
Exploration of a library of triazolothiadiazole and triazolothiadiazine compounds as a highly potent and selective family of cholinesterase and monoamine oxidase inhibitors: design, synthesis, X-ray diffraction analysis and molecular docking studies†
Imtiaz Khan‡
a,
Syeda Mahwish Bakhtb,
Aliya Ibrara,
Saba Abbasb,
Shahid Hameed*a,
Jonathan M. Whitec,
Usman Ali Ranad,
Sumera Zaibb,
Mohammad Shahide and
Jamshed Iqbal*b
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad-45320, Pakistan. E-mail: shameed@qau.edu.pk; Fax: +92-51-90642241; Tel: +92-51-90642133
bCentre for Advanced Drug Research, COMSATS Institute of Information Technology, Abbottabad-22060, Pakistan. E-mail: drjamshed@ciit.net.pk; Fax: +92-992-383441; Tel: +92-992-383591 Tel: +92-992-383596
cSchool of Chemistry and Bio-21 Institute, University of Melbourne, Parkville-3052, Australia
dSustainable Energy Technologies (SET) Center, College of Engineering, King Saud University, PO-Box 800, Riyadh 11421, Saudi Arabia
eGerman Center for Neurodegenerative Diseases (DZNE), Germany
First published on 13th February 2015
Abstract
There is a high demand for the collection of small organic molecules (especially N-heterocycles) with diversity and complexity in the process of drug discovery. This need for privileged scaffolds in medicinal research gives an impetus for the development of nitrogen-containing compounds which are widely encountered in natural products, drugs and pharmaceutically active compounds. In this context, a diverse library of new triazolothiadiazole (4a–l) and triazolothiadiazine (5a–p) compounds was designed, synthesized and evaluated as potent and selective inhibitors of electric eel acetylcholinesterase (EeAChE) and horse serum butyrylcholinesterase (hBChE) by Ellman's method using neostigmine and donepezil as standard inhibitors. Among the screened triazolothiadiazoles, 4j emerged as a lead candidate showing the highest inhibition with an outstanding IC50 value of 0.117 ± 0.007 μM against AChE, which is ∼139-fold greater inhibitory efficacy as compared to neostigmine, whereas 4k displayed ∼506-fold strong inhibition with IC50 of 0.056 ± 0.001 μM against BChE. In the triazolothiadiazine series, 5j and 5e depicted a clear selectivity towards EeAChE with IC50 values of 0.065 ± 0.005 and 0.075 ± 0.001 μM, respectively, which are ∼250- and ∼218-fold stronger inhibition as compared to neostigmine (IC50 = 16.3 ± 1.12 μM). In addition, the synthesized compounds were also tested for their monoamine oxidase (MAO-A and MAO-B) inhibition, where 4a from the triazolothiadiazole series delivered the highest potency against MAO-A with an IC50 value of 0.11 ± 0.005 μM which is ∼33-fold higher inhibition as compared to the standard inhibitor, clorgyline (IC50 = 3.64 ± 0.012 μM), whereas compound 5c from the triazolothiadiazine series turned out to be a lead inhibitor with an IC50 value of 0.011 ± 0.001 μM which is ∼330-fold stronger inhibition. Moreover, compounds 4b (triazolothiadiazole series) and 5o (triazolothiadiazine series) were identified as lead inhibitors against MAO-B. Molecular modelling studies were performed against human AChE and BChE to observe the binding site interactions of these compounds.
Introduction
Alzheimer's disease (AD) is a chronic, progressive, neurodegenerative disease1,2 and it currently afflicts 24 million people worldwide.1–3 It is the most common cause of senile dementia and is characterized by loss of both short term and long term memory due to neuronal loss, disorientation, difficulty in speaking or writing, loss of reasoning skills, and delusions, among other symptoms.4 A multitude of features, including metal ion dyshomeostasis and disruption of both lipid production and membrane structure, have been implicated in AD onset and progression.3 Recent research has suggested that some of these characteristics could be intertwined with other facets (i.e., protein aggregation) of the disease.2,3Two important enzymes from the group of serine hydrolases, acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.1.8; also known as pseudocholinesterase), are usually defined as cholinesterases (ChEs). Structurally, these serine hydrolases belong to the class of proteins known as the esterase/lipase family within the α/β-hydrolase fold superfamily.5 They differ in kinetics and substrate selectivity6 and the major role of AChE is to catalyze the hydrolysis of acetylcholine (ACh) in cholinergic synapses, whereas the function of BChE is less clearly defined because it can hydrolyze ACh as well as other esters.7,8 One of the major therapeutic strategies adopted for primarily symptomatic AD is based on the cholinergic hypothesis targeting cholinesterase enzymes (acetyl- and butyryl-cholinesterase).9 Inhibition of the hydrolysis of acetylcholine by blocking its metabolic enzyme acetylcholinesterase (AChE) increases the ACh concentration in cholinergic synapses and can subsequently affect a number of pathogenic processes and thereby provides a possible symptomatic treatment option for AD. On the other hand, butyrylcholinesterase (BChE) has recently been considered as a potential target because it also plays an important role in regulating ACh level.10 ChE inhibitors (ChEIs) are used in the treatment of various neuromuscular disorders and have provided the first generation of drugs for the treatment of Alzheimer's disease.11 AChE inhibitors currently approved as drugs for the treatment of Alzheimer's disease are donepezil, rivastigmine, galanthamine and tacrine. In view of the limited number of AChE inhibitors currently available on the market for the treatment of AD, the search for new ChEIs is of great interest and ongoing worldwide.
Monoamine oxidase (MAO; EC 1.4.3.4) is an important target to be considered for the treatment of AD, as catalyzes the oxidative deamination of a variety of biogenic and xenobiotic amines, with the concomitant production of hydrogen peroxide.12 Monoamine oxidase is a flavin adenine dinucleotide (FAD) containing enzyme, which is localized in the outer mitochondrial membranes of neuronal, glial, and other cells13 particularly abundant in the liver and brain.14 MAO exists as two isozymes: MAO-A and MAO-B, showing different substrate specificity, sensitivity to inhibitors, and amino acid sequences. MAO-A preferentially oxidizes norepinephrine and serotonine, and is selectively inhibited by clorgyline, while MAO-B preferentially deaminates β-phenylethylamine and is irreversibly inhibited by l-deprenyl.15 Selective MAO-A inhibitors are used clinically as antidepressants and anxiolytics, while MAO-B inhibitors are used for reduction of the progression of Parkinson's disease and of symptoms associated with Alzheimer's disease. Earlier MAO-inhibitors introduced into clinical practice for the treatment of depression were abandoned due to adverse side effects, such as ‘cheese effect’ characterized by hypertensive crises.16 For this reason, there is currently a need for compounds that are useful for enhancing cognitive function and for treating cognitive deterioration in AD, as well as compounds that can generally improve cognition in normal, diseased, and aging subjects,16 and presently the research has been directed towards the synthesis of new potential agents with possible clinical practice.
Nitrogen-containing heterocyclic compounds are the most abundant and integral scaffolds that occur ubiquitously in a variety of synthetic drugs, bioactive natural products, pharmaceuticals and agrochemicals. Owing to their widespread applications, these skeletons have long been a subject of immense interest, and substantial efforts have been made to the development of synthetic strategies which could lead to the discovery of new bioactive compounds in medicinal chemistry.17 Among them, conjugated heterocycles like triazolothiadiazoles and triazolothiadiazines are associated with a wide range of biological activities including antimicrobial,18 antitumor,19 antiviral,20 anticancer,21 CNS depressant,22 anti-inflammatory, analgesic,23 antioxidant,24 anti-HIV and anti-kinesin Eg5 activity.25 Numerous triazolothiadiazole derivatives have also revealed potent urease, phosphodiesterase and c-Met protein inhibitory potential.26,27 We have recently demonstrated the utility of these hybrid structures incorporating hetero-aromatic substituent (pyridine) at 3-position of the main skeleton displaying potential cytotoxic, anti-cholinesterase, alkaline phosphatase and anti-leishmanial activities.28,29 These N-bridgehead cholinesterase inhibiting lead structures were found to exhibit moderate activity and were used as a starting point for the development of highly potent and selective cholinesterase inhibitors. The cholinesterase inhibitory activities of these classes of compounds and their selectivity were dramatically improved by introducing a non-heteroaromatic substituents at 3-position. With this aim, we synthesized these hybrid fundamental templates with a variety of structural modifications and investigated SARs to find structures with high cholinesterase inhibitory potential. Additionally, the synthesized hybrids were also screened for their monoamine oxidase inhibition and were found to inhibit MAO-A selectively (Fig. 1). The results of this study are summarized in this paper.
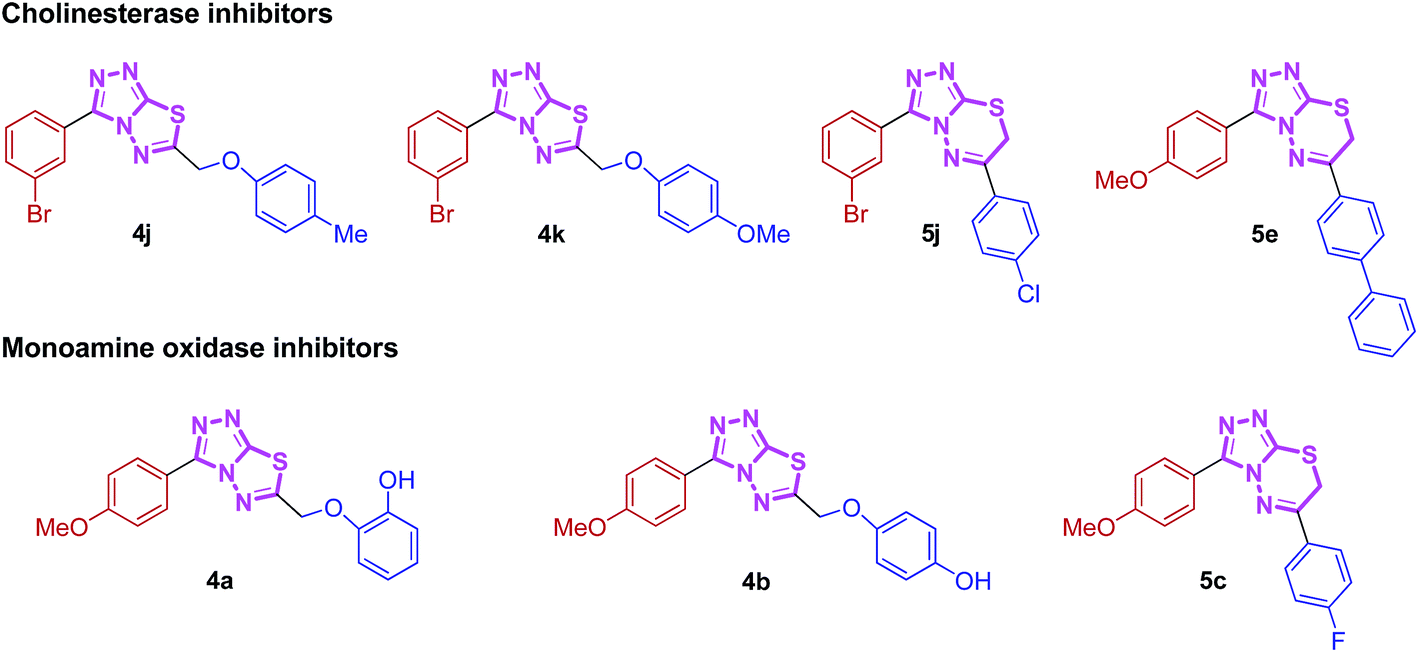 | ||
| Fig. 1 Synthesized heteroaromatic structures with remarkable cholinesterase and monoamine oxidase inhibitory potential. | ||
Results and discussion
Chemistry
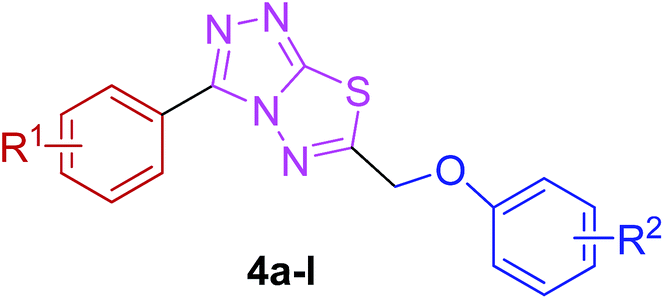
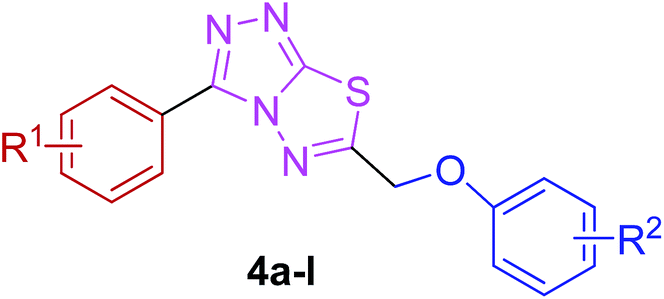
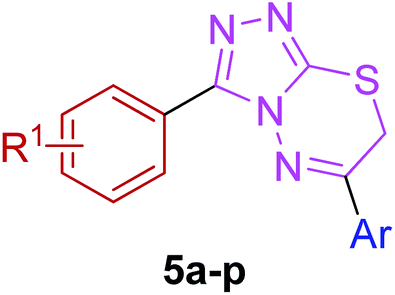
The target compounds were synthesized as outlined in Scheme 1. The key intermediate in the preparation of triazolothiadiazoles (4a–l) and triazolothiadiazines (5a–p) was 4-amino-1,2,4-triazole (3a,b) which in turn was prepared by a one-pot reaction of the corresponding benzoic acids (1a,b) with thiocarbohydrazide (2) at 190–200 °C.30 While 4-amino-3-mercapto-1,2,4-triazoles (3a,b) may exist in thione–thiol tautomeric forms, in the present study, the thiol structure was dominated in the solid state. Routine spectroscopic methods including FTIR and 1H NMR readily distinguished between these constitutional isomers.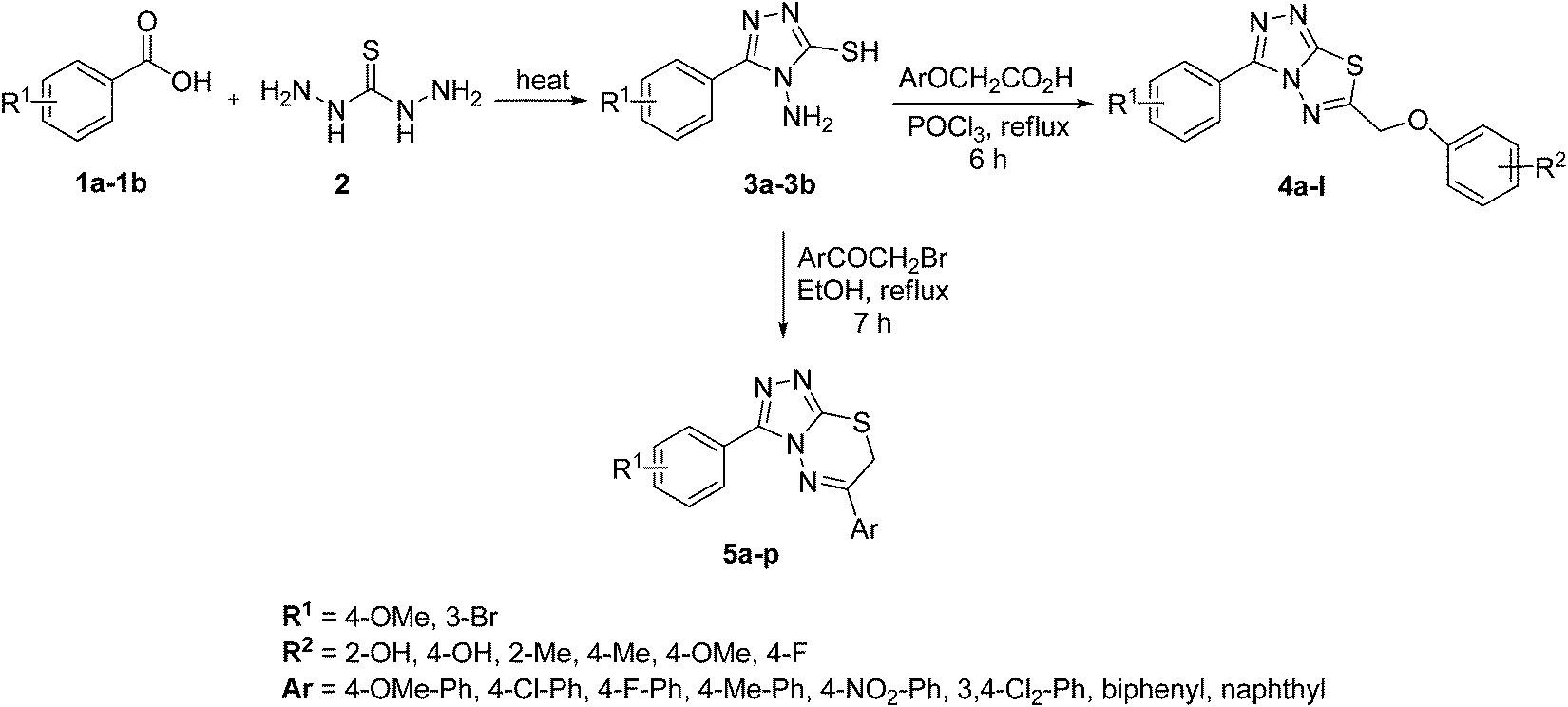 | ||
| Scheme 1 Synthesis of triazolothiadiazoles (4a–l)and triazolothiadiazines (5a–p). | ||
The FTIR spectra of 3a and 3b showed absorption bands at 2554 and 2563 cm−1, respectively, indicative of –SH group and no absorption band in the range 1300–1200 cm−1 attributable to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S group. The disappearance of the absorption bands for N–H in the range of 3400–3200 cm−1 also indicated the formation of thiol isomer. 1H NMR spectra of 4-amino-triazoles (3a,b) exhibited resonances for –NH2 protons as a singlet at 5.77 and 5.80 ppm (integrating two protons) and the –SH proton as a singlet at 13.83 and 14.03 ppm, respectively.
S group. The disappearance of the absorption bands for N–H in the range of 3400–3200 cm−1 also indicated the formation of thiol isomer. 1H NMR spectra of 4-amino-triazoles (3a,b) exhibited resonances for –NH2 protons as a singlet at 5.77 and 5.80 ppm (integrating two protons) and the –SH proton as a singlet at 13.83 and 14.03 ppm, respectively.
The reaction of corresponding 4-amino-1,2,4-triazole-3-thiol (3a,b) with aryloxy acids in the presence of phosphorous oxychloride delivered 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles (4a–l).28,29 Their proton NMR spectra showed new peaks in the range of 5.80–8.53 ppm (integration for two protons) attributed to methylene protons, and lacked signals characteristic of –SH and –NH2 protons, establishing that the –COOH group of the aryloxy acids reacted with –SH and –NH2 groups of the intermediate 3a,b to afford the corresponding conjugated products in efficient yields. Structural variations included electron-donating (–OH, –OMe, –Me) as well as electron-withdrawing (–F) substituents on the aryloxy ring attached to C-6 of triazolothiadiazole skeleton in addition to those present on aryl ring of benzoic acid attached to C-3 of the conjugated core.
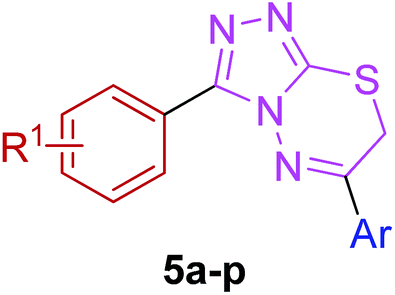
Moreover, 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (5a–p) were prepared through the reaction of corresponding 4-amino-1,2,4-triazole-3-thiol (3a,b) with substituted phenacyl bromides in absolute ethanol under reflux.28,29 Their proton 1H NMR spectra showed that the signals due to –SH and –NH2 protons had disappeared, and a new signal appeared at 4.58–4.40 ppm (integration for two protons) attributed to the methylene protons. This confirmed that the intermediate (3a,b) underwent ring closure to give the 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines in good yields. The generality of this method is validated by using a diverse range of substituents (electron-rich and electron-deficient) on the aryl ring affording corresponding hybrid products in high yields. The cyclocondensation reaction also proceeds efficiently when α-bromo ketones with bulky biphenyl and naphthyl groups were used as coupling partners affording desired products in good yields. Furthermore, purity of the synthesized compounds was ascertained by elemental analysis.
X-ray crystallography
For a full structural elucidation, compounds 4i and 5e were confirmed by X-ray diffraction analysis. The crystal and instrumental parameters used in the unit cell determination, the data collection, and structure refinement parameters are presented in Table 1.| Structural parameters | Compound 4i | Compound 5e |
|---|---|---|
| Empirical formula | C17H13BrN4OS | C23H18N4OS |
| Formula weight | 401.28 | 398.47 |
| Temperature (K) | 130(2) | 130(21) |
| Wavelength (Å) | 0.7107 | 0.7107 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| Unit cell dimensions (Å) | a = 7.5733(3), α = 105.441(5)° | a = 13.0300(3), α = 90° |
| b = 10.0581(7), β = 102.930(4)° | b = 17.3539(4), β = 98.990(2)° | |
| c = 11.2828(6), γ = 91.660(4)° | c = 8.6142(2), γ = 90° | |
| Volume (Å3) | 803.82(8) | 803.82(8) |
| Z | 2 | 4 |
| Density (calculated) (mg m−3) | 1.584 | 1.376 |
| Absorption coefficient (mm−1) | 0.285 | 0.191 |
| F(000) | 404 | 832 |
| Crystal size (mm3) | 0.59 × 0.39 × 0.18 | 0.35 × 0.2 × 0.15 |
| Theta range for data collection (°) | 2.973 to 29.121 | 2.903 to 29.188 |
| Index ranges | −9 ≤ h ≤ 10, −13 ≤ k ≤ 13, −14 ≤ l ≤ 12 | −15 ≤ h ≤ 17, −23 ≤ k ≤ 20, −11 ≤ l ≤ 11 |
| Reflections collected | 6521 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 392 392 |
| Independent reflections | 3736 [R(int) = 0.0331] | 4578 [R(int) = 0.0382] |
| Completeness to theta = 25.242° (%) | 99.7 | 99.8 |
| Absorption correction | Gaussian | Semi-empirical from equivalents |
| Max. and min. transmission | 0.639 and 0.328 | 1.00000 and 0.86346 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3736/0/218 | 4578/0/263 |
| Goodness-of-fit on F2 | 1.028 | 1.053 |
| Final R indices [I > 2σ(I)] | R1 = 0.0349, wR2 = 0.0729 | R1 = 0.0505, wR2 = 0.1156 |
| R Indices (all data) | R1 = 0.0448, wR2 = 0.0776 | R1 = 0.0736, wR2 = 0.1343 |
| Largest diff. peak and hole (e Å−3) | 0.472 and −0.512 | 0.316 and −0.400 |
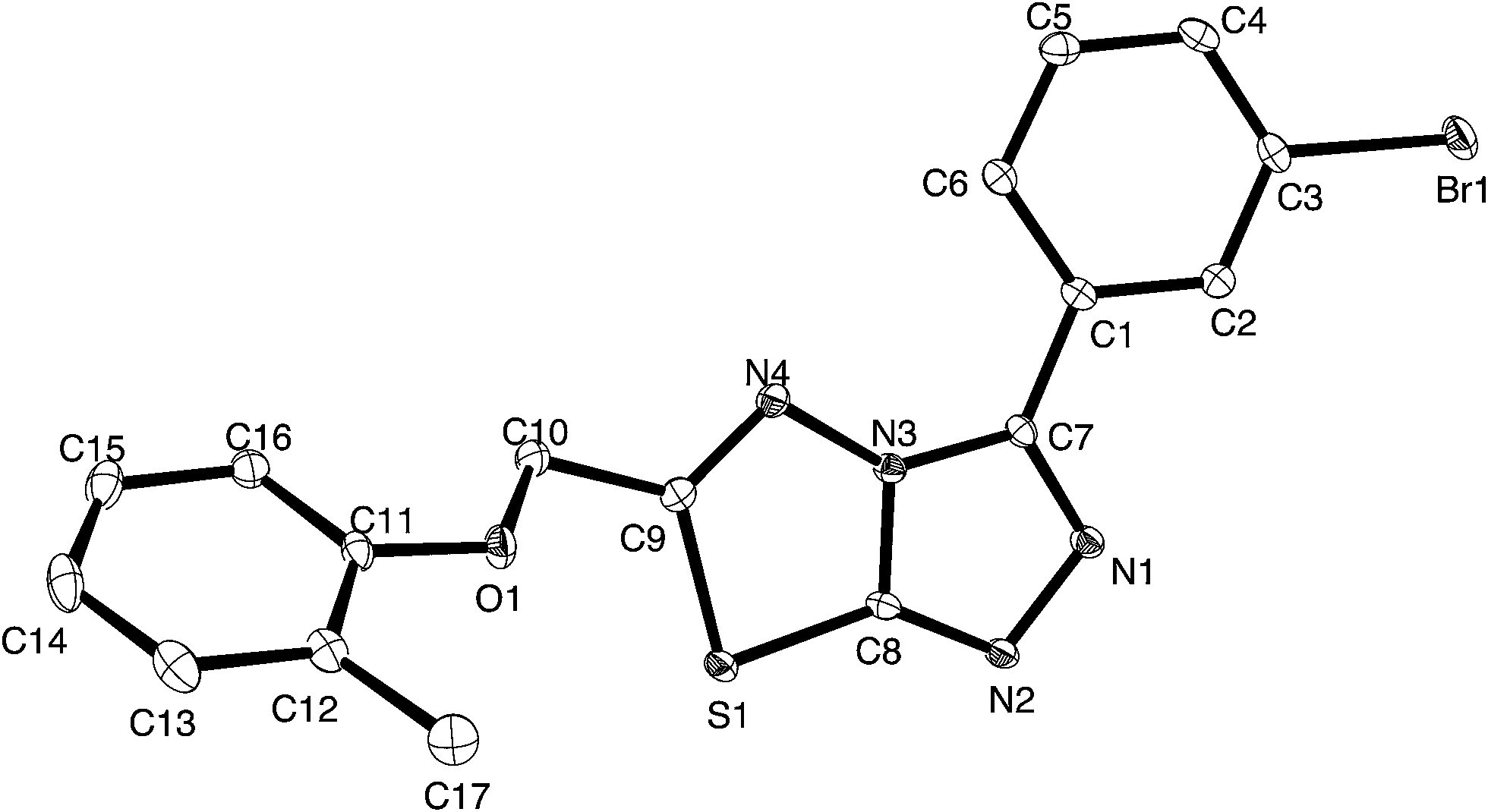 | ||
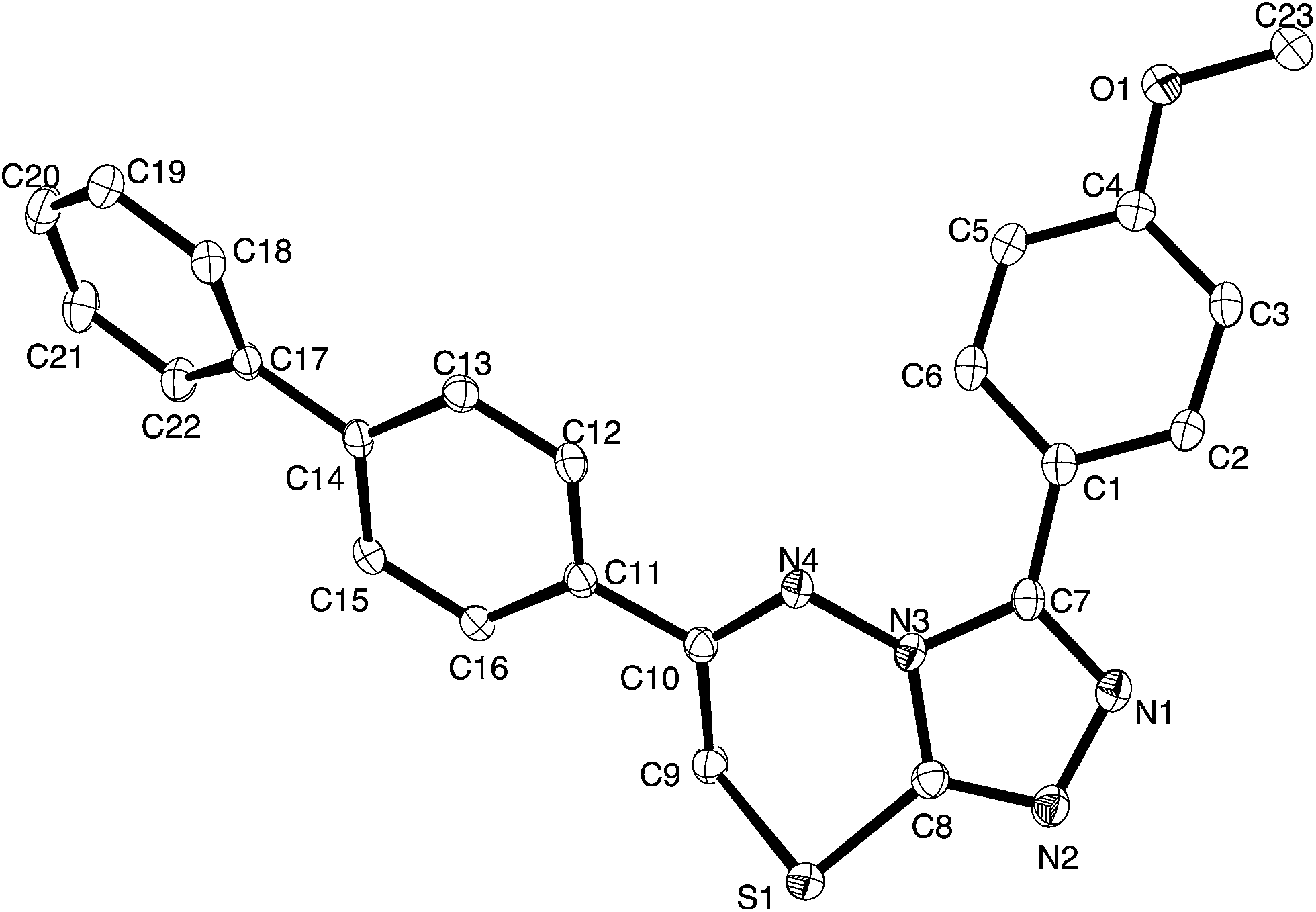
| Fig. 2 Thermal ellipsoid plot for 4i, ellipsoids are at the 30% probability level. | ||
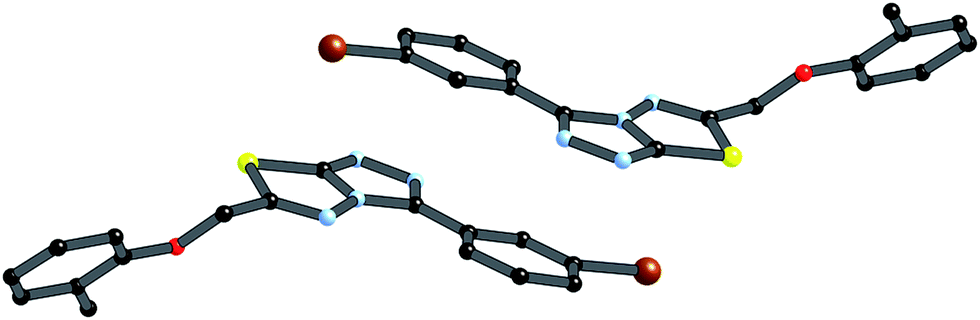
Crystals of 4i are densely packed as a result a number of strong intermolecular interactions. The most important interactions are summarized in Fig. 3–5. The first of these is a strong complementary interaction between a pair of triazolothiadiazole rings related by a centre of inversion. The S1⋯N2 distance is 2.795(2) Å, which is 0.555 Å shorter than the sum of the van der Waals radii for nitrogen and sulfur. The N2′⋯S1–C9 angle which is 163.9(2)° suggests that this may be an example of a ‘sigma-hole’ type interaction between the nitrogen lone pair and the sulfur atom. A search of the CSD for structures containing the triazolothiadiazole ring system, revealed no contacts between the S atom and N donors in the crystal which were significantly shorter than 3.0 Å, therefore this structure provides the shortest example of a N⋯S interaction yet observed in these types of compounds.32
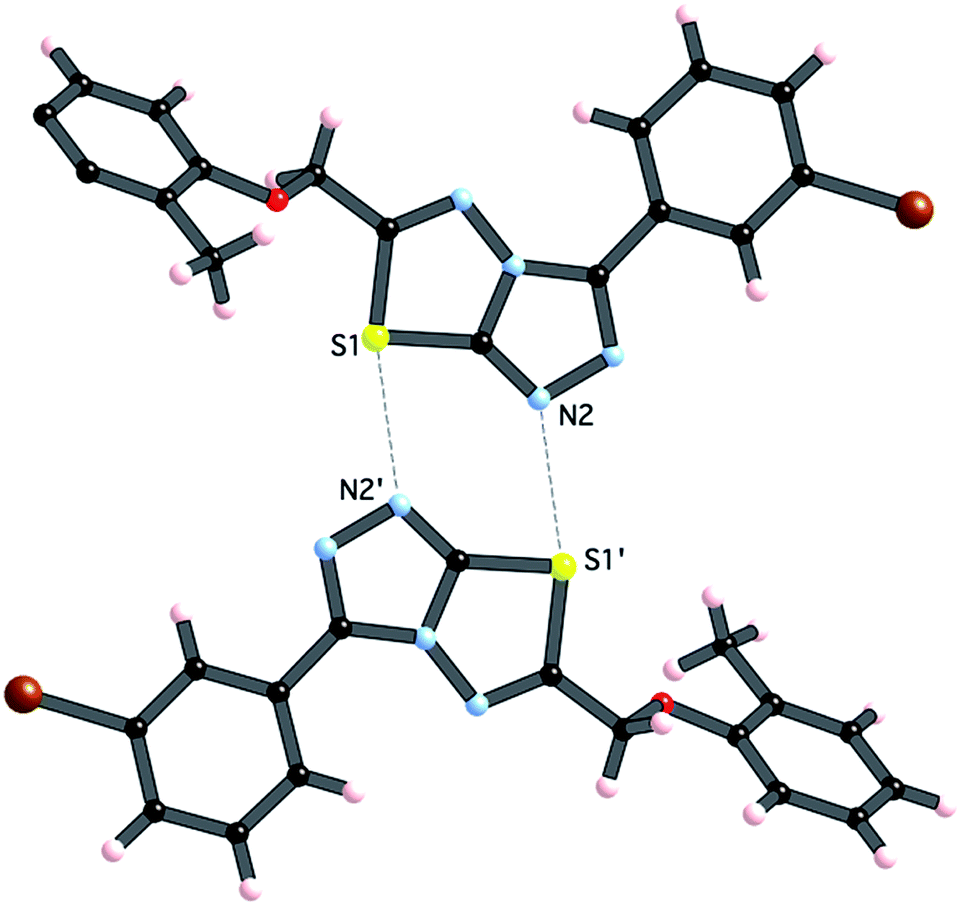 | ||
| Fig. 3 Interaction between [1,2,4]triazolothiadiazole rings. Atoms labeled with a (′) are related by the symmetry operation (−x, −y, 1 − z). | ||
 | ||
| Fig. 4 π-stacking between centrosymmetrically related pair of the 4i (symmetry operation is (−x, 1 − y, 1 − z). | ||
 | ||
| Fig. 5 C–H⋯π interaction between centrosymmetrically related pairs of 4i, symmetry operator (1 − x, −y, −z). | ||
The crystal packing of 4i is also stabilized by π-stacking interactions between pairs of the planar aryl-substituted triazolothiadiazole moieties. The distance between the mean planes of aryl-triazolothiadiazole is 3.342(2) Å while the centroid–centroid distance is 4.124 Å.
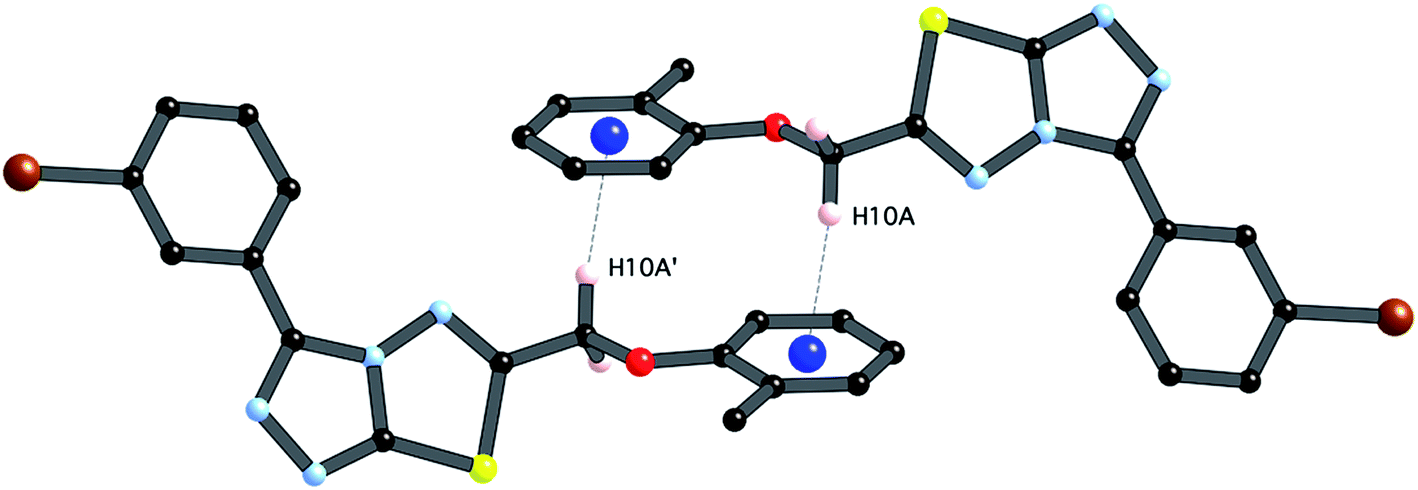
The third strong interaction evident in the packing of 4i is a complementary C–H⋯π interaction between centrosymmetrically related pairs of phenoxymethylene moieties. The distance between H10A and the mean plane defined by atoms C11–C16 is 2.753(2) Å while the distance of H10A to the centroid of this ring is 2.820 Å.
A partial packing diagram indicating all these interactions is presented in Fig. 6.
 | ||
| Fig. 6 Partial packing diagram illustrating the N⋯S sigma–hole interaction and the π–π and C–H⋯π interactions in crystals of 4i. | ||
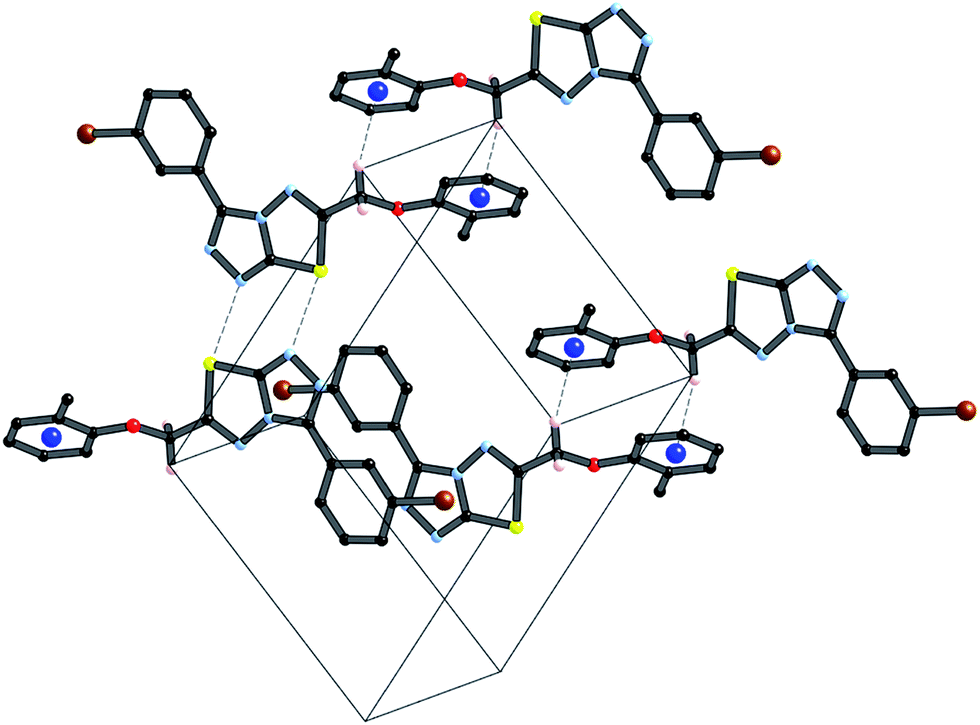
These three strong interactions, which extend in all 3-dimensions in the crystal (Fig. 7 and 8), result in a densely packed crystal which has a high crystal density of 1.658 g cm−3.
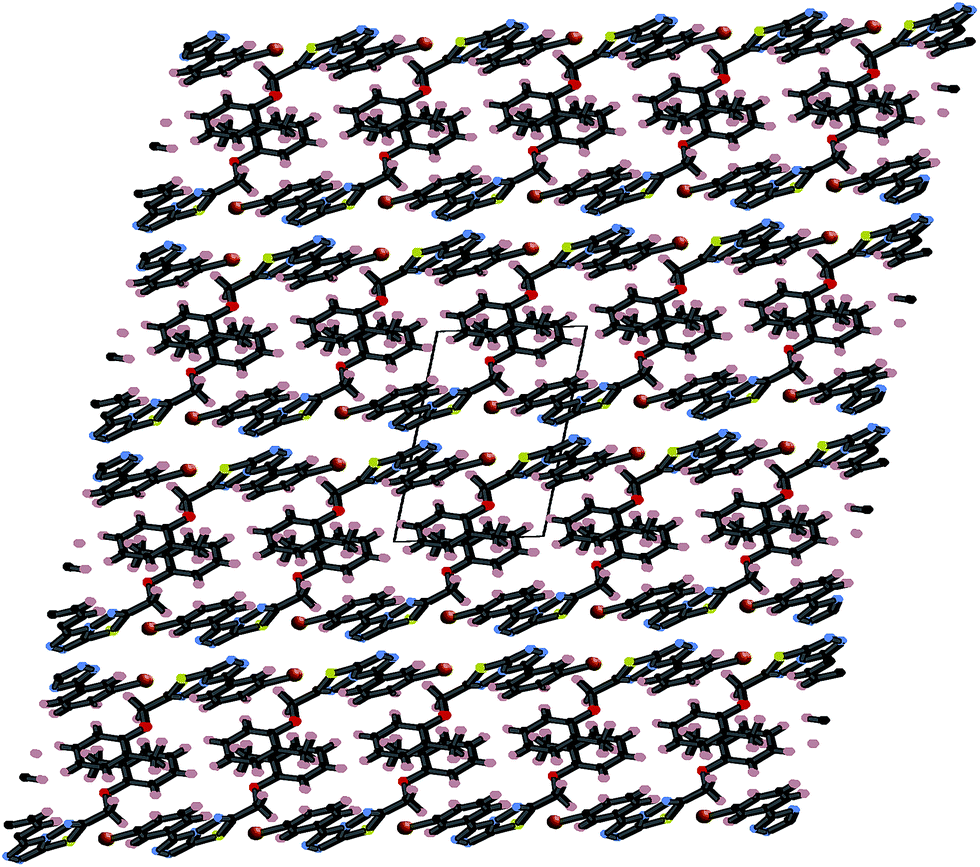 | ||
| Fig. 7 Packing diagram of 4i in the xz plane stabilized by π⋯π interactions. | ||
 | ||
| Fig. 8 Packing diagram of 4i in the yz plane stabilized by both N⋯S sigma–hole interactions and C–H⋯π interactions. | ||
N moiety in the thiadiazine ring (dihedral angles C16–C11–C10–C9 = −3.2(2)° and C16–C11–C10–N4 = 172.5(2)°, this conformation, which allows for conjugation between these two groups occurs at the cost of a close contact between hydrogen atoms attached to C16 and C9 (H16⋯H9b 1.94 Å). The biphenyl rings are twisted from coplanarity (C18–C17–C14–C13 dihedral angle = −26.5(2)°). The methoxyl substituent is near coplanar with the aromatic ring (dihedral angle C23–O1–C4–C3 = 10.5(2)°) this conformation allows for extensive delocalization of the oxygen lone-pair onto the ring through to the electron-deficient triazolothiadiazine moiety.
 | ||
| Fig. 9 Thermal ellipsoid plot for 5e. Ellipsoids are at the 30% probability level. | ||
In contrast to compound 4i, there are only relatively weak intermolecular interactions between molecules of 5e in the crystal. The shortest interactions are between hydrogen atoms H15 and H16 and the nitrogen atoms N1 and N2 respectively (distances N1⋯h16 and N2⋯H15 are 2.68 and 2.46 Å, respectively (Fig. 10). Consistent with the relatively weak intermolecular contacts in the structure of 5e, the crystal density 1.376 g cm−3 is significantly less than 4i.
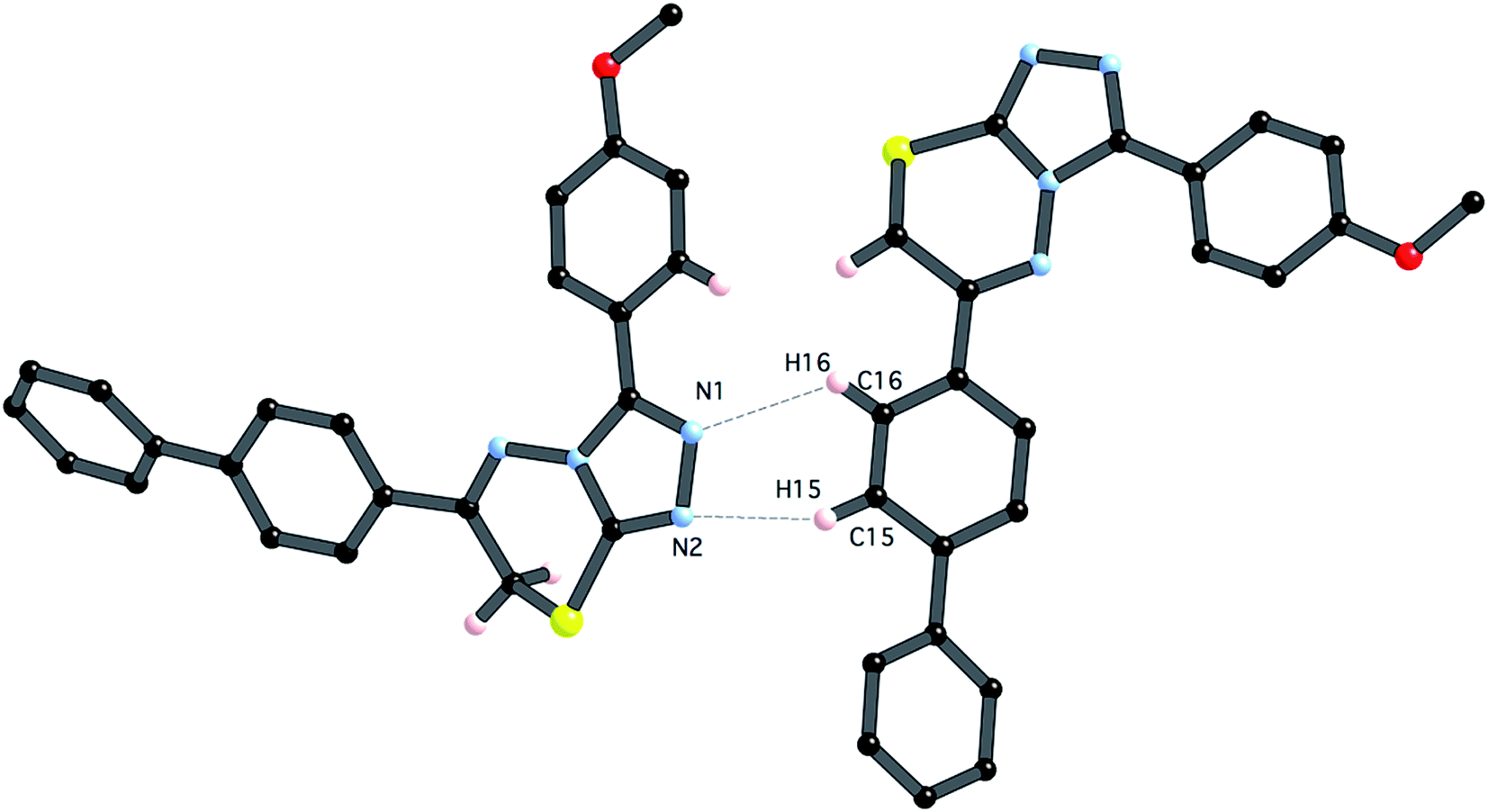 | ||
| Fig. 10 Close intermolecular contacts between molecules of 5e (symmetry code x, y − 0.5, 0.5 + z). | ||
Pharmacological studies
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | AChE | BChE |
| IC50 ± SEM (μM) | ||||
| a IC50 = concentration of inhibitor required for half-maximal enzyme inhibition and are reported as means of two independent experiments, each performed in triplicate (SEM, standard error of the mean). | ||||
| 4a | 4-OMe | 2-OH | 0.282 ± 0.017 | 4.287 ± 0.023 |
| 4b | 4-OMe | 4-OH | 0.538 ± 0.046 | 0.478 ± 0.011 |
| 4c | 4-OMe | 2-Me | 3.302 ± 0.142 | 4.987 ± 0.028 |
| 4d | 4-OMe | 4-Me | 0.564 ± 0.016 | 0.184 ± 0.007 |
| 4e | 4-OMe | 4-OMe | 0.245 ± 0.024 | 1.142 ± 0.013 |
| 4f | 4-OMe | 4-F | 0.272 ± 0.018 | 0.175 ± 0.003 |
| 4g | 3-Br | 2-OH | 0.213 ± 0.008 | 2.235 ± 0.014 |
| 4h | 3-Br | 4-OH | 0.152 ± 0.004 | 2.354 ± 0.051 |
| 4i | 3-Br | 2-Me | 0.311 ± 0.013 | 2.012 ± 0.018 |
| 4j | 3-Br | 4-Me | 0.117 ± 0.007 | 1.971 ± 0.032 |
| 4k | 3-Br | 4-OMe | 0.159 ± 0.014 | 0.056 ± 0.001 |
| 4l | 3-Br | 4-F | 0.227 ± 0.013 | 0.168 ± 0.003 |
| Neostigmine | — | — | 16.3 ± 1.12 | 28.3 ± 2.06 |
| Donepezil | — | — | 0.02 ± 0.003 | 7.23 ± 0.12 |
|
||||
|---|---|---|---|---|
| Entry | R1 | Ar | AChE | BChE |
| IC50 ± SEM (μM) | ||||
| a IC50 = concentration of inhibitor required for half-maximal enzyme inhibition and are reported as means of two independent experiments, each performed in triplicate (SEM, standard error of the mean). | ||||
| 5a | 4-OMe | 4-OMe-Ph | 0.870 ± 0.051 | 0.176 ± 0.013 |
| 5b | 4-OMe | 4-Cl-Ph | 1.09 ± 0.072 | 0.362 ± 0.015 |
| 5c | 4-OMe | 4-F-Ph | 0.365 ± 0.013 | 3.422 ± 0.008 |
| 5d | 4-OMe | 4-Me-Ph | 0.220 ± 0.005 | 0.066 ± 0.021 |
| 5e | 4-OMe | Biphenyl | 0.075 ± 0.001 | 3.936 ± 0.041 |
| 5f | 4-OMe | Naphthyl | 0.496 ± 0.016 | 3.174 ± 0.007 |
| 5g | 4-OMe | 4-NO2-Ph | 0.205 ± 0.013 | 2.382 ± 0.004 |
| 5h | 4-OMe | 3,4-Cl2-Ph | 0.301 ± 0.011 | 2.547 ± 0.016 |
| 5i | 3-Br | 4-OMe-Ph | 0.747 ± 0.042 | 0.748 ± 0.004 |
| 5j | 3-Br | 4-Cl-Ph | 0.065 ± 0.005 | 1.445 ± 0.0124 |
| 5k | 3-Br | 4-F-Ph | 0.259 ± 0.031 | 1.384 ± 0.016 |
| 5l | 3-Br | 4-Me-Ph | 0.950 ± 0.085 | 3.981 ± 0.0015 |
| 5m | 3-Br | Biphenyl | 1.303 ± 0.12 | 0.480 ± 0.028 |
| 5n | 3-Br | Naphthyl | 1.066 ± 0.76 | 0.343 ± 0.012 |
| 5o | 3-Br | 4-NO2-Ph | 0.854 ± 0.93 | 0.780 ± 0.062 |
| 5p | 3-Br | 3,4-Cl2-Ph | 0.248 ± 0.021 | 1.07 ± 0.084 |
| Neostigmine | — | — | 16.3 ± 1.12 | 28.3 ± 2.06 |
| Donepezil | — | — | 0.02 ± 0.003 | 7.23 ± 0.12 |
 | ||
| Fig. 11 The synthesized triazolothiadiazole structures (4a–l) investigated herein. | ||
 | ||
| Fig. 12 The synthesized triazolothiadiazine structures (5a–p) investigated herein. | ||
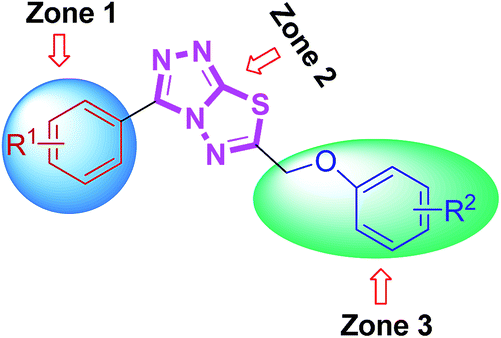
From the activity results presented in Table 2, it is evident that most of the hybrid compounds were significantly active for both AChE and BChE enzymes with more selectivity towards AChE. The variation of the substituents at aromatic rings (zone 1 and 3) significantly affected cholinesterase inhibitory potency and the selectivity as well. Remarkably, the behavior of these compounds is more selective for AChE than BChE inhibition having electron-rich (–OMe) group on phenyl ring attached at 3-position of conjugated skeleton. All the synthesized compounds (4a–l) were found to be significantly more potent against AChE than the standard drugs used. Among them, 4j was the lead candidate showing highest inhibition with IC50 value of 0.117 ± 0.007 μM, which is ∼139-fold stronger inhibition as compared to neostigmine. This compound possessed a bromo substituent at the meta-position of the phenyl ring (zone 1) attached at 3-position along with a 4-methyl substituent at the para-position of the aromatic ring (zone 3) attached at 6-position of triazolothiadiazole core. The same compound showed nearly 50% inhibition against BChE demonstrating the selective inhibitory potential towards AChE. Replacement of the zone 1 meta-bromo group with a para-methoxy substituent also afforded good inhibition against AChE (compound 4d; IC50 = 0.564 ± 0.016 μM; ∼29-fold strong inhibition) in addition to the ∼154-fold higher potency against BChE as compared to 4j. Among the same set of derivatives, the substituent electronic effect at various positions in zone 3 modulated the ChE inhibition as shown by compounds 4g–i. In general, all the synthesized triazolothiadiazole compounds were emerged as efficient inhibitors of AChE whereas compounds 4b, 4e, 4f, 4k, and 4l were found as potent candidates for BChE inhibition. It was also found that the combination of electron-rich and electron-deficient substituents in zone 1 and 3 resulted in stronger and selective inhibitors of AChE as compared to BChE where this combination is underlined in selected examples.
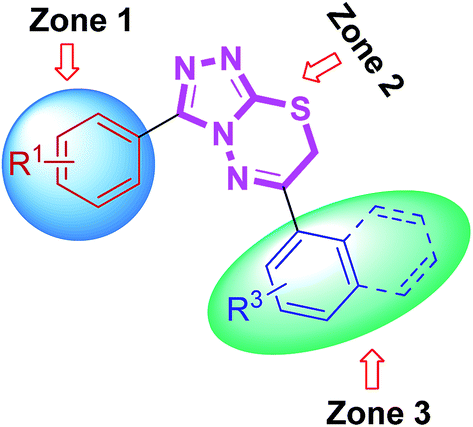
The newly synthesized triazolothiadiazine hybrids were also evaluated in vitro as inhibitors of electric eel acetylcholinesterase (EeAChE) and horse serum butyrylcholinesterase (hBChE). The inhibitory activities of novel compounds were compared to those of neostigmine and donepezil. All of the novel target analogues proved to be very potent and selective inhibitors of cholinesterases displaying inhibition abilities in micromolar range. A careful insight and analysis of the data listed in Table 3 led to several eventual structure–activity relationships (SAR) using a similar three zone approach (Fig. 12).
Some interesting considerations can be made by comparing enzyme inhibition data (Table 3) for the two sets of products bearing the same substituent in the zone 3 but variable groups (electron-rich and electron-deficient) on phenyl ring in zone 1. The majority of tested hybrids in the first set of compounds bearing para-methoxy group in zone 1 depicted a clear selectivity towards EeAChE highlighting 5e as the most potent AChE inhibitor in the series with IC50 value of 0.075 ± 0.001 μM which is ∼218-fold stronger inhibition as compared to neostigmine (IC50 = 16.3 ± 1.12 μM). This compound incorporates bulky biphenyl ring in zone 3 in addition to para-methoxy group in zone 1. Similar results were also obtained when the biphenyl ring at C-6 is replaced with another sterically demanding naphthyl group (compound 5f: IC50 = 0.496 ± 0.016 μM). The compounds incorporating electron-deficient substituents such as 5g (4-NO2) and 5h (3,4-Cl2) also revealed notable selective inhibition towards AChE with IC50 of 0.205 ± 0.013 μM (∼79-fold stronger inhibition) and 0.301 ± 0.011 μM (∼54-fold stronger inhibition), as compared to neostigmine. Within the first set of derivatives, compound 5d was the lead candidate against BChE with IC50 value of 0.066 ± 0.021 μM (∼427- and 109-fold stronger inhibition) as compared to neostigmine and donepezil, respectively. Other potent inhibitors of BChE are compounds 5a and 5b showing ∼160- and 78-fold stronger inhibition (IC50 = 0.176 ± 0.013 and 0.362 ± 0.015 μM, respectively) as compared to standard drug neostigmine.
In the second set of compounds where a meta-bromo substituent is present on the aromatic ring (zone 1), 5j emerged as a lead compound against AChE with IC50 value of 0.065 ± 0.005 μM showing ∼250-fold higher potency as compared to neostigmine in addition to ∼20-fold stronger inhibition against BChE (IC50 = 1.445 ± 0.0124 μM). This lead compound incorporates a combination of electron-withdrawing substituents (Br and Cl) on aryl ring in zone 1 and 3 at meta- and para-position, respectively. Similar results were acquired when another chloro substituent is introduced at 3-position (compound 5p; IC50 = 0.248 ± 0.021 μM against AChE and IC50 = 1.07 ± 0.084 μM against BChE). The replacement of electronically poor substituents with sterically encumbered substituents like biphenyl and naphthyl in zone 3 (compounds 5m and 5n; IC50 = 1.303 ± 0.12 and IC50 = 1.066 ± 0.76 μM, respectively) produced excellent inhibitions of both AChE and BChE with more selectivity towards BChE. In general, most of the compounds were found as potent and selective inhibitors of cholinesterases and the results obtained clearly indicated the influence of electronic and steric properties of substituents on the cholinesterase inhibition as well as selectivity.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | MAO-A | MAO-B |
| IC50 ± SEM (μM) | ||||
| a IC50 = concentration of inhibitor required for half-maximal enzyme inhibition and are reported as means of two independent experiments, each performed in triplicate (SEM, standard error of the mean). | ||||
| 4a | 4-OMe | 2-OH | 0.11 ± 0.005 | 14.24 ± 1.03 |
| 4b | 4-OMe | 4-OH | 0.25 ± 0.003 | 0.03 ± 0.001 |
| 4c | 4-OMe | 2-Me | 0.11 ± 0.007 | 0.33 ± 0.009 |
| 4d | 4-OMe | 4-Me | 0.24 ± 0.008 | 4.36 ± 0.21 |
| 4e | 4-OMe | 4-OMe | 1.85 ± 0.005 | 1.02 ± 0.003 |
| 4f | 4-OMe | 4-F | 4.77 ± 0.13 | 0.606 ± 0.014 |
| 4g | 3-Br | 2-OH | 14.65 ± 0.09 | 21.73 ± 0.027 |
| 4h | 3-Br | 4-OH | 7.42 ± 0.2 | 25.31 ± 0.02 |
| 4i | 3-Br | 2-Me | 1.23 ± 0.06 | 1.73 ± 0.09 |
| 4j | 3-Br | 4-Me | 1.23 ± 0.003 | 1.42 ± 0.005 |
| 4k | 3-Br | 4-OMe | 2.89 ± 0.007 | 2.97 ± 0.009 |
| 4l | 3-Br | 4-F | 6.90 ± 0.34 | 0.87 ± 0.006 |
| Clorgyline | — | — | 3.64 ± 0.012 | — |
| Deprenyl | — | — | — | 0.007 ± 0.001 |
|
||||
|---|---|---|---|---|
| Entry | R1 | Ar | MAO-A | MAO-B |
| IC50 ± SEM (μM) | ||||
| a IC50 = concentration of inhibitor required for half-maximal enzyme inhibition and are reported as means of two independent experiments, each performed in triplicate (SEM, standard error of the mean). | ||||
| 5a | 4-OMe | 4-OMe-Ph | 0.22 ± 0.004 | 0.18 ± 0.002 |
| 5b | 4-OMe | 4-Cl-Ph | 0.92 ± 0.007 | 3.66 ± 0.003 |
| 5c | 4-OMe | 4-F-Ph | 0.011 ± 0.001 | 5.92 ± 0.005 |
| 5d | 4-OMe | 4-Me-Ph | 5.64 ± 0.030 | 15.21 ± 0.170 |
| 5e | 4-OMe | Biphenyl | 0.09 ± 0.006 | 28.59 ± 2.070 |
| 5f | 4-OMe | Naphthyl | 1.23 ± 0.009 | 6.03 ± 0.370 |
| 5g | 4-OMe | 4-NO2-Ph | 0.82 ± 0.005 | 34.82 ± 0.041 |
| 5h | 4-OMe | 3,4-Cl2-Ph | 16.5 ± 1.070 | 42.78 ± 0.037 |
| 5i | 3-Br | 4-OMe-Ph | 2.08 ± 0.010 | 40.19 ± 0.025 |
| 5j | 3-Br | 4-Cl-Ph | 1.69 ± 0.004 | 32.09 ± 0.17 |
| 5k | 3-Br | 4-F-Ph | 3.92 ± 0.620 | 0.32 ± 0.017 |
| 5l | 3-Br | 4-Me-Ph | 2.17 ± 0.130 | 3.68 ± 0.170 |
| 5m | 3-Br | Biphenyl | 1.21 ± 0.004 | 3.86 ± 0.050 |
| 5n | 3-Br | Naphthyl | 3.51 ± 0.060 | 18.7 ± 0.100 |
| 5o | 3-Br | 4-NO2-Ph | 13.94 ± 0.900 | 0.08 ± 0.0003 |
| 5p | 3-Br | 3,4-Cl2-Ph | 0.02 ± 0.0005 | 0.67 ± 0.009 |
| Clorgyline | — | — | 3.64 ± 0.012 | — |
| Deprenyl | — | — | — | 0.007 ± 0.001 |
At a first glance, the inhibition data reported in Table 4 indicated that most of the examined triazolothiadiazole compounds exhibited a very high and selective inhibition of MAO-A as compared to MAO-B. Among the synthesized compounds, 4a delivered highest potency against MAO-A with IC50 value of 0.11 ± 0.005 μM which is ∼33-fold higher inhibition as compared to standard inhibitor, clorgyline (IC50 = 3.64 ± 0.012 μM). The previously mentioned SAR strategy was applied to explain the activity results (Fig. 11). According to that the most active analogue 4a possesses electron-donating groups at the para- and ortho-position of both aryl rings in zone 1 and 3, respectively. The replacement of the 2-hydroxy group with another electron-rich (–Me) group produced similar results (compound 4c; IC50 = 0.11 ± 0.007 μM, ∼33-fold strong inhibition). The change of substitution position on the aryl ring in zone 3 had a positive influence on the inhibitory activity. Compounds 4d and 4b were also found to exhibit significant inhibitions with IC50 values of 0.24 ± 0.008 (15-fold strong inhibition) and 0.25 ± 0.003 μM (14-fold strong inhibition), respectively. Similarly, compounds obtained by replacing bromo group with methoxy group (4i and 4j) also showed significant inhibition against MAO-A with IC50 values of 1.23 ± 0.06 and 1.23 ± 0.003 μM, respectively. The extent of this inhibition was found to be higher (∼3-fold) as compared to the standard clorgyline. The introduction of electron-withdrawing (–F) group at para-position of phenyl ring attached to C-6 of heteroaromatic core led to reduced activity as compared to the clorgyline (compound 4f; IC50 = 4.77 ± 0.13 μM). The combination of electron-deficient (–Br) group in zone 1 and electron-rich (–OH) group in zone 3 abolished the activity (compound 4g; IC50 = 14.65 ± 0.09 μM).
On the other hand, the synthesized triazolothiadiazoles showed reduced activity against MAO-B as compared to the standard deprenyl (IC50 = 0.007 ± 0.001 μM). The most active MAO-B inhibitor was compound 4b which showed an IC50 value of 0.03 ± 0.001 μM. This compound incorporates electron-rich functional groups at para-position of both aryl rings in zone 1 and 3. To sum up, MAO-A inhibitory potency of triazolothiadiazole compounds appeared to be mainly modulated by electronic effect which is clearly evidenced by the results presented herein whereas synthesized derivatives were inactive against MAO-B.
Similarly, all the synthesized triazolothiadiazine compounds were also evaluated for their ability to inhibit MAO-A and MAO-B. The corresponding IC50 values are shown in Table 5. The previously discussed SAR model (Fig. 12) was used to construct correlation between structural properties and biological activity results. Among the tested derivatives, compound 5c turned to be a lead candidate with IC50 value of 0.011 ± 0.001 μM which is ∼330-fold strong inhibition as compared to the standard inhibitor (clorgyline) used for comparison purpose. This compound incorporates a para-methoxy group at aromatic ring in zone 1 along with an electron-deficient (–F) functional group at aromatic ring in zone 3. The replacement of methoxy substituent with bromo group and introduction of two electron-withdrawing chloro substituents also produced similar results (compound 5p; IC50 = 0.02 ± 0.0005 μM, ∼182-fold strong inhibitory efficacy). Apart from electronic parameters, the steric effects were also found to contribute positively towards the inhibition of MAO-A. In this regard, compound 5e exhibited strong inhibition with an IC50 value of 0.09 ± 0.006 μM which is 40-fold higher as compared to the standard inhibitor (clorgyline), whereas, the introduction of bulky naphthyl group led to the reduction in activity (IC50 = 1.23 ± 0.009 μM). But, still this inhibitory potential is 3-fold higher as compared to clorgyline. The synthesized structures with a combination of para-methoxy group and para-nitro or -chloro were also emerged as potent inhibitors of MAO-A (compound 5g; IC50 = 0.82 ± 0.005 and 0.92 ± 0.007 μM; ∼4.4- and ∼4-fold stronger inhibition, respectively). On the other hand, prepared compounds were also tested for MAO-B inhibition but the activity profile was diminished for the screened derivatives. In the evaluated series, compound 5o was found to demonstrate a highest inhibitory potency towards MAO-B with an IC50 value of 0.08 ± 0.0003 μM as compared to the standard, deprenyl (IC50 = 0.007 ± 0.001 μM). This compound incorporates a meta-bromo substituent on aromatic ring in zone 1 in addition to the highly polarizable para-nitro group on phenyl ring in zone 3. In general, both electronic and steric factors were actively found to modulate the MAO inhibition and the presented derivatives (triazolothiadiazoles and triazolothiadiazines) are proved to be better and selective MAO-A inhibitors and these data are an important observation to follow up a rational design of more potent and selective inhibitors of monoamine oxidases (MAO).
Docking validation was carried out by re-docking the ligand extracted from the experimentally determined enzyme-ligand adduct. The docking method was able to reproduce the experimentally observed bound conformation of the co-crystallized ligand with rmsd <2 Å. By re-docking the crystallographic ligand huprine W in the active site of AChE, similar interactions were seen with the residues Tyr337, Trp86, Gly122, Ser203 as huprine W showed in the crystal structure of AChE. The ring system of both compound series adapts a comparable position to huprine W and it interacts with side chain atoms of several aromatic residues including Tyr337 and Trp86. Furthermore, residue Thr83 is found to be involved in hydrogen bonding with triazolothiadiazole and triazolothiadiazine ring of compounds. The phenyl rings having different substituents at both ends of the compounds are directed towards the hydrophobic pockets formed by the residues Gly121, Phe297 and Gly122. π–π stacking is also observed by most of the compounds in the active site.
Fig. 13 shows the interactions of amino acids in the active site of AChE by co-crystal ligand huprine W in the right side and potent compound 4k in the left side. The figure shows that Trp86 and Tyr337 are involved in π-interaction with the ligand as well as compound 4k. His447 is also showing π-interaction in the active pocket. It is therefore predicted that the docking of the compounds in the active site of enzyme showed the similar interactions as shown by co-crystal ligand.
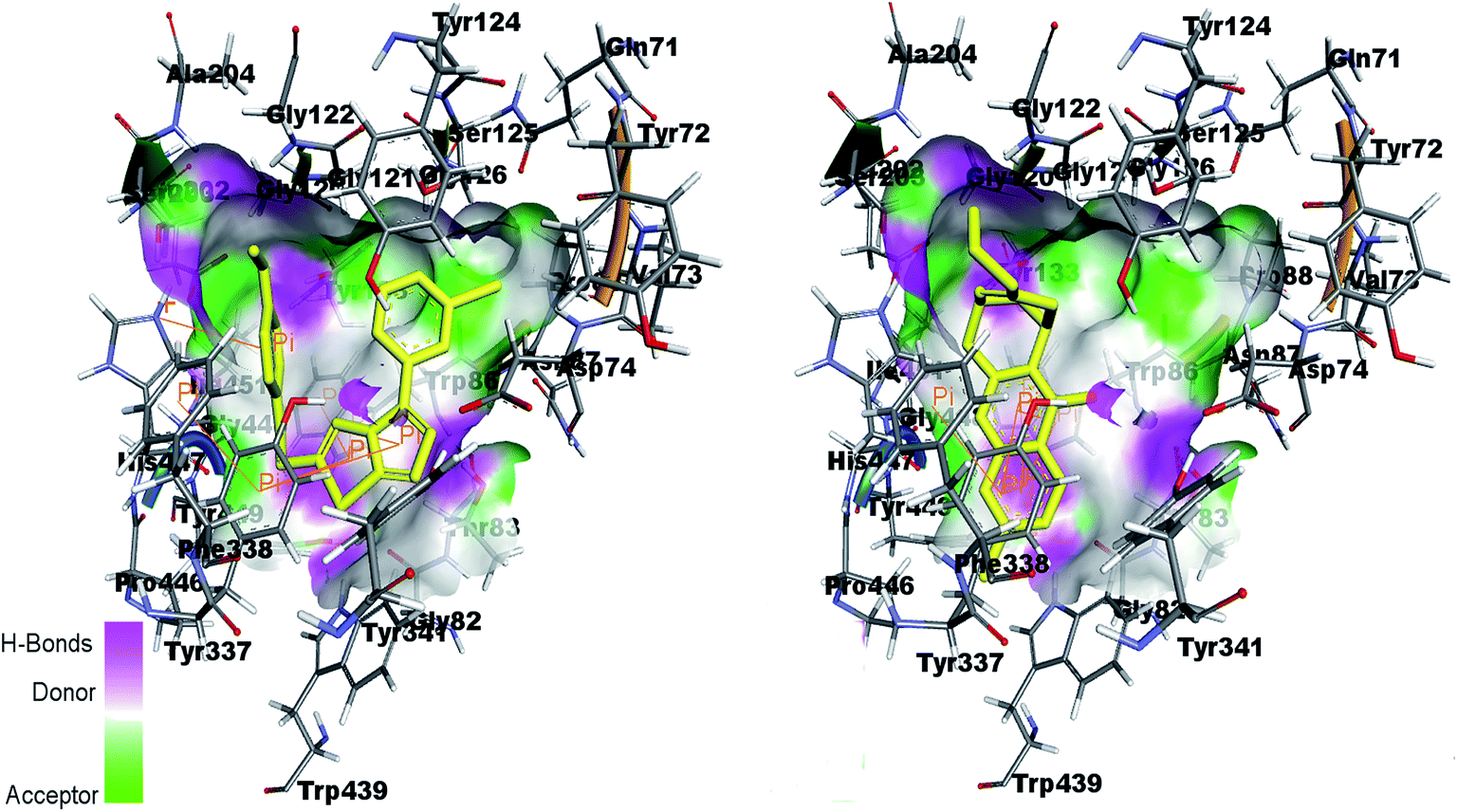 | ||
| Fig. 13 Binding site interactions of amino acids inside the active pockets of AChE for reference ligand huprine W (right) and the potent compound 4k (left). | ||
By re-docking the crystallographic ligand tacrine in the active site of BChE, the most important residue involved in the active site is Try82 showing π–π interaction with tacrine. The triazolothiadiazole (4a–l) and triazolothiadiazine (5a–p) ring system adapts a comparable position to tacrine and almost all the docked compounds interact with non-polar Try82 and shows hydrophobic interactions. Fig. 14 shows the similar binding interactions by reference ligand and compound 5d. Trp82 is involved in π-interaction as shown in figure.
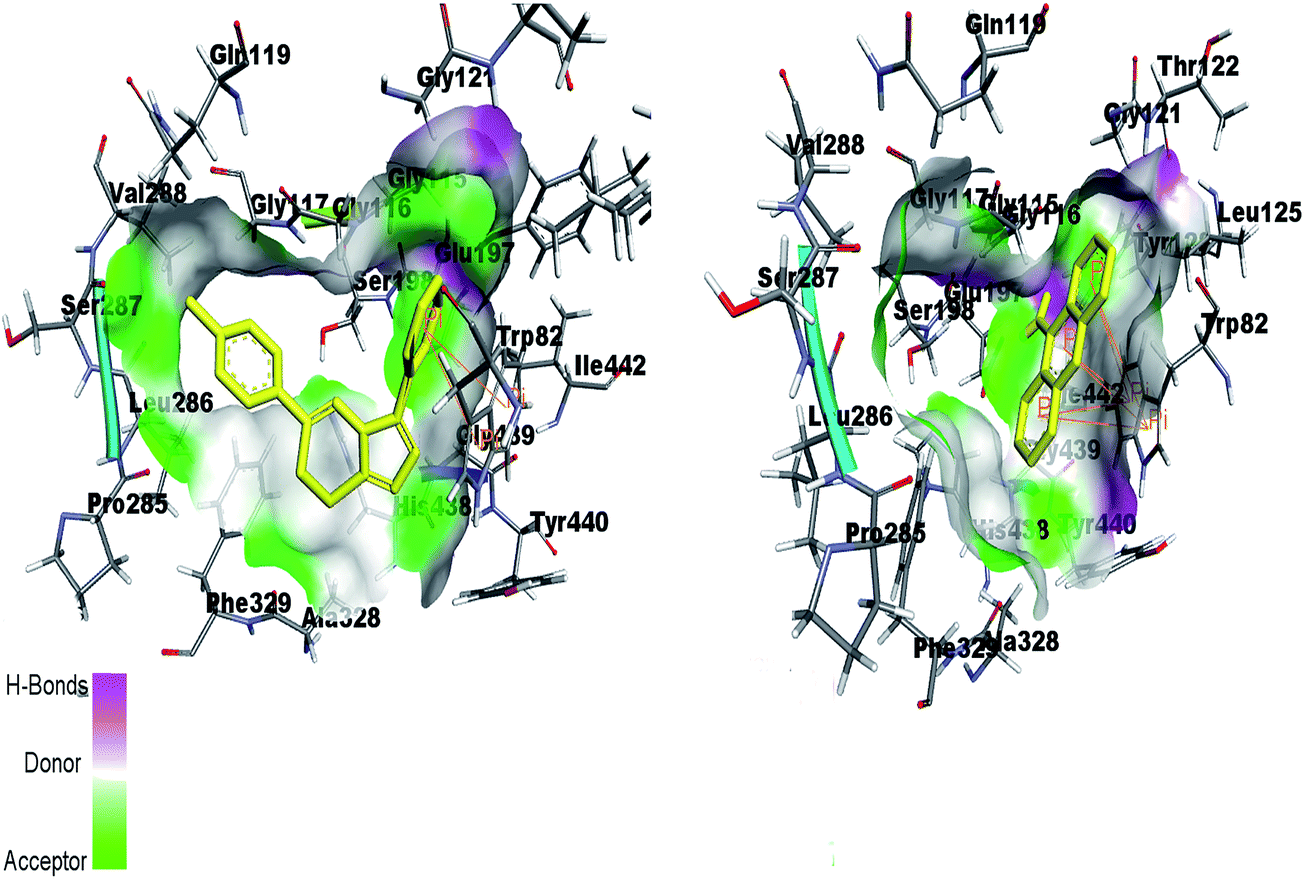 | ||
| Fig. 14 Binding site interactions of amino acids inside the active pockets of BChE for reference ligand tacrine (right) and the potent compound 5d (left). | ||
Fig. 15 (left) showed the predicted conformations of compounds 4a, 4i, 4j, 4k and 4l in the active site pocket of AChE. Similarly, Fig. 15 (right) represents the predicted conformations of compounds 5a, 5b, 5g, 5h, 5i, 5j, 5k, 5l, 5n, 5o and 5p in the active site pocket of AChE. For the 4a–l series of compounds, the central portion of triazolothiadiazole ring is directed towards the residues Gly82, Thr83, Tyr337 and Phe338 while the phenyl rings at the tails are directed towards the residues Trp86, Gly120, Gly121, Gly122, Asp74, Asn87, His447 and Gly448 and Gly82. In case of 5a–p, the tails having phenyl rings bearing different substituents attached are directed towards the residues Trp439, Asp74, Val73 and Ser125. Whereas, the triazolothiadiazine ring present at center is directed into pocket and residues Gly121, Gly126, Leu130, Gly448, His447, Tyr449 and Ser203 are involved in the interactions.
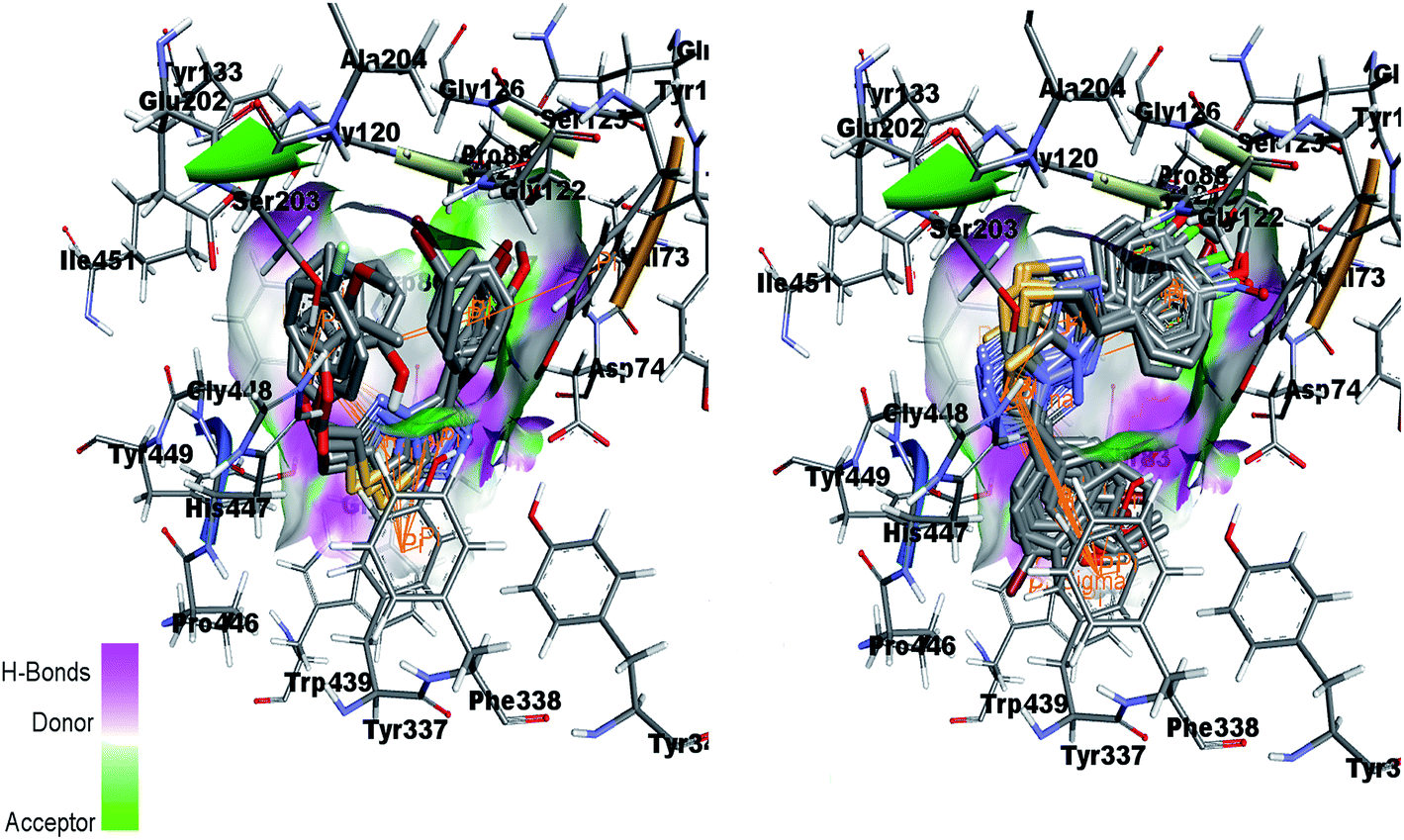 | ||
| Fig. 15 Overlap of 4a, 4i, 4j, 4k and 4l inside active site of AChE (left) and overlap of 5a, 5b, 5g, 5h, 5i, 5j, 5k, 5l, 5n, 5o and 5p in the active pocket of AChE (right). | ||
Fig. 16 (left) showed the predicted conformations of compounds 4a, 4c, 4d, 4e, 4g, 4i, 4j and 4l in the active site pocket of BChE. Similarly, Fig. 16 (right) represented the predicted conformations of compounds 5a, 5b, 5c, 5f, 5g, 5i, 5j, 5k, 5l, 5n, 5o and 5p in the active site pocket of BChE. In case of BChE active pocket, the residues Ser79, Trp82, Asn83, Gly121 and Gly78 are involved in the interaction with the phenyl rings at tails in the 4a–l series of compounds. Whereas, the central portion of triazolothiadiazole ring is directed towards the residues Gly115, Ser198, Gly439, Gly197 and Ala328. For 5a–p series, the triazolothiadiazine ring present at centre is directed into pocket and residues Asp70, Try86, Gly116, Ser198 and Try449 are involved in the interactions and the tails having phenyl rings with different substituents are directed towards the residues Phe398, Ser198, Trp430, Ala328.
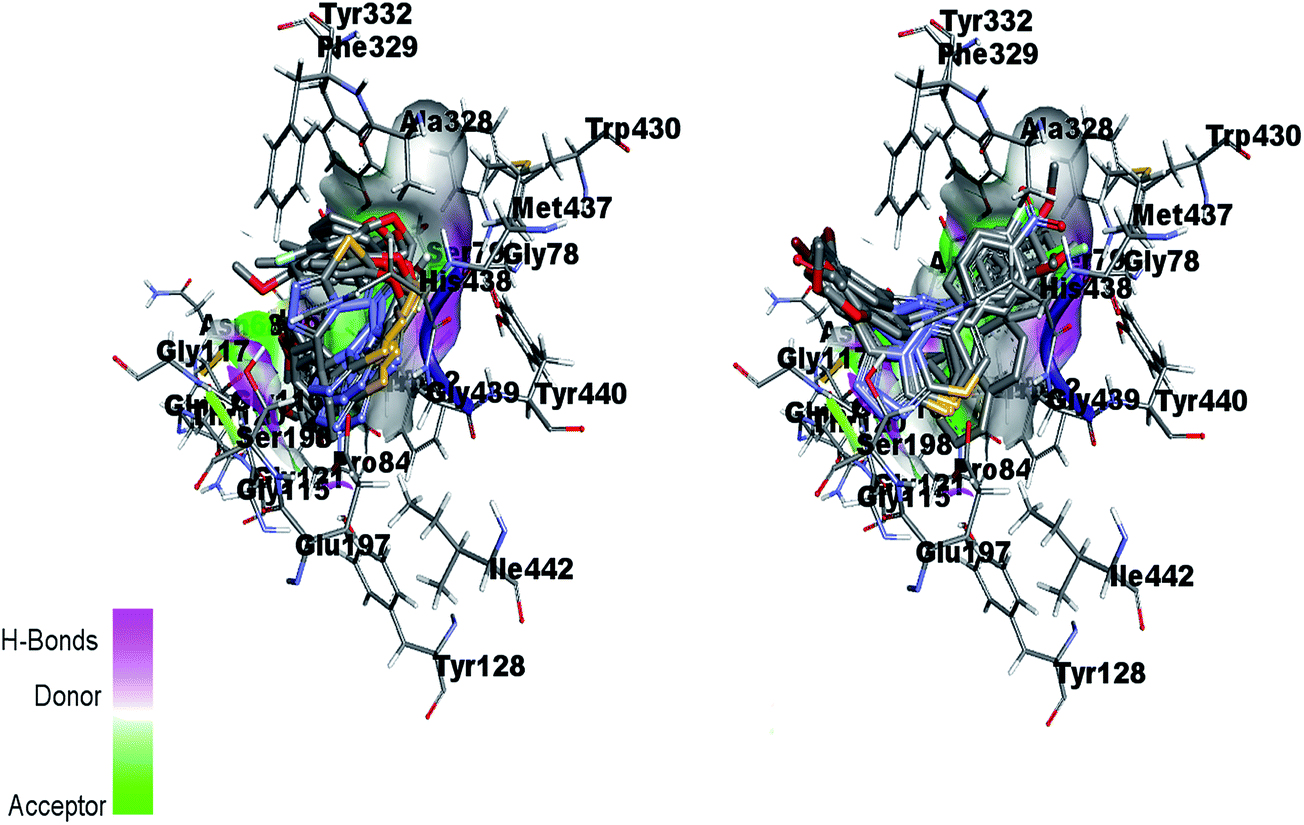 | ||
| Fig. 16 Overlap of 4a, 4c, 4d, 4e, 4g, 4i, 4j and 4l inside active site pocket of BChE (left) and overlap of 5a, 5b, 5c, 5f, 5g, 5i, 5j, 5k, 5l, 5n, 5o and 5p in the active pocket of BChE (right). | ||
Our docking scores demonstrated that most of the compounds of both the series possessed similar binding modes with different docking scores as shown in Tables 6 and 7. Along with other studies, when compounds were evaluated for drug likeness and oral bioavailability, the results were in accordance with Lipinski's rule of five parameters (Table 8). The TPSA values showed that compounds in the form of drugs can be easily penetrable as all compounds showed polar surface area in the range of 43–99 Å2. In general, for molecules to penetrate the blood–brain barrier and therefore to act as a receptor, an area less than 90 Å2 is usually needed and molecules with a polar surface area of greater than 120–140 Å2 tend to be poor in permeating cell membranes.35
| Codes | AChE | BChE |
|---|---|---|
| Docking scores (kcal mol−1) | ||
| 4a | −9.59 | −8.56 |
| 4b | −9.53 | −9.38 |
| 4c | −10.03 | −8.95 |
| 4d | −9.89 | −8.37 |
| 4e | −9.88 | −7.85 |
| 4f | −9.66 | −8.25 |
| 4g | −10.01 | −9.02 |
| 4h | −10.76 | −9.05 |
| 4i | −10.33 | −10.05 |
| 4j | −10.34 | −9.36 |
| 4k | −10.64 | −8.56 |
| 4l | −9.96 | −9.83 |
| Codes | AChE | BChE |
|---|---|---|
| Docking scores (kcal mol−1) | ||
| 5a | −10.13 | −9.16 |
| 5b | −10.21 | −9.49 |
| 5c | −10.43 | −8.65 |
| 5d | −10.85 | −9.07 |
| 5e | −9.47 | −6.91 |
| 5f | −11.92 | −10.18 |
| 5g | −10.08 | −8.31 |
| 5h | −10.15 | −9.53 |
| 5i | −11.01 | −9.86 |
| 5j | −11.02 | −10.17 |
| 5k | −10.38 | −9.48 |
| 5l | −10.86 | −10.17 |
| 5m | −10.1 | −8.11 |
| 5n | −11.77 | −11.08 |
| 5o | −11.14 | −9.45 |
| 5p | −11.16 | −10.78 |
| Codes | milogP |
TPSA | natoms | MW | nON | nOHNH | nviolations | nrotb | volume |
|---|---|---|---|---|---|---|---|---|---|
| a milogP = octanol–water partition coefficient, TPSA = Topological Polar Surface Area, natoms = number of atoms, MW = molecular weight, nON = number of hydrogen acceptor, nOHNH = number of hydrogen donor, nviolations = violations from Lipinski's rule, nrotb = number of rotatable bonds. |
|||||||||
| 4a | 3.026 | 81.787 | 25.0 | 354.391 | 7 | 1 | 0 | 5 | 293.812 |
| 4b | 3.24 | 81.787 | 25.0 | 354.391 | 7 | 1 | 0 | 5 | 293.812 |
| 4c | 4.12 | 61.559 | 25.0 | 352.419 | 6 | 0 | 0 | 5 | 302.355 |
| 4d | 4.168 | 61.559 | 25.0 | 352.419 | 6 | 0 | 0 | 5 | 302.355 |
| 4e | 3.776 | 70.793 | 26.0 | 368.418 | 7 | 0 | 0 | 6 | 311.34 |
| 4f | 3.883 | 61.559 | 25.0 | 356.382 | 6 | 0 | 0 | 5 | 290.726 |
| 4g | 3.754 | 72.553 | 24.0 | 403.261 | 6 | 1 | 0 | 4 | 286.152 |
| 4h | 3.968 | 72.553 | 24.0 | 403.261 | 6 | 1 | 0 | 4 | 286.152 |
| 4i | 4.848 | 52.325 | 24.0 | 401.289 | 5 | 0 | 0 | 4 | 294.695 |
| 4j | 4.896 | 52.325 | 24.0 | 401.289 | 5 | 0 | 0 | 4 | 294.695 |
| 4k | 4.504 | 61.559 | 25.0 | 417.288 | 6 | 0 | 0 | 5 | 303.68 |
| 4l | 4.611 | 52.325 | 24.0 | 405.252 | 5 | 0 | 0 | 4 | 283.065 |
| 5a | 3.95 | 61.55 | 25.0 | 352.419 | 6 | 0 | 0 | 4 | 302.355 |
| 5b | 4.571 | 52.316 | 24.0 | 356.838 | 5 | 0 | 0 | 3 | 290.345 |
| 5c | 4.057 | 52.316 | 24.0 | 340.383 | 5 | 0 | 0 | 3 | 281.741 |
| 5d | 4.342 | 52.316 | 24.0 | 336.42 | 5 | 0 | 0 | 3 | 293.371 |
| 5e | 5.688 | 52.316 | 29.0 | 398.491 | 5 | 0 | 1 | 4 | 348.218 |
| 5f | 5.052 | 52.316 | 27.0 | 372.453 | 5 | 0 | 1 | 3 | 320.801 |
| 5g | 3.852 | 98.14 | 26.0 | 367.39 | 8 | 0 | 0 | 4 | 300.144 |
| 5h | 5.177 | 52.316 | 25.0 | 391.283 | 5 | 0 | 1 | 3 | 303.881 |
| 5i | 4.702 | 52.316 | 24.0 | 401.289 | 5 | 0 | 0 | 3 | 294.695 |
| 5j | 5.323 | 43.082 | 23.0 | 405.708 | 4 | 0 | 1 | 2 | 282.685 |
| 5k | 4.809 | 43.082 | 23.0 | 389.253 | 4 | 0 | 0 | 2 | 274.081 |
| 5l | 5.094 | 43.082 | 23.0 | 385.29 | 4 | 0 | 1 | 2 | 285.71 |
| 5m | 6.441 | 43.082 | 28.0 | 447.361 | 4 | 0 | 1 | 3 | 340.558 |
| 5n | 5.805 | 43.082 | 26.0 | 421.323 | 4 | 0 | 1 | 2 | 313.141 |
| 5o | 4.604 | 88.906 | 25.0 | 416.26 | 7 | 0 | 0 | 3 | 292.483 |
| 5p | 5.929 | 43.082 | 24.0 | 440.153 | 4 | 0 | 1 | 2 | 296.221 |
Conclusion
In summary, to develop new drugs for the treatment of Alzheimer's disease, a small library of 28 new triazolothiadiazole and triazolothiadiazine compounds was designed and synthesized. The structural build-up was established on the basis of spectro-analytical methods and, in case of 4i and 5e, by X-ray crystallography. The newly synthesized triazolothiadiazoles (4a–l) and triazolothiadiazines (5a–p) were evaluated for their cholinesterase inhibition against EeAChE and hBChE by Ellman's method using neostigmine and donepezil as standard inhibitors. Among the screened derivatives, 4j emerged as a lead candidate showing highest inhibition with IC50 value of 0.117 ± 0.007 μM, which is ∼139-fold strong inhibition as compared to neostigmine. This compound incorporated a bromo substituent at the meta-position of the phenyl ring attached at C-3 along with 4-methyl substituent at the para-position of the aromatic ring attached at C-6 of triazolothiadiazole core. In the triazolothiadiazine series, 5j and 5e depicted a clear selectivity towards EeAChE with IC50 values of 0.065 ± 0.005 and 0.075 ± 0.001 μM, respectively, which is ∼250- and ∼218-fold stronger inhibition as compared to neostigmine (IC50 = 16.3 ± 1.12 μM), whereas, compound 5d was the lead candidate against BChE with IC50 value of 0.066 ± 0.021 μM (∼427- and 109-fold stronger inhibition) as compared to neostigmine and donepezil, respectively. Overall, these heteroaromatic structures with substituted aryl rings installed at 3- and 6-position have emerged as highly promising anti-Alzheimer drug candidates with potent inhibitory efficacy.In addition, the synthesized compounds were also tested for their monoamine oxidase (MAO) inhibition. Among triazolothiadiazole series, 4a delivered highest potency against MAO-A with IC50 value of 0.11 ± 0.005 μM which is ∼33-fold higher inhibition as compared to standard inhibitor, clorgyline (IC50 = 3.64 ± 0.012 μM), whereas, among triazolothiadiazine series, compound 5c turned to be a lead candidate with IC50 value of 0.011 ± 0.001 μM which is ∼330-fold strong inhibition as compared to the standard inhibitor, clorgyline. In general, the synthesized compounds were more potent and selective towards MAO-A, opening the possibility of rational design of more potent and selective inhibitors of monoamine oxidases (MAO). Computational analysis revealed plausible putative binding modes of the extended structure of 4k and 5d along the active site gorge of AChE and BuChE. These inhibitors can be considered as promising lead for further investigations for the treatment of AD.
Experimental
General
Unless otherwise noted, all materials were obtained from commercial suppliers (Aldrich and Alfa aesar companies) and used without further purification. Thin layer chromatography (TLC) was performed on Merck DF-Alufoilien 60F254 0.2 mm precoated plates. Product spots were visualized under UV light at 254 and 365 nm. Melting points were recorded on a Stuart melting point apparatus (SMP3) and are uncorrected. Infra-red (IR) spectra were recorded on FTS 3000 MX, Bio-Rad Merlin (Excalibur model) spectrophotometer. 1H NMR spectra were recorded on a Bruker Avance (300 MHz) spectrometer. Chemical shifts (δ) are quoted in parts per million (ppm) downfield of tetramethylsilane, using residual solvent as internal standard (DMSO-d6 at 2.50 ppm). Proton-decoupled 13CNMR spectra were recorded on a Bruker Avance (75 MHz) spectrometer using deuterated solvent as internal standard (DMSO-d6 at 39.52 ppm). The elemental analysis was performed on Leco CHNS-932 Elemental Analyzer, Leco Corporation (USA). Abbreviations used in the description of resonances are: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), Ar (aromatic).Preparation of 4-amino-1,2,4-triazole-3-thiol (3a,b)
A mixture of corresponding benzoic acid (1.0 mmol) and thiocarbohydrazide (1.5 mmol) was heated in a sand bath at 190–200 °C for 1 h. The product obtained on cooling was triturated with hot water, filtered, dried, and recrystallized (ethanol) to afford corresponding 4-amino-1,2,4-triazole-3-thiol.30N), 1546, 1511 (CC); 1H NMR (300 MHz, DMSO-d6): δ 13.83 (s, 1H, SH), 7.99 (d, 2H, J = 8.7 Hz, Ar-H), 7.07 (d, 2H, J = 8.7 Hz, Ar-H), 5.77 (s, 2H, NH2), 3.81 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 167.03, 161.28, 149.76, 130.05, 118.53, 114.40, 55.81. Analysis calcd for C9H10N4OS (222.06): C, 48.63; H, 4.53; N, 25.21; S, 14.43. Found: C, 48.54; H, 4.43; N, 25.18; S, 14.30.N), 1567, 1521 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.03 (s, 1H, SH), 8.28–8.27 (m, 1H, Ar-H), 8.01 (d, 1H, J = 8.1 Hz, Ar-H), 7.76–7.73 (m, 1H, Ar-H), 7.49 (t, 1H, J = 7.8 Hz, Ar-H), 5.80 (s, 2H, NH2); 13C NMR (75 MHz, DMSO-d6): δ 167.72, 148.49, 133.66, 131.22, 130.84, 128.33, 127.39, 122.08. Analysis calcd for C8H7BrN4S (269.96): C, 35.44; H, 2.60; N, 20.66; S, 11.83. Found: C, 35.29; H, 2.49; N, 20.57; S, 11.76.Preparation of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles (4a–l)
A mixture of corresponding 4-amino-1,2,4-triazole-3-thiol (3a,b) (1.0 mmol) and substituted aryloxy acids (1.1 mmol) in POCl3 (5 mL) was refluxed for 6 h. The reaction mixture was slowly poured into crushed ice with stirring and neutralized with sodium bicarbonate. Solid material was filtered, washed with cold water, dried, and recrystallized (ethanol) to afford 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles (4a–l).28,29N), 1565, 1502 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.13 (d, 2H, J = 8.7 Hz, Ar-H), 7.96 (d, 2H, J = 9.0 Hz, Ar-H), 7.17–7.04 (m, 3H, Ar-H), 6.93 (t, 1H, J = 5.7 Hz, Ar-H), 5.78 (s, 2H, OCH2), 5.56 (s, 1H, OH), 3.83 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 169.23, 166.87, 161.25, 152.55, 149.70, 145.84, 130.05, 129.91, 127.93, 125.71, 118.51, 118.28, 114.40, 65.85, 55.84. Analysis calcd for C17H14N4O3S (354.08): C, 57.62; H, 3.98; N, 15.81; S, 9.05. Found: C, 57.53; H, 3.74; N, 15.67; S, 8.90.N), 1547, 1502 (CC);1H NMR (300 MHz, DMSO-d6): δ 8.11 (d, 2H, J = 9.0 Hz, Ar-H), 7.15–7.01 (m, 6H, Ar-H), 5.55 (s, 2H, OCH2), 5.52 (s, 1H, OH), 3.82 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 168.50, 168.40, 161.23, 153.33, 147.49, 145.67, 127.93, 121.66, 121.26, 116.24, 115.05, 65.86, 55.87. Analysis calcd for C17H14N4O3S (354.08): C, 57.62; H, 3.98; N, 15.81; S, 9.05. Found: C, 57.54; H, 3.79; N, 15.70; S, 9.17.N), 1578, 1546 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.14 (d, 2H, J = 8.7 Hz, Ar-H), 7.23–7.11 (m, 5H, Ar-H), 6.94 (t, 1H, J = 7.2 Hz, Ar-H), 5.60 (s, 2H, OCH2), 3.84 (s, 3H, OCH3), 2.62 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 168.78, 161.25, 155.60, 154.11, 145.66, 131.28, 127.91, 127.62, 126.57, 122.28, 118.28, 115.05, 112.65, 65.38, 55.85, 16.37. Analysis calcd for C18H16N4O2S (352.10): C, 61.35; H, 4.58; N, 15.90; S, 9.10. Found: C, 61.17; H, 4.42; N, 15.73; S, 9.03.N), 1547, 1508 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.12 (d, 2H, J = 8.7 Hz, Ar-H), 7.16–7.12 (m, 4H, Ar-H), 7.01 (d, 2H, J = 8.7 Hz, Ar-H), 5.55 (s, 2H, OCH2), 3.83 (s, 3H, OCH3), 2.23 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 168.49, 161.23, 155.40, 154.16, 145.64, 131.44, 130.52, 127.90, 118.26, 115.39, 115.02, 65.40, 55.83, 20.54. Analysis calcd for C18H16N4O2S (352.10): C, 61.35; H, 4.58; N, 15.90; S, 9.10. Found: C, 61.53; H, 4.39; N, 15.74; S, 9.24.N), 1548, 1505 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.13 (d, 2H, J = 8.7 Hz, Ar-H), 7.15 (d, 2H, J = 8.7 Hz, Ar-H), 7.07 (d, 2H, J = 9.0 Hz, Ar-H), 6.90 (d, 2H, J = 9.0 Hz, Ar-H), 5.53 (s, 2H, OCH2), 3.83 (s, 3H, OCH3), 3.70 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 168.62, 161.24, 154.90, 154.18, 151.43, 145.65, 127.91, 118.25, 116.75, 115.21, 115.04, 66.01, 55.85, 55.83. Analysis calcd for C18H16N4O3S (368.09): C, 58.68; H, 4.38; N, 15.21; S, 8.70. Found: C, 58.53; H, 4.29; N, 15.32; S, 8.56.N), 1552, 1504 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.11 (d, 2H, J = 8.7 Hz, Ar-H), 7.46 (bs, 2H, Ar-H), 7.18–7.12 (m, 4H, Ar-H), 5.58 (s, 2H, OCH2), 3.82 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 168.03, 161.23, 159.37, 156.22, 154.20, 153.82, 145.66, 127.90, 118.22, 117.19, 117.08, 116.76, 116.45, 115.03, 65.97, 55.85. Analysis calcd for C17H13FN4O2S (356.07): C, 57.29; H, 3.68; N, 15.72; S, 9.00. Found: C, 57.43; H, 3.46; N, 15.56; S, 9.09.N), 1556, 1510 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.03 (s, 1H, OH), 8.28 (s, 1H, Ar-H), 8.01 (d, 1H, J = 7.8 Hz, Ar-H), 7.76 (d, 1H, J = 7.8 Hz, Ar-H), 7.53–7.47 (t, 2H, J = 7.8 Hz, Ar-H), 7.25–7.16 (m, 1H, Ar-H), 7.12–6.94 (m, 2H, Ar-H), 5.80 (s, 2H, OCH2); 13C NMR (75 MHz, DMSO-d6): δ 168.44, 161.70, 156.23, 151.54, 146.38, 142.87, 135.50, 132.65, 129.87, 128.54, 126.32, 125.12, 116.77, 112.68, 111.09, 65.38. Analysis calcd for C16H11BrN4O2S (401.98): C, 47.66; H, 2.75; N, 13.89; S, 7.95. Found: C, 47.50; H, 2.53; N, 13.71; S, 7.78.N), 1567, 1515 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.06 (s, 1H, OH), 8.33 (bs, 1H, Ar-H), 8.20 (d, 1H, J = 8.1 Hz, Ar-H), 7.76 (d, 1H, J = 8.1 Hz, Ar-H), 7.59–7.54 (m, 1H, Ar-H), 7.25–7.16 (m, 2H, Ar-H), 7.12–6.94 (m, 2H, Ar-H), 5.65 (s, 2H, OCH2); 13C NMR (75 MHz, DMSO-d6): δ 168.34, 161.87, 156.65, 151.76, 146.34, 142.87, 135.58, 132.98, 129.09, 128.06, 125.32, 125.19, 118.67, 65.38. Analysis calcd for C16H11BrN4O2S (401.98): C, 47.66; H, 2.75; N, 13.89; S, 7.95. Found: C, 47.53; H, 2.57; N, 13.72; S, 7.80.N), 1565, 1534 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.37 (bs, 1H, Ar-H), 8.22 (d, 1H, J = 8.1 Hz, Ar-H), 7.78 (d, 1H, J = 8.4 Hz, Ar-H), 7.59 (t, 1H, J = 8.1 Hz, Ar-H), 7.24–7.13 (m, 3H, Ar-H), 6.95 (t, 1H, J = 6.6 Hz, Ar-H), 5.66 (s, 2H, OCH2), 2.27 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 168.26, 161.77, 156.24, 150.54, 145.34, 142.87, 133.53, 132.57, 129.98, 128.45, 126.55, 125.72, 117.53, 112.68, 111.09, 65.38, 16.40. Analysis calcd for C17H13BrN4OS (400.00): C, 50.88; H, 3.27; N, 13.96; S, 7.99. Found: C, 50.69; H, 3.11; N, 13.77; S, 7.84.N), 1556, 1522 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.34 (bs, 1H, Ar-H), 8.22 (d, 1H, J = 8.1 Hz, Ar-H), 7.77 (d, 1H, J = 8.1 Hz, Ar-H), 7.56 (t, 1H, J = 7.8 Hz, Ar-H), 7.45–7.42 (m, 2H, Ar-H), 7.24–7.19 (m, 2H, Ar-H), 5.60 (s, 2H, OCH2), 2.24 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 168.87, 161.47, 155.29, 151.50, 145.87, 138.57, 133.80, 132.52, 128.18, 126.59, 124.62, 117.53, 112.60, 65.76, 16.49. Analysis calcd for C17H13BrN4OS (400.00): C, 50.88; H, 3.27; N, 13.96; S, 7.99. Found: C, 50.64; H, 3.36; N, 13.85; S, 7.79.N), 1543, 1527 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.35–8.32 (m, 1H, Ar-H), 8.22 (d, 1H, J = 8.1 Hz, Ar-H), 7.77 (d, 1H, J = 8.1 Hz, Ar-H), 7.57 (t, 1H, J = 7.8 Hz, Ar-H), 7.16–6.77 (m, 4H, Ar-H), 5.57 (s, 2H, OCH2), 3.70 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 168.60, 161.57, 156.45, 150.34, 145.87, 142.87, 133.12, 129.76, 126.38, 125.62, 118.51, 115.68, 111.08, 65.34, 55.42. Analysis calcd for C17H13BrN4O2S (415.99): C, 48.93; H, 3.14; N, 13.43; S, 7.68. Found: C, 48.73; H, 3.27; N, 13.31; S, 7.50.N), 1558, 1517 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.34–8.31 (m, 1H, Ar-H), 8.20 (d, 1H, J = 7.8 Hz, Ar-H), 7.78.7.75 (m, 1H, Ar-H), 7.57 (t, 1H, J = 8.1 Hz, Ar-H), 7.20–7.16 (m, 4H, Ar-H), 5.63 (s, 2H, OCH2); 13C NMR (75 MHz, DMSO-d6): δ 168.93, 159.39, 156.24, 155.26, 153.83, 153.80, 144.34, 133.51, 131.90, 128.43, 127.94, 125.17, 122.73, 117.18, 117.07, 116.78, 116.47, 65.92. Analysis calcd for C16H10BrFN4OS (403.97): C, 47.42; H, 2.49; N, 13.83; S, 7.91. Found: C, 47.53; H, 2.35; N, 13.74; S, 7.78.Preparation of 1,2,4-triazolo[3,4-b][1,3,4]-thiadiazines (5a–p)
A mixture of corresponding 4-amino-1,2,4-triazole-3-thiol (3a,b) (1.0 mmol) and substituted phenacyl bromides (1.2 mmol) was refluxed in absolute ethanol (10 mL) for 7 h. The reaction mass was poured into crushed ice and neutralized with sodium bicarbonate. Solid product obtained was filtered, washed with water, dried, and recrystallized (ethanol) to afford conjugated products (6a–p).28,29N), 1541, 1513 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.00–7.96 (m, 4H, Ar-H), 7.15–7.11 (m, 4H, Ar-H), 4.40 (s, 2H, SCH2), 3.85 (s, 3H, OCH3), 3.84 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 162.75, 161.22, 155.99, 151.65, 142.62, 129.93, 129.91, 125.96, 115.04, 114.71, 56.03, 22.93. Analysis calcd for C18H16N4O2S (352.10): C, 61.35; H, 4.58; N, 15.90; S, 9.10. Found: C, 61.26; H, 4.62; N, 15.78; S, 9.01.N), 1548, 1487 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.03–8.00 (m, 2H, Ar-H), 7.97–7.92 (m, 2H, Ar-H), 7.67–7.62 (m, 2H, Ar-H), 7.15–7.10 (m, 2H, Ar-H), 4.42 (s, 2H, SCH2), 3.83 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 161.21, 155.25, 151.96, 142.41, 137.23, 132.82, 129.96, 129.78, 129.67, 118.70, 114.71, 55.81, 23.07. Analysis calcd for C17H13ClN4OS (356.05): C, 57.22; H, 3.67; N, 15.70; S, 8.99. Found: C, 57.11; H, 3.54; N, 15.53; S, 9.08.N), 1532, 1515 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.08 (bs, 2H, Ar-H), 7.98 (d, 2H, J = 6.9 Hz, Ar-H), 7.42 (bs, 2H, Ar-H), 7.13 (d, 2H, J = 6.6 Hz, Ar-H), 4.42 (s, 2H, SCH2), 3.84 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 161.74, 161.17, 155.97, 151.69, 141.60, 133.76, 129.93, 125.96, 118.67, 115.34, 114.71, 55.83, 22.97. Analysis calcd for C17H13FN4OS (340.08): C, 59.99; H, 3.85; N, 16.46; S, 9.42. Found: C, 59.81; H, 3.76; N, 16.55; S, 9.31.N), 1589, 1512 (CC); 1H NMR (300 MHz, DMSO-d6): δ 7.98 (m, 4H, Ar-H), 7.39 (d, 2H, J = 8.7 Hz, Ar-H), 7.17 (d, 2H, J = 9.0 Hz, Ar-H), 4.45 (s, 2H, SCH2), 3.84 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 161.64, 157.12, 151.46, 143.22, 142.96, 130.90, 130.28, 130.22, 128.08, 117.62, 114.85, 55.92, 23.12, 21.54. Analysis calcd for C18H16N4OS (336.10): C, 64.26; H, 4.79; N, 16.65; S, 9.53. Found: C, 64.09; H, 4.66; N, 16.81; S, 9.40.N), 1573, 1509 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.11 (d, 2H, J = 8.7 Hz, Ar-H), 8.00 (d, 2H, J = 8.7 Hz, Ar-H), 7.89 (d, 2H, J = 8.4 Hz, Ar-H), 7.77 (d, 2H, J = 8.4 Hz, Ar-H), 7.50 (d, 1H, J = 7.2 Hz, Ar-H), 7.46–7.39 (m, 2H, Ar-H), 7.17 (d, 2H, J = 8.7 Hz, Ar-H), 4.52 (s, 2H, SCH2), 3.84 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 161.68, 156.86, 151.53, 143.98, 139.15, 132.54, 130.36, 130.22, 129.60, 128.87, 128.79, 127.74, 127.35, 117.51, 114.87, 55.92, 23.18. Analysis calcd for C23H18N4OS (398.12): C, 69.32; H, 4.55; N, 14.06; S, 8.05. Found: C, 69.17; H, 4.67; N, 13.94; S, 8.01.N), 1581, 1537 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.65 (bs, 1H, Ar-H), 8.13–8.06 (m, 3H, Ar-H), 8.02 (d, 3H, J = 9.0 Hz, Ar-H), 7.69–7.62 (m, 2H, Ar-H), 7.16 (d, 2H, J = 8.7 Hz, Ar-H), 4.58 (s, 2H, SCH2), 3.85 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 161.23, 156.08, 152.00, 142.53, 134.70, 132.91, 131.26, 129.96, 129.64, 129.46, 129.35, 128.79, 128.23, 127.67, 123.66, 118.86, 114.76, 55.83, 23.05. Analysis calcd for C21H16N4OS (372.10): C, 67.72; H, 4.33; N, 15.09; S, 8.61. Found: C, 67.64; H, 4.43; N, 14.96; S, 8.49.N), 1580, 1518 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.41 (d, 2H, J = 9.0 Hz, Ar-H), 8.25 (d, 2H, J = 9.0 Hz, Ar-H), 7.95 (d, 2H, J = 8.7 Hz, Ar-H), 7.15 (d, 2H, J = 9.0 Hz, Ar-H), 4.50 (s, 2H, SCH2), 3.84 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 161.43, 156.18, 152.55, 142.59, 140.27, 138.25, 134.78, 132.85, 129.65, 129.16, 128.65, 55.84, 23.35. Analysis calcd for C17H13N5O3S (367.07): C, 55.58; H, 3.57; N, 19.06; S, 8.73. Found: C, 55.53; H, 3.66; N, 18.85; S, 8.61.N), 1547, 1536 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.07–8.00 (m, 2H, Ar-H), 7.95 (d, 2H, J = 8.7 Hz, Ar-H), 7.90 (s, 1H, Ar-H), 7.15 (d, 2H, J = 9.0 Hz, Ar-H), 4.52 (s, 2H, SCH2), 3.84 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 161.46, 156.18, 152.55, 149.66, 145.84, 145.41, 142.76, 140.56, 138.47, 134.73, 132.87, 129.57, 124.65, 55.83, 23.21. Analysis calcd for C17H12Cl2N4OS (390.01): C, 52.18; H, 3.09; N, 14.32; S, 8.20. Found: C, 52.34; H, 3.02; N, 14.38; S, 8.04.N), 1586, 1514 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.21 (d, 1H, J = 1.5 Hz, Ar-H), 8.04 (d, 1H, J = 7.8 Hz, Ar-H), 7.99 (d, 2H, J = 9.0 Hz, Ar-H), 7.75 (d, 1H, J = 8.7 Hz, Ar-H), 7.54 (t, 1H, J = 8.1 Hz, Ar-H), 7.13 (d, 2H, J = 8.7 Hz, Ar-H), 4.41 (s, 2H, SCH2), 3.85 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 162.86, 156.28, 150.43, 143.53, 133.26, 131.46, 130.69, 129.91, 128.63, 127.11, 125.80, 122.25, 115.06, 56.06, 23.00. Analysis calcd for C17H13BrN4S (400.00): C, 50.88; H, 3.27; N, 13.96; S, 7.99. Found: C, 50.69; H, 3.12; N, 13.80; S, 7.78.N), 1587, 1556 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.21–8.20 (m, 1H, Ar-H), 8.13–8.10 (m, 2H, Ar-H), 8.04 (d, 1H, J = 7.8 Hz, Ar-H), 7.75 (d, 1H, J = 8.7 Hz, Ar-H), 7.67–7.62 (m, 2H, Ar-H), 7.54 (t, 1H, J = 8.1 Hz, Ar-H), 4.46 (s, 2H, SCH2); 13C NMR (75 MHz, DMSO-d6): δ 162.88, 155.28, 151.40, 142.59, 137.66, 137.28, 132.47, 130.59, 129.99, 126.64, 124.82, 122.76, 115.67, 23.08. Analysis calcd for C16H10BrClN4S (403.95): C, 47.37; H, 2.48; N, 13.81; S, 7.90. Found: C, 47.24; H, 2.27; N, 13.96; S, 7.74.N), 1567, 1513 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.21–8.20 (m, 1H, Ar-H), 8.08 (m, 2H, Ar-H), 8.04 (d, 1H, J = 7.8 Hz, Ar-H), 7.75 (d, 1H, J = 8.7 Hz, Ar-H), 7.42 (m, 2H, Ar-H), 7.54 (t, 1H, J = 8.1 Hz, Ar-H), 4.45 (s, 2H, SCH2); 13C NMR (75 MHz, DMSO-d6): δ 162.67, 155.88, 151.49, 142.87, 137.65, 137.28, 132.47, 130.45, 129.95, 126.84, 125.11, 124.82, 124.78, 122.76, 121.54, 115.65, 23.44. Analysis calcd for C16H10BrFN4S (387.98): C, 49.37; H, 2.59; N, 14.39; S, 8.24. Found: C, 49.24; H, 2.67; N, 14.26; S, 8.12.N), 1562, 1521 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.19–8.17 (m, 1H, Ar-H), 8.02 (d, 1H, J = 7.8 Hz, Ar-H), 7.92 (d, 2H, J = 8.7 Hz, Ar-H), 7.74 (d, 1H, J = 8.4 Hz, Ar-H), 7.53 (t, 1H, J = 7.8 Hz, Ar-H), 7.39 (d, 2H, J = 8.7 Hz, Ar-H), 4.48 (s, 2H, SCH2), 2.38 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 161.76, 155.65, 151.96, 142.34, 136.39, 134.18, 130.08, 129.17, 125.10, 124.72, 122.46, 121.54, 115.63, 23.07, 21.32. Analysis. calcd for C17H13BrN4S (384.00): C, 53.00; H, 3.40; N, 14.54; S, 8.32. Found: C, 52.88; H, 3.62; N, 14.43; S, 8.17.N), 1589, 1549 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.23–8.21 (m, 1H, Ar-H), 8.11–8.04 (m, 3H, Ar-H), 7.88 (d, 2H, J = 8.4 Hz, Ar-H), 7.78–7.75 (m, 3H, Ar-H), 7.56 (d, 2H, J = 7.8 Hz, Ar-H), 7.50 (t, 1H, J = 7.5 Hz, Ar-H), 7.42 (t, 1H, J = 6.9 Hz, Ar-H), 4.48 (s, 2H, SCH2); 13C NMR (75 MHz, DMSO-d6): δ 156.34, 150.64, 143.93, 143.56, 139.14, 133.37, 132.63, 131.49, 130.77, 129.56, 128.84, 128.67, 128.53, 127.70, 127.36, 127.21, 122.30, 23.17. Analysis calcd for C22H15BrN4S (446.02): C, 59.07; H, 3.38; N, 12.52; S, 7.17. Found: C, 58.93; H, 3.54; N, 12.34; S, 7.03.N), 1567, 1519 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.65 (bs, 1H, Ar-H), 8.23–8.21 (m, 2H, Ar-H), 8.13–8.06 (m, 3H, Ar-H), 8.02 (d, 1H, J = 9.0 Hz, Ar-H), 7.77 (d, 1H, J = 8.4 Hz, Ar-H), 7.69–7.62 (m, 2H, Ar-H), 7.54 (t, 1H, J = 7.8 Hz, Ar-H), 4.58 (s, 2H, SCH2); 13C NMR (75 MHz, DMSO-d6): δ 161.23, 156.08, 152.09, 145.87, 142.53, 134.56, 132.91, 131.37, 129.96, 129.56, 129.46, 129.35, 128.84, 128.67, 128.23, 127.70, 123.66, 118.30, 114.76, 23.05. Analysis calcd for C20H13BrN4S (420.00): C, 57.02; H, 3.11; N, 13.30; S, 7.61. Found: C, 56.89; H, 3.26; N, 13.18; S, 7.47.N), 1525, 1494 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.41 (d, 2H, J = 7.5 Hz, Ar-H), 8.24 (d, 2H, J = 7.5 Hz, Ar-H), 8.16 (bs, 1H, Ar-H), 8.01 (d, 1H, J = 6.9 Hz, Ar-H), 7.77 (d, 1H, J = 6.9 Hz, Ar-H), 7.56 (t, 1H, J = 7.2 Hz, Ar-H), 4.51 (s, 2H, SCH2); 13C NMR (75 MHz, DMSO-d6): δ 161.87, 156.29, 150.47, 145.66, 142.50, 133.76, 131.48, 130.69, 128.99, 128.67, 127.16, 125.80, 122.25, 23.42. Analysis calcd for C16H10BrN5O2S (414.97): C, 46.17; H, 2.42; N, 16.82; S, 7.70. Found: C, 45.98; H, 2.27; N, 16.97; S, 7.54.N), 1556, 1508 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.24 (bs, 1H, Ar-H), 8.16 (d, 1H, J = 8.1 Hz, Ar-H), 7.95 (d, 2H, J = 6.9 Hz, Ar-H), 7.90 (s, 1H, Ar-H), 7.78 (d, 1H, J = 8.4 Hz, Ar-H), 7.54 (t, 1H, J = 7.2 Hz, Ar-H), 4.51 (s, 2H, SCH2); 13C NMR (75 MHz, DMSO-d6): δ 161.46, 156.28, 152.47, 149.65, 145.60, 140.59, 138.71, 135.74, 130.69, 129.43, 128.67, 126.32, 125.88, 122.25, 118.54, 23.12. Analysis calcd for C16H9BrCl2N4S (437.91): C, 43.66; H, 2.06; N, 12.73; S, 7.29. Found: C, 43.53; H, 1.96; N, 12.85; S, 7.10.Pharmacological protocols
Molecular modelling studies of cholinesterases
Before studying the proteins in molecular modeling experiments, the structures of enzymes and compounds were prepared. The enzyme structures were protonated with Protonate3D37 algorithm implemented in the molecular modeling tool MOE.38 The structures were energy minimized using Amber99 force field with all the heteroatoms and solvent molecules present in the protein. The backbone atoms were restrained with a small force in order to avoid collapse of the binding pockets during energy minimization calculations. After minimization, the co-crystallized ligands and solvent molecules were removed.
Acknowledgements
The authors would like to sincerely appreciate the Deanship of Scientific Research at King Saud University for its funding through the Research Group Project no RGV-VPP-345.References
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- P. Faller and C. Hureau, Chem.–Eur. J., 2012, 18, 15910–15920 CrossRef CAS PubMed.
- T. Hartmann, J. Kuchenbecker and M. O. W. Grimm, J. Neurochem., 2007, 103, 159–170 CrossRef CAS PubMed.
- M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8, 856–865 CrossRef CAS PubMed.
- M. Cygler, J. D. Schrag, J. L. Sussman, M. Harel, I. Silman, M. K. Gentry and B. P. Doctor, Protein Sci., 1993, 2, 366–382 CrossRef CAS PubMed.
- E. Giacobini, Pharmacol. Res., 2004, 50, 433–440 CrossRef CAS PubMed.
- E. Groner, Y. Ashani, D. Schorer-Apelbaum, J. Sterling, Y. Herzig and M. Weinstock, Mol. Pharmacol., 2007, 71, 1610–1617 CrossRef CAS PubMed.
- S. Y. Chiou, C. F. Huang, M. T. Hwang and G. Lin, J. Biochem. Mol. Toxicol., 2009, 23, 303–308 CrossRef CAS PubMed.
- J. L. Cummings, N. Engl. J. Med., 2004, 351, 56–67 CrossRef CAS PubMed.
- M. M. Mesulam, A. Guillozet, P. Shaw, A. Levey, E. G. Duysen and O. Lockridge, Neuroscience, 2002, 110, 627–639 CrossRef CAS.
- (a) H. M. Greenblatt, H. Dvir, I. Silman and J. L. Sussman, J. Mol. Neurosci., 2003, 20, 369–383 CrossRef CAS; (b) R. Cacabelos, J. Mol. Biol., 2008, 448, 213–357 CAS; (c) A. Contestabile, Behav. Brain Res., 2011, 221, 334–340 CrossRef CAS PubMed.
- M. B. H. Youdim, J. P. M. Finberg and K. F. Tipton, in Monoamine Oxidase, ed. U. Tredelenburg and N. Weiner, Springer-Verlag, Berlin, 1988, pp. 119–192 Search PubMed.
- K. N. Westlund, R. M. Denney, R. M. Rose and C. W. Abell, Neuroscience, 1988, 25, 439–456 CrossRef CAS.
- A. S. Kalgutkar, D. K. Dalvie, N. Castagnoli and T. J. Taylor, Chem. Res. Toxicol., 2001, 14, 1139–1162 CrossRef CAS.
- T. P. Singer, in Chemistry and Biochemistry of Flavoenzymes, ed. F. Müller, CRC Press, Boca Ratón, FL, USA, 1990, vol. III, pp. 437–470 Search PubMed.
- (a) M. B. H. Youdim, D. Edmondson and K. F. Tipton, Nat. Rev. Neurosci., 2006, 7, 295–309 CrossRef CAS PubMed; (b) M. Bortolato, K. Chen and J. C. Shih, Adv. Drug Delivery Rev., 2008, 60, 1527–1533 CrossRef CAS PubMed.
- (a) C. W. Muir, A. R. Kennedy, J. M. Redmond and A. J. B. Watson, Org. Biomol. Chem., 2013, 11, 3337–3340 RSC; (b) I. Khan, S. Ali, S. Hameed, N. H. Rama, M. T. Hussain, A. Wadood, R. Uddin, Z. Ul-Haq, A. Khan, S. Ali and M. I. Choudhary, Eur. J. Med. Chem., 2010, 45, 5200–5207 CrossRef CAS PubMed; (c) I. Khan, M. Hanif, A. A. Khan, N. H. Rama, M. T. Hussain, M. A. S. Aslam and J. Iqbal, Aust. J. Chem., 2012, 65, 1413–1419 CrossRef CAS; (d) I. Khan, A. Ibrar, M. Waqas and J. M. White, Phys. Rev. Res. Int., 2013, 3, 10–17 Search PubMed; (e) A. Ibrar, I. Khan and N. Abbas, Arch. Pharm. Chem. Life Sci., 2013, 346, 423–446 CrossRef CAS PubMed; (f) I. Khan, A. Ibrar and N. Abbas, Arch. Pharm. Chem. Life Sci., 2014, 347, 1–20 CrossRef CAS PubMed; (g) M. Hanif, I. Khan, N. H. Rama, S. Noreen, M. I. Choudhary, P. G. Jones and M. Iqbal, Med. Chem. Res., 2012, 21, 3885–3896 CrossRef CAS; (h) I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193–244 CrossRef CAS PubMed; (i) I. Khan, A. Ibrar, W. Ahmed and A. Saeed, Eur. J. Med. Chem., 2015, 90, 124–169 CrossRef CAS PubMed.
- S. N. Swamy, Basappa, B. S. Priya, B. Prabhuswamy, B. H. Doreswamy, J. S. Prasad and K. S. Rangappa, Eur. J. Med. Chem., 2006, 41, 531–538 CrossRef CAS PubMed.
- (a) B. S. Holla, K. N. Poojary, B. S. Rao and M. K. Shivananda, Eur. J. Med. Chem., 2002, 37, 511–517 CrossRef; (b) I. Khan, A. Ibrar and N. Abbas, Eur. J. Med. Chem., 2013, 63, 854–868 CrossRef CAS PubMed.
- J. T. Witkoaski, R. K. Robins, R. W. Sidwell and L. N. Simon, J. Med. Chem., 1972, 15, 150–154 Search PubMed.
- K. S. Bhat, B. Poojary, D. J. Prasad, P. Naik and B. S. Holla, Eur. J. Med. Chem., 2009, 44, 5066–5070 CrossRef CAS PubMed.
- R. Gupta, S. Paul, A. K. Gupta and P. L. Kachroo, Indian J. Chem., 1994, 33B, 888–891 CAS.
- (a) V. Mathew, J. Keshavayya and V. P. Vaidya, Eur. J. Med. Chem., 2006, 41, 1048–1058 CrossRef CAS PubMed; (b) V. Mathew, D. Giles, J. Keshavayya and V. P. Vaidya, Arch. Pharm., 2009, 342, 210–222 CrossRef CAS PubMed; (c) P. Karegoudar, D. J. Prasad, M. Ashok, M. Mahalinga, B. Poojary and B. S. Holla, Eur. J. Med. Chem., 2008, 43, 808–815 CrossRef CAS PubMed; (d) M. S. Karthikeyan, B. S. Holla, B. Kulkuraya and N. S. Kumari, Monatsh. Chem., 2007, 138, 1309–1316 CrossRef CAS.
- D. Sunil, A. M. Isloor, P. Shetty, K. Satyamoorthy and A. S. B. Prasad, Arabian J. Chem., 2010, 3, 211–217 CrossRef CAS PubMed.
- I. Khan, S. Hameed, N. A. Abdul-Reda, N. A. Al-Masoudi and J. Simpson, Z. Naturforsch., B: Chem. Sci., 2015, 70, 47–58 Search PubMed.
- (a) M. Hanif, M. Saleem, M. T. Hussain, N. H. Rama, S. Zaib, M. A. M. Aslam, P. G. Jones and J. Iqbal, J. Braz. Chem. Soc., 2012, 23, 854–860 CrossRef CAS PubMed; (b) A. P. Skoumbourdis, C. A. LeClair, E. Stefan, A. G. Turjanski, W. Maguire, S. A. Titus, R. Huang, D. S. Auld, J. Inglese, C. P. Austin, S. W. Michnick, M. Xia and C. J. Thomas, Bioorg. Med. Chem. Lett., 2009, 19, 3686–3692 CrossRef CAS PubMed.
- D. Lauffer, P. Li and K. Mcginty, EP 2 435 443 B1, July 31, 2013.
- I. Khan, S. Zaib, A. Ibrar, N. H. Rama, J. Simpson and J. Iqbal, Eur. J. Med. Chem., 2014, 78, 167–177 CrossRef CAS PubMed.
- I. Khan, A. Ibrar, S. Zaib, S. Ahmad, N. Furtmann, S. Hameed, J. Simpson, J. Bajorath and J. Iqbal, Bioorg. Med. Chem., 2014, 22, 6163–6173 CrossRef CAS PubMed.
- S. M. I. Badr and R. M. Barwa, Bioorg. Med. Chem., 2011, 19, 4506–4512 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., 2002, B58, 380–388 CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- G. L. Ellman, K. D. Courtney, V. Andres Jr and R. M. Feather-Stone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS.
- A. Stössel, M. Schlenk, S. Hinz, P. Küppers, J. Heer, M. Gütschow and C. E. Müller, J. Med. Chem., 2013, 56(11), 4580–4596 CrossRef PubMed.
- S. A. Hitchcock and L. D. pennington, J. Med. Chem., 2006, 49(26), 7559–7583 CrossRef CAS PubMed.
- D. S. Goodsell, Computational docking of biomolecular complexes with AutoDock, Cold Spring Harb Protoc, 2009, pdb prot5200 Search PubMed.
- P. D.h.w.c.c.j.p.h. P. Labute, http://www.chemcomp.com/journal/proton.htm.
- C.C.G.I.h.w.c.c. MOE (The Molecular Operating Environment) Version 2010.10. Chemical Computing Group Inc, http://www.chemcomp.com.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- Accelrys Software Inc., Discovery Studio Modeling Environment, Release 4.0, Accelrys Software Inc., San Diego, CA; 2013 Search PubMed.
- V. N. Wouatsa, L. Misra, S. Kumar, O. Prakash, F. Khan, F. Tchoumbougnang and R. K. Venkatesh, Chem. Cent. J., 2013, 7, 125–139 CrossRef PubMed.
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714–3717 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1033196 and 1033197. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra00906e |
| ‡ Present address: School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. |
| This journal is © The Royal Society of Chemistry 2015 |