
Scalable, template-free synthesis of conducting polymer microtubes†
Kryssia P. Díaz-Orellana and
Mark E. Roberts*
Department of Chemical and Biomolecular Engineering, Clemson University, 204 Earle Hall, Clemson, SC 29634, USA. E-mail: mrober9@clemson.edu
First published on 4th March 2015
Abstract
The integration of new materials in commercial energy storage systems faces many challenges, such as scalable manufacturing, charge–discharge efficiency, long term cycle stability, and high power and high energy density. Compared with conventional high-surface area carbon nanomaterials, electroactive conducting polymers (ECPs) exhibit an increase in energy density, which is attractive for next generation supercapacitor electrode materials. When designed with micro- and nanoscale dimensions, ECP electrodes display an increase in power density by decreasing the ion diffusion length in bulk electrodes. Here, we describe a template-free method for synthesizing polypyrrole microtubes on various stainless steel meshes with a process inherently scalable to large-area substrates. Microtube growth is governed by the nucleation of hydrogen gas at the mesh joints as a result of proton reduction at the platinum counter electrode. Depending on the size and spacing of the mesh wires and the substrate proximity to the working electrode, polypyrrole microtubes can be created with cylindrical and conical shapes with diameters ranging from 50–400 μm and heights up to 1400 μm. Polymer electrodes exhibiting cylindrical structures electrochemically grown with electrode potentials below 0.8 V exhibit excellent electrochemical performance comparable to thin polymer films. The process scalability is demonstrated using larger area substrates (up to 4 cm2) by carefully controlling the spacing between the working (substrate) and counter electrodes, which also provides an increase in microtube density from 350 cm−2 to 560 cm−2 without any loss in performance.
Introduction
The performance of portable power systems is currently limited by the properties of the active electrode materials, and further advances in energy storage will require materials that exhibit rapid and efficient charge transfer processes, long term cycle stability, low-cost and natural abundance. Emerging energy storage devices, such as supercapacitors, have the potential to provide high energy-density (charge-storage capacity) and high power-density (fast charge–discharge rates) to bridge the gap between traditional batteries and high-power capacitors.1–7 Currently, supercapacitor technologies have attracted interest in four niche applications: renewable energy generation sources to stabilize the variable incoming power; transportation with electric vehicles for regenerative braking systems and rapid acceleration;1,2,6,7 load leveling to prevent power disruptions due to short time fluctuations; and for portable electronics. The general aim of developing new electrical energy storage technology is a positive impact in the world's economy and ecology.8In supercapacitors, many types of electrode materials can be used, ranging from high surface area, inert carbon nanomaterials to Faradaic metal oxides and conducting polymers. Porous carbon nanomaterials rely upon electrical double layer capacitance (EDLC), where the charge is stored physically at the electrode interfaces (non-Faradaic process).9 Cells comprising high surface area, inactive electrode materials are referred to as Electrochemical Double-Layer Capacitors (EDLC) and are characterized by high power densities, but are limited by low energy densities. Electroactive conducting polymers (ECPs) and metals oxides10,11 have been widely investigated as materials for pseudocapacitors, where the bulk of the material undergoes rapid redox reactions to provide the capacitive response (Faradaic process),1 providing a higher energy density compared to EDLCs.12,13 The trade off, however, is during charging and discharging, pseudocapacitive materials experience a volume change (swelling–contraction process) due to the ion exchange with the electrolyte, causing poor cycle life (few thousand cycles) compared to carbon-based materials – which undergo ion adsorption–desorption processes – that exhibit stability over hundreds of thousands of cycles.1,12
ECPs (e.g. polypyrrole, polyaniline and polythiophene derivatives) are highly conductive, easily processable, flexible, have a low environmental impact, and very low cost, especially in comparison to metal oxides,12 such as ruthenium oxide (RuO2) – the benchmark standard in pseudocapacitance.1,14 Typically, ECPs are stable in the oxidized (p-doped) state, and therefore function as a cathode material in a supercapacitor. During oxidation–reduction reactions, counter ions migrate into and out of the polymer matrix to maintain charge neutrality, resulting in a continual change in volume.15–17 Although, the potential applications of conducting polymers are wide, the main challenges in the last many years have been creating well-defined shapes,16 which can help mitigate the cycling degradation caused by volumetric changes or increase power densities by providing shorter ion-diffusion lengths.2,12
ECPs can be synthesized by oxidative polymerization, which is easily scalable and produces materials that require subsequent processing into electrode films, or by electrochemical polymerization, which leads to chemically-bound films on conductive substrates.15 The latter is limited by the size of the substrate; however, precise control of the potential, current and state of charge of the resulting polymer is easily achievable.17 Controlling the structure of ECPs electrodes can lead to improved properties for applications in electrochemistry, electroanalysis, electrocatalysis, energy conversion and storage, chemical, optical and biosensors, drug delivery, protein purification and actuators, among others.18–20 A variety of synthesis methods have been developed to generate ECP micro-/nanostructures, such as nanowires, nanotubes, nanonetworks, nanospheres, microcontainers19,21–25 and hollow spheres.26 The most commonly utilized methods are categorized as either hard-template or soft-template approaches, and other more complex techniques, such as nanoimprinting lithography, have also been studied.18,27
In hard-template methods, a pre-formed template with a precisely defined structure is used to guide the growth, morphology and size of the material, which limits the size, morphology, and large scale production of tailored ECP structures.28 Common templates used are anodic aluminum oxide membranes, track etched polycarbonate membranes, porous silica, mesoporous zeolites, carbon nanotubes, and highly oriented hydrophilic graphite.15,16,28,29 This method is effective for synthesizing arrays of aligned polymer micro-/nanotubes and wires with controllable length, thickness and diameter;18 the first two of which are controlled by the polymerization time and the latter are controlled by the size of the pores or channels in the membrane. In chemical polymerization, the membrane is immersed in the solution with the monomer, dopant and/or oxidant allowing the polymerization within the pores.29 For electrochemical polymerization a metallic interface is required, which increases the complexity and cost of the process.15,16,29 In order to obtain the micro-/nanostructures, post-synthesis treatments are required to remove the template, which can damage the structures.
On the other hand, soft-template synthesis, also referred to as template-free or self-assembly,29 is simple and cheap and does not required a template or potentially harsh post-synthesis treatment.26 Nanostructure formation is achieved by self-assembly during polymerization, and is driven by selective control of non-covalent interactions, such as hydrogen bonds, van der Waals forces, π–π stacking and electrostatic interactions.18,29 Templates used include surfactant micelles, colloidal particles, structure-directing molecules,28 oligomers, bubbles,19,21–23,30–34 interfacial polymerization, dilute polymerization, reverse emulsion polymerization, among others.18,35–37 During oxidative polymerization, micelles are formed by dopant and/or monomer-dopant acting as a soft-template to guide the formation of micro-/nanotubes, -wires or -spheres,18 whereas in electrochemical polymerization the surface potential, reaction rate (applied current), and concentration of surfactants, dopants, all influence the morphology of the final structures. The advantage over the oxidative process is improved control of the shapes; however, the quantity of product is limited by the size of the working electrode.29 The main challenge is the control of the morphology, orientation, and diameter of the 1D structures.18
Various research efforts are underway to improve the controllability of soft-template methods for synthesizing ECP nanostructures. Shi and co-workers19,21–23 demonstrated the synthesis of polypyrrole (PPy) microcontainers using bubbles as templates in the presence of various surfactants (β-naphthalenesulfonic acid, (+) & (−) camphorsulfonic acid, sodium dodecylbenzenesulfonate and polystyrene sulfonic acid). Hydrogen gas formed by reducing protons at the counter electrode led to the formation of gas bubbles on the working electrode, which were stabilized by the anionic surfactant molecules and the growing polypyrrole structures. Compared to planar films, the structured electrodes exhibited improved redox activity and ion transport;15,16 however, this method utilizes high potentials that lead to overoxidization of the polypyrrole. The upper voltage limit where polypyrrole overoxidizes depends on the pH of the electrolyte solution; higher pH solutions lower the potential at which overoxidation occurs.38–42 For this reason Bajpai and co-workers30,31 synthesized polypyrrole micro-containers at relatively low potential, 0.8 V, which shows low overoxidation rates for pH < 0.1 V. The general consensus from these reports was that microstructure formation required the use of surfactants to stabilize the bubbles in the solution.
Large scale production of well aligned arrays of conducting polymers with controllable morphologies and sizes is still a challenge. In this article, a simple approach is demonstrated to prepare large quantities of ECP microtubes utilizing low potentials without the need for a surfactant or substrate based template. Due to its capacity for high power and energy density in supercapacitors, polypyrrole is studied as the electrode material for microtube-based devices. However, since the mechanism for electrochemical synthesis is similar for various monomers, it is expected that this approach can be applied to other ECPs with relative ease. A discussion of how to control the polymer assembly and microtubes synthesis by changing the substrate geometry is presented, along with the electrochemical properties of the created structures.
Results and discussion
A general schematic of the setup and process for electropolymerizing polypyrrole microtubes on stainless steel mesh is shown on Fig. 1. Chronopotentiometry was used to control the polymerization because setting the applied current maintains a constant polymerization rate.16 Stainless steel (SS) mesh substrates were immersed in a solution containing 0.09 M pyrrole and 0.5 M H2SO4 along with a reference (Ag/AgCl 3 M NaCl) and counter electrode, and polymer electrodes were grown to a specified charge. Initially, a thin polymer coating forms on the SS mesh wires until a charge of 3 C cm−2 is achieved, after which gas bubbles begin to nucleate on the surface on the mesh substrate. Between a deposition charge of 3 and 5 C cm−2 the film continues to grow as the surface become saturated with gas bubbles. Beyond, a charge of 8 C cm−2 only microtubes grow perpendicular to the working electrode surface guided by the presence of the gas bubbles. The growth of the microtubes was examined on M200 and M400 substrates to visualize the initial growth process and provide insight into the bubble-guided microtube growth mechanism.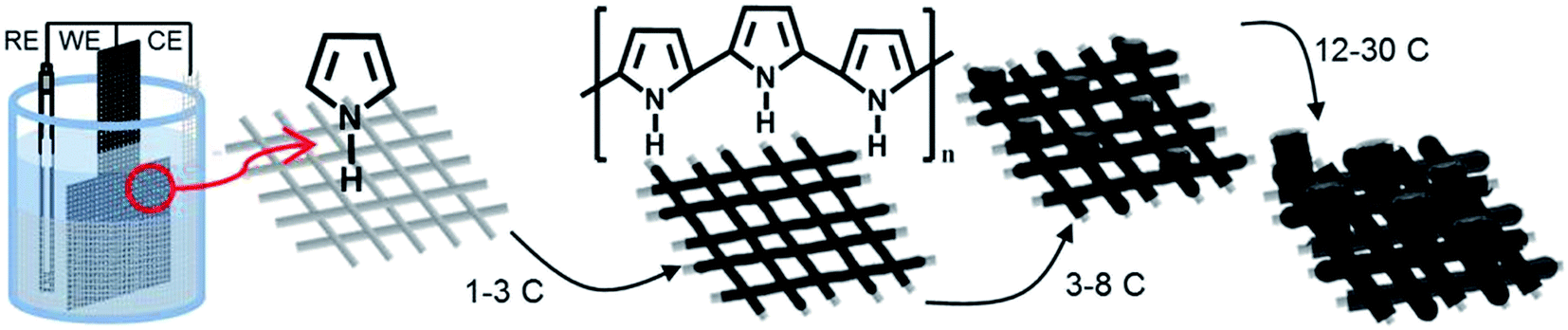 | ||
| Fig. 1 Electropolymerization process for synthesizing polypyrrole microtubes on stainless steel mesh. | ||
Scanning electron micrograph (SEM) images are shown in Fig. 2 for microtubes grown on M200 substrates at 16 mA cm−2 to a final charge of 1, 3, 5, 8, 15 C cm−2. When the polymer coating is forming on the working electrode, protons from the electrolyte are reduced to form hydrogen gas on the Pt counter electrode, which continuously bubble out of solution. At the same time, the hydrogen gas concentration in solution increases and eventually gas bubbles begin to nucleate on the mesh surface, specifically at the joints between two wires. Polymer clusters then begin to form around these bubbles on the surface of the SS mesh wires (Fig. 2a). Above a deposition charge of 3 C cm−2, hydrogen gas bubbles visibly appear (with the naked eye) on the working electrodes and the clusters that had previously formed begin taking the form of a flower (Fig. 2b). As the tubes continue to grow out of the flower-like structures, the bubbles remain at the tip of the tube through the course of the polymerization process. As shown if Fig. 2c–e, the microtubes continue to grow in diameter and length from 5 to 15 C cm−2 until the polymerization is stopped. The tube height and diameter a presented as a function of deposition charge in Fig. 3, along with the film thickness on the mesh wires. As shown, the tube height and diameter grow nearly linearly with deposition charge. At 15 C cm−2 the microtubes are already well formed; therefore, with increased deposition charge – from 15 C cm−2 up to 30 C cm−2 – the microtubes continue to grow with little change to their diameter to the final dimensions shown in Tables 1 and S2.† Below, we show that for some substrates, microtubes grow with uniform diameter after about 15 C cm−2.
 | ||
| Fig. 2 Polypyrrole microtubes growth on M200. SEM image are shown for polymers grown with total charges of (a) 1 C cm−2, (b) 3 C cm−2, (c) 5 C cm−2, (d) 8 C cm−2, (e) 15 C cm−2 at 16 mA cm−2. | ||
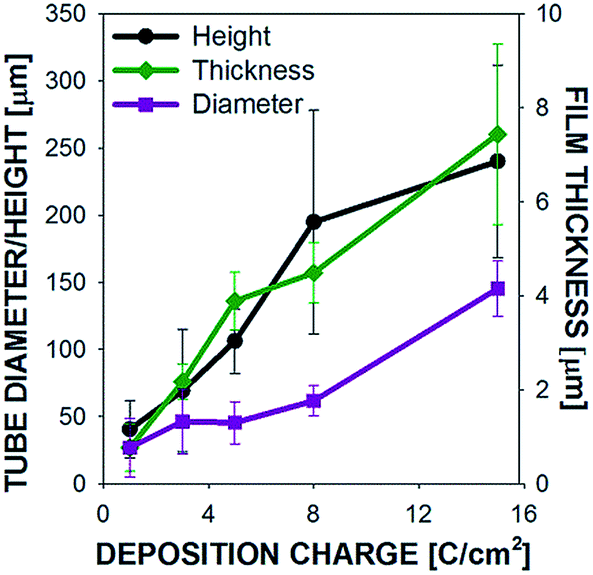 | ||
| Fig. 3 Physical dimensions of the polypyrrole microtubes grown on M400 are shown as a function of polymerization charge (1–15 C cm−2), in addition to the thickness of the polymer film on the mesh wires. | ||
| Substrate [mesh] | Film [μm] | Height [μm] | Diameter top [μm] | Diameter middle [μm] | Diameter bottom [μm] | Ratio [DT to DM] |
|---|---|---|---|---|---|---|
| M40 | 12.1 | 700 | 200 | 200 | 140 | 2.2 |
| M60 | 12.0 | 720 | 180 | 220 | 120 | 2.7 |
| M100 | 9.1 | 720 | 410 | 330 | 160 | 5.6 |
| M200 | 15.8 | 940 | 210 | 220 | 190 | 1.2 |
| M250 | 12.6 | 340 | 240 | 210 | 130 | 3.0 |
| M325 | 14.5 | 620 | 270 | 260 | 180 | 2.2 |
| M400 | 17.1 | 1300 | 180 | 160 | 120 | 1.9 |
To further explain the microtube growth mechanism, a set of experiments were carried out in a two-compartment system where the counter electrode was in a separate container from the reference and the working electrode and both containers were joined by a salt bridge. All other conditions were kept similar to previous experiments for growing PPy microtubes. When the counter is separated from the working electrode, microtubes did not grow; which supports the hypothesis that the generation of hydrogen at the counter electrode (from the reduction of protons in solution) leads to formation of gas bubbles on the counter electrode and also the nucleation of gas bubbles on the working electrode. Furthermore, we showed that when counter electrodes other than Pt were utilized, microtubes could not be achieved.
After demonstrating that the nucleation of gas bubbles on the working electrode were fundamental for the formation of microtubes, a series of experiments adapted from the so-called bubble method19,21–23,30,31 were performed. As opposed to previous work, our approach did not utilize surfactant molecules, which were previously thought to be required to produce microstructures. These experiments utilized a voltammetric cycle at negative potentials (−0.3 V to −0.8) prior to the addition of the monomer to create H2 bubbles on the working electrode. Electropolymerization was then performed after monomer addition to generate structures with the shape of balls or bowls, which support previous observations reported in literature.19,21–23,30,31 However, this method could not be utilized to create reproducible microtubes (Fig. S3†).
The analysis of the bubbled-guided growth mechanism presented above was carried out on M200 substrates, where fairly uniform growth (with respect to the tube diameter) was observed after the microtube was formed. The dimensions of the stainless steel mesh, however, were shown to have a significant influence on the growth of the polymers structures. While microtubes could be achieved on any mesh, the mesh wire diameter and spacing seemed to affect the growth of the tubes. The microstructure of polymer tubes obtained on various stainless steel substrates are shown in Fig. 4. A generalized presentation of the microtube density and long range order is shown in Fig. 4a shows for microtubes grown on M400 using 10 mA cm−2 for a total charge of 30 C cm−2, which is typical for each mesh. As shown, many tubes are vertically positioned while others appear to collapse on themselves, likely due to the stress upon drying the aqueous solvent. In Fig. 4b–i, SEM images are displayed for selected microtubes on various stainless steel meshes and SS foil, which are grown as previously described. Generally, two kinds of microstructures were found: either a cone-like, which are narrow at the base, or cylinder-like microtubes exhibiting a fairly uniform diameter throughout. Stainless steel foil was used as a control to compare the microstructure of polypyrrole electrodes grown on typically used planar substrates and various SS meshes.19 Several disadvantages have been described for using foil substrates. Most importantly, when relatively thick films are grown, volume changes that occur upon ion insertion and removal during oxidation and reduction processes cause the film to delaminate from the electrode resulting in poor adhesion and poor electrochemical performance. From our experiments, we found poor adhesion on SS foils for films grown with charges greater than 7.23 C cm−2.
 | ||
| Fig. 4 Polypyrrole microtubes structures grown on various stainless steel meshes at 10 mA cm−2 for 30 C cm−2. SEM images are shown for polypyrrole microtubes grown on (a) M400, (b) M40, (c) M60, (d) M100, (e) M200, (f) M250, (g) M325, (h) M400, and (i) foil for 7.23 C cm−2. | ||
Tabulated data of various microtube properties are presented in Table 1 for polypyrrole microtubes grown on SS mesh substrates. Each parameter was calculated from measurements made on SEM images, which were taken on substrates positioned at 45° relative to the incident beam. Multiple measurements were recorded for each parameter and average values are presented. The film thickness was determined by subtracting the diameter of bare stainless steel wire from substrates with polypyrrole films. On average insignificant variation was observed in film thickness since the surface areas of each substrate were similar. Diameters were recorded at varying positions along the microtubes and are designated as bottom (near the substrate), middle and top to quantify the shape of the microtube structures. The ratio of diameter at the top to the middle (DT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DM) was calculated to reduce the shape to a single number, where a ratio of 1 (as is the case for polypyrrole tubes grown on M200 and M400) indicate cylindrical tubes, whereas a ratio larger than 1 (PPy tubes on M40, M60, M100, M250, M325), indicates conical structures. Tubes with a more pronounced conical shape are represented by larger ratios. On average, microtubes grown with a charge of 30 C cm−2 with a cylindrical shape exhibited a height around 1100 μm, while the conical tubes were significantly smaller, with a height dependent upon how conical the tubes were.
:DM) was calculated to reduce the shape to a single number, where a ratio of 1 (as is the case for polypyrrole tubes grown on M200 and M400) indicate cylindrical tubes, whereas a ratio larger than 1 (PPy tubes on M40, M60, M100, M250, M325), indicates conical structures. Tubes with a more pronounced conical shape are represented by larger ratios. On average, microtubes grown with a charge of 30 C cm−2 with a cylindrical shape exhibited a height around 1100 μm, while the conical tubes were significantly smaller, with a height dependent upon how conical the tubes were.
The electrochemical properties of polypyrrole microtubes (10 mA cm−2, 30 C cm−2) are presented in Fig. 5 for electrodes grown on M60, M100, M200, M400 SS meshes. Fig. 5a and b show the capacitance vs. potential plots at a relatively fast scan rate (100 mV s−1) and a slow scan rate (10 mV s−1), respectively, over a potential range between 0 to 0.8 V. Due to the synthesis of relatively thick films (11.98, 9.09, 15.82, 17.06 μm, respectively), a scan rate dependence is observed in the capacitance of each electrode, which is exemplified by the higher specific capacitance observed for low scan rates (10 mV s−1, Fig. 5b) compared to the higher scan rate (100 mV s−1, Fig. 5a). More on this aspect will be discussed later. Surprisingly, we found that PPy microtubes grown on coarser mesh substrates (larger wire diameters and larger wire spacing) exhibited a poor capacitance compared to electrodes grown on the finer mesh substrates (M200, M400). It is important to note that specific surface area of each substrate is similar (Table S1†); however, the electrochemical performance of the microtube electrodes strongly correlates with deposition potential profile (Fig. 5c). That is, when PPy films are grown on M60, M100 SS substrates a higher voltage (above 0.85 V) is required during the polymerization to achieve the target current density, which leads to overoxidization and interchain cross-linking, which reduces the electrochemical activity.38–41 Lower applied current densities, which maintained the polymerization potential below 0.8 V, did not produce microtube structures.
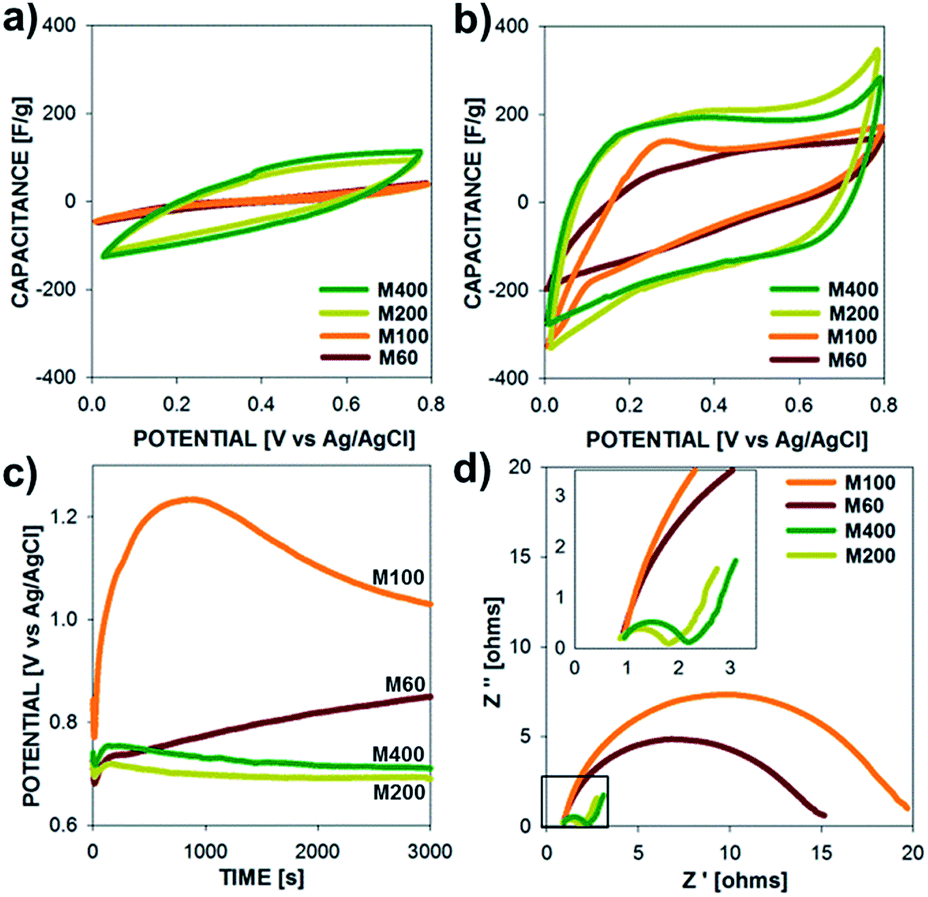 | ||
| Fig. 5 Electrochemical properties (specific capacitance vs. potential) of polypyrrole microtubes (10 mA cm−2, 30 C cm−2) on stainless steel meshes (M60, M100, M200, M400) recorded at scan rates of (a) 100 mV s−1 and (b) 10 mV s−1. (c) Potential required during deposition to maintain the constant current of 10 mA cm−2. (d) Nyquist plot of imaginary vs. real impedance from EIS in the frequency range of 10k Hz to 0.1 Hz. | ||
Overoxidation of ECPs produces films with lower conductivity and higher electrode resistances, which is evident by the Nyquist plot presented in Fig. 5d. Regardless of the substrate type, a similar Ohmic series resistance was observed (high frequency limit of EIS); however, PPy films grown at potentials above 1 V exhibited significantly higher charge transfer resistance (low frequency side of semi-circle in Fig. 5d), which is consistent with an over-oxidized polymer electrode. In addition to showing higher charge transfer resistances, PPy electrodes exhibit kinetic control over a much larger time scales; the low frequency intercepts on the Nyquist plots correspond to frequencies of 1.3, 3.2, 39.8, and 31.6 Hz for PPy electrodes on M60, M100, M200, and M400 SS substrates, respectively. Based on these results, the finer meshes substrates (M200 and M400) were used for subsequent measurements and device fabrication.
The electrochemical properties of the PPy microtubes grown with increasing deposition charge (structures presented in Fig. 2) are shown in Fig. 6. The electrode mass exhibits a linear correlation with deposition charge indicating a constant Coulombic efficiency during electrode synthesis, according to Fadaray's law. As shown in Fig. 6b, thin electrodes grown with charge densities of less than 5 C cm−2 showed a linear relationship between peak current and scan rate due to the absence of ion-diffusion limitations. Thick microtube electrodes grown with charge densities greater than 8 C cm−2 displayed a sublinear dependence of peak current with scan rate, due to the increased resistance to ion transport in thick films (tall microtubes). Structures grown with charge densities of 15 C cm−2 displayed a peak current plateau at scan rates above 50 mV s−1, illustrating the rate-limitations of thick electrodes. As shown in the insert, each electrode mass displays a linear current–scan rate dependence at low scan rates.
Ion-transport limitations are further clarified in Fig. 6c and d, which show the specific capacitance (current–scan rate) vs. potential plots at 100 mV s−1 and 10 mV s−1, respectively. At fast scan rates, specific capacitance of the films decreases as Q increases, while these profiles are constant at low scan rates. Data from electrochemical impedance spectroscopy are shown in Fig. 6e and f. As the electrode mass increases, the charge transfer resistance increases and the frequency at which the electrodes switch from kinetic control (high frequency) to mass transfer control (low frequency) decreases due to longer time scales required for ion transport within the electrode. The Bode plot of phase Z vs. frequency corroborates the time-scale dependence of the electrochemical processes. As the frequency decreases, the electrochemical behavior switches from resistive (0° phase angle) to capacitive (90° phase angle). From Fig. 6f, it is clear that the frequency at which the behavior switches decreases with electrodes of increasing mass due to the longer time scales required for ion-diffusion during the charge–discharge processes.
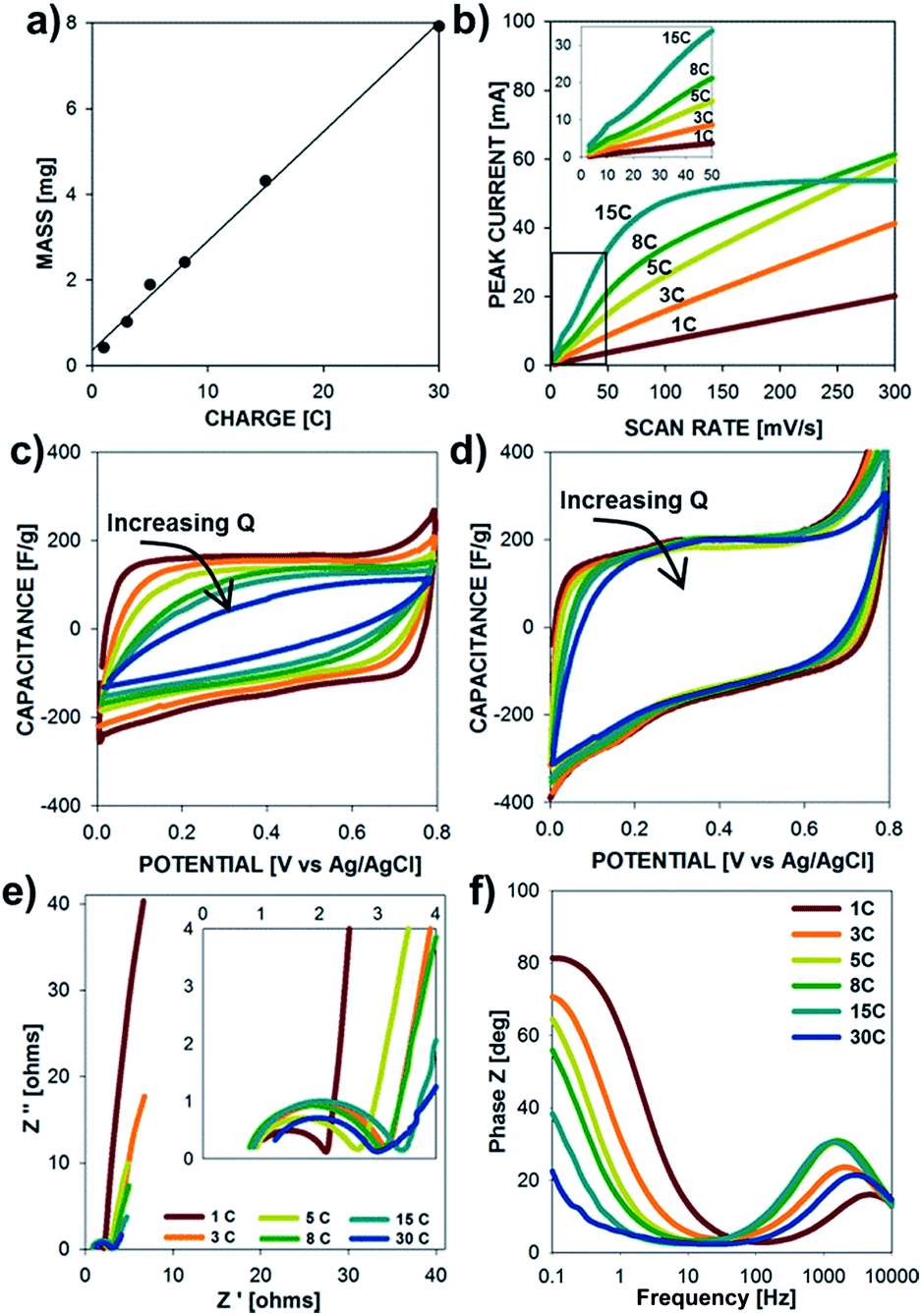 | ||
| Fig. 6 Electrochemical properties of polypyrrole microtubes with increasing charge deposited by chronopotentiometry at 16 mA cm−2. (a) Electrode mass vs. deposition charge. (b) Peak current vs. scan rate for electrodes with increasing deposition charge. Specific capacitances vs. voltage at a scan rate of (c) 100 mV s−1 and (d) 10 mV s−1. (e) Nyquist plot of imaginary vs. real impedance from EIS measurements for a frequency range of 10k Hz to 0.1 Hz at 0.7 V with an amplitude of 10 mV. (f) Bode plot of the phase in impedance with frequency. | ||
A symmetrical supercapacitor device was built with two polypyrrole microtubes electrodes deposited on M200 at 10 mA cm−2 for 30 C cm−2, and their performance is shown in Fig. 7. Fig. 7a and b show the cyclic voltammetry and specific capacitance plots for scan rates between 5–100 mV s−1, displaying a high specific capacitance (on a per mass of active material basis) of 50 F g−1, which is expected for a symmetric cell of electrodes with a single electrode capacitance of ∼200 F g−1. The Nyquist plot (Fig. 7c) shows that these cells exhibit a low Ohmic series resistance and charge transfer resistance in the given setup. The low frequency end of the kinetic control was found at 5 Hz and switch from Warburg impedance to nearly ideal capacitance occurred at 0.6 Hz. Fig. 7d shows the specific charge vs. cell voltage measured form 0 to 0.75 V for different currents (30, 15, 7.5, 3.75, 1.87 mA cm−2). For a 60 s discharge at 7.5 mA cm−2 the specific charge is 7.6 mA h g−1 up to a maximum of 12 mA h g−1 at 0.47 mA cm−2. The performance of the supercapacitor cell (Fig. 7b) is comparable to cells comprising thin PPy electrodes with low scan rate dependence.43
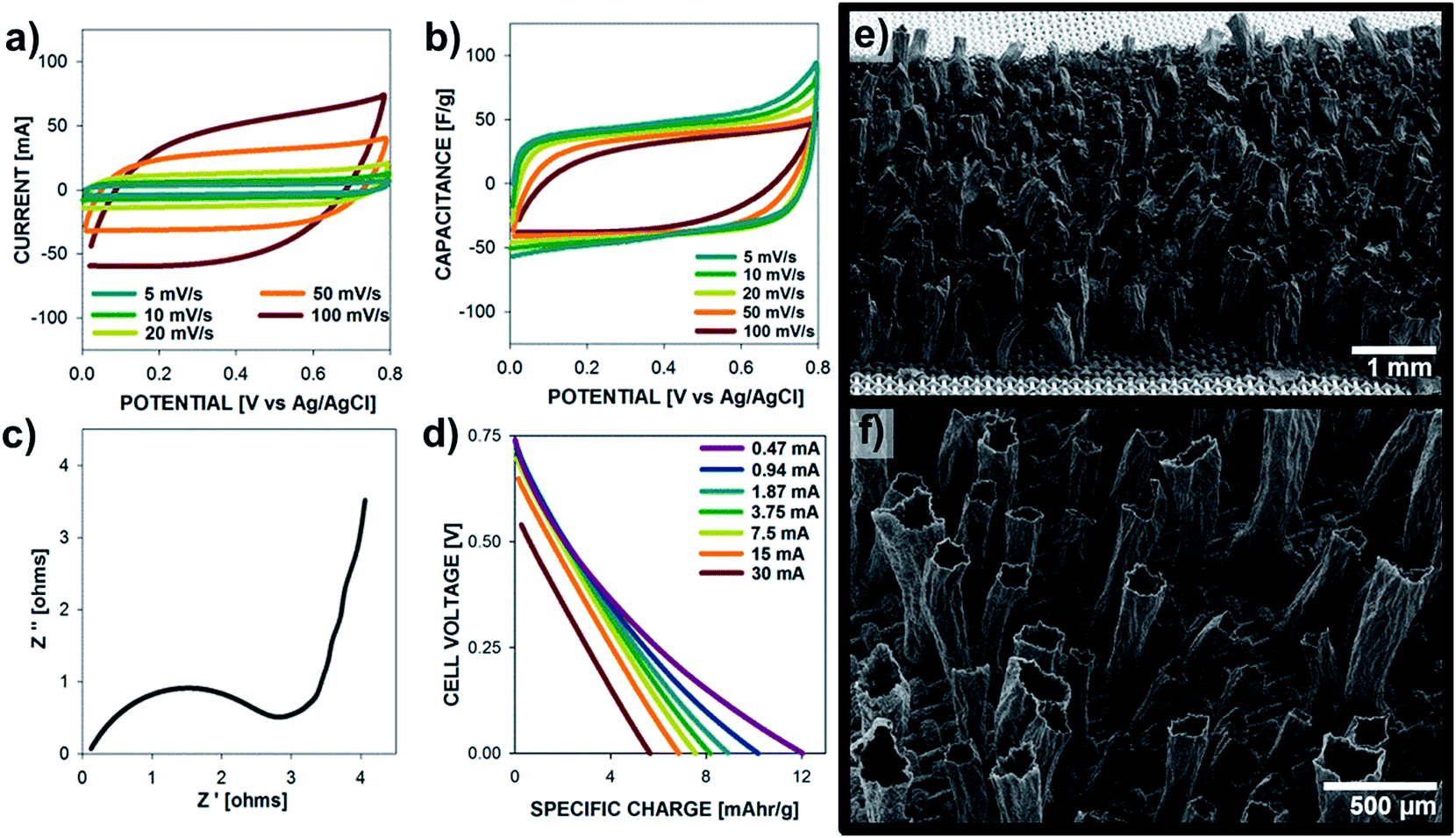 | ||
| Fig. 7 Supercapacitor polypyrrole microtubes device, electrodes deposited on M200 at 10 mA cm−2 for 30 C cm−2. (a) Cyclic voltammetry (current vs. voltage) at various scan rates (100, 50, 20, 10, 5 mV s−1), (b) specific capacitance vs. voltage from profiles shown in (a), (c) Nyquist plot of imaginary vs. real impedance from EIS measurements, (d) cell discharge profiles with increase current densities (30, 15, 7.5, 3.75, 1.87, 0.94, 0.47 mA cm−2), (e and f) SEM images of PPy microtubes grow on large-area M200 SS substrates for 60 C at 30 mA. | ||
After demonstrating that PPy microstructures grown on M200 and M400 SS substrates displayed superior electrochemical performance and more consistent microtube structures compared to those grown on coarse meshes and foils, microtube synthesis was investigated on larger scale substrates to demonstrate the scalability of our process. Using similar current densities and solution conditions as previously examined, PPy microtubes were grown on ca. 4 cm × 1 cm substrates assembled in a 3-electrode cell. The electrochemical performance of the electrodes were similar to behavior presented in Fig. 4; however, the microtubes surface density of ∼560 cm−2 obtained on these substrates was notably larger than that achieved on the smaller substrates (1 cm × 1 cm), ∼350 cm−2. In the larger substrate system, the distance between the working and counter electrodes was carefully controlled, which contributed to a higher nucleation density of gas bubbles, and therefore microtubes, on the substrate surface.
Conclusions
A template-free synthesis method has been demonstrated, capable of providing control of polypyrrole microtube size and structure by utilizing stainless steel mesh size and electrochemical polymerization conditions (e.g. current density). Polymer microtube synthesis is governed by the nucleation of hydrogen gas bubbles – generated by the reduction of protons on a platinum counter electrode – which guide the formation of polymer structures in a narrow current density range. The mesh size is shown to influence in the structure and electrochemical performance of the microtubes by confining the gas bubble to a specific size. Cylindrical tubes are found on substrates with fine mesh structures (M200–M400) while conical structures, with narrow bases near the mesh, are obtained on larger meshes (<M200). The electrochemical performance of polypyrrole microtubes in a single electrode configuration (∼200 F g−1), in addition the performance of their symmetrical supercapacitor cells (∼50 F g−1), were comparable to analogous polypyrrole thin film electrodes. The synthesis of polypyrrole microtubes can be scaled to larger systems by increasing the mesh and electrochemical bath size without any loss in electrochemical performance.Experimental
Materials
All chemicals were purchased from Sigma-Aldrich and Fischer Scientific and used as received unless otherwise noted. Pyrrole monomer was purified by distillation.Substrate preparation
Stainless steel meshes were prepared by cutting a 1.2 × 1.4 cm squares with a 1.3 × 0.4 cm neck to serve as connection (Fig. 1), sonicated for 10 min in ethanol, dried, weighed (Ohaus DV215CD Semi-micro balance, readability 0.01 mg) and treated 15 min (each side) in UV-ozone (Novascan PSD-UV). Parafilm was used to cover the substrates edges and make a 1 cm2 deposition area.Microtubes synthesis
Pyrrole (0.09 M solution) was electropolymerized at room temperature in a 0.5 M H2SO4 solution (previously degassed with N2 for 10 min) by chronopotentiometry using currents of 10–16 mA) and charge densities of 1–30 C cm−2. Electropolymerization was performed in a three electrode cell with a VersaSTAT 4 potentiostat/galvanostat and the VersaStudio v2.20.4631 Electrochemistry Software (Princenton Applied Research). The working electrode was either a stainless steel mesh (McMaster-Carr, Super-Corrosion Resistance Type 316 Stainless Steel Wire Cloth, 40 × 40, 60 × 60, 100 × 100, 200 × 200, 250 × 250, 325 × 325, 400 × 400, (Table S1†) or stainless steel foil; platinum gauze 52 mesh (Alfa Aesar) was used as the counter electrode, and a Ag/AgCl in 3 M NaCl solution as reference electrode. The platinum mesh was cleaned between each synthesis by rinsing with deionized water and acetone, and the remaining residue removed under the flame of a propane torch. In some cases a 3.3 wt% poly(4-styrene sulfonic) acid (PSSA) in 0.5 M H2SO4 solution was also used as dopant (Fig. S2†). All samples were dried overnight in a vacuum oven (101.5 kPa) at room temperature and weighed afterwards to determine film mass. In the two-container experiments, the counter electrode (Pt mesh) was setup in container separated from the working electrode (Stainless steel M200) and reference electrode (Ag/AgCl in 3 M NaCl). The cells each contained 0.09 M Py in 0.5 M H2SO4 and were connected by a salt bridge comprising a filter paper (Fisherbrand Qualitative P8-creped) soaked in the same solution. Chronopotentiometry at 10 mA for 30 C cm−2 was used to polymerize pyrrole.Bubble method
M200 mesh substrates were cycled form −0.3 to −0.8 V for 1 cycle at 0.1 V s−1 in neat 0.5 M H2SO4 that was previously degassed, leading to the formation of bubbles on the substrate surface. Electrolyte with pyrrole (0.18 M) was then added to the cell. Chronoamperometry at 10 or 16 mA for 30 C or 60 C was performed.Large-scale microtubes production
Mesh substrates (M200 and M400) were prepared with dimensions of 4 cm × 1 cm with 1.8 cm × 0.4 cm neck substrates and cleaned as described previously. One side was covered with parafilm and the substrates were gently rolled into a 1 cm radius of curvature cylinder, leaving the uncovered stainless steel facing the counter electrode at a uniform distance. The experiments were performed with chronoamperometry at 30 mA for 60 C.Electrochemical analysis
Electrochemical characterization was performed in a 0.5 M H2SO4 solution (previously degassed with nitrogen gas for 10 min) using cyclic voltammetry from 0 to 0.8 V at various scan rates (300, 100, 30, 10, 3 mV s−1), electrochemical impedance spectroscopy (EIS) at 0.7 V over a frequency range of 10000 to 0.1 Hz using a perturbation amplitude of 10 mV, and charge discharge measurements at current densities of 15, 7.5, 3.75 mA cm−2 (−15,−7.5,−3.75 mA cm−2) from 0 to 0.75 V unless otherwise stated.
Surface morphology
The film surface characterization was preform by scanning electron microscopy (SEM) in a Hitachi SU-6600 Analytical VP FE-SEM at a beam intensity of 5 kV; the samples were placed on 45° aluminum sample holders with carbon tape. Images were captured at magnifications of 35×, 90×, 200×, and 300×. Quartz PCI software was used to make all the measurements, this software is calibrated to the microscope. Averages were taken for the diameters and heights presented (between 5–10 specimens depending on the sample).Supercapacitor testing
A symmetric supercapacitor was assembled in MTI Corp coin cell apparatus using polypyrrole microtubes electrodes deposited on M200 at 10 mA cm−2 for 30 C cm−2, and a Whatman glass microfiber type A filter soaked in 1 M H2SO4 as the separator/electrolyte. Each electrode was charged at 0.4 V prior to analysis. Electrochemical characterization was similar to the single electrode measurements, unless otherwise stated. Cyclic voltammetry was perform at scan rates of 100, 50, 20, 10 and 5 mV s−1 and charge–discharge measurements were performed at currents of 30, 15, 7.5, 3.75, 1.87 mA cm−2 (−30, −15, −7.5, −3.75, −1.87 mA cm−2), from 0 to 0.75 V.Acknowledgements
M.E.R acknowledges support from NSF 1246800 and 3M Non-Tenured Faculty Grant Award.References
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- R. A. Fisher, M. R. Watt and W. J. Ready, ECS J. Solid State Sci. Technol., 2013, 2, M3170–M3177 CrossRef CAS PubMed.
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213–234 CrossRef CAS PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- A. Evans, V. Strezov and T. J. Evans, Renewable Sustainable Energy Rev., 2012, 16, 4141–4147 CrossRef PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS.
- S. Bose, T. Kuila, A. K. Mishra, R. Rajasekar, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 767–784 RSC.
- C. D. Lokhande, D. P. Dubal and O.-S. Joo, Curr. Appl. Phys., 2011, 11, 255–270 CrossRef PubMed.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- A. K. Shukla, A. Banerjee, M. K. Ravikumar and A. Jalajakshi, Electrochim. Acta, 2012, 84, 165–173 CrossRef PubMed.
- J.-Y. Kim, K.-H. Kim, H.-K. Kim, S.-H. Park, K. Y. Chung and K.-B. Kim, RSC Adv., 2014, 4, 16115–16120 RSC.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef PubMed.
- Y.-Z. Long, M.-M. Li, C. Gu, M. Wan, J.-L. Duvail, Z. Liu and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef CAS PubMed.
- L. Qu, G. Shi, F. E. Chen and J. Zhang, Macromolecules, 2003, 36, 1063–1067 CrossRef CAS.
- Z. Yin and Q. Zheng, Adv. Energy Mater., 2012, 2, 179–218 CrossRef CAS.
- L. Qu, G. Shi, J. Yuan, G. Han and F. E. Chen, J. Electroanal. Chem., 2004, 561, 149–156 CrossRef CAS PubMed.
- J. Yuan, L. Qu, D. Zhang and G. Shi, Chem. Commun., 2004, 994–995, 10.1039/b400614c.
- L. Qu and G. Shi, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3170–3177 CrossRef CAS.
- M. J. Antony and M. Jayakannan, J. Phys. Chem. B, 2010, 114, 1314–1324 CrossRef CAS PubMed.
- A. L. M. Reddy, S. R. Gowda, M. M. Shaijumon and P. M. Ajayan, Adv. Mater., 2012, 24, 5045–5064 CrossRef CAS PubMed.
- M. Wan, Adv. Mater., 2008, 20, 2926–2932 CrossRef CAS.
- K. Kumar, V. Luchnikov, B. Nandan, V. Senkovskyy and M. Stamm, Eur. Polym. J., 2008, 44, 4115–4121 CrossRef CAS PubMed.
- Y. Zhao, B. Liu, L. Pan and G. Yu, Energy Environ. Sci., 2013, 6, 2856–2870 CAS.
- L. Xia, Z. Wei and M. Wan, J. Colloid Interface Sci., 2010, 341, 1–11 CrossRef CAS PubMed.
- V. Bajpai, P. He and L. Dai, Adv. Funct. Mater., 2004, 14, 145–151 CrossRef CAS.
- V. Bajpai, P. He, L. Goettler, J. H. Dong and L. Dai, Synth. Met., 2006, 156, 466–469 CrossRef CAS PubMed.
- S. Gupta, J. Raman Spectrosc., 2008, 39, 1343–1355 CrossRef CAS.
- F. Hui, B. Li, P. He, J. Hu and Y. Fang, Electrochem. Commun., 2009, 11, 639–642 CrossRef CAS PubMed.
- M. Mazur, J. Phys. Chem. C, 2008, 112, 13528–13534 CAS.
- J. Huang, B. Quan, M. Liu, Z. Wei and L. Jiang, Macromol. Rapid Commun., 2008, 29, 1335–1340 CrossRef CAS.
- K. Wang, J. Huang and Z. Wei, J. Phys. Chem. C, 2010, 114, 8062–8067 CAS.
- S. Percec, C. Bolas, L. Howe, D. J. Brill and J. Li, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4966–4976 CrossRef CAS.
- T. W. Lewis, G. G. Wallace, C. Y. Kim and D. Y. Kim, Synth. Met., 1997, 84, 403–404 CrossRef CAS.
- Y. Li and R. Qian, Electrochim. Acta, 2000, 45, 1727–1731 CrossRef CAS.
- D. S. Park, Y. B. Shim and S. M. Park, J. Electrochem. Soc., 1993, 140, 609–614 CrossRef CAS PubMed.
- A. Kumar, R. K. Singh, K. Agarwal, H. K. Singh, P. Srivastava and R. Singh, J. Appl. Polym. Sci., 2013, 130, 434–442 CrossRef CAS.
- C. C. Bof Bufon, T. Heinzel, P. Espindola and J. Heinze, J. Phys. Chem. B, 2009, 114, 714–718 CrossRef PubMed.
- D. Qu and H. Shi, J. Power Sources, 1998, 74, 99–107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI contains stainless steel mesh specifications (Table S1), the electrochemical properties of the microtubes at different current densities (Fig. S1 and Table S2), PPy/PSSA microtubes (Fig. S2) and experimental data from the so-called bubble method (Fig. S3). See DOI: 10.1039/c4ra16000b |
| This journal is © The Royal Society of Chemistry 2015 |