
Stereocontrolled synthesis of propionate motifs from L-lactic and L-alanine aldehydes. A DFT study of the hydrogen transfer under endocyclic control†‡
François
Godin
ab,
Martin
Duplessis
ab,
Cindy
Buonomano
ab,
Thao
Trinh
ab,
Karine
Houde
ab,
Daniel
Chapdelaine
a,
Jacques
Rodrigue
ab,
André
Boutros
a and
Yvan
Guindon
*abc
aInstitut de recherches cliniques de Montréal (IRCM), Bio-organic Chemistry Laboratory, 110 avenue des Pins Ouest, Montréal, Québec, Canada H2W 1R7. E-mail: yvan.guindon@ircm.qc.ca
bDépartement de chimie, Université de Montréal, C.P. 6128, succursale Centre-ville, Montréal, Québec, Canada H3C 3J7
cDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montréal, Québec, Canada H3A 2K6
First published on 22nd July 2014
Abstract
Reported herein is the synthesis of all possible stereotriad propionates derived from L-lactic acid and L-alanine. The approach is based on a stereocontrolled Mukaiyama aldol reaction followed by a free radical-mediated hydrogen transfer, for which the choice of Lewis acid (monodentate or bidentate) dictates the stereochemical outcome in each reaction. The sequential process has been applied iteratively for the synthesis of eight stereopentad motifs derived from β,γ-alkoxyaldehydes. New experimental data and DFT calculations (BHandHLYP/TZVP) of the transition states involved in the radical reduction of aluminate intermediates support the proposed model of the endocyclic effect leading to high 2,3-syn selectivities for the synthesis of propionates and subunits thereof.
Introduction
Polyketide natural products are biosynthesized in response to environmental stimuli by various organisms (bacteria, fungi and plants).1 They display a dense array of consecutive stereogenic centers identified as polypropionates.These structural subunits present an alternating sequence of methyl and hydroxyl groups, such as in zincophorin 1 (Fig. 1),2 and can be flanked by a stereogenic center bearing a hydroxyl (e.g. aflastatin A, 2)3 or an amine (e.g. superstolide A, 3).4 The important biological activities of this class of compounds5 have stimulated significant research efforts for the development of different approaches to access acyclic polypropionates starting from α-methyl aldehydes.6 Some of these strategies were also applied to α-alkoxy6c,7 or α-amino aldehydes8 derived, respectively, from L-lactic acid or L-alanine.
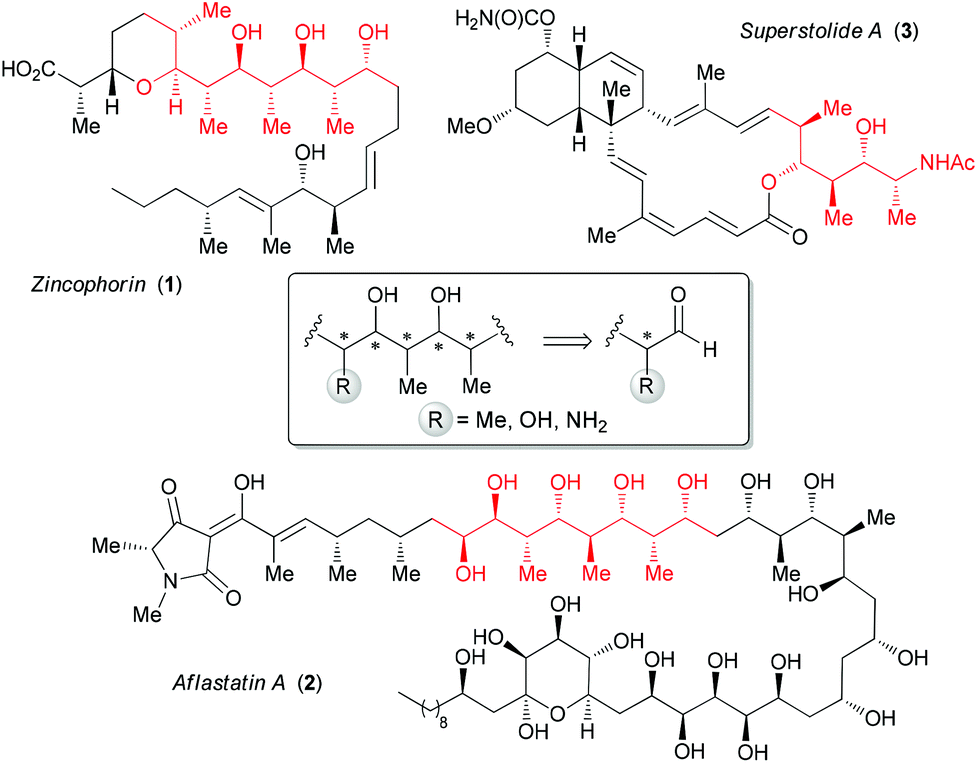 | ||
| Fig. 1 Representative polyketides. | ||
Our group previously reported a reaction sequence leading to propionate units by treating β-benzyloxy-α-methyl aldehyde 4 in the presence of a bidentate Lewis acid (such as TiCl4) with a mixture of tetrasubstituted enoxysilanes, bearing either a bromide 5 or a selenoether 6 (Scheme 1).9 The resulting C3–C4 anti adducts, bearing tertiary bromides (7a,b) or selenides (8a,b), were obtained with high diastereoselectivity. The complementary 3,4-syn inductions were also generated selectively by monodentate activation, using Lewis acids such as BF3·OEt, in the Mukaiyama aldol reaction.9b
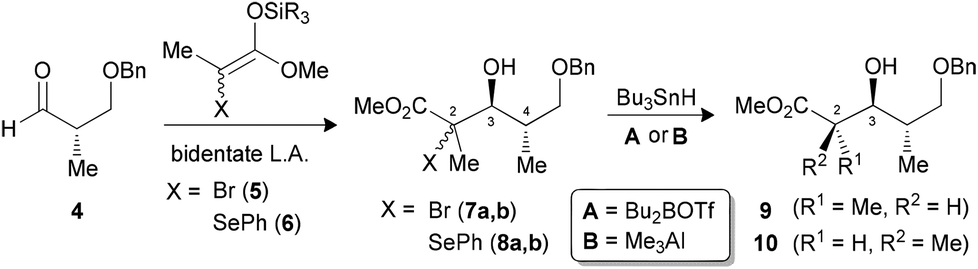 | ||
| Scheme 1 Synthesis of propionates using a reaction sequence of Mukaiyama aldol and radical reduction. | ||
It was demonstrated subsequently that the radical precursors at C2 could be reduced to the corresponding acyclic propionates by a stereocontrolled hydrogen transfer, where the stereochemistry is dictated by the nature of the Lewis acid (Scheme 1).10 Indeed, by forming a borinate intermediate prior to the initiation of the radical, the C2–C3 anti product 9 could be obtained selectively. This reaction was recently examined in silico by DFT calculation.11 The 2,3-syn product 10 could alternatively be generated by treating the mixture of precursors with Me3Al to likely form an aluminate intermediate linking the oxygen at C3 with the ester (endocyclic effect).10a,12
Herein, we examined the applicability of our methodology to generate the four propionate motifs, as well as different polypropionate fragments, using the aldehyde derived from L-lactic acid. The mechanistic hypothesis at the origin of the 2,3-syn stereocontrol of the free radical reduction mediated by aluminates is also examined by DFT calculations. The potential of our methodology to generate propionates bearing amine stereogenic centers will also be demonstrated by synthesizing the four stereotriads derived from L-alanine.
Results and discussion
Synthesis of the stereotriads from L-lactic aldehyde
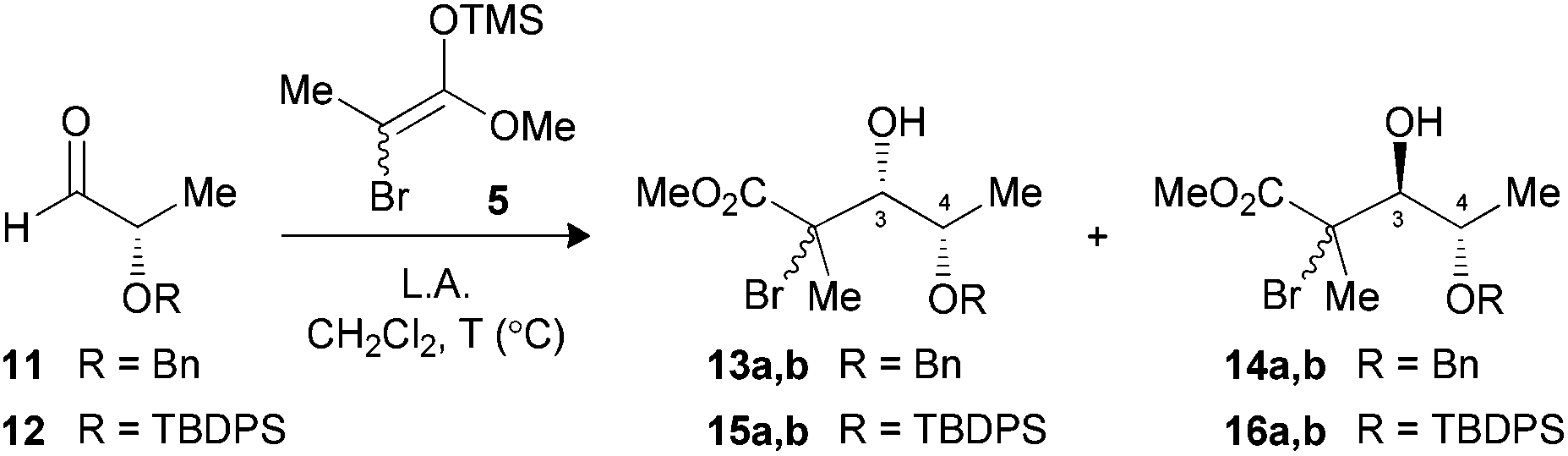
The O-benzylated L-lactic aldehyde 11 was first considered for the Mukaiyama aldol reaction. The substrate was precomplexed with TiCl4 at −78 °C followed by the addition of enoxysilane 5, leading exclusively to the Cram-chelate adduct 13a,b (3,4-syn) in excellent yield (Table 1, entry 1).13 Other bidentate Lewis acid, such as MgBr2·OEt2, afforded similar results in terms of diastereoselectivity, albeit with a slightly lower yield (entry 2). Low selectivity was then noted using BF3·OEt2 (entry 3). Previous studies suggested that the Felkin–Anh facial selectivity can be improved by increasing the steric hindrance of substituents proximal to the aldehyde.9a,b,14 The α-alkoxy protecting group was hence replaced by a TBDPS (12) to give an excellent yield and ratio favoring the 3,4-anti product 16a,b with BF3·OEt2 (entry 4).|
|
||||
|---|---|---|---|---|
| Entry (substrate) | LA (equiv.) | T (°C) | Yielda (%) | Ratiob (3,4-syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3,4-anti) :3,4-anti) |
| a Yields of isolated products. b Product ratios were determined by 1H NMR analysis of the crude reaction mixture. c The aldehyde was pretreated with TiCl4 for 2 min, prior to the addition of enoxysilane (2.0 equiv.). d Lewis acid added to a mixture of aldehyde and enoxysilane (2.0 equiv.). | ||||
| 1 (11) | TiCl4c (1.3) | −78 | 89 | >20:1 (13,14) |
| 2 (11) | MgBr2·OEt2d (5.0) | −60 | 73 | >20:1 (13,14) |
| 3 (11) | BF3·OEt2d (1.3) | −78 | 88 | 3:1 (13,14) |
| 4 (12) | BF3·OEt2d (1.3) | −78 | 92 | 1:>20 (15,16) |
The hydrogen transfer reaction with Bu3SnH was then evaluated on the Mukaiyama adducts obtained. Reduction of tertiary bromides 13a,b and 16a,b pretreated with Me3Al at −78 °C led to excellent syn selectivities and yield (Table 2, entries 1 and 3). Under the same conditions, substrate 19a,b (X = Br) or 20a,b (X = SePh), bearing an isopropyl chain at C3, also provided the exclusive formation of the 2,3-syn product (entries 5 and 6). The complementary 2,3-anti selectivity was generated by in situ formation of a borinate at C3 prior to the addition of Bu3SnH (entries 2 and 4).
| Entry (substrate) | LA (equiv.) | Yielda (%) | Ratiob (2,3-syn:2,3-anti) |
|---|---|---|---|
| a Yields of isolated products. b Product ratios were determined by 1H NMR analysis of the crude reaction mixture. c Substrates were pretreated with Me3Al (3.0 equiv.) for 1 h at −78 °C, prior to the addition of Bu3SnH, Et3B and air. d Substrates were pretreated with iPr2NEt (1.5 equiv.) and Bu2BOTf (1.3 equiv.) for 1 h at −78 °C, prior to the addition of Bu3SnH, Et3B and air. e Complete conversion was observed by 1H NMR spectroscopy. | |||
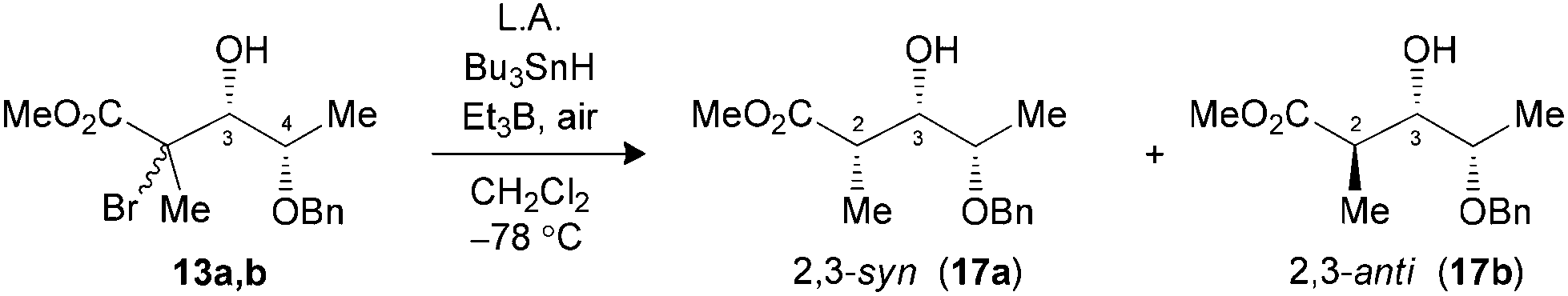
|
|||
| 1 (13) | Me3Alc (3.0) | 82 | >20:1 (17) |
| 2 (13) | Bu2BOTfd (1.3) | 90 | 1:>20 (17) |
|
|||
| 3 (16) | Me3Alc (3.0) | 69 | >20:1 (18) |
| 4 (16) | Bu2BOTfd (1.3) | 66 | 1:>20 (18) |

|
|||
| 5 (19) | Me3Alc (3.0) | —e | >20:1 (21) |
| 6 (20) | Me3Alc (3.0) | 74 | >20:1 (21) |
DFT study of the hydrogen transfer reaction
Our group recently reported a transition state DFT study (BHandHLYP/TZVP) for the radical reduction of borinate radical intermediates, such as 22 (generated from precursor 19a,b or 20a,b).11 Convincing correlations between the experimental and calculated difference in Gibbs free energy (ΔΔG‡) were obtained (Fig. 2). In addition to a minimization of the steric interactions with the incoming hydride, the anti-predictive transition state (TS A) is proposed to be favored because dipole–dipole interactions and allylic-1,3 strain are alleviated, whereas only the latter (i.e. A1,3) is relieved in the transition state leading to the product 2,3-syn (TS B).15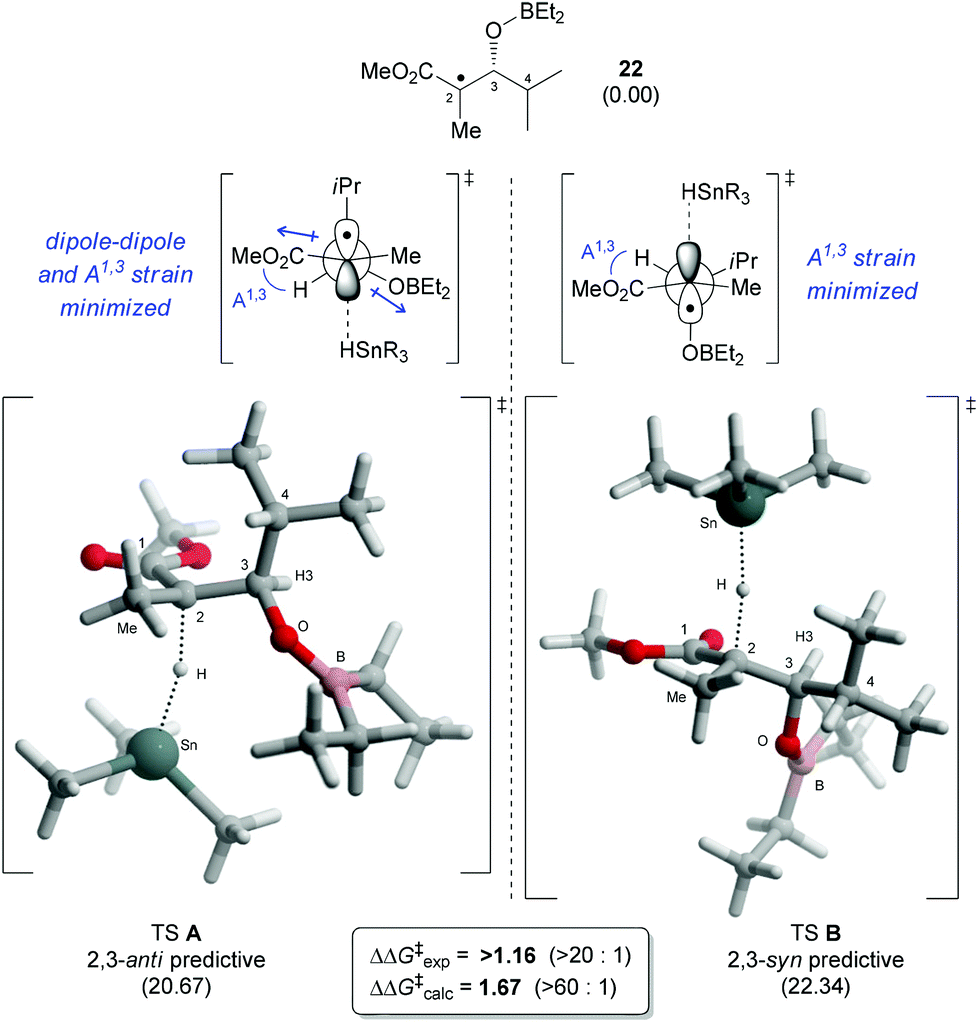 | ||
| Fig. 2 Lowest energy transition states 2,3-anti (TS A) and 2,3-syn (TS B) for the hydrogen transfer of borinate radical intermediate 22 identified by DFT (BHandHLYP/TZVP). Gibbs free energies (kcal mol−1) in DCM are reported in parentheses relative to the energy of 22. | ||
A similar computational methodology was employed in the present study16 for the hydrogen transfer of aluminate free radical intermediates. The radical intermediate bearing an isopropyl chain at C3 was first examined by DFT using Gaussian 0917 at the BHandHYLP/TZVP level. The ground state analysis of 23 revealed that the aluminate prefers to adopt cyclic conformations involving a chelation with the ester (Fig. 3b–d). The acyclic conformation (23-1-E,18Fig. 3a), similar to the one identified for the corresponding borinate in TS A (Fig. 2), was significantly higher in energy. In the different six-membered half-chair complexes, the aluminate can interact with the electron lone pairs of the OMe once the radical at C2 is delocalized in a Z-enol conformation (23-2-Z). As expected, the empty orbitals of aluminium prefers to complex with the carbonyl of the ester (E-enol radical). Indeed, shorter O1–Al bond distances and lower Gibbs free energies were noted for these conformations (Fig. 3c and d). Moreover, NBO analysis19 of structure 23-2-E indicated that the important stabilizing interaction between the carbonyl and the aluminium (nO1 → n*Al = 54.7 kcal mol–1)20 is mainly responsible for the important decrease in the energy of the SOMO, and the reduced electron density of the radical at C2. Conformation 23-3-E, minimizing the allylic-1,2 strain between the methyl at C2 and H3, was calculated to be slightly higher in energy than 23-2-E for which the iPr chain is positioned in a pseudo-equatorial orientation.
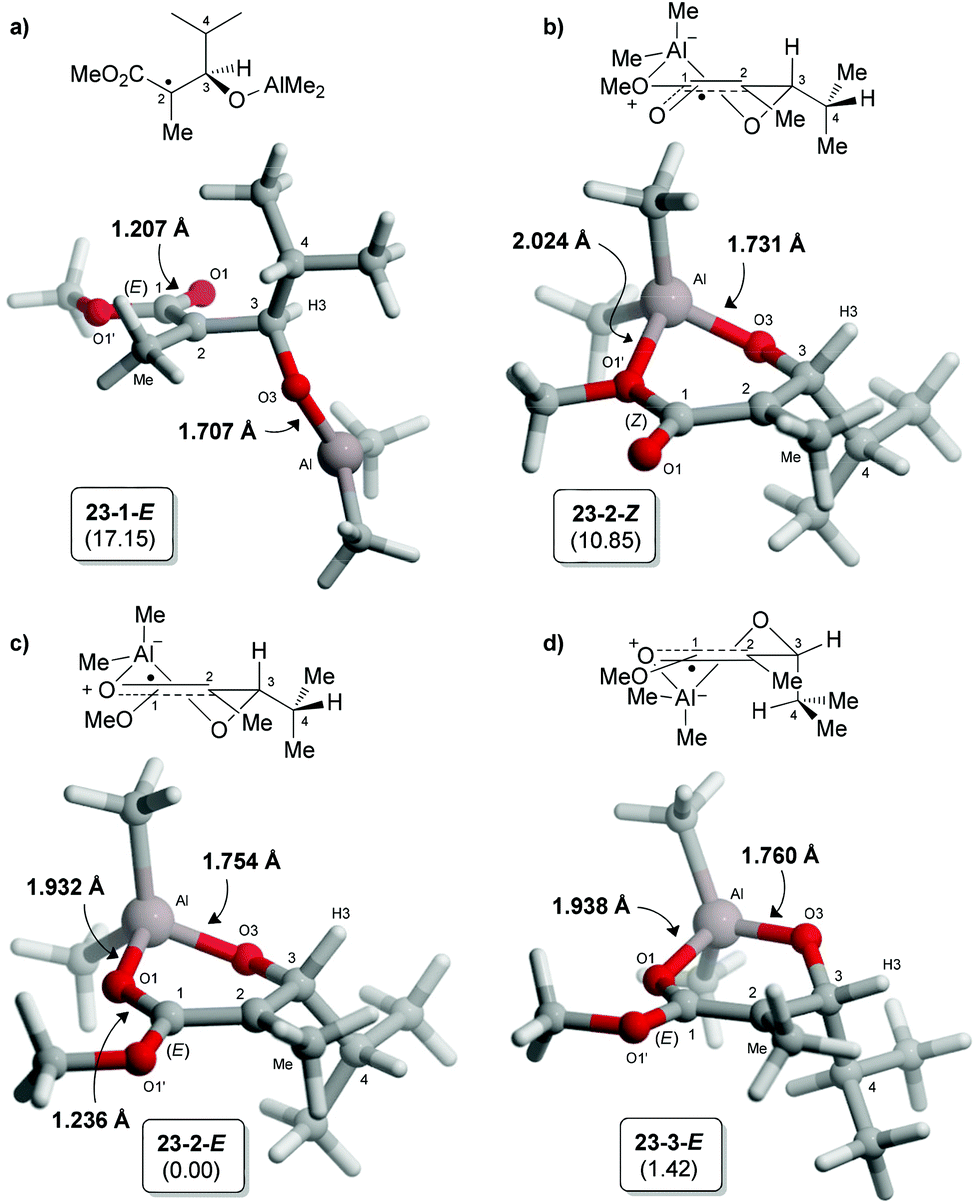 | ||
| Fig. 3 Ground state conformations of aluminate radical intermediate 23 identified at the BHandHLYP/TZVP level of theory. Gibbs free energies (kcal mol−1) in DCM are reported in parentheses relative to the energy of 23-2-E. | ||
The analysis of hydrogen transfer performed on the radical intermediate 23 with Me3SnH revealed the preferred conformations in transition states C and D, respectively, leading to the 2,3-syn and 2,3-anti products (Fig. 4). The lowest energy TS C validates the model previously proposed for the endocyclic effect.9a,10a Moreover, the calculated difference in energy (ΔΔG‡) with TS D, favoring 2,3-syn product 21a, correlates with the experimental selectivity. We can suggest that the higher energy of anti-predictive TS D possibly arises from a minimization of the steric interaction between the iPr chain and the incoming hydride, causing the methyl at C2 and the C3–C4 bonds to be eclipsed. This hypothesis is further supported by a decomposition of the activation energy,21 a model which allows a separation of the different contributions at the transition state. The analysis revealed that a larger strain energy (ΔE‡d) for the radical is noted in TS D (5.85 kcal mol–1) than in TS C (4.70 kcal mol–1), while the energy of interaction with the incoming hydride (ΔE‡int) is similar in both (Fig. 4). Since the conformation adopted in TS C is similar to its lowest energy ground state (23-2-E, Fig. 3c), less distortion (ΔE‡d) is needed for the radical intermediate to achieve its reactive conformation at the transition state, possibly the main contribution at the origin of the endocyclic effect.
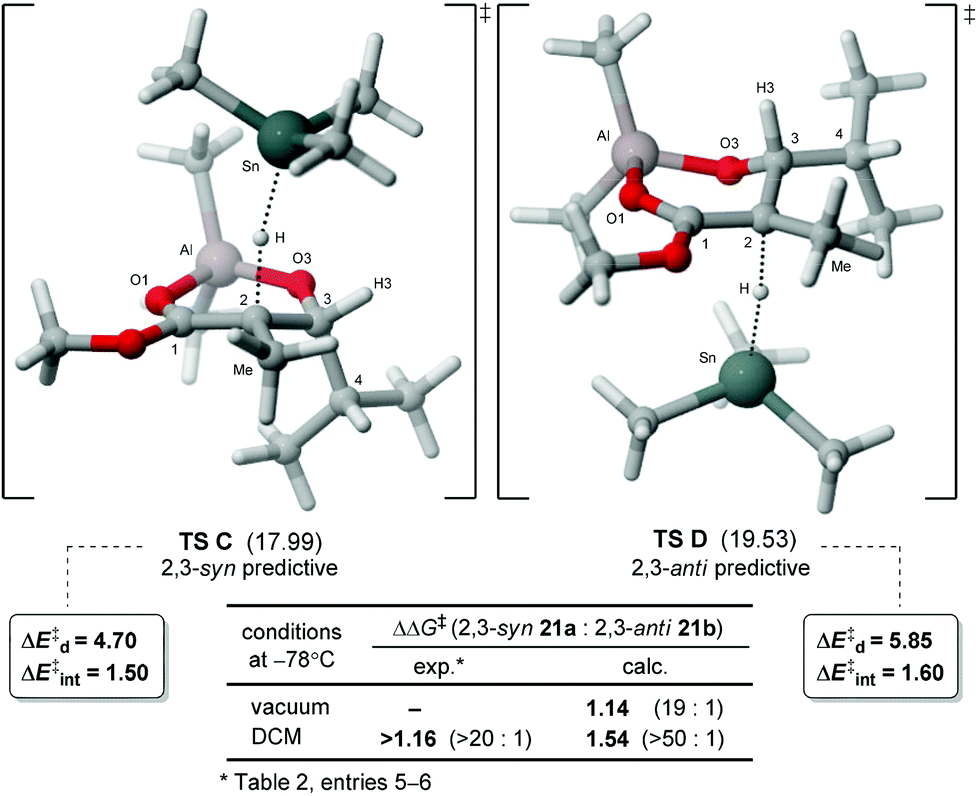 | ||
| Fig. 4 Transition states 2,3-syn (TS C) and 2,3-anti (TS D) for the hydrogen transfer of aluminate radical intermediate 23. Gibbs free energies (kcal mol−1) in DCM are reported in parentheses relative to the ground state energy of 23-2-E. | ||
We then turned our attention to the hydrogen transfer reaction involving an aluminate intermediate bearing an alkoxy group at C4 by modelling the radical 24 (Fig. 5).22 Not surprisingly, the analysis revealed that the lowest energy transition state (TS E) was leading to the syn-product and displayed a conformation similar to the corresponding isopropyl analogue (TS C, Fig. 4). The analysis of anti-predictive transition states for 24 was also interesting. Indeed, the lowest energy structure identified, TS F1, is similar to TS D (Fig. 4) and for which the OMe is pointing away from the cycle. Our group previously reported an amplification of the 2,3-anti ratios for the reduction of the carbon-centered radical adjacent to a cycle bearing an oxygen, which was termed the exocyclic effect.11,23 In the analysis of 24, another structure identified was TS F2 mimicking an exocycle by the chelation of the aluminate (O3–Al) with the oxygen O4. Although the energy of TS F2 was calculated to be slightly higher than TS F1, its relevance for other similar reactions will have to be further investigated. After consideration of solvent effects using the PCM method,24 the difference in energy ΔΔG‡ between the lowest energy transition states E and F1 correlated with the exclusive formation of the 2,3-syn product 17a.
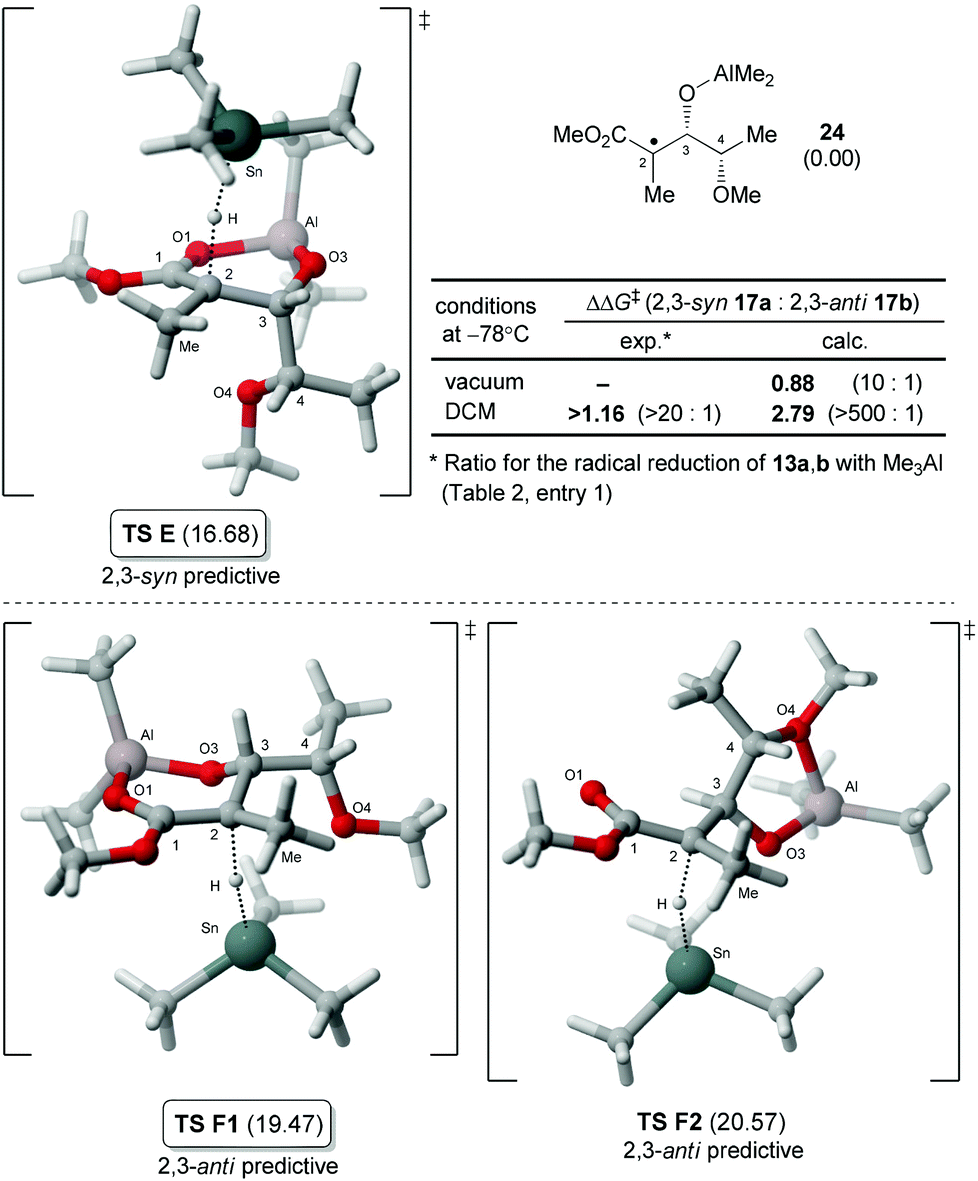 | ||
| Fig. 5 Transition states 2,3-syn (TS E) and 2,3-anti (TS F1, F2) for the hydrogen transfer of aluminate radical intermediate 24. Gibbs free energies (kcal mol−1) in DCM are reported in parentheses relative to the ground state energy of 24. | ||
Synthesis of stereopentads from L-lactic aldehydes
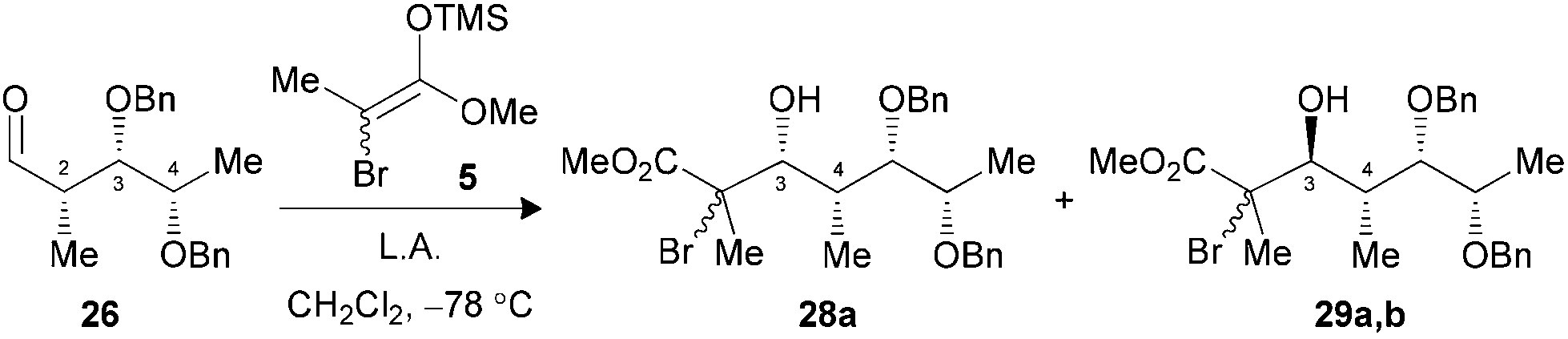
We have previously demonstrated that our reaction sequence can be applied iteratively to elongate the propionates obtained from β-alkoxy-α-methyl aldehydes to access all possible stereopentads.25 Moreover, this previous study revealed the difficulty of forming a Cram-chelate complex at the aldolization step with aldehyde 25 bearing a C2–C3 syn relationship, as poor ratio and yield were observed. These problems were solved by the use of 2.5 equiv. of TiCl3OiPr, for which a bicyclic [3.3.1]-type complex was suggested (Fig. 6).26 However, the situation would be slightly different in the present study if such intermediates are allowed. Indeed, the aldol reaction on a propionate motif derived from L-lactic acid, such as 26, would lead to the formation of a [3.2.1]-complex with the chelation of alkoxy groups at C3, C4 and the carbonyl. We decided to investigate if the formation of these titanium complexes could lead to an increase in diastereoselectivity by preparing aldehydes 26 and 27 from the corresponding esters 17a and 17b.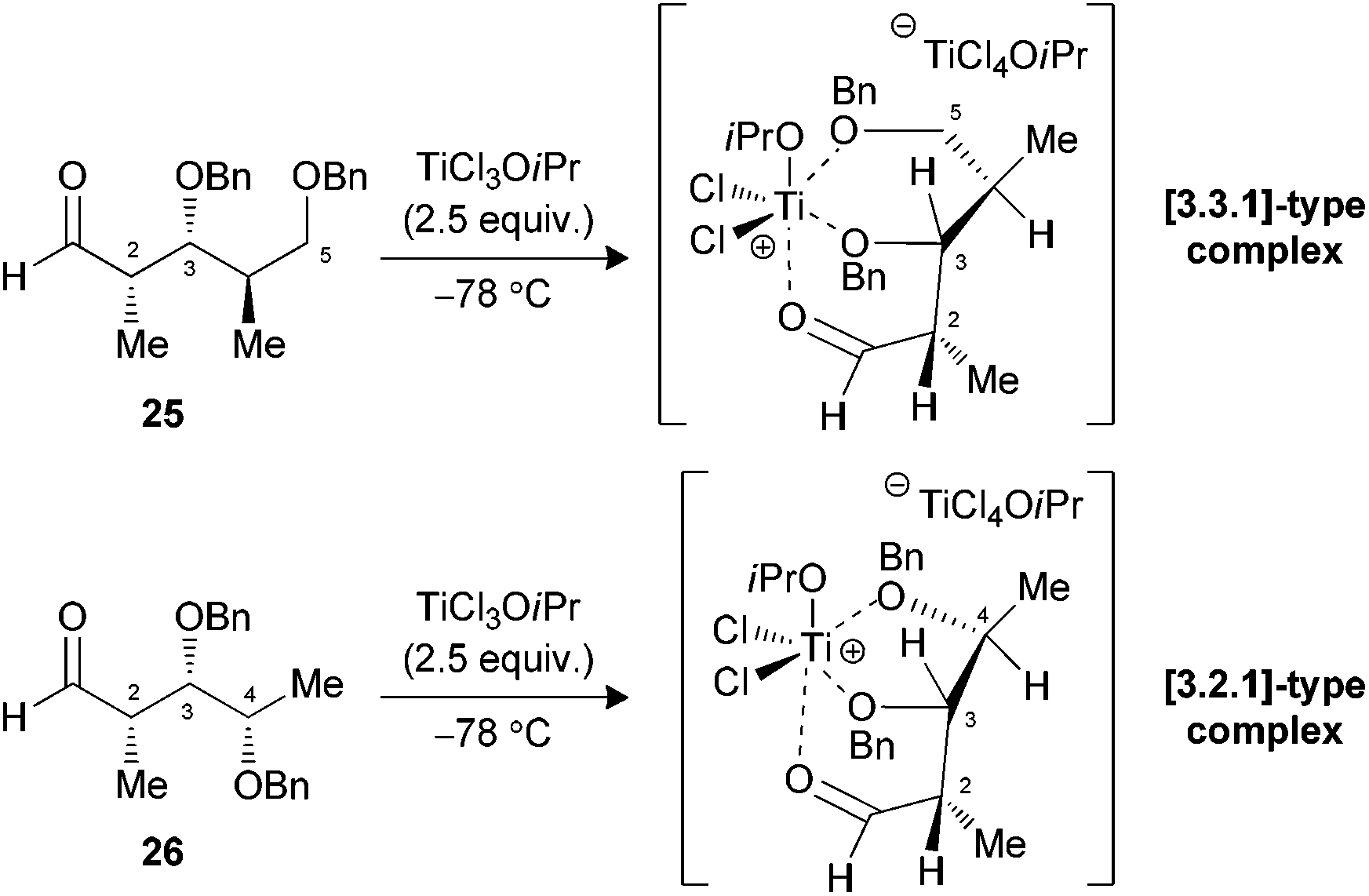 | ||
| Fig. 6 Proposed titanium complexes of aldehydes 25 and 26 with TiCl3OiPr. | ||
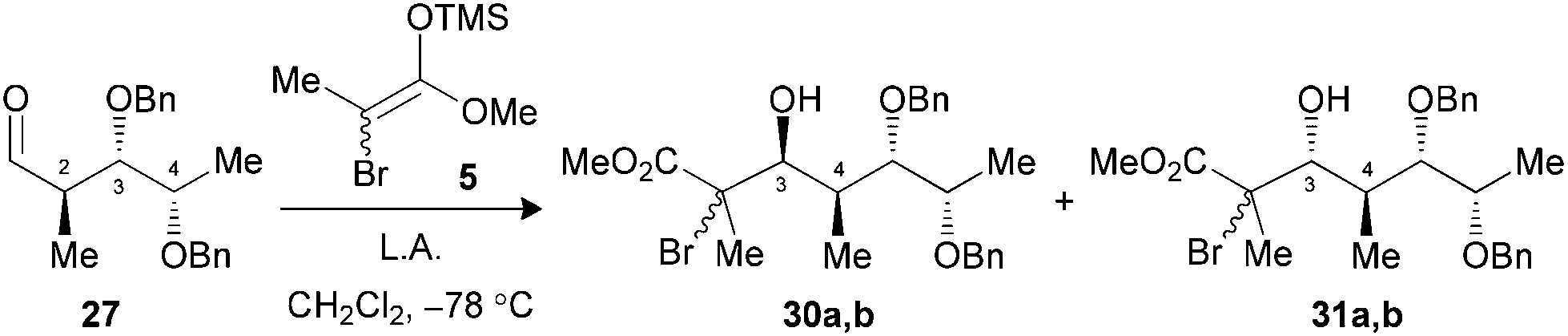
Under the conditions evaluated, C2–C3 syn aldehyde 26 only led to decomposition products when TiCl4 was used to promote the reaction (Table 3, entry 1). Modification of the Lewis acid to TiCl3OiPr provided an acceptable yield of tertiary bromides with, however, poor diastereoselectivity using 1.3 equiv. (entry 2). An increase in the stoichiometry of the Lewis acid significantly improved the selectivity, with the exclusive formation of 3,4-anti adducts 29a,b (entry 3). In our previous study, aldehydes bearing a 2,3-anti relationship did not require the formation of an ate complex.25b The analysis of the results presented herein with substrate 27 revealed the necessity to increase TiCl3OiPr to 2.5 equiv. to achieve exclusive formation of the Cram-chelate product 31a,b (entries 5–7). Seemingly, the presence of the OBn group at C4 could alter the formation of a bidendate complex and explain the variations in terms of reactivity. As noted for similar substrates,9c,25 activation of either aldehyde 26 or 27 with BF3·OEt2 led to an excellent ratio in favor of the 3,4-syn product (entries 4 and 8).
| Entry (substrate) | LA (equiv.) | Yielda (%) | Ratiob (3,4-syn:3,4-anti) |
|---|---|---|---|
| a Yields of isolated products. b Product ratios were determined by 1H NMR analysis of the crude reaction mixture. c The aldehyde was pretreated with a Lewis acid for 5 min, prior to the addition of enoxysilane (2.0 equiv.). d Lewis acid added to a mixture of aldehyde and enoxysilane (2.0 equiv.). | |||
|
|||
| 1 (26) | TiCl4c (1.3) | Decomp. | — (28,29) |
| 2 (26) | TiCl3OiPrc (1.3) | 73 | 1:2.4 (28,29) |
| 3 (26) | TiCl3OiPrc (2.5) | 81 | 1:>20 (28,29) |
| 4 (26) | BF3·OEt2d (1.5) | 85 | >20:1 (28,29) |
|
|||
| 5 (27) | TiCl4c (1.3) | Decomp. | — (30,31) |
| 6 (27) | TiCl3OiPrc (1.3) | 78 | 1:1.6 (30,31) |
| 7 (27) | TiCl3OiPrc (2.5) | 82 | 1:>20 (30,31) |
| 8 (27) | BF3·OEt2d (1.5) | 86 | >20:1 (30,31) |
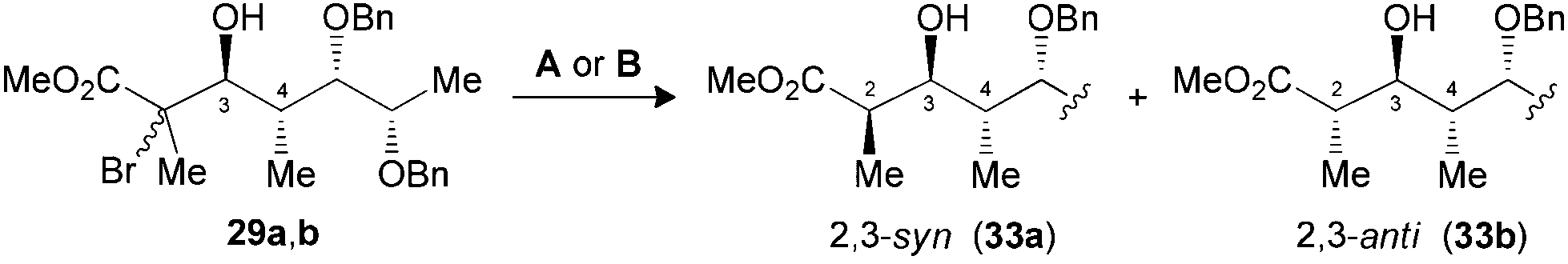
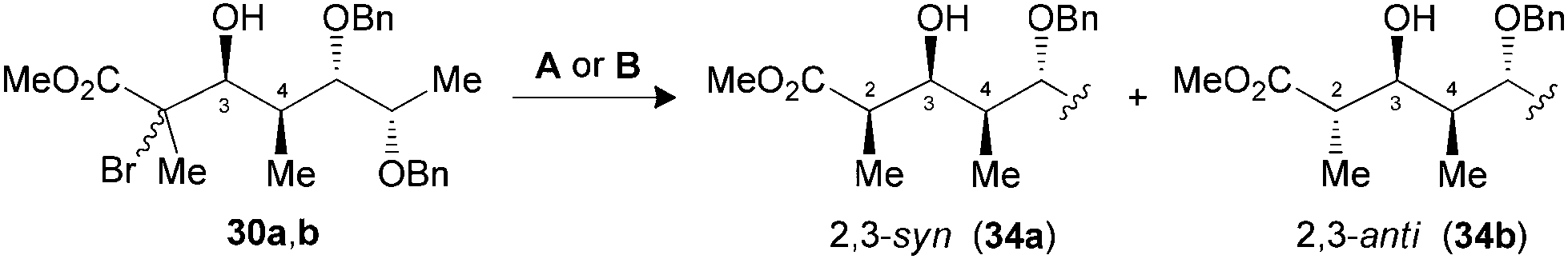
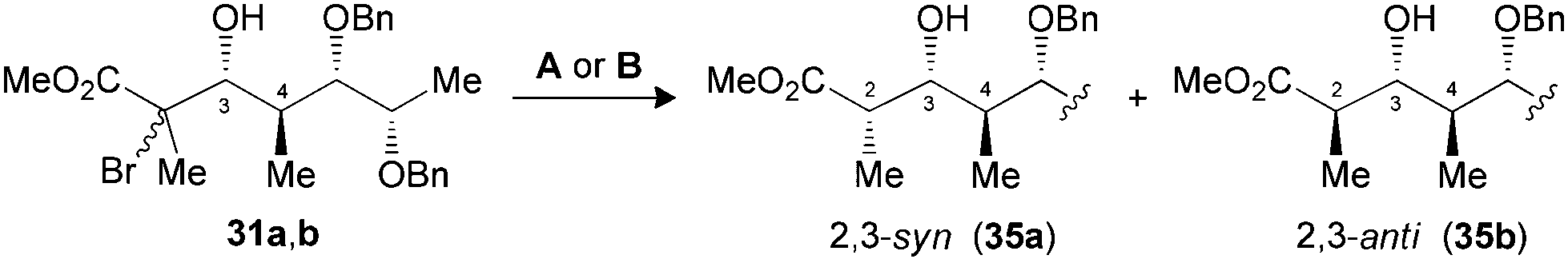
The synthesis of polypropionate units was completed by performing hydrogen transfer reactions on precursors 28–31, precomplexed either with Bu2BOTf (conditions A) or Me3Al (conditions B). Excellent selectivities were observed in favor of 2,3-anti products 32–35b for the reduction of borinate intermediates (Table 4, entries 1, 3, 5 and 7), while the complementary 2,3-syn products 32–35a was achieved exclusively when Me3Al was used as the Lewis acid (entries 2, 4, 6 and 8). For all the propionate motifs considered herein, the outcome of the reaction is dictated by the configuration at C3 and is not influenced by the additional stereocenters distal to the radical, which highlights the robustness of our methodology to accommodate different substrates. Based on the previous study of borinates11 and the DFT analysis of aluminate intermediates herein, we suggest that the steric hindrance provided by the non-hydrogen substituent at C3 is mainly responsible for the excellent selectivities noted for the reduction of propionates and subunits thereof.
| Entry (substrate) | Conditionsa | Yieldb (%) | Ratioc (2,3-syn:2,3-anti) |
|---|---|---|---|
| a Condition A: substrates were pretreated with iPr2NEt (1.5 equiv.) and Bu2BOTf (1.3 equiv.) for 1 h at −78 °C, prior to the addition of Bu3SnH, Et3B and air. Condition B: substrates were pretreated with Me3Al (3.0 equiv.) for 1 h at −78 °C, prior to the addition of Bu3SnH, Et3B and air. b Yields of isolated products. c Product ratios were determined by 1H NMR analysis of the crude reaction mixture. | |||

|
|||
| 1 (28) | A | 72 | 1:>20 (32) |
| 2 (28) | B | 74 | >20:1 (32) |
|
|||
| 3 (29) | A | 89 | 1:>20 (33) |
| 4 (29) | B | 79 | >20:1 (33) |
|
|||
| 5 (30) | A | 83 | 1:>20 (34) |
| 6 (30) | B | 71 | >20:1 (34) |
|
|||
| 7 (31) | A | 78 | 1:>20 (35) |
| 8 (31) | B | 73 | >20:1 (35) |
Stereotriads from L-alanine
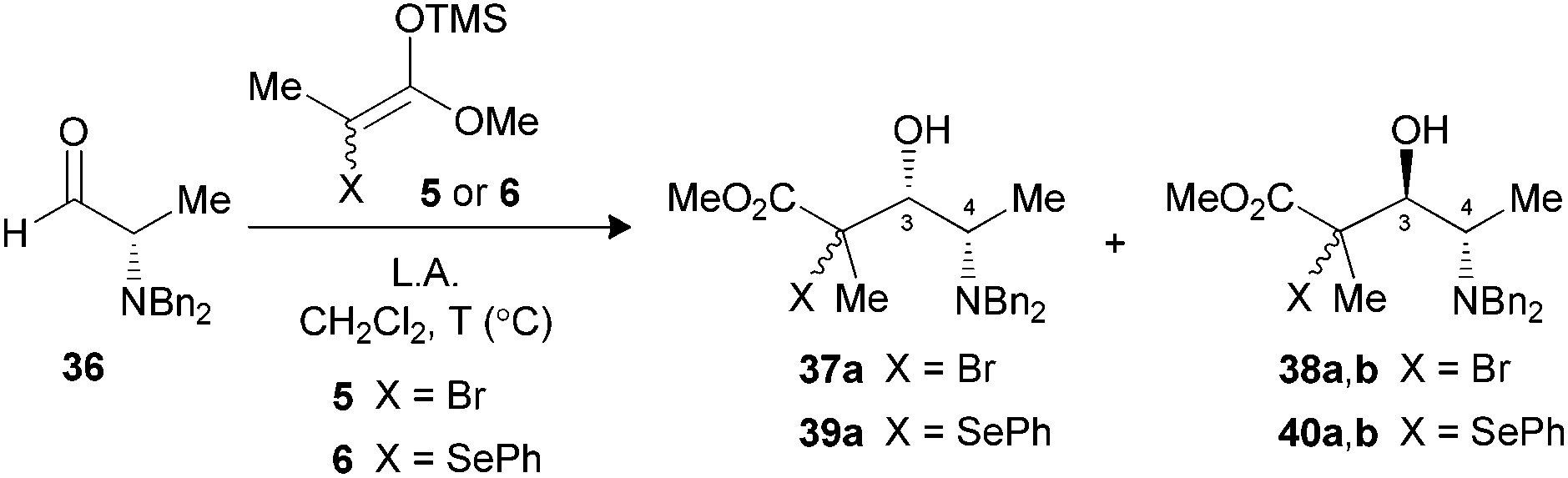
We were also interested to investigate if the methodology could be applied to α-amino aldehydes derived from L-alanine. Our first attempts with N,N-dibenzylaldehyde 36 using different Lewis acids at −78 °C did not allow the formation of the expected product (Table 5, entry 1). Warming the reaction mixture to 0 °C led to the decomposition of the starting material (entry 2). Since no starting aldehyde was recovered, we hypothesized that raising the temperature might have destroyed the aldol product formed, or could have participated in the decomposition of the substrate. In order to examine if the stability of tertiary bromides contributed to the problem, we replaced the bromoenoxysilane 5 with the silylated phenylselenoether 6. This latter silylated nucleophile displayed a reduced reactivity, as compared to their bromo analogues, but the resulting tertiary selenides were more stable.9b When aldehyde 36 was submitted to the same conditions with reagent 6, an excellent selectivity and yield favoring the 3,4-anti isomer 40a,b was observed (entry 3), suggesting that the bromides were indeed a part of the problem at 0 °C. A variety of bidentate Lewis acids were then considered to promote the aldol reaction under Cram-chelate conditions and generate 3,4-syn product 39a. We were surprised to note that Me2AlCl was 3,4-syn selective (entry 4), since this Lewis acid was reported to form bidentate complexes that should provide 3,4-anti products.27 When the aldehyde was precomplexed for two minutes with MgBr2·OEt2 prior to the addition of 6, a moderate selectivity was noted favoring the desired 3,4-syn product (entry 5). Increasing the precomplexation time (entry 6) or changing the order of addition unfortunately led to significant decomposition of the substrate. Seemingly, the steric hindrance caused by benzyl protecting groups increased significantly the energy required to form the bidentate complex.28 Moreover, these results suggest that the transition states under Felkin–Anh or Cram-chelate conditions are very close in energy at that temperature.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | LA (equiv.) | Enolate | T (°C) | Timea (min) | Yieldb (%) | Ratioc (3,4-syn:3,4-anti) |
| a Precomplexation time of the mixture of aldehyde and Lewis acid. b Yields of isolated products. c Product ratios were determined by 1H NMR analysis of the crude reaction mixture. d Lewis acid added to a mixture of aldehyde and enoxysilane (2.0 equiv.). e No product observed. f Aldehyde was pretreated with MgBr2·OEt2, prior to the addition of enoxysilane (2.0 equiv.). | ||||||
| 1 | BF3·OEt2d (1.5) | 5 | −78 | — | —e | — (37,38) |
| 2 | BF3·OEt2d (1.5) | 5 | 0 | — | Decomp. | — (37,38) |
| 3 | BF3·OEt2d (1.5) | 6 | 0 | — | 89 | 1:>20 (39,40) |
| 4 | Me2AlCld (2.5) | 6 | −40 | — | 68 | 1:>20 (39,40) |
| 5 | MgBr2·OEt2f (5.0) | 6 | 0 | 2 | 55 | 4.5:1 (39,40) |
| 6 | MgBr2·OEt2f (5.0) | 6 | 0 | 60 | Decomp. | — (39,40) |
The use of N-CBz-protected alaninal 41 was next envisaged, since this substrate was previously reported to form chelates and give rise to 3,4-syn Mukaiyama adducts.29 When the aldehyde was precomplexed for two minutes with either MgBr2·OEt2 or TiCl4 prior to the addition of 5, modest ratios favoring 3,4-syn products were noted (Table 6, entries 1 and 2). Optimal conditions both in terms of yield and selectivity were obtained with Me2AlCl by increasing the precomplexation time (entries 3 and 4) or using BF3·OEt2 in reverse addition (entry 5), where a ratio of 10:1 favoring the unexpected 3,4-syn product 42a in both cases. These results suggest that an intramolecular hydrogen bond between the amine and the carbonyl had possibly occurred, thus leading to a Cram-chelate like reaction.
|
|
||||
|---|---|---|---|---|
| Entry | LA (equiv.) | Timea (min) | Yieldb (%) | Ratioc42:43 (3,4-syn:3,4-anti) |
| a Precomplexation time of the mixture of aldehyde and Lewis acid. b Yields of isolated products. c Product ratios were determined by 1H NMR analysis of the crude reaction mixture. d The aldehyde was pretreated with a Lewis acid, prior to the addition of enoxysilane (2.0 equiv.). e Lewis acid added to a mixture of aldehyde and enoxysilane (2.0 equiv.). | ||||
| 1 | MgBr2·OEt2d (5.0) | 2 | 69 | 4:1 |
| 2 | TiCl4d (1.1) | 2 | 70 | 2:1 |
| 3 | Me2AlCld (1.2) | 15 | 47 | 6:1 |
| 4 | Me2AlCld (1.2) | 60 | 66 | 10:1 |
| 5 | BF3·OEt2e (1.5) | — | 65 | 10:1 |
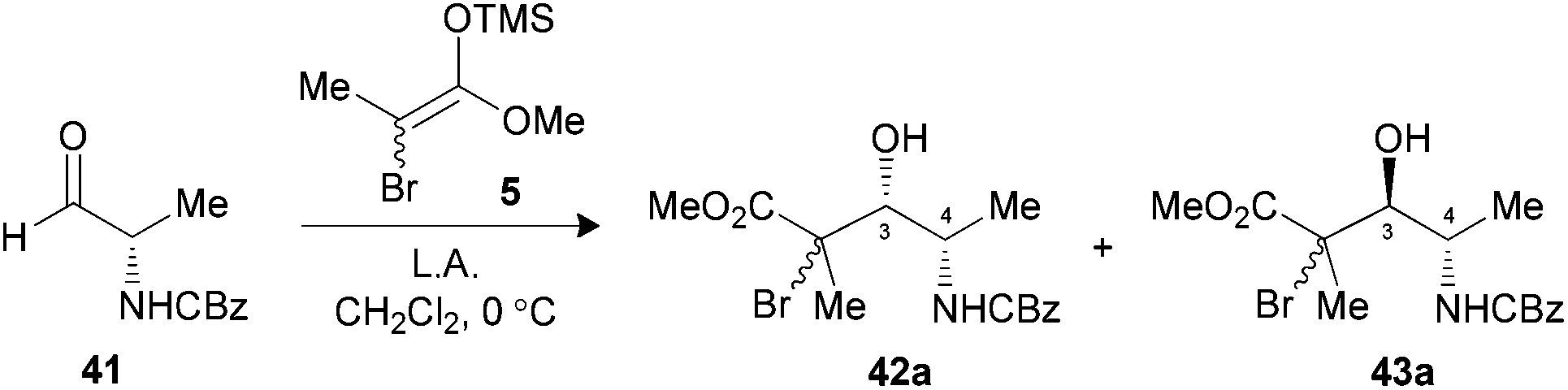
Radical reductions of the N,N-dibenzylated precursors 40a,b were first performed under the conditions reported to provide endocyclic control.10a Unfortunately, the use of Me3Al resulted in recovery of the starting phenylselenide, whereas Me2AlCl afforded only modest levels of diastereoselectivity (Table 7, entries 1 and 2). Changing to the other Lewis acid (MgBr2·OEt2) slightly improved the selectivity of the hydrogen transfer reaction in favor of the 2,3-syn product 44a (entry 3). The best results in terms of selectivity were obtained for the reduction of N-CBz precursor 42a using Me3Al (entry 5). Alternatively, both substrates 40a,b and 42a furnished the exclusive formation of their respective 2,3-anti products 44b and 45b, when the radical reduction was performed on their borinate intermediates (entries 4 and 6).
| Entry (substrate) | LA (equiv.) | Yielda (%) | Ratiob (2,3-syn:2,3-anti) |
|---|---|---|---|
| a Yields of isolated products. b Product ratios were determined by 1H NMR analysis of the crude reaction mixture. c Substrates were pretreated with a Lewis acid for 1.5 h at −78 °C, prior to the addition of Bu3SnH, Et3B and air. d Starting aldehyde recovered. e Addition of iPr2NEt (2.0 equiv.) before the Lewis acid. f Substrates were pretreated with iPr2NEt (1.5 equiv.) and Bu2BOTf for 1 h at −78 °C, prior to the addition of Bu3SnH, Et3B and air. | |||
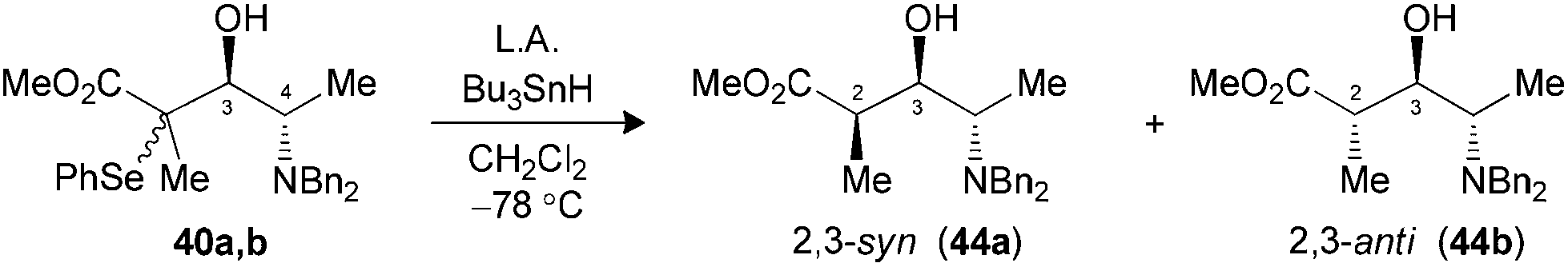
|
|||
| 1 (40) | Me3Alc (3.0) | —d | — (44) |
| 2 (40) | Me2AlClc,e (2.5) | 64 | 2:1 (44) |
| 3 (40) | MgBr2·OEt2c (5.0) | 56 | 5:1 (44) |
| 4 (40) | Bu2BOTff (1.3) | 88 | 1:>20 (44) |
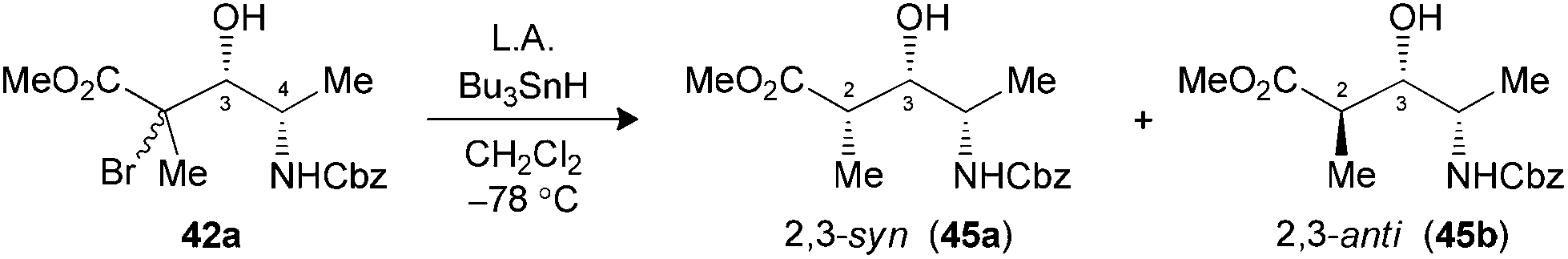
|
|||
| 5 (42) | Me3Al3c (3.0) | 65 | >20:1 (45) |
| 6 (42) | Bu2BOTff (1.3) | 83 | 1:>20 (45) |
In summary, we have reported a Mukaiyama aldol reaction combined with a radical reduction to generate all four stereotriad propionates using the aldehydes derived from L-lactic acid and L-alanine, judiciously tuning the nature of the Lewis acid. This study also emphasized the robustness and the predictability of the hydrogen transfer reactions for carbon-centered free radicals adjacent to an ester. DFT calculations of the transition states involving aluminate intermediates supported the model of the radical reduction under endocyclic control to generate 2,3-syn products. Moreover, we have demonstrated herein that the reaction sequence is applicable iteratively to generate efficiently polypropionates from β,γ-alkoxyaldehydes. The selective formation of Cram-chelate adducts at the aldol step, for the synthesis of the stereopentads, suggest the possibility of a [3.2.1]-type complex by TiCl3OiPr with 2.5 equiv. Finally, our study also underlined that some α-aminoaldehydes behave differently at the aldolization step than other protected alkoxy substrates, thereby requiring minor adjustments in the experimental protocol. The iterative application of the methodology to γ-amino-β-alkoxyaldehydes and further extensions of this work are in progress, and will be reported in due course.
Experimental
Representative procedure for the radical reduction using Me3Al
To a cold (−78 °C) solution of a mixture of bromides 13a,b9a (65 mg, 0.196 mmol) in dry CH2Cl2 (0.1 M) was added a 2 M solution of Me3Al in hexanes (2.5 equiv., 245 μL). After stirring for 1 h at −78 °C, the mixture was successively treated with Bu3SnH (1.5 equiv., 80 μL), a 1 M solution of Et3B in CH2Cl2 (0.2 equiv., 39 μL) and dry air via a syringe (2 mL). Supplementary addition of Et3B solution (0.2 equiv., 39 μL) and air (2 mL) was done every 30 min until the reaction was judged completed by TLC (3–4 h). The reaction mixture was treated with the addition of 1,4-dinitrobenzene (0.2 equiv., 6 mg) and stirring for 15 min at −78 °C. To the mixture was added MeOH dropwise until gas evolution ceased, followed by a saturated aqueous solution of potassium sodium tartrate (Rochelle's salt) and stirring overnight at room temperature, before separation of the organic phase. The aqueous layer was extracted with Et2O (3×) and combined organic fractions were washed (2×) with a saturated aqueous solution of KF and brine, then dried (MgSO4), filtered and concentrated in vacuo.Representative procedure for the Mukaiyama aldol reaction using TiCl3OiPr
To a cold (−78 °C) solution of aldehyde 26 (168 mg, 0.538 mmol) in dry CH2Cl2 (0.1 M) was added a 1 M solution of freshly prepared TiCl3OiPr in CH2Cl2 (2.5 equiv., 1.35 mL), followed by stirring for 5 min at −78 °C. To the mixture was added the silylated enol ether 5 (3 equiv., 300 μL), followed by stirring for 1 h at −78 °C or until the aldehyde was completely consumed, as verified by TLC. The reaction mixture was treated with a saturated aqueous solution of NH4Cl, followed by separation of the organic phase at room temperature. The aqueous layer was extracted with CH2Cl2 (3×) and combined organic fractions were dried (MgSO4), filtered and concentrated in vacuo.Acknowledgements
The authors wish to express their gratitude to the Natural Sciences and Engineering Research Council of Canada (NSERC) for its financial support. Fellowship support (B2) from Fonds Québécois de la Recherche sur la Nature et les Technologies (FQRNT) to F.G. is also gratefully acknowledged.Notes and references
- (a) D. O'Hagan, Nat. Prod. Rep., 1992, 9, 447 RSC; (b) J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380 RSC; (c) B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285 CrossRef CAS PubMed; (d) A. M. P. Koskinen and K. Karisalmi, Chem. Soc. Rev., 2005, 34, 677 RSC.
- For the isolation of the compound, see: (a) H. A. Brooks, D. Gardner, J. P. Poyser and T. J. King, J. Antibiot., 1984, 37, 1501 CrossRef CAS PubMed; (b) U. Gräfe, W. Schade, M. Roth, L. Radics, M. Incze and K. Ujszaszy, J. Antibiot., 1984, 37, 836 CrossRef; For the total syntheses, see: (c) S. J. Danishefsky, H. G. Selnick, R. E. Zelle and M. P. Deninno, J. Am. Chem. Soc., 1988, 110, 4368 CrossRef CAS; (d) M. Defosseux, N. Blanchard, C. Meyer and J. Cossy, J. Org. Chem., 2004, 69, 4626 CrossRef CAS PubMed; (e) K. Komatsu, K. Tanino and M. Miyashita, Angew. Chem., Int. Ed., 2004, 43, 4341 CrossRef CAS PubMed; (f) T. J. Harrison, S. Ho and J. L. Leighton, J. Am. Chem. Soc., 2011, 133, 7308 CrossRef CAS PubMed.
- For the isolation of the compound, see: (a) S. Sakuda, M. Ono, K. Furihata, J. Nakayama, A. Suzuki and A. Isogai, J. Am. Chem. Soc., 1996, 118, 7855 CrossRef CAS; (b) H. Ikeda, N. Matsumori, M. Ono, A. Suzuki, A. Isogai, H. Nagasawa and S. Sakuda, J. Org. Chem., 2000, 65, 438 CrossRef CAS PubMed; For the synthesis of various fragments, see: (c) D. A. Evans, W. C. Trenkle, J. Zhang and J. D. Burch, Org. Lett., 2005, 7, 3335 CrossRef CAS PubMed; (d) S. B. Narute and C. V. Ramana, Tetrahedron, 2013, 69, 1830 CrossRef CAS.
- For the isolation of the compound, see: (a) M. V. Dauria, C. Debitus, L. G. Paloma, L. Minale and A. Zampella, J. Am. Chem. Soc., 1994, 116, 6658 CrossRef CAS; (b) M. V. Dauria, L. G. Paloma, L. Minale, A. Zampella and C. Debitus, J. Nat. Prod., 1994, 57, 1595 CrossRef CAS; For the synthesis of various fragments, see: (c) A. Zampella and M. V. D'Auria, Tetrahedron: Asymmetry, 2001, 12, 1543 CrossRef CAS; (d) W. S. Yu, Y. Zhang and Z. D. Jin, Org. Lett., 2001, 3, 1447 CrossRef CAS PubMed; (e) I. Paterson and A. C. Mackay, Synlett, 2004, 1359 CrossRef CAS; For a total synthesis, see: (f) M. Tortosa, N. A. Yakelis and W. R. Roush, J. Am. Chem. Soc., 2008, 130, 2722 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed.
- For examples of different approaches to access polypropionates, see: (a) S. Danishefsky and D. F. Harvey, J. Am. Chem. Soc., 1985, 107, 6647 CrossRef CAS; (b) H. C. Brown and K. S. Bhat, J. Am. Chem. Soc., 1986, 108, 293 CrossRef CAS; (c) W. R. Roush, M. A. Adam, A. E. Walts and D. J. Harris, J. Am. Chem. Soc., 1986, 108, 3422 CrossRef CAS; (d) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2012, 134, 3925 CrossRef CAS PubMed; (e) M. Miyashita, M. Hoshino and A. Yoshikoshi, J. Org. Chem., 1991, 56, 6483 CrossRef CAS; (f) I. Paterson and J. A. Channon, Tetrahedron Lett., 1992, 33, 797 CrossRef CAS; (g) D. A. Evans, M. G. Yang, M. J. Dart, J. L. Duffy and A. S. Kim, J. Am. Chem. Soc., 1995, 117, 9598 CrossRef CAS; (h) N. F. Jain, N. Takenaka and J. S. Panek, J. Am. Chem. Soc., 1996, 118, 12475 CrossRef CAS; (i) J. A. Marshall, Chem. Rev., 1996, 96, 31 CrossRef CAS PubMed; (j) J. A. Marshall, Chem. Rev., 2000, 100, 3163 CrossRef CAS PubMed; (k) O. Arjona, R. Menchaca and J. Plumet, J. Org. Chem., 2001, 66, 2400 CrossRef CAS PubMed; (l) M. Lautens, J. T. Colucci, S. Hiebert, N. D. Smith and G. Bouchain, Org. Lett., 2002, 4, 1879 CrossRef CAS PubMed; (m) R. Tirado, G. Torres, W. Torres and J. A. Prieto, Tetrahedron Lett., 2005, 46, 797 CrossRef CAS; (n) D. E. Ward, Chem. Commun., 2011, 47, 11375 RSC and references therein; (o) H. Kim, S. Ho and J. L. Leighton, J. Am. Chem. Soc., 2011, 133, 6517 CrossRef CAS PubMed; For recent reviews of the different methodologies, see: (p) J. Li and D. Menche, Synthesis, 2009, 2293 CAS; (q) M. Turks, S. Laclef and P. Vogel, in Stereoselective Synthesis of Drugs and Natural Products, John Wiley & Sons, Inc., 2013, pp. 271–318 Search PubMed.
- (a) H. C. Brown, K. S. Bhat and R. S. Randad, J. Org. Chem., 1989, 54, 1570 CrossRef CAS; (b) J. S. Panek and R. Beresis, J. Org. Chem., 1993, 58, 809 CrossRef CAS; (c) J. A. Marshall and H. R. Chobanian, J. Org. Chem., 2000, 65, 8357 CrossRef CAS PubMed; (d) J. G. Solsona, J. Nebot, P. Romea and F. Urpi, J. Org. Chem., 2005, 70, 6533 CrossRef CAS PubMed; (e) S. Díaz-Oltra, J. Murga, E. Falomir, M. Carda, G. Peris and J. A. Marco, J. Org. Chem., 2005, 70, 8130 CrossRef PubMed; For recent applications of the methodologies, see: (f) L. C. Dias and M. A. B. Ferreira, J. Org. Chem., 2012, 77, 4046 CrossRef CAS PubMed; (g) A. Gille and M. Hiersemann, Org. Lett., 2010, 12, 5258 CrossRef CAS PubMed; (h) R. Villa, A. L. Mandel, B. D. Jones, J. J. La Clair and M. D. Burkart, Org. Lett., 2012, 14, 5396 CrossRef CAS PubMed; (i) I. E. Wrona, A. Gozman, T. Taldone, G. Chiosis and J. S. Panek, J. Org. Chem., 2010, 75, 2820 CrossRef CAS PubMed.
- (a) J. S. Panek and P. Liu, Tetrahedron Lett., 1997, 38, 5127 CrossRef CAS; (b) A. Giordano, A. Spinella and G. Sodano, Tetrahedron: Asymmetry, 1999, 10, 1851 CrossRef CAS; (c) I. Paterson and A. C. Mackay, Tetrahedron Lett., 2001, 42, 9269 CrossRef CAS; (d) N. A. Yakelis and W. R. Roush, J. Org. Chem., 2003, 68, 3838 CrossRef CAS PubMed; (e) Q. B. Pan, B. L. Zou, Y. J. Wang and D. W. Ma, Org. Lett., 2004, 6, 1009 CrossRef CAS PubMed; (f) J. A. Marshall and J. J. Mulhearn, Org. Lett., 2005, 7, 1593 CrossRef CAS PubMed; (g) S. Hanessian, Y. H. Hou, M. Bayrakdarian and M. Tintelnot-Blomley, J. Org. Chem., 2005, 70, 6735 CrossRef CAS PubMed; For a review, see: (h) M. T. Reetz, Chem. Rev., 1999, 99, 1121 CrossRef CAS PubMed; For a recent application, see: (i) C. J. Leitheiser, K. L. Smith, M. J. Rishel, S. Hashimoto, K. Konishi, C. J. Thomas, C. H. Li, M. M. McCormick and S. M. Hecht, J. Am. Chem. Soc., 2003, 125, 8218 CrossRef CAS PubMed.
- (a) Y. Guindon, K. Houde, M. Prévost, B. Cardinal-David, S. R. Landry, B. Daoust, M. Bencheqroun and B. Guérin, J. Am. Chem. Soc., 2001, 123, 8496 CrossRef CAS PubMed; (b) Y. Guindon, M. Prévost, P. Mochirian and B. Guérin, Org. Lett., 2002, 4, 1019 CrossRef CAS PubMed; (c) P. Mochirian, F. Godin, I. Katsoulis, I. Fontaine, J.-F. Brazeau and Y. Guindon, J. Org. Chem., 2011, 76, 7654 CrossRef CAS PubMed.
- (a) Y. Guindon, J. F. Lavallée, M. Llinas-Brunet, G. Horner and J. Rancourt, J. Am. Chem. Soc., 1991, 113, 9701 CrossRef CAS; (b) Y. Guindon and J. Rancourt, J. Org. Chem., 1998, 63, 6554 CrossRef CAS; (c) J. P. Bouvier, G. Jung, Z. P. Liu, B. Guérin and Y. Guindon, Org. Lett., 2001, 3, 1391 CrossRef CAS PubMed.
- F. Godin, M. Prévost, S. I. Gorelsky, P. Mochirian, M. Nguyen, F. Viens and Y. Guindon, Chem. – Eur. J., 2013, 19, 9308 CrossRef CAS PubMed.
- (a) Y. Guindon, B. Guérin, J. Rancourt, C. Chabot, N. Mackintosh and W. W. Ogilvie, Pure Appl. Chem., 1996, 68, 89 CrossRef CAS; (b) B. Guérin, C. Chabot, N. Mackintosh, W. W. Ogilvie and Y. Guindon, Can. J. Chem., 2000, 78, 852 CrossRef.
- For a detailed article on 5-membered ring chelates in Mukaiyama aldol reactions, see: M. T. Reetz, Acc. Chem. Res., 1993, 26, 462 CrossRef CAS.
- (a) D. A. Evans, S. J. Siska and V. J. Cee, Angew. Chem., Int. Ed., 2003, 42, 1761 CrossRef CAS PubMed; (b) D. A. Evans, V. J. Cee and S. J. Siska, J. Am. Chem. Soc., 2006, 128, 9433 CrossRef CAS PubMed.
- (a) B. Giese, W. Damm, F. Wetterich and H. G. Zeitz, Tetrahedron Lett., 1992, 33, 1863 CrossRef CAS; (b) K. Durkin, D. Liotta, J. Rancourt, J. F. Lavallée, L. Boisvert and Y. Guindon, J. Am. Chem. Soc., 1992, 114, 4912 CrossRef CAS; (c) B. Giese, W. Damm, F. Wetterich, H. G. Zeitz, J. Rancourt and Y. Guindon, Tetrahedron Lett., 1993, 34, 5885 CrossRef.
- For a complete description of the computational methodology, see the ESI.‡.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- The geometry of the enol radical (E or Z) is indicated for the different conformations.
- For a description and application of the natural bonding orbital (NBO) analysis, see: (a) E. D. Glendening, C. R. Landis and F. Weinhold, WIREs: Comp. Mol. Sci., 2012, 2, 1 CrossRef CAS; (b) F. Weinhold, J. Comput. Chem., 2012, 33, 2440 CrossRef CAS PubMed.
- This value corresponds to the sum of all contributions (nO1 → n*Al) for α-spin orbitals, as calculated by the NBO analysis. See the ESI.‡.
- The activation strain model separates the activation energy of a transition state (ΔE‡) into the sum of the strain energy (ΔE‡d) and the interaction energy (ΔE‡int), such that ΔE‡ = ΔE‡d[radical] + ΔE‡d[hydride] + ΔE‡int in the model studied herein. For a description of the model, see: (a) A. Krapp, F. M. Bickelhaupt and G. Frenking, Chem. – Eur. J., 2006, 12, 9196 CrossRef CAS PubMed; (b) W. J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118 RSC; (c) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646 CrossRef CAS PubMed; (d) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187 CrossRef CAS PubMed.
- The methoxy surrogate was employed in the calculations.
- (a) Y. Guindon, C. Yoakim, V. Gorys, W. W. Ogilvie, D. Delorme, J. Renaud, G. Robinson, J. F. Lavallée, A. Slassi, G. Jung, J. Rancourt, K. Durkin and D. Liotta, J. Org. Chem., 1994, 59, 1166 CrossRef CAS; (b) Y. Guindon, A. Slassi, J. Rancourt, G. Bantle, M. Bencheqroun, L. Murtagh, E. Ghiro and G. Jung, J. Org. Chem., 1995, 60, 288 CrossRef CAS; (c) Y. Guindon, A.-M. Faucher, É. Bourque, V. Caron, G. Jung and S. R. Landry, J. Org. Chem., 1997, 62, 9276 CrossRef CAS; (d) Y. Guindon, Z. P. Liu and G. Jung, J. Am. Chem. Soc., 1997, 119, 9289 CrossRef CAS.
- For a description of the polarizable continuum model (PCM) of solvation, see: (a) M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43 CrossRef CAS; (b) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed; (c) J. Tomasi, WIREs: Comp. Mol. Sci., 2011, 1, 855 CrossRef CAS.
- (a) Y. Guindon and J.-F. Brazeau, Org. Lett., 2004, 6, 2599 CrossRef CAS PubMed; (b) J.-F. Brazeau, P. Mochirian, M. Prévost and Y. Guindon, J. Org. Chem., 2009, 74, 64 CrossRef CAS PubMed.
- The terms [3.3.1]- and [3.2.1]-complexes for the description of titanium intermediates is derived from the IUPAC nomenclature of saturated alicyclic systems.
- D. A. Evans, B. D. Allison, M. G. Yang and C. E. Masse, J. Am. Chem. Soc., 2001, 123, 10840 CrossRef CAS PubMed.
- M. T. Reetz, Pure Appl. Chem., 1992, 64, 351 CrossRef CAS.
- S. Kiyooka, K. Suzuki, M. Shirouchi, Y. Kaneko and S. Tanimori, Tetrahedron Lett., 1993, 34, 5729 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, physical characterization and NMR spectra for all new compounds. For DFT calculations, the optimized geometries, energies and Cartesian coordinates for all ground and transition states structures are provided. CCDC 1001298 and 1001299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00142g |
| This journal is © the Partner Organisations 2014 |