Synthesis of β-methyl-α-methylene-γ-butyrolactone from biorenewable itaconic acid†
Ravikumar R.
Gowda
and
Eugene Y.-X.
Chen
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523-1872, USA. E-mail: eugene.chen@colostate.edu
First published on 5th March 2014
Abstract
An efficient, high-yielding route to β-methyl-α-methylene-γ-butyrolactone (βMMBL)—a monomer for the production of high-performance engineering bioplastics—from biorenewable and inexpensive itaconic acid has been developed. The monomer prepared by this new route is of high purity as isolated without purification, as evidenced by NMR as well as small and large-scale polymerization tests.
Green chemistry principle #7—Use of Renewable Feedstocks—states: a raw material or feedstock (for fine chemicals or materials) should be renewable rather than depleting whenever technically and economically practicable.1 Currently, however, depleting fossil reserves are predominant resources employed for the manufacture of synthetic polymer materials that are essential to modern life and the global economy. With fossil fuels rapidly depleting, increasing attention has been paid to using bio-based or naturally occurring monomers for the production of polymers.2 In this context, plant biomass-derived renewable methylene butyrolactones, such as the parent α-methylene-γ-butyrolactone (MBL)3 and its γ-methyl derivative, γ-methyl-α-methylene-γ-butyrolactone (γMMBL),4 are of particular interest in exploring the prospects of replacing the petroleum-based acrylic monomers for the production of specialty chemicals and polymers.5 Not only are such monomers biorenewable, their derived polymers, poly(α-methylene-γ-butyrolactone) (PMBL) and poly(γ-methyl-α-methylene-γ-butyrolactone) (PγMMBL), Scheme 1, also exhibit enhanced thermal, physical and mechanical properties, as compared to petroleum-based linear acrylic polymers. For example, PMBL has good durability, optical properties, a high refractive index of 1.540, and a high glass-transition temperature (Tg) of 195 °C (for atactic polymer).5,6 The Tg of PγMMBL is even higher, up to 227 °C (for atactic polymer).7,8
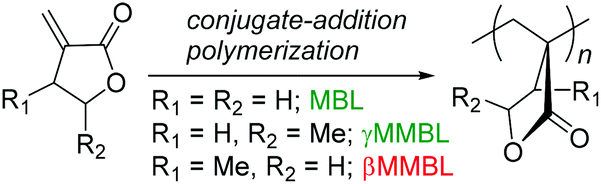 | ||
| Scheme 1 The three most common members of the α-methylene-γ-butyrolactone family and their derived renewable polymers as bioplastics. | ||
A more intriguing and attractive member of the α-methylene-γ-butyrolactone family is the β-methyl derivative, β-methyl-α-methylene-γ-butyrolactone (βMMBL). Its derived atactic polymer by free-radical polymerization,9 PβMMBL, has been shown to display much superior materials properties, such as excellent optical properties and heat resistance as well as a high refractive index, as compared to linear acrylic polymers, thus rendering PβMMBL and its copolymers excellent candidates as plastic optical fibers.10 Furthermore, metal-catalyzed stereospecific coordination polymerization of βMMBL leads to stereo-defect-free11 or highly stereoregular12,13 thermally robust PβMMBL materials, which exhibit remarkable solvent resistance (resistant to all common organic solvents at RT as well as under refluxing conditions) and an extremely high Tg of up to 300 °C, thus qualifying as high-performance engineering plastics. Unfortunately, the currently adopted synthetic route9,12,13 to the βMMBL monomer (Scheme 2) is neither sustainable nor efficient and economical. It uses petroleum-based starting materials (acetaldehyde and methyl acrylate) and stoichiometric quantities of toxic reagents in several steps, involves long reaction times (∼15 days in total), and requires tedious separation and purification procedures (distillation and column chromatography), giving rise to the monomer in low overall yield (typically ∼20%). Overall, the current route to βMMBL is laborious and time-consuming, as well as non-sustainable and economical. As such, future application and development of these otherwise highly intriguing βMMBL-based polymeric materials are severely hampered as a result of the limited availability of the monomer. Accordingly, the central objective of this research was to develop an efficient, user friendly, and economical synthesis of the βMMBL monomer starting from a biorenewable feedstock, altogether designing a more sustainable or greener process. Ultimately, the new route should produce this important monomer in large commercial quantities suitable for large-scale polymerization reactions.
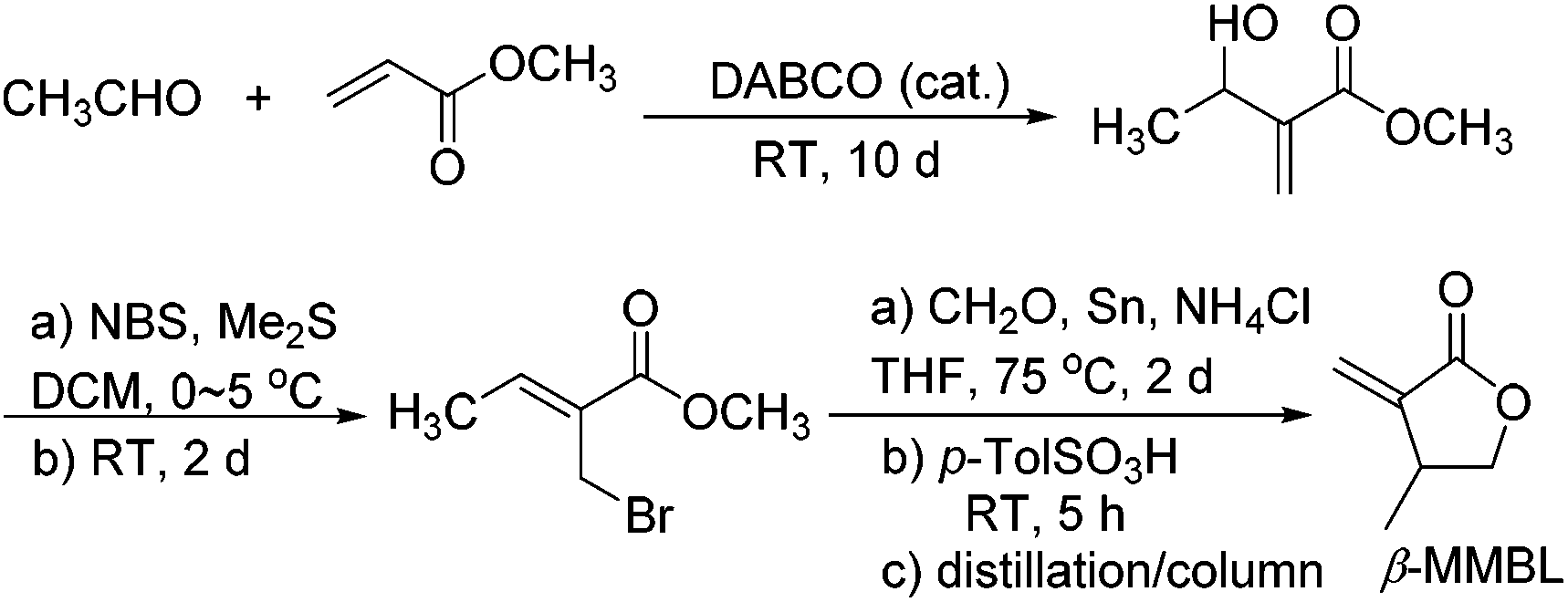 | ||
| Scheme 2 Current synthetic route to βMMBL, which takes ∼15 days and affords the monomer in a low overall yield of ∼20%.9,12,13 | ||
Our approach en route to the synthesis of the βMMBL monomer has focused on the use of the sugar-based itaconic acid (IA), or methylenesuccinic acid, because IA was identified by a DOE study as one of the twelve molecules that are potential new platform chemicals derived from biomass sugars.14 IA has also been utilized as a precursor (to the anhydride monomer) for the copolymerization of anhydrides and epoxides.15 IA is produced industrially by fermentation of carbohydrates such as glucose with the fungus Aspergillus terreus.16 Moreover, it is rather inexpensive (currently priced at about $30 per kilogram) and commercially available in large quantities.
The first sustainable route, outlined in Scheme 3, uses molecular hydrogen gas and consists of three steps. The first step involves reduction of IA with ruthenium(III) acetylacetonate, Ru(acac)3, and a bidentate phosphine ligand, 1,4 bis(diphenylphosphino)-butane (dppb), in dry THF at 195 °C, under 1450 psi H2 pressure for 20 h, which resulted in a mixture of γ-butyrolactone regio-isomers, 2-methyl-γ-butyrolactone and 3-methyl-γ-butyrolactone (the desired isomer) in a 1.15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, plus 2-methylbutane-1,4-diol, in 74% combined yield. This reduction step was adapted from the recently reported preparation of 3-methyltetrahydrofuran by Geilen and co-workers.17 The use of the dry THF is necessary to produce the lactones. However, the reaction is not regio- and chemoselective, producing the two lactone isomers and also reducing partially both the carboxylic groups of IA. As such, the yield of 3-methyl-γ-butyrolactone desired for the βMMBL preparation was less than 35%, and it was also inseparable by typical techniques (vide infra). The reaction was also tested with itaconic anhydride instead of IA, but the lactone formation was not observed. We also tested reduction of itaconic anhydride with RANEY® nickel to produce lactones18 but without success in our hands. Fractional distillation of the regioisomer mixture did not noticeably alter the ratio; hence, this mixture, without further purification, was directly treated with sodium methoxide and diethyl oxalate in the next step to generate methyl oxalyl β-methyl-γ-butyrolactone sodium salt,18 followed by work-up with water and tbutylmethyl ether, the aqueous layer of which was separated and used in the final step without any purification. The resulting sodium salt was treated with potassium carbonate and aqueous formaldehyde in tbutylmethyl ether to afford βMMBL18 after purifications using distillation followed by column chromatography. The overall yield was found to be low, only 14.5% based on the starting IA. Overall, although this route employs the biomass-based IA and the unit operation is reduced to about 2 days, the low overall yield and the use of the high pressure and expensive H2 limit its applicability in large-scale production of this monomer.
:1 ratio, plus 2-methylbutane-1,4-diol, in 74% combined yield. This reduction step was adapted from the recently reported preparation of 3-methyltetrahydrofuran by Geilen and co-workers.17 The use of the dry THF is necessary to produce the lactones. However, the reaction is not regio- and chemoselective, producing the two lactone isomers and also reducing partially both the carboxylic groups of IA. As such, the yield of 3-methyl-γ-butyrolactone desired for the βMMBL preparation was less than 35%, and it was also inseparable by typical techniques (vide infra). The reaction was also tested with itaconic anhydride instead of IA, but the lactone formation was not observed. We also tested reduction of itaconic anhydride with RANEY® nickel to produce lactones18 but without success in our hands. Fractional distillation of the regioisomer mixture did not noticeably alter the ratio; hence, this mixture, without further purification, was directly treated with sodium methoxide and diethyl oxalate in the next step to generate methyl oxalyl β-methyl-γ-butyrolactone sodium salt,18 followed by work-up with water and tbutylmethyl ether, the aqueous layer of which was separated and used in the final step without any purification. The resulting sodium salt was treated with potassium carbonate and aqueous formaldehyde in tbutylmethyl ether to afford βMMBL18 after purifications using distillation followed by column chromatography. The overall yield was found to be low, only 14.5% based on the starting IA. Overall, although this route employs the biomass-based IA and the unit operation is reduced to about 2 days, the low overall yield and the use of the high pressure and expensive H2 limit its applicability in large-scale production of this monomer.
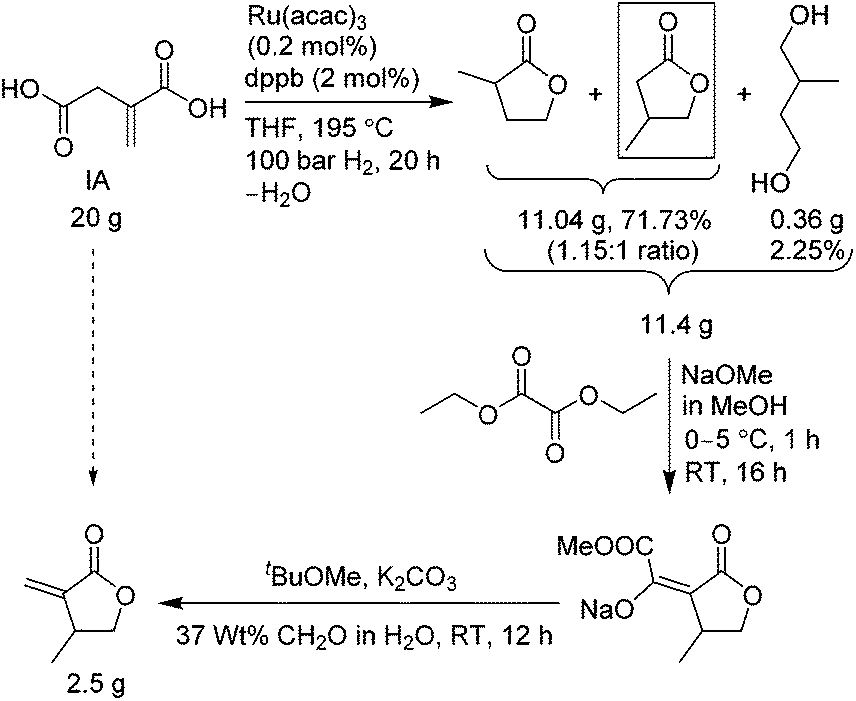 | ||
| Scheme 3 Synthesis of βMMBL from itaconic acid (IA) with molecular hydrogen gas. | ||
The second greener route, outlined in Scheme 4, was designed to improve the overall yield and avoid using H2 by introducing regioselective reduction of the carboxyl group of IA and catalytic hydrogen transfer reduction. Specifically, this route consists of the following six steps. The first step is a simple regioselective partial esterification reaction of IA with methanol in the presence of a catalytic amount of acetyl chloride,19 affording the corresponding monomethyl ester 1 as colorless crystals in 80% isolated yield from a 100 g scale reaction. The second step involves catalytic hydrogen transfer reduction20 (as an alternative method to the classical hydrogenation reaction) of 1 with the Pd/C catalyst and ammonium formate, the latter of which acts as the hydrogen source in methanol. Under the conditions outlined in Scheme 4, 1 was selectively hydrogenated to 219,21 as a viscous oil in 95% crude yield. The amount of ammonium formate was varied from 1 to 10 equiv.; although all the ratios tested produced the product 2, the use of 10 equiv. of ammonium formate yielded the product with the least amount of impurities. Noteworthy here is that the Pd/C catalyst can be recycled by simple filtration and the excess ammonium formate added can be recycled by precipitating in chloroform. The hydrogenation reaction was tested with the recycled Pd/C and ammonium formate, and the product 2 was formed in comparable yields.
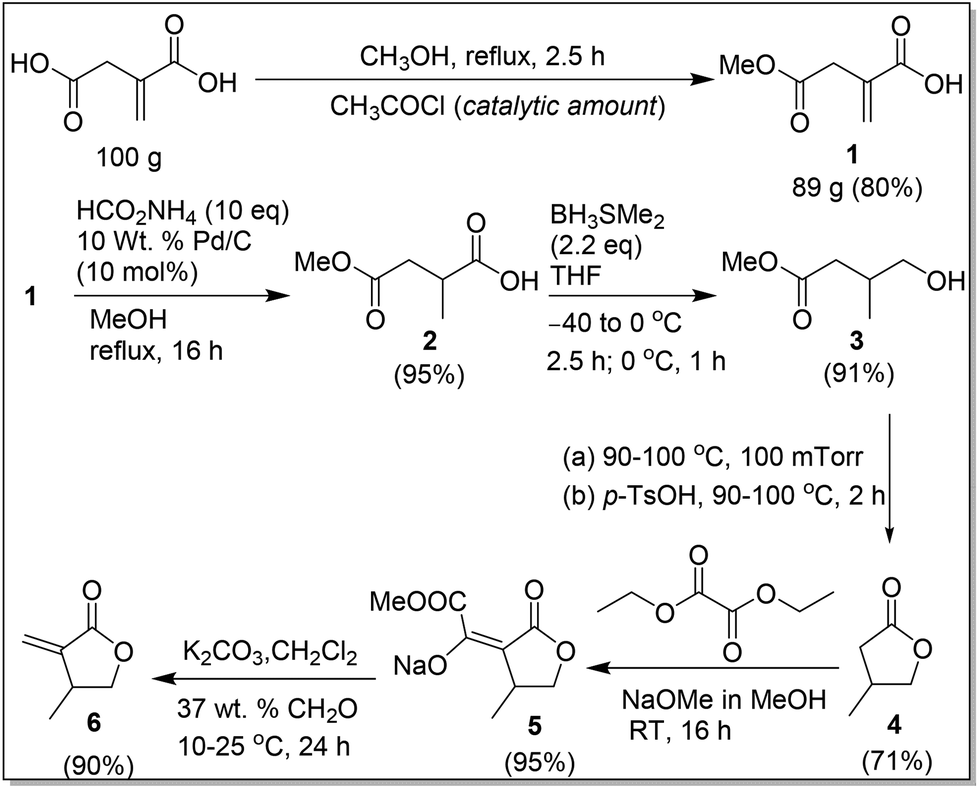 | ||
| Scheme 4 Greener synthesis of βMMBL from itaconic acid. | ||
The third step of the second route is reduction of the carboxylic group of 2 to a primary alcohol with the BH3·SMe2 complex in dry THF according to the conditions outlined in Scheme 4. This reaction was quenched with methanol, followed by a simple work-up with removal of methanol to afford 319b,21b,22 as a colorless liquid in 95% yield; this crude product was used in the next step without further purification. This reaction was tested with 1 and 1.2 equiv. of BH3·SMe2 under otherwise identical conditions; however only 30% conversion of 2 was achieved. The reaction was also tested with 2.0 M of BH3·SMe2 in THF according to the conditions outlined in Scheme 4 and an additional 6 h stirring with a gradual increase of temperature to RT, which afforded 61% of 3 and, interestingly, also 39% of 4.21b However, we were not successful in producing 4 exclusively, upon stirring the reaction mixture at RT for an extended period, which in turn resulted in the formation of byproducts.
The fourth step involves distillation of crude 3 to give a mixture of 3 (35%) and 4 (65%). This mixture was further treated with a catalytic amount of p-TsOH·H2O, followed by distillation to afford 4 exclusively in 71% yield; removal of the byproduct methanol is mandatory, prior to distillation at 100 mTorr, in order to give the pure 4. The direct treatment of the crude 3 with a catalytic amount of p-TsOH·H2O resulted in a mixture of 52% of 3, 48% of 4, and methanol; when methanol was removed from this mixture prior to vacuum distillation, a mixture of 18% of 3 and 82% of 4 was formed.
The fifth step is isolation of intermediate 5, which is essential for producing the pure βMMBL directly. Compound 4 was treated with diethyl oxalate and sodium methoxide in methanol to afford 5 in 95% isolated yield after washing with hexanes. The final step is condensation of formaldehyde with 5 in water and dichloromethane according to the conditions outlined in Scheme 4. This step was straightforward, and a simple work-up procedure, consisting of separation of the organic layer and removal of all volatiles under reduced pressure, directly resulted in the formation of the spectroscopically pure βMMBL (6) (by 1H NMR)23 as a colorless liquid in 90% isolated yield. Overall, the second greener route not only starts with the renewable, inexpensive building block IA, operates in a short unit operation time (∼2.5 days), and avoids the use of the expensive and high-pressure H2, but also it is a relatively high-yielding process, with an overall yield of 53% based on the starting IA, and produces βMMBL in high purity without the need for extensive purification procedures such as distillation and column chromatography. Hence, it can be considered as a more sustainable, economical, and user friendly route to βMMBL.
To examine whether the βMMBL monomer synthesized from the second greener route outlined in Scheme 4 is suitable for both small and large-scale polymerization reactions or not, we have performed several polymerization runs with βMMBL, along with γMMBL as a control. For this purpose, we chose a recently developed polymerization method of organocatalytic conjugate-addition polymerization24,25 mediated by N-heterocyclic carbenes in DMF, particularly 1,3-di-tert-butylimidazolin-2-ylidene (ItBu). The polymerization results are summarized in Table 1. It can be clearly seen from the table that polymerization of γMMBL in the presence of even a small amount of ItBu (100 ppm) was rapid (in 5 min) and quantitative (99% isolated polymer yield), regardless of the monomer scale employed herein (from 0.525 g in run 1 to 10 g in run 2 to 30 g in run 3), producing high molecular weight PγMMBL (Mn = 7.79 × 104 to 1.08 × 105 g mol−1). Similarly, quantitative conversion of βMMBL (0.525 g scale in run 4 and 10 g scale in run 5) by ItBu (100 ppm) was achieved, although the reaction time was extended to 15 to 17 min. The observed slower rate of the βMMBL polymerization relative to the γMMBL polymerization can be attributed to the difference in monomer sterics, with βMMBL being bulkier as the methyl is placed next to the double bond where the conjugate-addition event takes place. Overall, these polymerization results demonstrated the suitability of the βMMBL monomer prepared by the current new greener route for both small and large-scale polymerization reactions.
| Run no. | Catalyst | Monomer | Monomer scale | Monomer/catalyst | Solvent | Amount of solvent | Time (min) | Yield (%) | M n (kg mol−1) | PDIb (Mw/Mn) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: ItBu was added to the monomer at RT; n.d. = not determined. b Number-average molecular weight (Mn) and polydispersity index (PDI = Mw/Mn) determined by gel-permeation chromatography (GPC) relative to poly(methyl methacrylate) standards. | ||||||||||
| 1 | ItBu | γMMBL | 0.525 g | 10000 |
DMF | 4.5 mL | 5 | >99 | 77.924 | 1.9724 |
| 2 | ItBu | γMMBL | 10 g | 10000 |
DMF | 45 mL | 4 | >99 | 108 | 2.25 |
| 3 | ItBu | γMMBL | 30 g | 10000 |
DMF | 70 mL | 4 | >99 | n.d. | n.d. |
| 4 | ItBu | βMMBL | 0.525 g | 10000 |
DMF | 4.5 mL | 17 | >99 | n.d. | n.d. |
| 5 | ItBu | βMMBL | 10 g | 10000 |
DMF | 45 mL | 15 | 99 | n.d. | n.d. |
In summary, we have developed an efficient and economical route to the βMMBL monomer using the inexpensive, biomass-derived itaconic acid. This new route is readily scalable, thus offering the important βMMBL monomer in large commercial quantities suitable for both small and large-scale polymerization reactions. As compared to the existing acetaldehyde + methyl acrylate route to this monomer, the new route reported herein not only uses the renewable, inexpensive building block IA and operates in a much shorter unit operation time period of ∼2.5 days (vs. 15 days), but also it is a much higher yielding process, with an overall yield of 53% based on the starting IA (vs. 20%), and produces βMMBL in high purity without the need for extensive purification procedures. Hence, this new route can be justified as a more sustainable, economical, and user friendly process. This new greener process could also be potentially extended to the synthesis of enantiomerically pure βMMBL by controlling stereochemistry of intermediate 2 through asymmetric hydrogen transfer reduction; this route could then replace the current, multistep process starting from the expensive, enantiomerically pure methyl β-hydroxyisobutyrate.26 A study addressing this aspect of asymmetric monomer synthesis is currently underway.
Acknowledgements
This work was supported by the US National Science Foundation (NSF-1300267) and the Colorado Office of Economic Development and International Trade through a BDEGP grant (13BGF-27) administrated by CSURF-CSU Ventures.Notes and references
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- Selected reviews: (a) M. J.-L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Synthesis of biodegradable polymers from renewable resources, Polym. Chem., 2012, 3, 836–851 RSC; (b) G. W. Coates and M. A. Hillmyer, A virtual issue on “Polymers from Renewable Resources”, Macromolecules, 2009, 42, 7987–7989 CrossRef CAS; (c) A. Gandini, Polymers from Renewable Resources: a Challenge for the Future of Macromolecular Materials, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS; (d) C. K. Williams and M. A. Hillmyer, Polymers from Renewable Resources: A Perspective for a Special Issue of Polymer Reviews, Polym. Rev., 2008, 48, 1–10 CrossRef CAS; (e) M. A. R. Meier, J. O. Metzger and U. S. Schubert, Plant Oil Renewable Resources as Green alternatives in Polymer Science, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- (a) R. R. A. Kitson, A. Millemaggi and R. J. K. Taylor, Angew. Chem., Int. Ed., 2009, 48, 9426–9451 CrossRef CAS PubMed; (b) H. M. R. Hoffmann and J. Rabe, Angew. Chem., Int. Ed. Engl., 1985, 24, 94–110 CrossRef.
- (a) L. E. Manzer, ACS Symp. Ser., 2006, 921, 40–51 CrossRef CAS PubMed; (b) L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS PubMed.
- (a) R. R. Gowda and E. Y.-X. Chen, Encyclo. Polym. Sci. Tech., 2013 DOI:10.1002/0471440264.pst606; (b) S. Agarwal, Q. Jin and S. Maji, ACS Symp. Ser., 2012, 1105, 197–212 CrossRef CAS PubMed; (c) R. Mullin, C&E News, 2004, 82(45), 29–37 Search PubMed.
- (a) J. E. Pickett and Q. Ye, to General Electric Company, US Pat0122625 A1, 2007 Search PubMed; (b) C. J. Brandenburg, to E. I. du Pont de Nemours and Company, US Pat6841627 B2, 2005 Search PubMed; (c) M. K. Akkapeddi, Macromolecules, 1979, 12, 546–551 CrossRef CAS.
- Y. Hu, X. Xu, Y. Zhang, Y. Chen and E. Y.-X. Chen, Macromolecules, 2010, 43, 9328–9336 CrossRef CAS.
- G. M. Miyake, S. E. Newton, W. R. Mariott and E. Y.-X. Chen, Dalton Trans., 2010, 39, 6710–6718 RSC.
- C. U. Pittman Jr. and H. Lee, J. Polym. Sci., Polym. Chem., 2003, 41, 1759–1777 CrossRef.
- K. Sakashita, K. Iwasaka, Y. Tsukamoto and A. Aoyagi, EP1834968 A1, 2007 Search PubMed.
- X. Chen, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2012, 134, 7278–7281 CrossRef CAS PubMed.
- Y. Hu, G. M. Miyake, B. Wang, D. Cui and E. Y.-X. Chen, Chem.–Eur. J., 2012, 18, 3345–3354 CrossRef CAS PubMed.
- Y. Hu, X. Wang, Y. Chen, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Organometallics, 2013, 32, 1459–1465 CrossRef CAS.
- Top Value Added Chemicals from Biomass, ed. T. Werpy and G. Petersen, U.S. Department of Energy (DOE) report: DOE/GO-102004-1992, 2004 Search PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2(586), 1596/1–1596/6 CAS.
- (a) T. Klement and J. Büchs, Bioresour. Technol., 2013, 135, 422–431 CrossRef CAS PubMed; (b) A. Jarry and Y. Seraudie, US Pat5457040 A, 1995 Search PubMed.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed.
- (a) K. Sakashita and H. Nogami, to Mitsubishi Rayon Co., Ltd, JP2007217388 A 20070830, 2007 Search PubMed; (b) H. Nogami and K. Sakashita, to Mitsubishi Rayon Co., Ltd, JP2007254293 A 20071004, 2007 Search PubMed; (c) H. Nogami, K. Sakashita and H. Okada, to Mitsubishi Rayon Co., Ltd, JP2009007300 A 20090115, 2009 Search PubMed.
- (a) B. R. Baker, R. E. Schaub and J. H. Williams, J. Org. Chem., 1952, 17, 116–131 CrossRef; (b) M. Candy, L. Tomas, S. Parat, V. Heran, H. Bienaymé, J.-M. Pons and C. Bressy, Chem.–Eur. J., 2012, 18, 14267–14271 CrossRef CAS PubMed.
- (a) G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS; (b) Z. Paryzek, H. Koenig and B. Tabaczka, Synthesis, 2003, 2023–2026 CrossRef CAS.
- (a) J. Almena, A. Monsees, R. Kadyrov, T. H. Riermeier, B. Gotov, J. Holz and A. Börner, Adv. Synth. Catal., 2004, 346, 1263–1266 CrossRef CAS; (b) M. Ostermeier, B. Brunner, C. Korff and G. Helmchen, Eur. J. Org. Chem., 2003, 3453–3459 CrossRef CAS.
- A. Carpita, D. Neri and R. Rossi, Gazz. Chim. Ital., 1987, 117, 481–489 CAS.
- See ESI† for experimental and characterization details.
- Y. Zhang, M. Schmitt, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2013, 135, 17925–17942 CrossRef CAS PubMed.
- Y. Zhang and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2012, 51, 2465–2469 CrossRef CAS PubMed.
- H. Mattes, K. Hamada and C. Benezra, J. Med. Chem., 1987, 30, 1948–1951 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterizations. See DOI: 10.1039/c3qo00089c |
| This journal is © the Partner Organisations 2014 |