
Ionic liquid-based random copolymers: a new type of polymer electrolyte with low glass transition temperature
Heyi Hu†
a,
Wen Yuan†*ab,
Zhe Jiaa and
Gregory L. Baker‡
a
aDepartment of Chemistry, Michigan State University, East Lansing 48824, USA. E-mail: yuanwen@msu.edu; yuanwen@lbl.gov; Tel: +1-517-355-9715
bEnvironmental Energy Technologies Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Rd., Berkeley, CA 94720, USA
First published on 28th November 2014
Abstract
A new type of polymer electrolyte has been prepared from the side-chains of ionic liquids (IL) and an analogue of ethylene oxide (EO) directly grafted on a polyethylene oxide backbone. By tuning the two types of monomer composition during polymerization, a series of copolymers with different monomer ratios between IL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EO = 8:1 to 1:4 have been synthesized. All copolymerized polyionic liquids showed higher ionic conductivity than an IL homopolymer. By choosing a composition with IL:EO = 1:1, the corresponding polymer gave the highest conductivity, 1.2 × 10−4 S cm−1 at room temperature, compared to 9.3 × 10−6 S cm−1 from a homopolymer. We attribute this to one order of magnitude improvement of conductivity from a low glass transition temperature (Tg) below −40 °C.
:EO = 8:1 to 1:4 have been synthesized. All copolymerized polyionic liquids showed higher ionic conductivity than an IL homopolymer. By choosing a composition with IL:EO = 1:1, the corresponding polymer gave the highest conductivity, 1.2 × 10−4 S cm−1 at room temperature, compared to 9.3 × 10−6 S cm−1 from a homopolymer. We attribute this to one order of magnitude improvement of conductivity from a low glass transition temperature (Tg) below −40 °C.
1. Introduction
Compared to ionic liquids polyionic liquids (PILs) exhibit improved mechanical stability, processability and durability.1–3 The corresponding polymer electrolytes are suitable for use in electrochemical devices due to their chemical and physical stability and negligible vapor pressure, preventing electrolyte leakage.2,4–10 However, a critical drawback of PILs is that after polymerization the conductivity usually suffers a huge decrease in comparison to the monomers.11–14It is well known that a lower glass transition temperature (Tg) leads to higher conductivity.1–3 One way to lower the Tg is to introduce anionic side-chains into a polymer such as bis(trifluoromethanesulfonyl)imide (TFSI). Baker's group has successfully grafted TFSI anions onto a polyethylene oxide (PEO) backbone using anionic ring-opening polymerization. The PILs obtained have a low glass transition temperature, about −14 °C. As polymer electrolytes they have moderate conductivity, around 10−5 S cm−1 at 30 °C and close to 10−3 S cm−1 at 90 °C.11
Another way to reduce the Tg of polymers is to introduce a low Tg component to the polymer backbone, forming a copolymer.15–17 Elabd and coworkers have studied the effect of introducing a low Tg component on the ionic conductivity of PILs.16 The copolymer was made by free-radical polymerization of a nonionic monomer, n-hexyl methacrylate (HMA) and an ionic monomer 1-(2-methacryloyl)ethyl-3-butylimidazolium tetrafluoroborate (MEBIMBF4). A series of copolymers with different levels of HMA were obtained by tuning the HMA concentration during the free radical polymerization. The results indicated that the Tg of the copolymer decreased with increasing HMA composition, and the conductivity increased correspondingly.
In the present study, a second component chosen for copolymer synthesis was an ethylene oxide (EO) analogue. PEO is synthesized by the polymerization of ethylene oxide and has Tg ranging from −50 to −57 °C. However, ethylene oxide is a gas at room temperature, making it hard to process under normal polymerization conditions. In view of this, a liquid analogue of ethylene oxide, 2-((2-(2-(2-methoxyethoxy)ethoxy)ethoxy)methyl)oxirane (ME3MO), was synthesized and used as monomer.18 The random copolymer polyEPCH-co-polyME3MO was synthesized by cationic ring-opening polymerization by an activated monomer mechanism using trifluoroborate etherate, and 1,4-butanediol as initiator.19 After quaternarization and ion exchange reaction, the PIL copolymer electrolyte showed a conductivity more than one order of magnitude greater than that of a PIL homopolymer.
2. Experimental
2.1 Materials
Unless otherwise specified, all chemicals and solvents were of ACS reagent grade and were used as received without further purification. Epichlorohydrin (EPCH, 99%, Aldrich) was purified over CaH2, distilled under vacuum and stored in a glove box until use. 1-Butylimidazole was distilled under vacuum and stored in a refrigerator. Toluene was dried by refluxing with sodium p-benzylphenol, distilled under nitrogen and stored in a glove box. Boron trifluoride diethyl etherate (BF3OEt2) (≥46% based on BF3, Aldrich) and 1,4-butanediol (99%, Aldrich) were distilled under nitrogen and also stored in a glove box. Lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, >98%, TCI) was kept in a glove box and used without further purification.2.2 Synthesis of 2-((2-(2-(2-methoxyethoxy)ethoxy)ethoxy)methyl)oxirane (ME3MO)
ME3MO was synthesized according to the literature method.20 NaH (60 wt% in mineral oil, 9 g, 0.23 mol) was rinsed with toluene and added to 200 mL THF. Triethylene glycol monomethyl ether (32.8 g, 0.2 mol) was then added and stirred for 2 h. The solution was cooled to 0 °C in an ice bath, followed by the gradual addition of epichlorohydrin (18.4 g, 0.2 mol). The solution was stirred between 0 °C and room temperature overnight. After reaction, the solution was neutralized using 30% methanolic H2SO4. The solution was filtered, concentrated using a rotavap and distilled under vacuum. Yield, 31.2 g (71%).1H NMR (CDCl3, 500 MHz), δ (ppm) 3.66 (dd, J = 11.7, 3.0 Hz, 1H), 3.61–3.49 (br, 10H), 3.44–3.40 (m, 2H), 3.29 (dd, J = 11.7, 5.9 Hz, 2H), 3.25 (s, 3H), 3.05–3.00 (m, 1H), 2.66 (dd, J = 5.0, 4.2 Hz, 1H), 2.48 (dd, J = 5.0, 2.7 Hz, 1H). 13C NMR (CDCl3, 500 MHz) δ (ppm) 71.55, 71.50, 70.28, 70.16 (overlapping signals), 70.06, 58.50, 50.32, 43.66. MS (TOF MS ES+): calculated for C10H20O5Na+ (m/z), 243.1208; found, 243.1213.
2.3 Synthesis of polyEPCH by cationic ring-opening polymerization
PolyEPCH was synthesized by cationic ring-opening polymerization using the literature method.19 In a dry Schlenk flask, 1,4-butanediol (0.44 g, 4.8 mmol) was added to 22.5 mL dry toluene. This solution was degassed by a freeze–pump–thaw procedure three times and then protected by a nitrogen gas flow. To the solution, BF3OEt2 (0.12 g, 0.81 mmol) was slowly added under nitrogen and stirred 60 min at room temperature. The solution was cooled to 0 °C in an ice bath, followed by the dropwise addition of EPCH (7.5 g, 81 mmol). The polymerization was terminated after 8 h by adding a small amount of distilled water, and the polymer solution washed with sodium bicarbonate solution (5% w/v), followed by further washing with distilled water to neutral pH. The solvent was removed by vacuum distillation and the product dried overnight at 60 °C under vacuum. Yield, 6 g (80%).1H NMR (CDCl3, 500 MHz) δ 3.78–3.66 (4H), 3.66–3.59 (1H).
2.4 Synthesis of polyME3MO
PolyME3MO was synthesized in a similar manner to polyEPCH. In a dry Schlenk flask, 1,4-butanediol (0.44 g, 4.8 mmol) was added to 22.5 mL dry toluene. The solution was degassed three times by a freeze–pump–thaw procedure and then protected by a nitrogen gas flow. To this solution BF3OEt2 (0.12 g, 0.81 mmol) was slowly added under a nitrogen atmosphere and stirred 60 min at room temperature. The solution was cooled to 0 °C in an ice bath, followed by the dropwise addition of ME3MO (17.8 g, 81 mmol). The polymerization was stopped after 18 h by adding a small amount of distilled water. The purification method was in this case different, however, since polyME3MO is soluble in water. The polymer solution was first washed with sodium bicarbonate solution (5% wt/v), and the polymer extracted using CHCl3 and precipitated from hexane. Yield, 13.4 g (75%).1H NMR (CDCl3, 500 MHz) δ 3.74–3.22 (20H).
2.5 Synthesis of polyEPCH-co-polyME3MO random copolymer
PolyEPCH-co-polyME3MO random copolymer was synthesized in a similar manner to polyEPCH. A series of copolymers were obtained by changing the molar concentration of ME3MO monomer, keeping the concentration of EPCH monomer constant. For example, the polyEPCH-co-polyME3MO random copolymer with a 1:1 monomer molar ratio (polyEPCH-co-polyME3MO-1/1) was prepared as follows.
In a dry Schlenk flask, 1,4-butanediol (0.44 g, 4.8 mmol) was added to 22.5 mL dry toluene. The solution was degassed three times by a freeze–pump–thaw procedure and then protected by a nitrogen gas flow. To this solution, BF3OEt2 (0.12 g, 0.81 mmol) was added slowly under a nitrogen atmosphere and stirred 60 min at room temperature. The solution was cooled to 0 °C in an ice bath, followed by the dropwise addition of pre-mixed EPCH (7.5 g, 81 mmol) and ME3MO (17.8 g, 81 mmol). The copolymer was purified similarly to polyME3MO. Yield, 18.5 g (73%). For copolymers with different monomer molar ratios, the yield varied from 60 to 80%.
1H NMR (CDCl3, 500 MHz) δ 3.66–3.44 (22H), 3.39–3.36 (3H).
2.6 Synthesis of polyGBIMTFSI-co-polyME3MO
A series of polyGBIMTFSI-co-polyME3MO were synthesized based on different polyEPCH-co-polyME3MO samples. PolyGBIMTFSI-co-polyME3MO-1/1 was synthesized as follows.PolyEPCH-co-polyME3MO-1/1 (12.5 g, 40 mmol based on its repeating unit) was first placed in a 100 mL Schlenk flask, followed by the addition of 1-butylimidazole (14.9 g, 120 mmol) and a magnetic stirrer bar. The mixture was stirred until homogeneous. 1-Butylimidazole was again used both as reactant and solvent. The solution was degassed three times using the freeze–pump–thaw procedure, then placed in an oil bath at 115 °C and refluxed overnight under nitrogen. After reaction, the polyGBIMCl-co-polyME3MO polymer was precipitated from ethyl ether, collected and dried under vacuum for 8 h. The polymer was dissolved in 100 mL Milli-Q water, followed by an excess of LiTFSI (17.2 g, 60 mmol). The product, polyGBIMTFSI-co-polyME3MO-1/1, was collected by 30 min centrifugation, washed three times with Milli-Q water and dried overnight at 100 °C in vacuo. Yield, 23.7 g (87%).
1H NMR (d6-DMSO, 500 MHz) δ 9.20–8.98 (1H), 7.88–7.50 (2H), 4.50–3.00 (27H), 1.82–1.72 (2H), 1.28–1.14 (2H), 0.92–0.80 (3H).
2.7 Characterization
The 1H and 13C NMR spectra were recorded on a Varian UnityPlus-500 spectrometer in CDCl3 or DMSO-d6 at room temperature, using the residual solvent proton signals as chemical shift standards. gHSQC and gHMBC 2D NMR spectra were recorded using an Agilent DDR2 500 MHz NMR spectrometer equipped with 7600AS 96 autosamplers. Polymer molecular weight was measured by gel permeation chromatography (GPC) at 35 °C using two PL-gel 10μ mixed-B columns in series. DMF was used as the eluting solvent at a flow rate of 1 mL min−1, with a Waters 2410 differential refractometer as detector. Monodispersed poly(ethylene oxide) was used as a standard for calibrating molecular weight. Thermogravimetric analysis (TGA) was performed using a PerkinElmer TGA 7 at a heating rate of 10 °C min−1 over a temperature range of 30 to 850 °C in air. TGA samples were dried in vacuo overnight before use. After increasing the temperature to 120 °C, samples were held 30 min in the TGA 7 before continuing the heating. Differential scanning calorimetry (DSC) was performed using a TA DSC Q100 instrument at a heating/cooling rate of 10 °C min−1 under nitrogen.2.8 Ionic conductivity
Ionic conductivity measurements were carried out in a home-made cell by impedance spectroscopy measurements, using an AC impedance analyzer HP 4192A over a frequency range 5 to 13 MHz with an applied voltage of 10 mV. The sample cell contained two steel disks serving as symmetrical electrodes, separated by a Teflon collar. All samples were equilibrated at a predetermined temperature 15 min before measurement. The resistance of the samples was estimated from a Nyquist plot, and the conductivity calculated from the electrode distance and cell cross-sectional area, as shown in eqn (1):
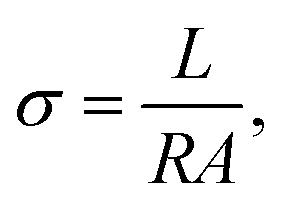 | (1) |
3. Results and discussion
The synthesis of 2-((2-(2-(2-methoxyethoxy)ethoxy)ethoxy)methyl)oxirane (ME3MO) is shown in Fig. 1(a). Triethylene glycol monomethyl ether was first deprotonated by sodium hydride and then reacted with epichlorohydrin (EPCH). The product ME3MO was obtained in 71% yield and characterized by 1H NMR, shown in Fig. 1(b). Peak 7 in the 1H NMR spectrum was assigned to the terminal CH3 group in the PEO oligomer chain, due to its distinct chemical shift and integral value of 3.03. The remaining peaks were assigned according to 2D NMR, gHSQC and gHMBC (Fig. 2(a) and (b)). From gHSQC, most of the peaks could be assigned, apart from peak 6 in Fig. 1(b). The peaks at 44 and 51 ppm in the 13C spectrum were assigned to the C of the CH2O group in the oxirane ring, and that at 72 ppm to the C in the CH2O group adjacent to the oxirane ring. Based on the correlation between the carbons and protons, the peaks from 1 to 5 in 1H NMR were assigned as follows. Peak 6 was assigned according to gHMBC. Since the protons of peak 6 interacted with the carbon of peak 7, peak 6 was assigned to the protons attached to the C closest to peak 7 in the 13C spectrum. Finally, the broad peak with an integral of 10.66 in 1H NMR was assigned to the remaining five CH2 protons in the PEO oligomer chain.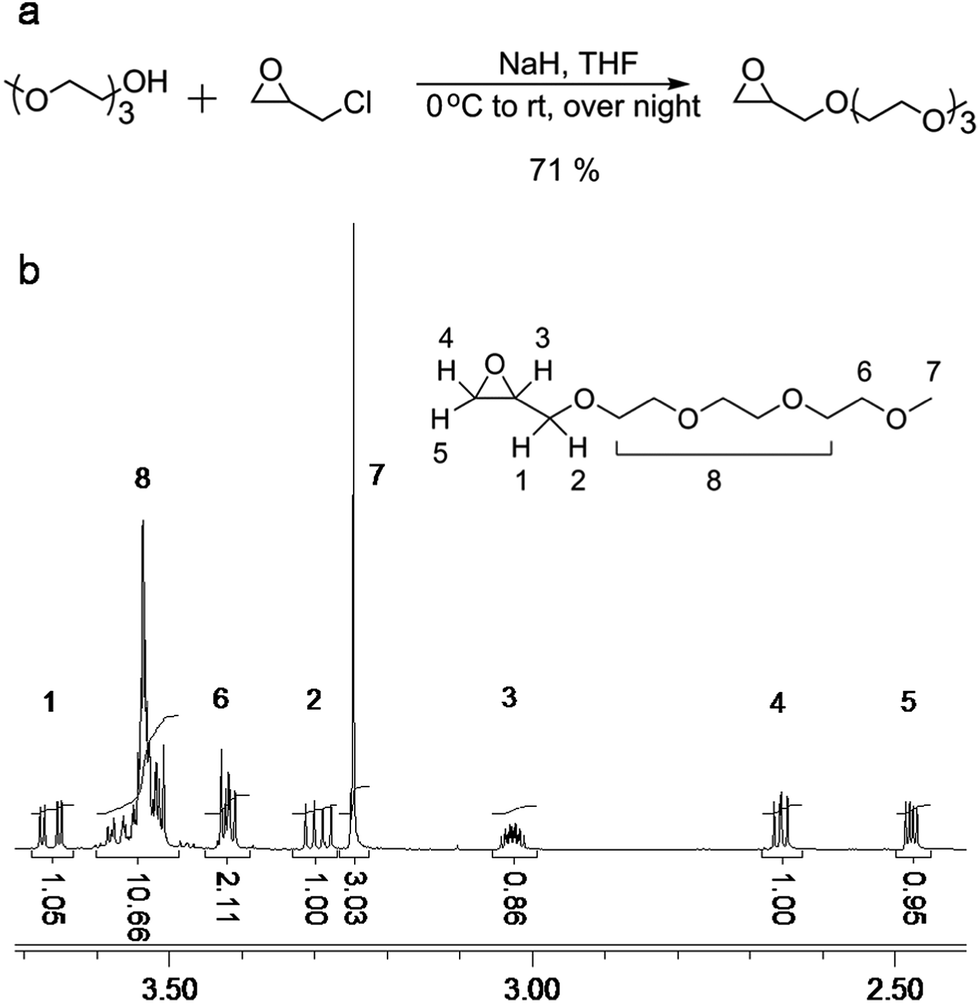 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of ME3MO monomer, and (b) the corresponding 1H NMR spectrum. | ||
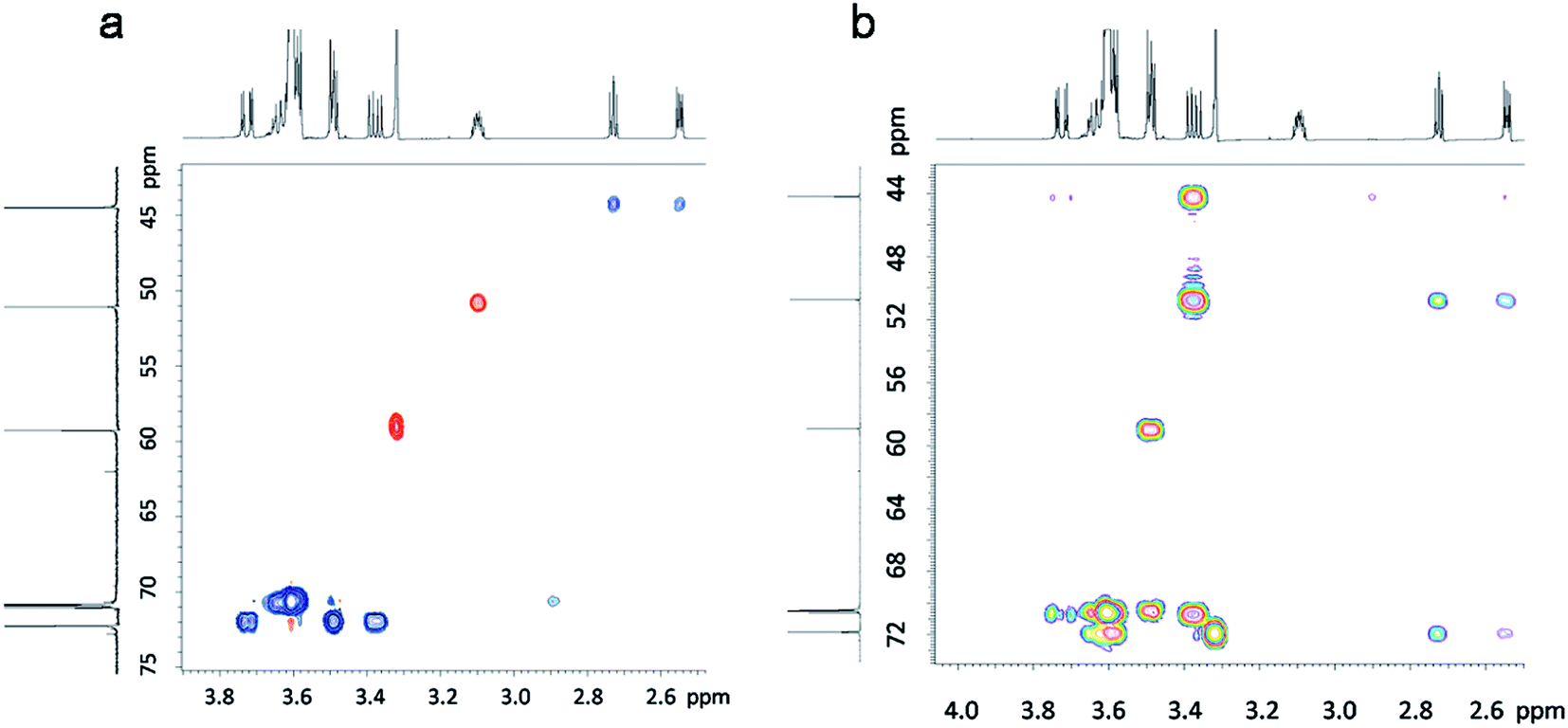 | ||
| Fig. 2 (a) gHSQC and (b) gHMBC of the ME3MO monomer. | ||
Using BF3OEt2 and 1,4-butanediol as initiator, cationic ring-opening polymerization was used to synthesize polyepichlorohydrin (polyEPCH), polyME3MO and polyEPCH-co-polyME3MO, as shown in Fig. 3(a). Six copolymer samples of different monomer composition, and polyEPCH and polyME3MO homopolymer, were synthesized. The molecular weight data are summarized in Table 1. PolyEPCH homopolymer had a Mn of 2.2 × 103 g mol−1, and polyME3MO homopolymer had Mn of 3.4 × 103 g mol−1. The Mn of copolymer increased with the increase in polyME3MO composition.
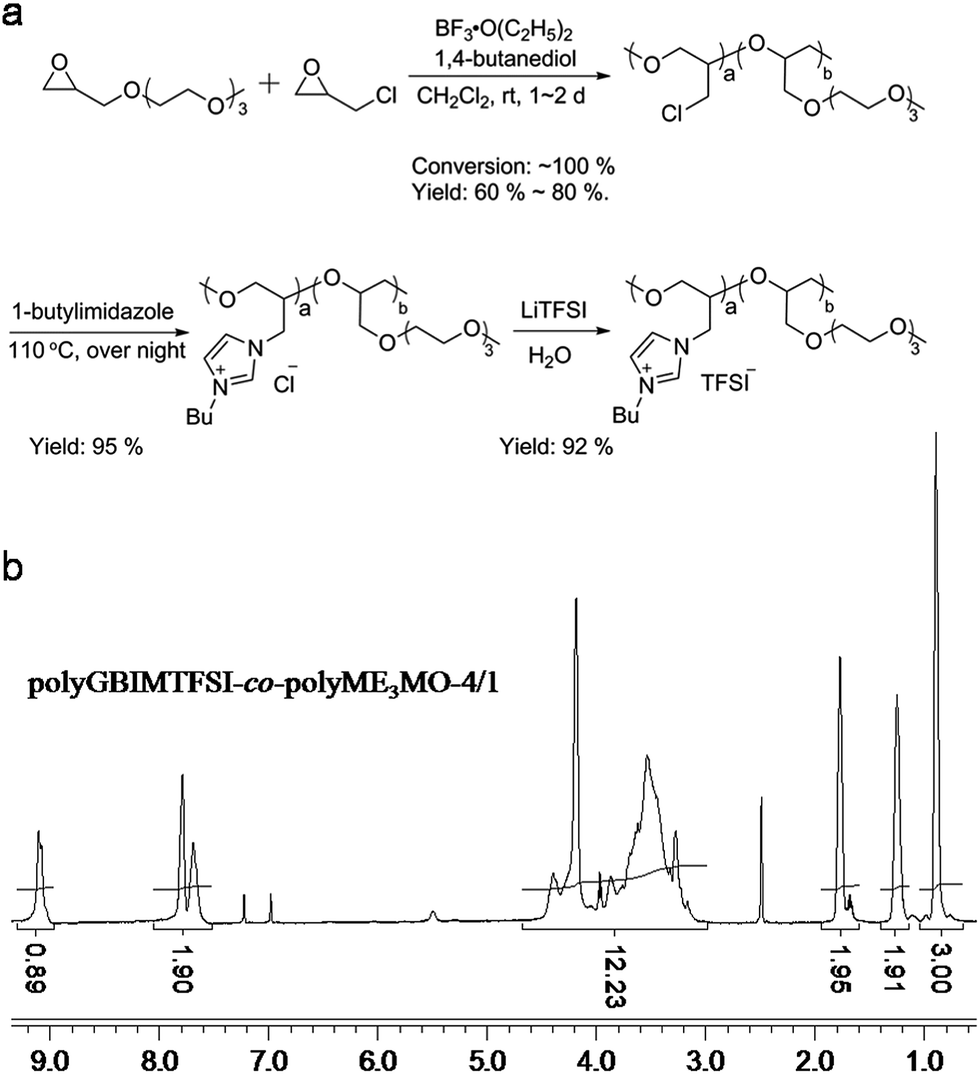 | ||
| Fig. 3 (a) Schematic illustration of the synthesis of polyGBIMTFSI-co-polyME3MO, and (b) its corresponding HNMR spectrum. | ||
| [M]1/[M]2/[diol]/[ BF3OEt2]a | Mn × 10−3b g mol | Mw × 10−3b g mol | Mp × 10−3b g mol | PDIb |
|---|---|---|---|---|
| a [M]1, [M]2, [diol] and [BF3OEt2] represent molar concentrations of EPCH, ME3MO, 1,4-butanediol and BF3OEt2, respectively.b Mn is the number average molecular weight, Mw the weight average molecular weight, Mp the peak molecular weight, and PDI is the polydispersity index. Data were obtained by GPC, using PEO as standard. | ||||
| 100/0/6/1 | 2.2 | 3.3 | 2.8 | 1.50 |
| 0/100/6/1 | 3.4 | 4.8 | 3.6 | 1.41 |
| 100/12.5/6/1 | 3.9 | 5.8 | 4.0 | 1.49 |
| 100/25/6/1 | 4.4 | 6.4 | 5.0 | 1.45 |
| 100/50/6/1 | 4.9 | 7.4 | 6.0 | 1.51 |
| 100/100/6/1 | 5.4 | 8.7 | 7.1 | 1.61 |
| 100/200/6/1 | 7.3 | 11.7 | 9.4 | 1.60 |
| 100/400/6/1 | 12.0 | 19.8 | 15.4 | 1.65 |
PolyGBIMTFSI-co-polyME3MO was synthesized by quaternarizing polyEPCH-co-polyME3MO with 1-butylimidazole, followed by an ion exchange reaction. Only the polyEPCH component in the copolymer was quaternarized, whereas polyME3MO was left intact. A high yield was achieved for all the samples. The structure of the polyGBIMTFSI-co-polyME3MO was characterized by 1H NMR, as shown in Fig. 3(b). The peaks between 3.0 and 4.5 ppm represented protons from the polyGBIMTFSI backbone, all the protons from polyME3MO and one CH2 from the butyl group. The three peaks above 7.4 ppm were assigned to the three protons from the imidazolium ring, and the three peaks below 1.8 ppm were the protons from CH2CH2CH3 of the butyl group on the imidazolium ring. The integral of CH3 group was set as 3, and the integral of broad peaks between 3.0 and 4.5 ppm was 12.23. The integrals corresponded very well to the composition of the copolymer, polyGBIMTFSI-co-polyME3MO-4/1.
The thermal stability of polyGBIMTFSI and its random copolymers was studied using TGA, as shown in Fig. 4. PolyGBIMTFSI and polyGBIMTFSI-co-polyME3MO with a higher molar ratio of polyGBIMTFSI (increased from 8/1 to 2/1) showed similar excellent thermal stability, with thermal degradation temperatures above 375 °C. On the other hand, polyGBIMTFSI-co-polyME3MO, with a higher molar ratio of polyME3MO (increased from 1/1 to 1/4) showed relatively poor thermal stability, and began to lose weight at about 200 °C. As shown in a previous study, polyEPCH also started to lose weight around 200 °C.11 The improvement in the thermal stability of polyGBIMTFSI arose from quaternarization, replacing the weak bonding between the pendant CH2–Cl and CH2–O(CH2CH2O)3CH3.
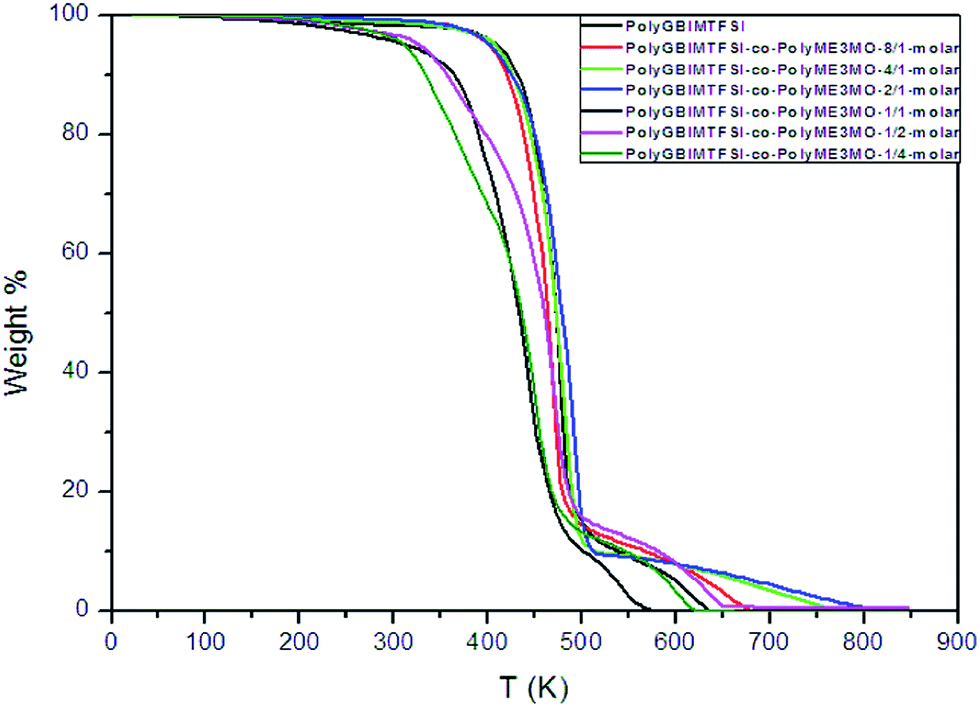 | ||
| Fig. 4 TGA traces of polyGBIMTFSI, polyME3MO and their random copolymers of varying molar composition. | ||
The glass transition temperatures (Tg) of polyGBIMTFSI, polyME3MO and their random copolymers were also studied by DSC. As shown in Table 2, polyGBIMTFSI had the highest Tg (−25 °C), and polyME3MO the lowest (−52 °C). All the random copolymers showed Tg between these values. With its higher molar composition of polyME3MO, the copolymer had a lower Tg.
| Sample | Tg (°C) |
|---|---|
| PolyGBIMTFSI | −25 |
| PolyGBIMTFSI-co-polyME3MO (8/1) | −29 |
| PolyGBIMTFSI-co-polyME3MO (41) | −33 |
| PolyGBIMTFSI-co-polyME3MO (2/1) | −36 |
| PolyGBIMTFSI-co-polyME3MO (1/1) | −41 |
| PolyGBIMTFSI-co-polyME3MO (1/2) | −43 |
| PolyGBIMTFSI-co-polyME3MO (1/4) | −45 |
| PolyME3MO | −52 |
The ionic conductivities of polyGBIMTFSI and polyGBIMTFSI-co-polyME3MO were measured over a temperature range from 25 to 90 °C, as shown in Fig. 5. The ionic conductivity data agreed well with the Tg data. PolyGBIMTFSI, with the highest Tg, showed the lowest ionic conductivity. With increasing polyME3MO the conductivity increased up to polyGBIMTFSI-co-polyME3MO-1/1. However, at higher compositions of polyME3MO the conductivity began to decrease, and polyGBIMTFSI-co-polyME3MO-1/4 had lower conductivity than its 1/1 analogue. This phenomenon is readily understood, and similar results have been reported in the literature.16 Introduction of polyME3MO lowered the Tg, which in turn had a positive effect on the ionic conductivity of the copolymer. On the other hand, increasing the polyME3MO composition meant a decrease in the polyGBIMTFSI composition, resulting in lower concentration of charge carriers and a negative effect on the ionic conductivity of the copolymer. Initially the conductivity of the copolymers increased with increasing polyME3MO composition, since the reduction in Tg had a more significant effect on ionic conductivity than the reduction in charge carrier concentration. However, in the case of polyGBIMTFSI-co-polyME3MO-1/2 and polyGBIMTFSI-co-polyME3MO-1/4, as the concentration of charge carriers became very low the negative effect overwhelmed the positive effect.
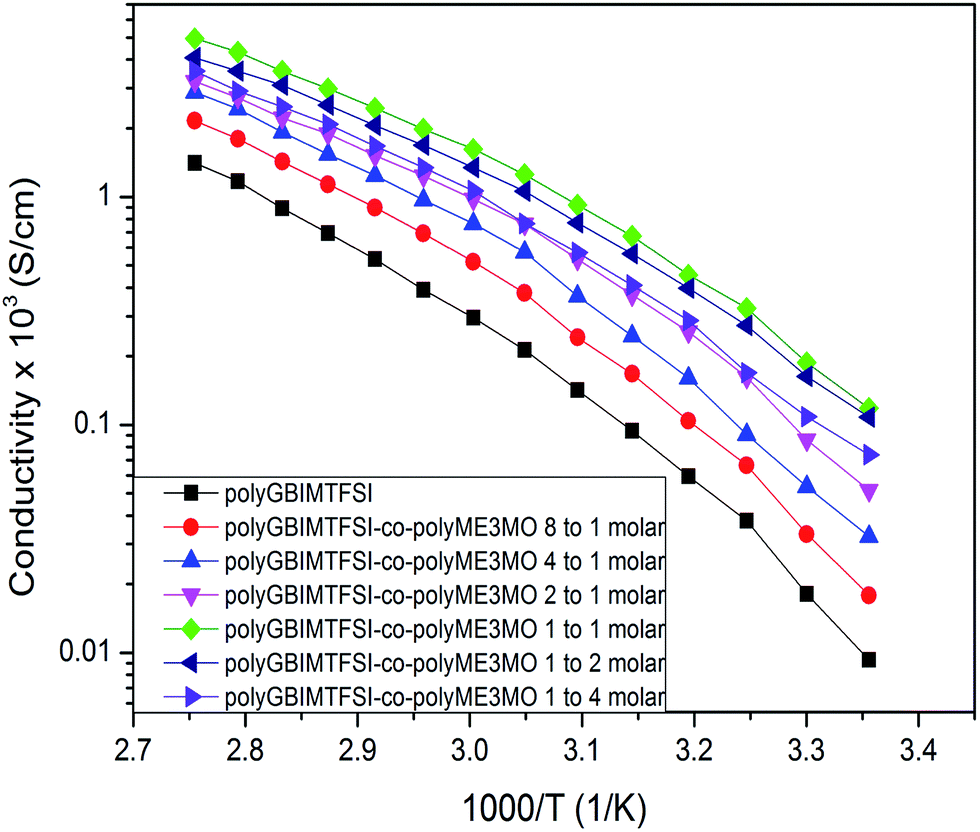 | ||
| Fig. 5 Ionic conductivity of polyGBIMTFSI and its random copolymers of varying molar composition. | ||
PolyGBIMTFSI-co-polyME3MO-1/1 showed the highest conductivity (around 1.2 × 10−4 S cm−1 at 25 °C), which was more than one order of magnitude higher than that of the polyGBIMTFSI homopolymer (9.3 × 10−6 S cm−1 at 25 °C). On the other hand, the conductivity of a common ionic liquid, 1-methyl-3-propylimidazolium iodide (MPII), was 1.1 × 10−3 S cm−1 at 25 °C.10,11 The conductivity of polyGBIMTFSI-co-polyME3MO-1/1 was less than one order of magnitude lower than that of MPII. Introducing a low Tg component to form a PIL copolymer proved to be very effective in improving the ionic conductivity of polyionic liquids.
4. Conclusions
2-((2-(2-(2-Methoxyethoxy)ethoxy)ethoxy)methyl)oxirane (ME3MO), a liquid analogue of ethylene oxide, was synthesized and copolymerized with epichlorohydrin. The resulting copolymer (polyEPCH-co-polyME3MO) was quaternarized with 1-butylimdazole, followed by an ion exchange reaction, to form a PIL copolymer (polyGBIMTFSI-co-polyME3MO). By tuning the monomer composition, a series of PIL copolymers from polyGBIMTFSI-co-polyME3MO-8/1 to polyGBIMTFSI-co-polyME3MO-1/4 were synthesized. All the monomers and polymers were characterized by 1D and 2D NMR. The polymerized ionic liquids in the study showed a degradation temperature above 350 °C. All PIL copolymer samples showed higher ionic conductivity than the PIL homopolymer up to 1.2 × 10−4 S cm−1 at room temperature, which was more than one order of magnitude higher than the PIL homopolymer (9.3 × 10−6 S cm−1 at 25 °C). It was found that the introduction of a component such as ethylene oxide, with low Tg or a PEO backbone, to form a PIL copolymer proved very effective in improving the ionic conductivity of PIL. Ongoing research will include potential applications such as lithium polymer batteries, solid supercapacitors or solid-state dye-sensitized solar cells.Acknowledgements
This study was supported financially by the National Science Foundation (DMR–0934568) and the MSU Center for Alternative Energy Storage Research and Technology (CAESRT). W.Y. is also indebted to Dr James McCusker, Dr Keith Promislow and Dr Lawrence Drzal for valuable discussions.References
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS PubMed.
- J. Y. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS PubMed.
- O. Green, S. Grubjesic, S. W. Lee and M. A. Firestone, Polym. Rev., 2009, 49, 339–360 CrossRef CAS.
- H. Ohno and K. Ito, Chem. Lett., 1998, 1, 751–752 CrossRef.
- M. Yoshizawa and H. Ohno, Chem. Lett., 1999, 1, 889–890 CrossRef.
- H. Ohno, Electrochim. Acta, 2001, 46, 1407–1411 CrossRef CAS.
- M. Yoshizawa and H. Ohno, Electrochim. Acta, 2001, 46, 1723–1728 CrossRef CAS.
- S. Washiro, M. Yoshizawa, H. Nakajima and H. Ohno, Polymer, 2004, 45, 1577–1582 CrossRef CAS PubMed.
- W. Yuan, H. Zhao, H. Y. Hu, S. Wang and G. L. Baker, ACS Appl. Mater. Interfaces, 2013, 5, 4155–4161 CAS.
- H. Y. Hu, W. Yuan, H. Zhao and G. L. Baker, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 121–127 CrossRef CAS.
- H. Y. Hu, W. Yuan, L. S. Lu, H. Zhao, Z. Jia and G. L. Baker, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2104–2110 CrossRef CAS.
- M. Dobbelin, I. Azcune, M. Bedu, A. R. de Luzuriaga, A. Genua, V. Jovanovski, G. Cabanero and I. Odriozola, Chem. Mater., 2012, 24, 1583–1590 CrossRef CAS.
- H. Ohno, M. Yoshizawa and W. Ogihara, Electrochim. Acta, 2004, 50, 255–261 CrossRef CAS PubMed.
- Y. Zhang, Y. W. Zhu, G. Lin, R. S. Ruoff, N. P. Hu, D. W. Schaefer and J. E. Mark, Polymer, 2013, 54, 3605–3611 CrossRef CAS PubMed.
- Y. S. Ye, J. H. Choi, K. I. Winey and Y. A. Elabd, Macromolecules, 2012, 45, 7027–7035 CrossRef CAS.
- H. Chen, J. H. Choi, D. Salas-de La Cruz, K. I. Winey and Y. A. Elabd, Macromolecules, 2009, 42, 4809–4816 CrossRef CAS.
- W. Yuan, H. Zhao and G. L. Baker, Org. Electron., 2014, 15, 3362–3369 CrossRef CAS PubMed.
- Z. Jia, W. Yuan, H. Zhao, H. Y. Hu and G. L. Baker, RSC Adv., 2014, 4, 41087–41098 RSC.
- A. U. Francis, S. Venkatachalam, M. Kanakavel, P. V. Ravindran and K. N. Ninan, Eur. Polym. J., 2003, 39, 831–841 CrossRef CAS.
- S. J. Jungk, J. A. Moore and R. D. Gandour, J. Org. Chem., 1983, 48, 1116–1120 CrossRef CAS.
Footnotes |
| † These authors contributed equally to this study. |
| ‡ Professor Gregory L. Baker passed away on 18 October 2012. |
| This journal is © The Royal Society of Chemistry 2015 |