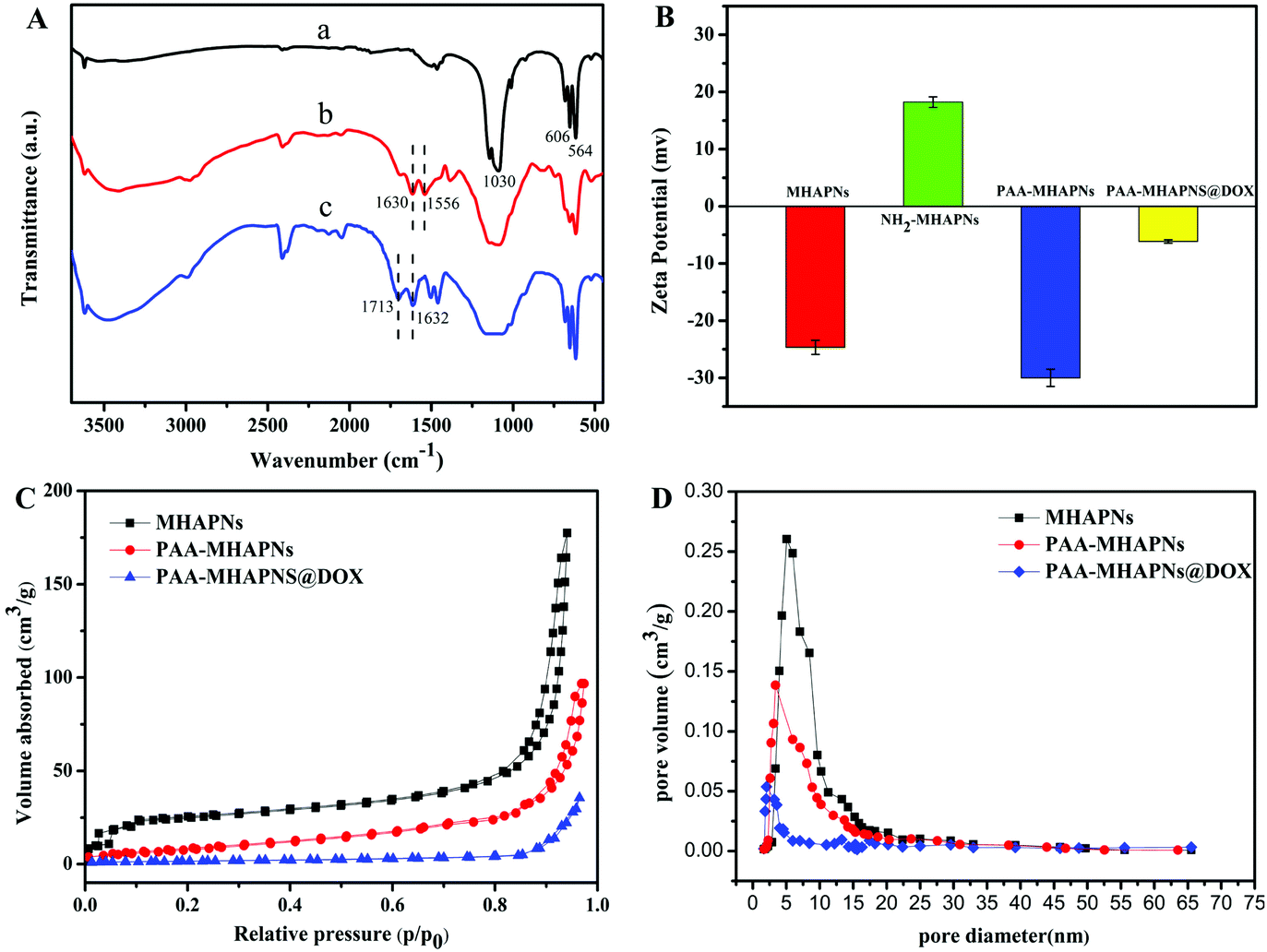
Fig. 4A displays the FTIR spectra of the MHAPNs (a), NH2-MHAPNs (b) and PAA–MHAPNs (c). The absorption peak at 1030 cm−1 was ascribed to the stretching vibration of the phosphate (PO43−) groups, and the absorption peaks at 563 and 608 cm−1 belonged to the bending vibration of the phosphate (PO43−) groups of hydroxyapatite (Fig. 4A-a). The bands which appeared at 1630 and 1556 cm−1 were attributed to the N–H stretching vibrations and bending vibrations of NH2-MHAPNs (Fig. 4A-b), clearly showing that the amino groups have been grafted onto the surface of the MHAPNs. Meanwhile in Fig. 4A-c, the new adsorption peaks which appeared at 1713 and 1632 cm−1 could be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and N–H bending vibration in the amide bonds, respectively, which indicated the successful grafting of PAA onto the MHANPs.
O stretching vibration and N–H bending vibration in the amide bonds, respectively, which indicated the successful grafting of PAA onto the MHANPs.
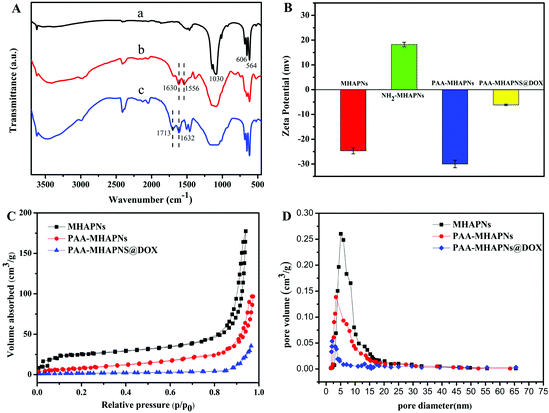 |
| | Fig. 4 (A) FTIR spectra of (a) MHAPNs, (b) NH2-MHAPNs and (c) PAA–MHAPNs; (B) zeta potential measured at each step in deionized water; (C) nitrogen adsorption–desorption isotherms and (D) BJH pore size distributions for MHAPNs, PAA–MHAPNs and DOX@PAA–MHAPNs. | |
This was also further confirmed by zeta potential measurements (Fig. 4B) in deionized water at each step. Due to the existence of OH− groups on the MHAPNs, the zeta potential value of the MHAPNs is negative (−25.53 mV). The potential value of the MHAPNs was increased to +15.56 mV when amino groups were grafted onto their surface, since amine groups can be protonated. After grafting with PAA, the zeta potential of PAA–MHAPNs was decreased to −30 mV, which indicated the existence of a large number of carboxyl groups. Then, when DOX was loaded into PAA–MHAPNs, the potential value was increased to −6.8 mV due to the number of deprotonized carboxyl groups being reduced by electrostatic interactions. There is no doubt that the variation of the surface charge property in each step suggests the successful conjugation of the functional groups onto the surface of the MHAPNs.
The N2 adsorption/desorption isotherms of the MHAPNs, PAA–MHAPNs, and DOX@PAA–MHAPNs are shown in Fig. 4C. All the samples showed a typical Type IV isotherm cycle for mesoporous materials under the BDDT (Branauer–Deming–Deming–Teller) system with a typical H1 hysteresis loop (in accordance with the IUPAC classification) and a well-defined step at approximately P/P0 = 0.80–0.98. The curve for the MHAPNs indicated the properties of typical mesoporous materials with a specific surface area of 183.46 m2 g−1, and an average pore diameter of 5.03 nm with a narrow pore distribution. After grafting with PAA, the surface area and pore volume decreased to 98.6 m2 g−1 and 0.143 cm3 g−1, respectively. This was due to the fact that some of the mesoporous channels had been encapsulated by the PAA. When DOX was loaded into the MHAPNs, the surface area and pore volume were reduced further. The textural parameters of the corresponding materials are summarized in Table 1. The results demonstrated that DOX@PAA–MHAPNs had been successfully prepared.
Table 1 Textural parameters of the MHAPNs, PAA–MHAPNs and PAA–MHAPNs@DOX
| Samples |
S
BET (m2 g−1) |
Vp (cm3 g−1) |
BJH (nm) |
| MHAPNs |
183.46 |
0.262 |
5.03 |
| PAA–MHAPNs |
98.6 |
0.143 |
3.96 |
| PAA–MHAPNs@DOX |
48.3 |
0.054 |
2.21 |
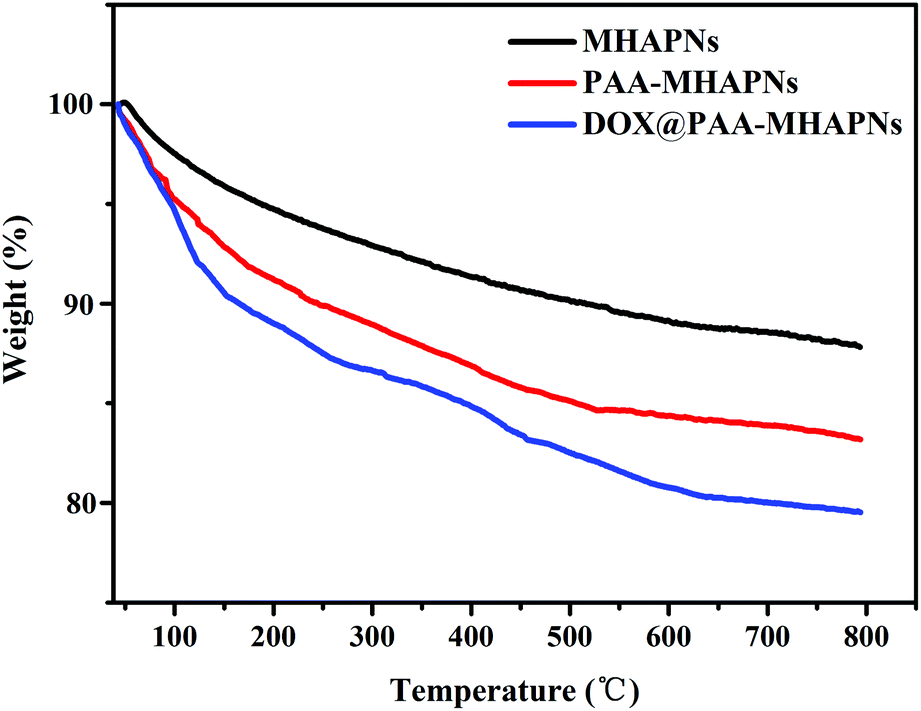
The grafted amount of PAA and loading content of DOX on the MHAPNs was estimated by TGA. As shown in Fig. 5, the weight loss was slow from 10 °C to 100 °C, which was due to the desorption of weakly-bound water. In the region of 200–800 °C, PAA–MHAPNs and DOX@PAA–MHAPNs showed a weight loss of 16.7 wt% and 20 wt%, respectively. This can be attributed to the decomposition of PAA and the loss of DOX. Thus, the graft ratio of PAA in the PAA–MHAPNs nanoparticles could be calculated to be approximately 6.2 wt% and the drug loading amount could reach about 3.3 wt%. These results indicate that the PAA–MHAPNs system has a high drug loading capacity, and could be applied as a drug carrier.
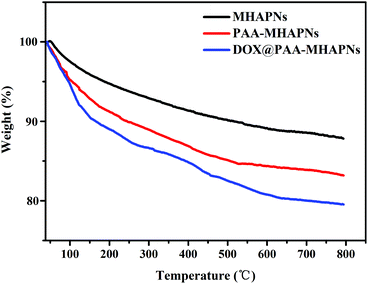 |
| | Fig. 5 TGA curves of the MHAPNs, PAA–MHAPNs and DOX@PAA–MHAPNs. | |
3.2 Drug loading and pH-responsive release properties
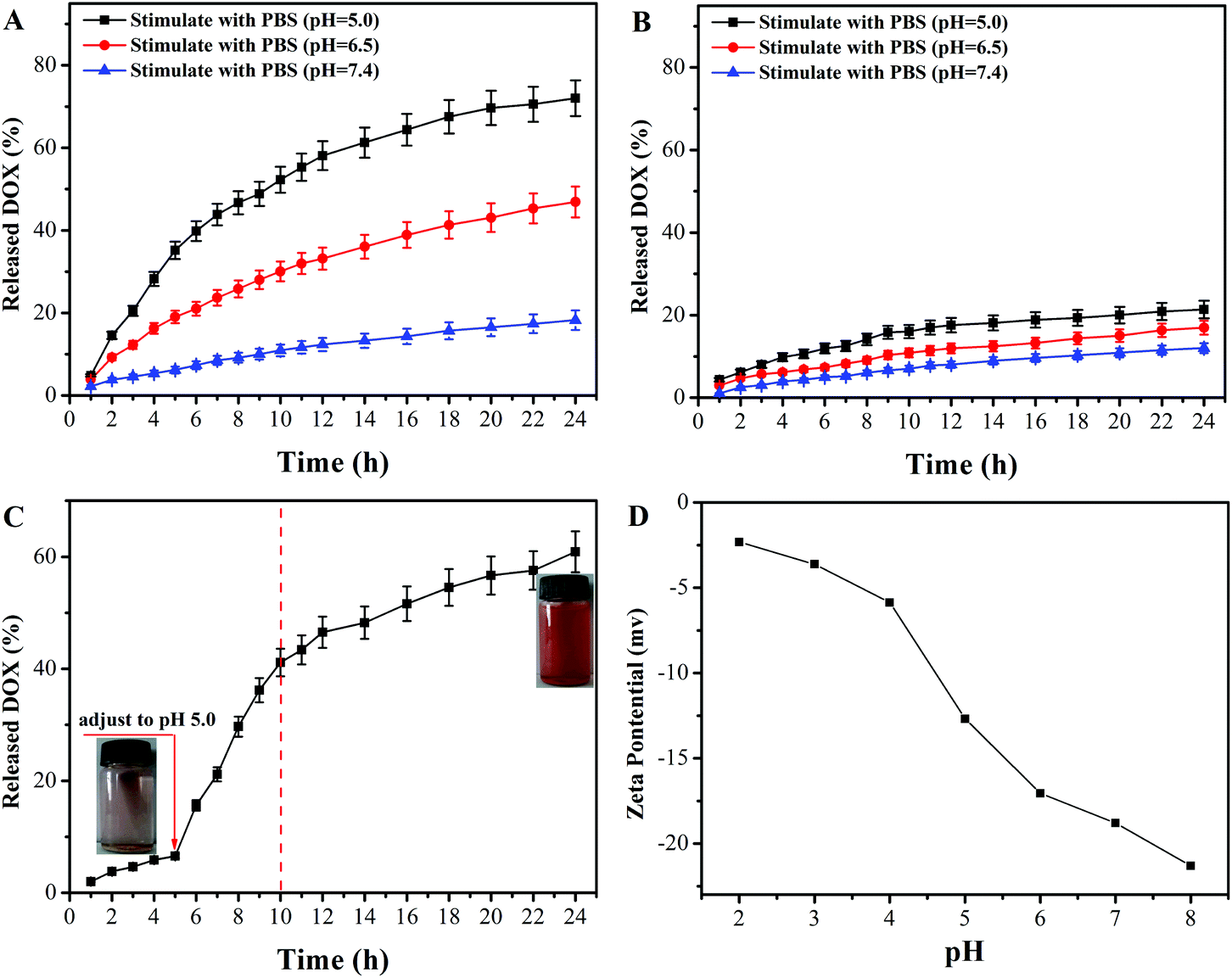
The in vitro controlled drug release behavior of DOX@PAA–MHAPNs and DOX@MHAPNs was studied in PBS buffered solutions at pH 7.4, 6.5, and 5.0, mimicking the physiological pH in normal tissue and blood, the tumor extracellular environment, and subcellular endosomes, respectively. The molybdenum blue method confirmed the degradability of MHAPNs and PAA–MHAPNs at a low pH value of 5.0, so they could be applied as intracellular delivery vehicles (Fig. S2†). It can be seen from Fig. 6A that the drug release rate of DOX@PAA–MHAPNs was obviously pH dependent and increased with the decrease of pH. As shown in Fig. 6A, in normal physiological medium (pH 7.4), about 15% of the DOX was released from the DOX@PAA–MHAPNs system after 24 h. However, the cumulative release amount of DOX could increase up to 48% and 72% after 24 h when the pH value was decreased to 6.5 and 5.0, respectively. The obviously improved release amount of DOX could be attributed to the fact that with the decrease of pH more of the PAA was protonized, which would lead to the dissociation of electrostatic interactions between PAA and DOX, such that more of the incorporated DOX was released. Fig. 6B demonstrates that the drug release of DOX@MHAPNs was also slightly pH dependent. This is due to the fact that the zeta-potential of the MHAPNs is slightly pH dependent (Fig. 6D). However, compared to DOX@PAA–MHAPNs, the drug release amount for DOX@MHAPNs is much lower, at only 21%, 13.35% and 8.86% in PBS buffer solutions of different pH after 24 h. This result indicates that DOX@PAA–MHAPNs exhibited a more pronounced pH-dependent drug release behavior than DOX@MHAPNs. In order to further investigate the pH-triggered drug release of the DOX@PAA–MHAPNs system, the pH value of the incubation solution was adjusted to 5.0 when the system was incubated with a medium of pH 7.4 after 5 h. As shown in Fig. 6C, only 8% of DOX was released from the DOX@PAA–MHAPNs system during the initial 5 h (pH 7.4), whereas the cumulative amount of released DOX could reach 42% in the next 5 h when the pH was adjusted to 5.0. The released DOX could be clearly visualized from the change in the color of the solution. The mechanism relies on the fact that the electrostatic interactions between the DOX and PAA–MHAPNs are stable under physiological conditions (pH 7.4), leading to the inhibition of drug release from DOX@PAA–MHAPNs. Upon exposure to an acidic environment, the electrostatic interactions are destroyed, resulting in the drug being released.
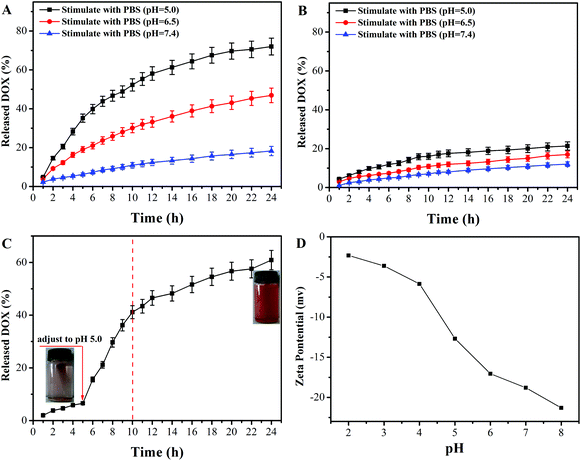 |
| | Fig. 6 Cumulative release profiles of DOX from (A) DOX@PAA–MHAPNs and (B) DOX@MHAPNs at different pH values, (C) delayed release of DOX from DOX@PAA–MHAPNs when the pH value is adjusted to 5.0 after incubation for 5 h, and (D) pH dependence of the zeta potential of the MHAPNs. | |
3.3 Cytotoxicity assay
The in vitro cell cytotoxicity of the MHAPNs, PAA–MHAPNs, DOX@MHAPNs, DOX@PAA–MHAPNs, and free DOX to HepG2 cells was investigated by MTT assay. Fig. 7A shows that the MHAPNs and PAA–MHAPNs did not have obvious cytotoxic effects on the HepG2 cells at 1–320 μg mL−1 after incubation for 24 h. This result demonstrated that the MHAPNs and PAA–MHAPNs were nontoxic at low concentrations and only slightly toxic at high concentrations. Therefore, PAA–MHAPNs should be suitable to use as the drug carrier in a drug delivery system. However, the growth of cells was inhibited after incubation with DOX@MHAPNs, free DOX and DOX@PAA–MHAPNs at a series of DOX concentrations. As shown in Fig. 7B, the killing efficiency of DOX@MHAPNs, free DOX and DOX@PAA–MHAPNs was increased significantly along with the increasing amount of DOX. Among them, DOX@MHAPNs showed remarkably higher cell viability than that of free DOX or DOX@PAA–MHAPNs. The reduced toxicity is mainly due to the fact that the DOX@MHAPNs system has little DOX loaded into it, leading to a less efficient cellular uptake of DOX compared with that of the free DOX or DOX@PAA–MHAPNs. It can also be seen that the cytotoxicity of DOX@PAA–MHAPNs is almost the same as the free DOX at most of the tested concentrations. Meanwhile, in the acidic endosome environment, an enhancement of the killing efficiency was demonstrated in the DOX@PAA–MHAPNs system due to the higher release of DOX. In general, the triggered drug release and the nature of the vehicle itself resulted in an enhanced killing efficacy compared with that of conventional chemotherapy using DOX only. Summarizing the results, we confirmed that the PAA–MHAPNs nanoparticles were capable of the controlled delivery of drug molecules in response to cancer cells.
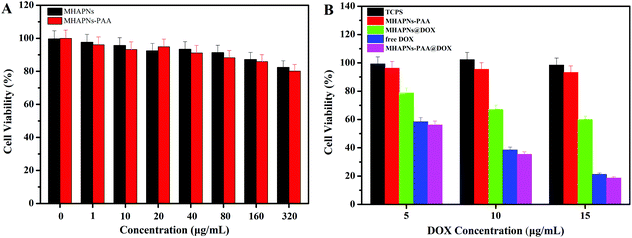 |
| | Fig. 7
In vitro viability of HepG2 cells incubated with different concentrations of MHAPNs and PAA–MHAPNs for 24 h (A) and different concentrations of PAA–MHAPNs, DOX@MHAPNs, free DOX and DOX@PAA–MHAPNs for 24 h (B). | |
3.4 Cellular uptake assay
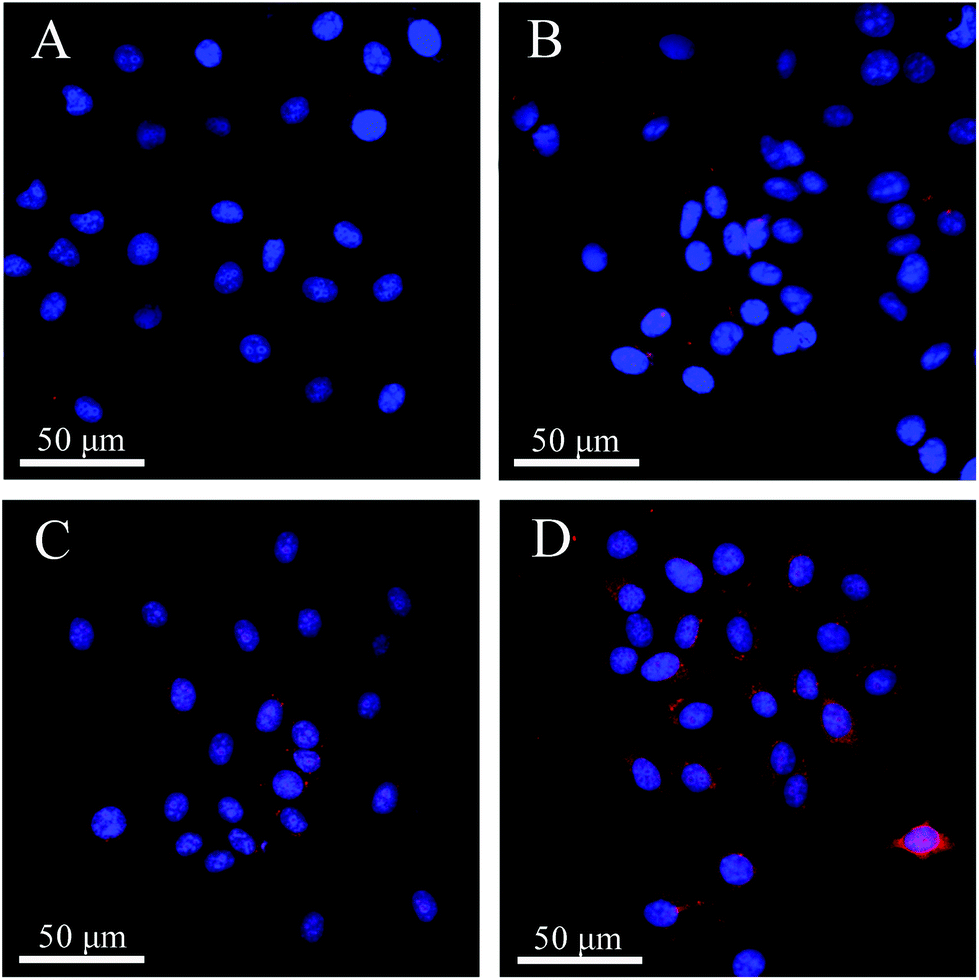
The different uptake efficiencies of the as-prepared DOX@MHAPNs and DOX@PAA–MHAPNs were investigated by confocal laser scanning microscopy (CLSM). Fig. 8 shows the confocal images of HepG2 cells incubated with the DOX@MHAPNs and DOX@PAA–MHAPNs for 12 h and 24 h. The red fluorescence from internalized DOX illustrated in the confocal image clearly indicated that drug molecules were transported into the HepG2 cells by the MHAPNs and PAA–MHAPNs carriers. It was found that prolonging the time from 12 h to 24 h is beneficial to the internalization of both the unmodified MHAPNs and PAA–MHAPNs into the cells. However, the fluorescence intensity of the HepG2 cells incubated with DOX@PAA–MHAPNs, which was modified with PAA, was markedly higher than that of the HepG2 cells incubated with MHAPNs. This result implied that DOX@PAA–MHAPNs with high drug loading could be effectively internalized into HepG2 cells, with triggered drug release into the cells.
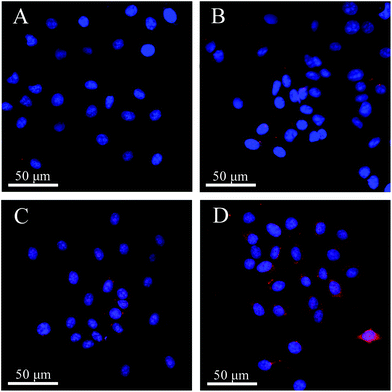 |
| | Fig. 8 LCSM images of HepG2 cells incubated with DOX@MHAPNs (A and B) and DOX@PAA–MHAPNs (C and D) at a concentration of 20 μg mL−1 for 12 h (A and C) and 24 h (B and D). | |
4 Conclusions
In summary, we have successfully designed and synthesized a MHAPN-based therapeutic drug delivery system with great potential for synergetic cancer therapy. PAA was utilized to act as both a pH-responsive switch to modulate the release of the loaded DOX in relation to pH, and as a binding site to enhance the drug loading efficiency due to the strong electrostatic interactions between PAA and DOX. After cell uptake, DOX release was triggered by the dissociation of the electrostatic interactions in acidic subcellular compartments. Moreover, we have successfully demonstrated that the DOX@PAA–MHAPNs system showed a remarkably enhanced efficiency for killing cancer cells, and the low cytotoxicity, efficient intracellular pH-stimulated drug release, and the nature of the cancer cell anti-proliferative effect induced by the MHAPN based vehicle afford a promising strategy for designing a controlled response, low dosage drug delivery system for potential in vivo cancer therapy.
Acknowledgements
This work was financially supported by NSFC (21474025), Open Project of SKLSSM (sklssm2015010), Heilongjiang Province Postdoctoral Foundation of China (LBH-Z13086), China Postdoctoral Science Foundation (2013M541372), Fundamental Research Funds for the Central Universities (HIT. NSRIF2015047), and the Weihai Science and Technology Development Plan project (2013GNS028) from Weihai city, Shandong province of China.
Notes and references
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805–6817 CrossRef CAS PubMed.
- Z. Zhou, X. Ma, C. J. Murphy, E. Jin, Q. Sun, Y. Shen, E. A. Van Krik and J. Murdoch, Angew. Chem., Int. Ed., 2014, 53, 10949–10955 CrossRef CAS PubMed.
- L. Hosta-Rigau, R. Chandrawati, E. Saveriades, P. D. Odermatt, A. Postman, F. Ercole, K. Breheney, K. L. Wark, B. Stadler and F. Caruso, Biomacromolecules, 2008, 29, 3548–3555 Search PubMed.
- B. Sahoo, K. S. P. Devi, S. K. Sahu, S. Nayak, T. K. Maiti, D. Dhara and P. Pramanik, Biomater. Sci., 2013, 1, 647–657 RSC.
- T. Panczyk, T. P. Warzocha and P. J. Camp, J. Phys. Chem. C, 2010, 114, 21299–21308 CAS.
- C. Xu, D. Yang, L. Min, Q. Li, H. Zhu and T. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12911–12920 CAS.
- H. C. Shum, A. Bandyopadhyay, S. Bose and D. A. Weitz, Chem. Mater., 2009, 21, 5548–5555 CrossRef CAS.
- P. Yang, Z. Quan, C. Li, X. Kang, H. Lian and J. Lin, Biomaterials, 2008, 29, 4341–4347 CrossRef CAS PubMed.
- W. Amer, K. Abdelouahdi, H. R. Ramananarivo, M. Zahouily, A. Fihri, K. Djessas, K. Zahouily, R. S. Varma and A. Solhy, CrystEngComm, 2014, 16, 543–549 RSC.
- G. Bharath, A. J. Kumar, K. Karthick, D. Mangalaraj, C. Viswanathan and N. Ponpandian, RSC Adv., 2014, 4, 37446–37457 RSC.
- W. L. Suchanek, K. Byrappa, P. Shuk, R. E. Riman, V. F. Janas and K. S. TenHuisen, Biomaterials, 2004, 25, 4647–4657 CrossRef CAS PubMed.
- C. Zhou, Y. Hong and X. Zhang, Biomater. Sci., 2013, 1, 1012–1028 RSC.
- Y. F. Zhao and J. Ma, Microporous Mesoporous Mater., 2005, 87, 110–117 CrossRef CAS.
- Z. Xia, L. Liao and S. Zhao, Mater. Res. Bull., 2009, 44, 1626–1629 CrossRef CAS.
- T. Long, Y. Guo, Y. Liu and Z. Zhu, RSC Adv., 2013, 3, 24169–24176 RSC.
- Z. Li, Z. Liu, M. Yin, X. Yang, Q. Yuan, J. Ren and X. Qu, Biomacromolecules, 2012, 13, 4257–4263 CrossRef CAS PubMed.
- D. Li, J. He, W. Cheng, Y. Wu, Z. Hu, H. Tian and Y. Huang, J. Mater. Chem. B, 2014, 2, 6089–6096 RSC.
- R. K. Singh, T. H. Kim, K. D. Patel, J. J. Kim and H. W. Kim, J. Mater. Chem. B, 2014, 2, 2039–2050 RSC.
- J. S. Son, M. Appleford, J. L. Ong, J. C. Wenke, J. M. Kim, S. H. Choi and D. S. Oh, J. Controlled Release, 2011, 153, 133–140 CrossRef CAS PubMed.
- Z. Zhou, S. Zhu and D. Zhang, J. Mater. Chem., 2007, 17, 2428–2433 RSC.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- C. H. Lee, S. H. Cheng, I. P. Huang, J. S. Sours, C. S. Yang, C. Y. Mou and L. W. Lo, Angew. Chem., Int. Ed., 2010, 49, 8214–8219 CrossRef CAS PubMed.
- V. Cauda, C. Arygo, A. Schlossbauer and T. Bein, J. Mater. Chem., 2010, 20, 4305–4311 RSC.
- R. Casasus, E. Climent, D. Marcos, F. Sancenon, J. Soto, J. Cano and E. Ruiz, J. Am. Chem. Soc., 2008, 130, 1903–1917 CrossRef CAS PubMed.
- Y. Zhu, J. Shi, W. Shen, X. Dong, J. Feng, M. Ruan and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 5083–5087 CrossRef CAS PubMed.
- X. Wu, Y. Tian, M. Yu, J. Han and S. Han, Biomater. Sci., 2014, 2, 972–979 RSC.
- C. R. Thomas, D. P. Ferris, J. H. Lee, E. Choi, M. H. Cho, J. S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed.
- T. D. Nguyen, K. C. F. Leung, M. Liong, Y. Liu, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2101–2110 CrossRef CAS.
- C. Park, K. Lee and C. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275–1278 CrossRef CAS PubMed.
- D. P. Ferris, Y. L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686–1688 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, Z. Li, L. Peng, D. Lin and W. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef CAS PubMed.
- R. Liu, X. Zhao, T. Wu and P. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS PubMed.
- Y. Wang, Q. Luo, L. Gao, C. Gao, H. Du, G. Zha, X. Li, Z. Shen and W. Zhu, Biomater. Sci., 2014, 2, 1367–1376 RSC.
- H. J. Kim, H. Matsuda, H. Zhou and I. Honma, Adv. Mater., 2006, 18, 3083–3088 CrossRef CAS.
- J. Xuan, M. Pelletier, H. Xia and Y. Zhao, Macromol. Chem. Phys., 2008, 212, 498–506 CrossRef.
- A. Schlossbayer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef PubMed.
- D. S. Chu, D. L. Sellers, M. J. Bocek, A. E. Fischedick, P. J. Horner and S. H. Pun, Biomater. Sci., 2015, 3, 41–45 RSC.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- J. Su, F. Chen, V. L. Cryns and P. B. Messersmith, J. Am. Chem. Soc., 2011, 133, 11850–11853 CrossRef CAS PubMed.
- L. E. Gerweck, Semin. Radiat. Oncol., 1998, 8, 176–182 CrossRef CAS PubMed.
- Y. L. Zhao, Z. Li, S. Kabehie, Y. Y. Botros, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 13016–13025 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, B. Zhang and D. Xu, Adv. Healthcare Mater., 2012, 1, 321–325 CrossRef CAS PubMed.
- X. Chen, X. Cheng, A. H. Soeriyadi, S. M. Sagnella, X. Lu, J. A. Scott, S. B. Lowe, M. Kavallaris and J. Justin Gooding, Biomater. Sci., 2014, 2, 121–130 RSC.
- H. Peng, R. Dong, S. Wang, Z. Zhong, M. Luo, C. Bai, Q. Zhao, J. Li, L. Chen and H. Xiong, Int. J. Parasitol., 2013, 446, 153–159 CAS.
- L. Yuan, Q. Tang, D. Yang, J. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5bm00228a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.