Mechanical stabilities of silicene
Qing Peng*a,
Xiaodong Wenb and
Suvranu Dea
aDepartment of Mechanical, Aerospace and Nuclear Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180, USA. E-mail: qpeng.org@gmail.com; Tel: 1-518-279-6669
bTheoretical Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
First published on 16th May 2013
Abstract
The mechanical stabilities of planar (g) and low-buckled (b) honeycomb monolayer structures of silicon under various large strains are investigated using density functional theory (DFT). The mechanical properties, including the ultimate stresses, ultimate strains, and high order elastic constants of silicene are predicted, as well as the structure evolutions. Both g-Si and b-Si can sustain large strains (η ≥ 0.15) for armchair, zigzag, and biaxial deformation. The third, fourth, and fifth order elastic constants are indispensable for accurate modeling of the mechanical properties under strains larger than 0.03, 0.06, and 0.08 respectively. The second order elastic constants, including in-plane stiffness, are predicted to monotonically increase with pressure while the Poisson ratio monotonically decreases with increasing pressure. Our results on the positive ultimate strengths and strains, second order elastic constants, and the in-plane Young's modulus indicate that both g-Si and b-Si are mechanically stable.
1 Introduction
The silicon-based counterpart of graphene, namely “silicene”,1–4 arises intense interest due to the promising technological applications, as well as fundamental research. With Dirac cones in its electronic structure, the charge carriers in silicene would behave as massless relativistic particles as in graphene, which make it straightforward to transfer all the expectations of graphene including use in high speed electronic devices based on ballistic transport at room temperature to this innovative material. Thus silicene is expected to provide an easily implementable way to improve the performance and scalability of electronic silicon devices without departure from the silicon-based status quo, which is a crucial advantage and a large cost reduction. In addition, as a one-atom thick silicon sheet arranged in a honeycomb lattice (Fig. 1), silicene exhibits photoelectronic effects, lower thermal conductivity, and higher chemical activities than bulk silicon materials.5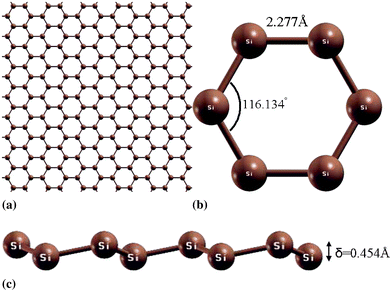 | ||
| Fig. 1 (a) Silicene plane. (b) Overview and (c) side-view of low-buckled silicene. | ||
Even before the isolation of graphene, LDA-DFT predicted that in contrast with the planar honeycomb lattice of graphene, a buckled honeycomb structure of Si might be formed.6–8 On the basis of the first-principles calculations of structural optimization, phonon dispersions, and finite temperature molecular dynamics, the low-buckled (LB) honeycomb structures is predicted to be stable with an equilibrium buckling height of 0.044 nm, opposite to planar (PL) and high-buckled (HB) structures.9 We denote the low-buckled, planar, and high-buckled honeycomb structures as b, g, and B structures respectively in this paper. To the best of our knowledge, the B structure has not been confirmed experimentally. As a result, we only focus on the g and b structures of silicene.
The band structures of LB silicene are ambipolar, and its charge carrier can behave like a massless Dirac fermion at the K point owing to the π and π* bands linearly crossing at the Fermi level. It also was found that the electronic and magnetic properties of silicene nanoribbons show size and geometry dependence.9,10 Using ABINIT software (a DFT package with pseudo potentials and a plane wave basis set), the Fermi velocities in the vicinity of the Dirac point were estimated to be 6.3 × 105 m s−1 and 5.1 × 105 m s−1 for graphene and silicene, respectively.11 Most of the other known features of silicene resemble those of graphene. However, the unique features of silicene exhibit the potential to provide a new future for the electronics industry, which is currently Si-based.
The sp3 hybridization in silicon leads to the common covalent Si–Si bonds, the favorable configuration with respect to the sp2 or the mixed sp2–sp3 orbitals.2,12,13 The most stable form of a 2D silicon sheet is buckled due to the larger atom–atom distance-induced weakening of the π–π overlaps.14 Having explored group 14 structure from 1D to 2D to 3D, it was proposed that C not only favors 4-coordination, but also is happy with π-bonding, allowing 3 or 2-coordination, while Si is less biased in its coordination than C, allowing 5 or 6-coordination, but tending towards 4-coordination. The π-bonding plays an important role in the stabilization of graphene structures; the low-lying π*, the high-lying π orbitals are likely to make Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si highly reactive towards bases and acids. To put it another way, to use surfactants might be another way to stabilize silicene.15,16 However, such an approach for synthesizing silicene has not been confirmed in experiments.
Si highly reactive towards bases and acids. To put it another way, to use surfactants might be another way to stabilize silicene.15,16 However, such an approach for synthesizing silicene has not been confirmed in experiments.
Although the dynamics stabilities of b-Si and g-Si were studied by DFT phonon calculations,9 their mechanical stabilities are still unknown. Two-dimensional (2D) nanomaterials are the basic “building blocks” for all other structures: buckyballs (0D) by wrapping, nanotubes (1D) by rolling, bulk (3D) by stacking.17 However, due to the quantum confinement resulting from the reduction of the third dimension, 2D nanomaterials present different properties from bulk. For example, the mechanical strengths are an order of magnitude larger than those in bulk. To understand mechanical properties is critical in designing parts or structures with silicene regarding practical applications. Strain engineering is a common and important approach tailoring the functional and structural properties of nanomaterials.18,19 One can expect that the properties of silicene will be affected by applied strain too. In addition, silicene is vulnerable to be strained with or without intent because of its monatomic thickness. For instance, there are strains because of the mismatch of lattices constants or surface corrugation with substrates.20,21 Therefore, knowledge of the mechanical properties of silicene is highly desired.
Depending on the loading, the mechanical properties are divided into four strain domains: linear elastic, nonlinear elastic, plastic, and fracture. Materials in the first two strain domains are reversible, i.e., they can restore to equilibrium status after the release of the loads. On the contrary, the last two domains are non-reversible. Defects are nucleated and accumulated with the increase of the strain, until rupture. As in graphene, the nonlinear mechanical properties are prominent since it remained elastic until the intrinsic strength was reached.22,23 Thus it is of great interest to examine the nonlinear elastic properties of silicene, which is necessary to understand the strength and reliability of structures and devices made of silicene.
Several previous studies have shown that 2D monolayers present a large nonlinear elastic deformation during the tensile strain up to the ultimate strength of the material, followed by a strain softening until fracture.23–26 We expect that silicene behaves in a similar manner. Under large deformation, the strain energy density needs to be expanded as a function of strain in a Taylor series to include quadratic and higher order terms. The higher order terms account for both nonlinearity and strain softening of the elastic deformation. They can also express other anharmonic properties of 2D nanostructures including phenomena such as thermal expansion, phonon–phonon interaction, etc.22
The goal of this paper is to study the mechanical stabilities and mechanical behaviors of silicene at large strains and to find an accurate continuum description of the elastic properties from ab initio density functional theory calculations. The total energies of the system, forces on each atom, and stresses on the simulation boxes are directly obtained from DFT calculations. The response of silicene under the nonlinear deformation and fracture is studied, including ultimate strength and ultimate strain. The high order elastic constants are obtained by fitting the stress–strain curves to analytical stress–strain relationships that belong to the continuum formulation.24 We compared silicene with graphene and graphane which have sp2 and sp3 type bonds respectively. Based on our result of the high order elastic constants, the pressure dependence properties, such as sound velocities and the second order elastic constants, including the in-plane stiffness, are predicted. Our results for the continuum formulation could also be useful in finite element modeling of the multiscale calculations for mechanical properties of silicene at the continuum level. The organization of this paper is as follows. Section II presents the computational details of DFT calculations. The results and analysis are in section III, followed by conclusions in section IV.
2 Density functional theory calculations
We consider a conventional unit cell containing 6 atoms with periodic boundary conditions (Fig. 2). The 6-atom conventional unit cell is chosen to capture the “soft mode”, which is a particular normal mode exhibiting an anomalous reduction in its characteristic frequency and leading to mechanical instability. This soft mode is a key factor in limiting the strength of monolayer materials and can only be captured in unit cells with hexagonal rings.27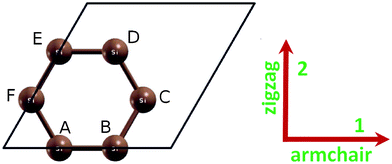 | ||
| Fig. 2 Atomic structure of silicene in the conventional unit cell (6 atoms, marked as A–F) in the undeformed reference configuration. | ||
The total energies of the system, forces on each atom, stresses, and stress–strain relationships of silicene under the desired deformation configurations are characterized via DFT. The calculations were carried out with the Vienna Ab initio Simulation Package (VASP)28 which is based on the Kohn–Sham Density Functional Theory (KS-DFT)29 with the generalized gradient approximations as parameterized by Perdew, Burke, and Ernzerhof (PBE) for exchange–correlation functions.30 The electrons explicitly included in the calculations are the 3s23p2 electrons for silicon atoms. The core electrons are replaced by the projector augmented wave (PAW) and pseudo-potential approach.31 A plane-wave cutoff of 500 eV is used in all the calculations. The calculations are performed at zero temperature.
The criterion to stop the relaxation of the electronic degrees of freedom is set by the total energy change to be smaller than 0.000001 eV. The optimized atomic geometry is achieved through minimizing Hellmann–Feynman forces acting on each atom until the maximum forces on the ions were smaller than 0.001 eV Å−1.
The atomic structures of all the deformed and undeformed configurations were obtained by fully relaxing a 6-atom unit cell where all atoms were placed in one plane. The simulation invokes periodic boundary conditions for the two in-plane directions.
The irreducible Brillouin zone is sampled with a Gamma-centered 21 × 21 × 1 k-mesh. Such a large k-mesh was used to reduce the numerical errors caused by the strain of the systems. The initial charge densities were taken as a superposition of atomic charge densities. There is a 15 Å thick vacuum region to reduce the inter-layer interaction to model the single layer system. To eliminate the artificial effect of the out-of-plane thickness of the simulation box on the stress, we use the second Piola–Kirchhoff stress24 to express the 2D forces per length with units of N m−1.
For a general deformation state, the number of independent components of the second, third, fourth, and fifth order elastic tensors are 21, 56, 126, and 252 respectively. However, there are only fourteen independent elastic constants that need to be explicitly considered due to the symmetries of the atomic lattice point group D6h, which consists of a six-fold rotational axis and six mirror planes.23
The fourteen independent elastic constants of g- and b-Si are determined by a least-squares fit to the stress–strain results from DFT calculations in two steps, detailed in our previous work,24 which had been well used to explore the mechanical properties of 2D materials.32–35 A brief introduction is that, in the first step, we use a least-squares fit of five stress–strain responses. Five relationships between stress and strain are necessary because there are five independent fifth-order elastic constants (FFOEC). We obtain the stress–strain relationships by simulating the following deformation states: uniaxial strain in the zigzag direction (zigzag); uniaxial strain in the armchair direction (armchair); and equibiaxial strain (biaxial). From the first step, the components of the second-order elastic constants (SOEC), the third-order elastic constants (TOEC), and the fourth-order elastic constants (FOEC) are over-determined (i.e., the number of linearly independent variables are greater than the number of constraints), and the fifth-order elastic constants are well-determined (the number of linearly independent variables are equal to the number of constraints). Under such circumstances, the second step is needed: least-squares solution to these over- and well-determined linear equations.
3 Results and analysis
3.1 Atomic structure
The equilibrium lattice constant for silicene was optimized by finding the minima of the energy as a function of the lattice constant. The total energy as a function of lattice spacing is obtained by varying lattice constants, with full relaxations of all the atoms. A least-squares fit of the energies versus lattice constants with a fourth-order polynomial function yields the equilibrium lattice constant as a = 3.901 and 3.865 Å for g and b structures respectively. The most energetically favorable structure is set as the strain-free structure in this study and the atomic structure, as well as the conventional cell, is illustrated in Fig. 2 for b-Si. Specifically, the bond length of the Si–Si bond is 2.277 Å in b-Si and 2.252 Å in g-Si. The six atoms in the conventional cell are marked as A to F, shown in Fig. 2. The bond lengths of AB, BC, CD are denoted as d1, d2, d3, respectively, the bond angles ABC and BCD for α1 and α2, respectively. Both the bond angles are 116.1°. The dihedral angle ABCD (γ) is 38.1°. The buckling height is the distance of atom B to the plane formed by atom A, C, and E, as illustrated in Fig. 1(c). Our atomic structure is in good agreement with previous DFT calculations9,13,36–38 and experiments.3,39–41When strain is applied, all the atoms are allowed full freedom of motion. A quasi-Newton algorithm is used to relax all atoms into equilibrium positions within the deformed unit cell that yields the minimum total energy for the imposed strain state of the super cell. Both compression and tension are considered with Lagrangian strains ranging from −0.1 to 0.4 with an increment of 0.01 in each step for all three deformation modes. It is important to include the compressive strains since they are believed to be the cause of the rippling of the free standing atomic sheet.42 It was observed that a graphene sheet experiences biaxial compression after thermal annealing,43 which could also happen on g- and b-Si. Such an asymmetrical range was chosen due to the non-symmetric mechanical responses of the material, as well as its mechanical instability,44 to the compressive strains and the tensile strains, as illustrated in the next subsection.
For b-Si, the bond lengths (d1, d2, and d3), bond angles (α1,α2), dihedral angle γ, and the buckling height Δ are a function of the applied strains, as plotted in Fig. 3. When the strain is applied along the armchair directions, the bond AB is parallel to the direction of the strain. The bond length d1 varies nonlinearly with respect to the applied strain. The bonds BC and CD are inclined to the armchair strain. The bond lengths d2 = d3 increase with the applied strain, and reach the maximum value of 2.371 Å at the armchair strain of 0.23. All the bond lengths are monotonically increasing with the increment of the zigzag and biaxial strains.
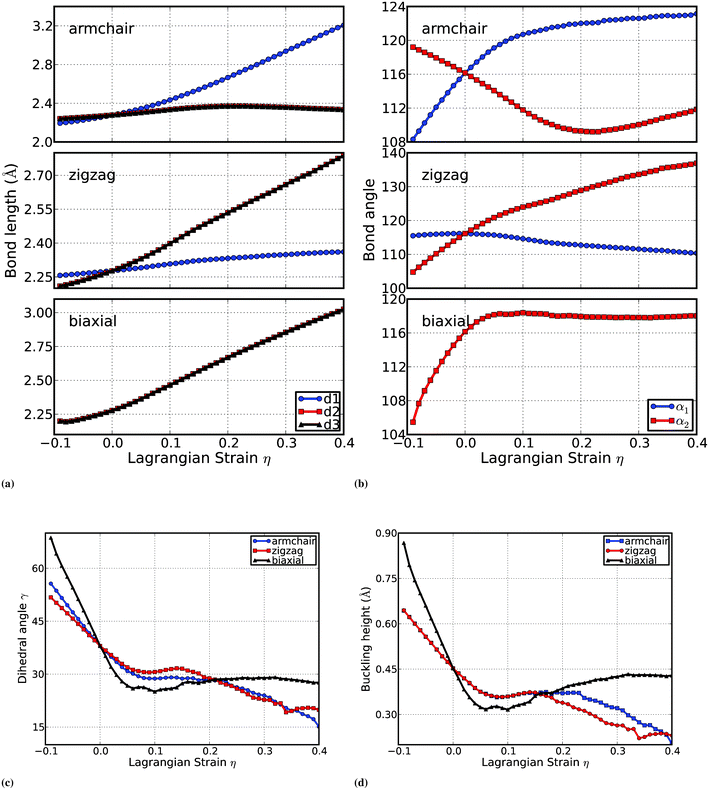 | ||
| Fig. 3 Evolution of the geometries of b-Si under the armchair, zigzag, and biaxial strains: (a) Bond lengths; (b) Bond angels; (c) Dihedral angles; (d) Buckling height. | ||
Two bond angles are examined explicitly. The bond angle α1 monotonically increases with increasing armchair strain and decreases with increasing zigzag strain. There is a maximum of the bond angle 118.38° under the biaxial strain of 0.1. The bond angle α2 has a minimum value of 109.201° at the armchair strain of 0.23, correlated to the maximum of bond length. It monotonically increases with the increment of zigzag strain.
The response of the dihedral angle to the applied strains is more complicated as shown in Fig. 3(c). The compressive strain increases the dihedral angle in all three tested deformation modes. As the tensile strain is applied, the dihedral angle decreases within a small strain, which is 0.1, 0.09, and 0.06 for armchair, zigzag, and biaxial strain, respectively, with the minima of 28.74, 30.51, and 25.94°, respectively. When the applied strain increases, the dihedral angle will increase to the maxima of 28.86° at an armchair strain of 0.16, 31.67° at a zigzag strain of 0.14, and 29.1° at a biaxial strain of 0.32. It is interesting to note that the dihedral angle as well as the rate of its decrement with respect to the strain are the same for both uniaxial strains (armchair and zigzag) when η > 0.2. There are two special points of the dihedral angle curves where three deformations reach the same value of γ: at zero strain and η = 0.21, with the corresponding values of 38.0° and 28.45° respectively.
The buckling height is an important parameter to characterize the corrugation of the b-Si surfaces. The compressive strains increases the buckling height. The tensile strains decrease the buckling height, as expected, but in a complex function. The tensile uniaxial strain has a minimum of 0.358 at η = 0.09, and a maximum of 0.373 at η = 0.14. The tensile biaxial strain has a minimum of 0.317 Å at η = 0.06, and a maximum of 0.433 Å at η = 0.32. There are two special points of the buckling height curves where three deformations reach the same value of Δ: at zero strain and η = 0.16, with the corresponding values of Δ = 0.454 Å and 0.371 Å respectively. It is worth pointing out that the buckling height is the same for both uniaxial deformations (armchair and zigzag) when η < 0.16.
One could notice that the dihedral angle γ is correlated to the buckling height Δ to some degree, but they are independent of each other. The different behavior of the dihedral angle to the armchair and zigzag strains indicates that neither the 2-atom unit cell nor 4-atom unit cell is sufficient to model the mechanical stabilities of b-Si; a 6-atom unit cell or larger unit cells are required.
There are points of intersection at zero strains in Fig. 3. These intersections signify the fact that silicene is non-isotropic. The bond lengths, bond angles, dihedral angle, and buckling height respond differently to the strains along different directions.
For g-Si, both the tensile and the compressive strains do not change the coplanar structure. The evolution of the geometry is much simpler, as an affine transformation according to the applied strains.
3.2 Strain energy
We define the strain energy per atom Es = (Etot − E0)/n, where Etot is the total energy of the strained system, E0 is the total energy of the strain-free system, and n = 6 is the number of atoms in the unit cell. This size-independent quantity is used for comparison between different systems. Fig. 4 shows the Es of silicene as a function of strain in uniaxial armchair, uniaxial zigzag, and biaxial deformation. Es is seen to be anisotropic with strain direction. Es is non-symmetrical for compression (η < 0) and tension (η > 0) for all three modes. This non-symmetry indicates the anharmonicity of the silicene structures. The harmonic region where the Es is a quadratic function of applied strain can be taken between −0.02 < η < 0.02. The stresses, derivatives of the strain energies, are linearly increasing with the increase of the applied strains in the harmonic region.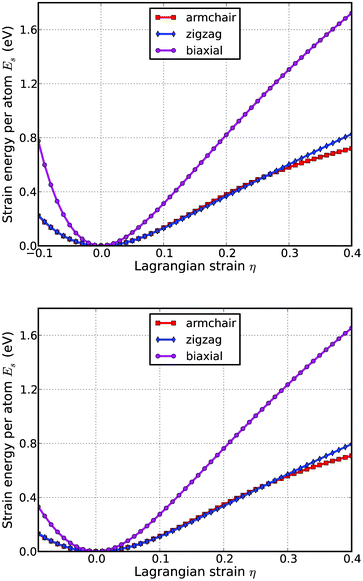 | ||
| Fig. 4 Energy-strain responses of g-Si (top) and b-Si (bottom) under armchair, zigzag, and biaxial strain. | ||
The anharmonic region is the range of strain where the linear stress–strain relationship is invalid and higher order terms are not negligible. With even larger loading of strains, the systems will undergo irreversible structural changes, and the systems are in a plastic region where they may fail. The maximum strain in the anharmonic region is the critical strain. The critical strain is 0.18 under armchair deformation. However, for the other two directions, the critical strains are not observed. The ultimate strains are determined as the corresponding strain of the ultimate stress, which is the maxima of the stress–strain curve, as discussed in the following section.
It is worth noting that in general the compressive strains will cause rippling of the free-standing thin films, membranes, plates, and nanosheets.42 The critical compressive strain for rippling instability is much less than the critical tensile strain for fracture, for example, 0.0001% versus 2% in graphene sheets.44 However, the rippling can be suppressed by applying constraints, such as embedding (0.7%),45 substrate (0.4% before heating),43 thermal cycling on SiO2 substrate (0.05%)46 and BN substrate (0.6%),47 and sandwiching.48 Our study of compressive strains is important in understanding the mechanics of these non-rippling applications. The rippling phenomena are interesting and important, which is, however, out the scope of this study.
3.3 Stress–strain curves
The second P–K stress versus Lagrangian strain relationship for uniaxial strains along the armchair and zigzag directions, as well as biaxial strains, are shown in Fig. 5 for both g-Si (left) and b-Si (right). The results show that the g-Si is stiffer than b-Si. However, b-Si can sustain larger strains than g-Si, mainly due to the buckling bonds which could be stretched longer. The evolution of the buckling structures will be further analyzed in the following subsections. The ultimate strength is the maximum stress that a material can withstand while being stretched, and the corresponding strain is the ultimate strain. Under ideal conditions, the critical strain is larger than the ultimate strain. The systems of perfect silicene under strains beyond the ultimate strains are in a metastable state, which can be easily destroyed by long wavelength perturbations and vacancy defects, as well as high temperature effects.49 The ultimate strain is determined by the intrinsic bonding strengths and acts as a lower limit of the critical strain. Thus it has a practical meaning in consideration for its applications.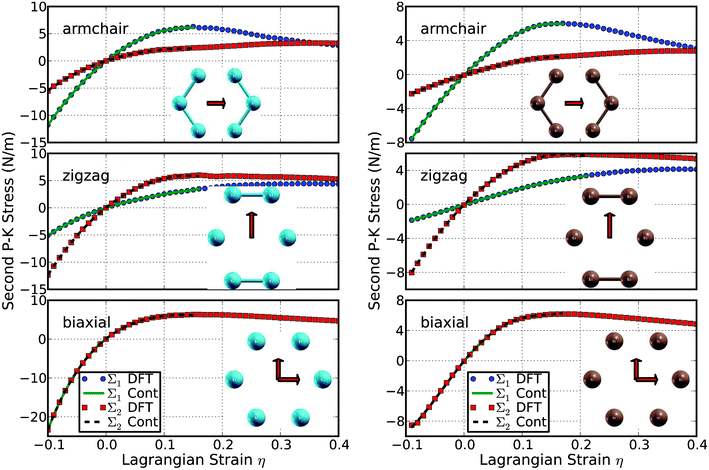 | ||
| Fig. 5 Stress–strain responses of g-Si (left) and b-Si (right) under the armchair, zigzag, and biaxial strain. Σ1 (Σ2) denotes the x (y) component of stress. “Cont” stands for the fitting of DFT calculations (“DFT”) to continuum elastic theory. The insets are geometry under ultimate strains. | ||
The ultimate strengths and strains corresponding to the different strain conditions of b and g structures of silicene are summarized in Table 1, compared with those of g-BN,24,50,51 graphene, and graphane.33 The g-BN and graphene are compared to the g-Si for their planar structure. The graphane is compared here for its buckling structure, similar to b-Si. The material behaves in an asymmetric manner with respect to compressive and tensile strains. With increasing strains, the Si–Si bonds are stretched and eventually rupture. The critical strains are not spotted in either g-Si or b-Si in the tested strain range under the three strain modes. This is different from graphene and graphane, which has a critical strain of 0.31 and 0.22 under armchair deformation respectively.33 This might be due to the increasing size of the atoms as well as bond lengths in silicene.
When the tensile strain is applied in the armchair direction, the bonds of those parallel with this direction are more severely stretched than those in other directions. The ultimate strain in armchair deformation is 0.15 (0.17) for g-Si (b-Si), smaller than that of g-BN, graphene, and graphane. Under the zigzag deformation, in which the strain is applied perpendicular to the armchair, there is no bond parallel to this direction. The bonds that are at an incline to the zigzag direction with an angle of 30° are more severely stretched than those in the armchair direction. The ultimate strain in this zigzag deformation is 0.16 (0.21) for g-Si (b-Si), smaller than that of g-BN, graphene, and graphane. At this ultimate strain, the bonds that are at an incline to the armchair direction appear to be ruptured (Fig. 5 middle panel). Under the biaxial deformation, the ultimate strain is ηbm = 0.15 (0.17) for g-Si (b-Si), which is smaller than that of g-BN, graphene, and graphane. At this applied ultimate strain, all the Si–Si bonds are observed to be ruptured (Fig. 5 bottom).
All the ultimate stresses and ultimate strains of both g-Si and b-Si are smaller than those of g-BN, graphene, and graphane. This could be owing to the strength of the bonds in that Si–Si bonds are much weaker than the B–N bond and C–C bonds. The bond strength of a chemical bond can be measured by the bond energy, which is 256 kJ mol−1 for Si–Si, 230 kJ mol−1 for SiSi, 346 kJ mol−1 for C–C, 602 kJ for CC, and 389 kJ mol−1 for B–N.52 Thus our findings agree with the bond strength calculations.
It should be noted that the softening of the perfect silicene under strains beyond the ultimate strains only occur for ideal conditions. The systems under this circumstance are in a metastable state, which can be easily destroyed by long wavelength perturbations and vacancy defects, as well as high temperature effects, and enter a plastic state.49 Thus only the data within the ultimate strain has physical meaning and was used in determining the high order elastic constants in the following subsection.
3.4 Elastic constants
The elastic constants are critical parameters in finite element analysis models for mechanical properties of materials. Our results of these elastic constants provide an accurate continuum description of the elastic properties of silicene from ab initio density functional theory calculations. They are suitable for incorporation into numerical methods such as the finite element technique.The second order elastic constants model the linear elastic response. The higher (>2) order elastic constants are important to characterize the nonlinear elastic response of silicene using a continuum description. These can be obtained using a least squares fit of the DFT data and are reported in Table 2. Corresponding values for graphene are also shown.
| g-Si | b-Si | Graphene33 | g-BN24 | Graphane26 | |
|---|---|---|---|---|---|
| a | 3.901 | 3.865 | 2.468 | 2.512 | 2.540 |
| Y s | 71.2 | 63.8 | 340.8 | 278.3 | 246.7 |
| ν | 0.401 | 0.325 | 0.178 | 0.225 | 0.078 |
| C 11 | 84.8 | 71.3 | 352.0 | 293.2 | 248.2 |
| C 12 | 34.1 | 23.2 | 62.6 | 66.1 | 19.4 |
| C 111 | −696.5 | −397.6 | −3089.7 | −2513.6 | −2374.1 |
| C 112 | −281.6 | −14.1 | −453.8 | −425.0 | −95.4 |
| C 222 | −617.2 | −318.9 | −2928.1 | −2284.2 | −2162.8 |
| C 1111 | 1951 | −830 | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 927 927 |
16547 |
−19492 |
| C 1112 | 1683 | −309 | 2731 | 2609 | 819 |
| C 1122 | 2549 | −5091 | 3888 | 2215 | 68 |
| C 2222 | 1108 | −629 | 18779 |
12288 |
14823 |
| C 11111 | −19595 |
20614 |
−118791 |
−65265 |
−103183 |
| C 11112 | −11405 |
6923 | −19173 |
−8454 | −816 |
| C 11122 | −7628 | 11681 |
−15863 |
−28556 |
−16099 |
| C 12222 | −16955 |
−7593 | −27463 |
−36955 |
−10151 |
| C 22222 | −21326 |
−29735 |
−134752 |
−100469 |
−134277 |
The in-plane Young's modulus Ys and Poison's ratio ν may be obtained from the following relationships: Ys = (C211 − C212)/C11 and ν = C12/C11. We have Ys = 71.2 and 63.8 N m−1 for g-Si and b-Si respectively, and ν = 0.401 and 0.325 respectively. The in-plane stiffness of both silicene structures (g/b) are very small compared to g-BN (26/23%), graphene (21/19%), and graphane (29/26%). Our results are in good agreement with previous DFT calculations.9,36
Higher order (>2) elastic constants are important quantities53 and can be determined by measuring the changes of sound velocities under the application of hydrostatic and uniaxial stresses.54 The high order elastic constants can be utilized to study the nonlinear elasticity, thermal expansion (through the Grüneisen parameter), temperature dependence of elastic constants, harmonic generation, phonon–phonon interactions, photon–phonon interactions, lattice defects, phase transitions, echo phenomena, and strain softening, and so on.33 Using the higher order elastic continuum description, one can calculate the stress and deformation state under uniaxial stress, rather than uniaxial strain.23 Explicitly, when pressure is applied, the pressure-dependent second-order elastic moduli can be obtained from the high order elastic continuum description.24 The third-order elastic constants are important in understanding the nonlinear elasticity of materials, such as changes in acoustic velocities due to finite strain. As a consequence, nano devices (such as nano surface acoustic wave sensors and nano waveguides) could be synthesized by introducing local strain.26,55
Stress–strain curves in the previous section show that they will soften when the strain is larger than the ultimate strain. From the view of electron bonding, this is due to the bond weakening and breaking. This softening behavior is determined by the TOECs and FFOECs in the continuum aspect. The negative values of TOECs and FFOECs ensure the softening of the silicene monolayer under large strain.
The hydrostatic terms (C11, C22, C111, C222, and so on) of both g and b silicene monolayers are smaller than those of g-BN and graphene, consistent with the conclusion that the silicene is “softer”. The shear terms (C12, C112, C1122, etc.) in general are smaller than those of g-BN, graphene, and graphane, which contributes to its high compressibility.
A good way to check the importance of the high order elastic constants is to consider the case when they are missing. With the elastic constants, the stress–strain response can be predicted from elastic theory.24 When we only consider the second-order elasticity, the stress varies with strain linearly. Let's take the biaxial deformation as an example. As illustrated in Fig. 6, the linear behaviors are only valid within a small strain range, about −0.03 ≤ η ≤ 0.03; the same result obtained from the energy versus strain curves in Fig. 4. With the knowledge of the elastic constants up to the third order, the stress–strain curve can be accurately predicted within the range of −0.06 ≤ η ≤ 0.06. Using the elastic constants up to the fourth order, the mechanical behaviors can be well treated up to a strain as large as 0.08. For the strains beyond 0.08, the fifth order elastic are required for an accurate modeling. The analysis of the uniaxial deformations comes to the same results. Further analysis on the g-Si and b-Si (Fig. 5 bottom) also confirms the results.
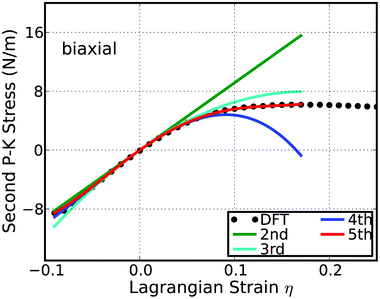 | ||
| Fig. 6 The predicted stress–strain responses from different orders: second, third, fourth, and fifth order, and compared to the DFT calculations in the biaxial deformation in b-Si. | ||
Our results illustrate that the monatomic layer structures possess different mechanical behaviors in contrast to the bulk or multi-layered structures, where the second-order elastic constants are sufficient in most cases. The second-order elastic constants are relatively easier to be calculated from the strain energy curves,49,56 however, they are not sufficient for monatomic layer structures. The high order elastic constants are required for an accurate description of the mechanical behaviors of monatomic layer structures since they are vulnerable to strain due to the geometry confinements.
Our results of mechanical properties of g-Si and b-Si are limited to zero temperature due to current DFT calculations. Once the finite temperature is considered, the thermal expansions and dynamics will in general reduce the interactions between atoms. As a result, the longitudinal mode elastic constants will decrease with respect to the temperature of the system. The variation of shear mode elastic constants should be more complex in responding to the temperature. A thorough study will be interesting, which is, however, beyond the scope of this study.
3.5 Pressure effect on the elastic moduli
With third-order elastic moduli, we can study the effect of the second-order elastic moduli on the pressure p acting in the plane of silicene. Explicitly, when pressure is applied, the pressure-dependent second-order elastic moduli (![[C with combining tilde]](https://www.rsc.org/images/entities/i_char_0043_0303.gif) 11, 12, 22) can be obtained from C11, C12, C22, C111, C112, C222, Ys, and ν as:
11, 12, 22) can be obtained from C11, C12, C22, C111, C112, C222, Ys, and ν as:
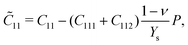 | (1) |
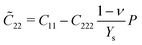 | (2) |
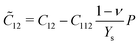 | (3) |
The second-order elastic moduli of silicene (both g and b structures) are seen to increase linearly with the applied pressure (Fig. 7), except the shear mode 12 of b-Si. The rates of the change of the second-order elastic moduli of the g-Si are larger than those of b-Si. Poisson's ratio decreases monotonically with the increase of pressure. The Poisson's ratio of the b-Si is more sensitive to the in-plane pressure than the g-Si. 11 is asymmetrical to 22 unlike the zero pressure case. 11 = 22 = C11 only occurs when the pressure is zero. This anisotropy could be the outcome of anharmonicity.
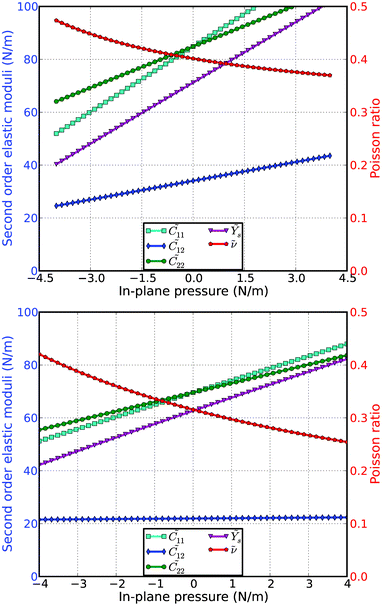 | ||
| Fig. 7 Second-order elastic moduli and Poisson ratio as function of the pressure for the g-Si (top) and b-Si (bottom) from DFT predictions. | ||
4 Conclusions
In summary, we studied the mechanical stabilities of planar and low-buckled silicene under various strains using DFT calculations. It is observed that both g and b-silicene exhibit a nonlinear elastic deformation up to an ultimate strain, which is 0.17, 0.21, and 0.17 for armchair, zigzag, and biaxial deformation of b-Si, respectively, and 0.15, 0.16, and 0.15 for g-Si, respectively; all of them are smaller than those of graphene, g-BN, and graphane. The deformation and failure behavior and the ultimate strength are anisotropic. Furthermore, we examined the evolution of the geometries, including bond lengths, bond angels, dihedral angles, and buckling height of b-Si under various loadings. The in-plane stiffness of both silicene structures (g/b) are very small compared to g-BN (26/23%), graphene (21/19%), and graphane (29/26%). However, silicene has a large Poisson ratio, 0.325 for b-Si and 0.401 for g-Si.The nonlinear elasticity of silicene was investigated. We found an accurate continuum description of the elastic properties of silicene by explicitly determining the fourteen independent components of high order (up to fifth order) elastic constants from the fitting of stress–strain curves obtained from DFT calculations. This data is useful to develop a continuum description which is suitable for incorporation into a finite element analysis model for its applications at a large scale. We also find that the harmonic elastic constants are only valid with a small range of −0.03 ≤ η ≤ 0.03. With the knowledge of the elastic constants up to the third order, the stress–strain curve can be accurately predicted within the range of −0.06 ≤ η ≤ 0.06. Using the elastic constants up to the fourth order, the mechanical behaviors can be accurately predicted up to a strain as large as 0.08. For the strains beyond 0.08, the fifth order elastic constants are required for an accurate modeling. The high order elastic constants reflect the high order nonlinear bond strength under large strains.
We predicted that both the second-order elastic constants and the in-plane stiffness monotonically increase with elevating pressure, while Poisson's ratio reverses. According to the results of the positive ultimate strengths and strains, second-order elastic constants, and the in-plane Young's modulus, we propose that both g-Si and b-Si are mechanically stable.
Acknowledgements
The authors would like to acknowledge the generous financial support from the Defense Threat Reduction Agency (DTRA) Grant # BRBAA08-C-2-0130 and # HDTRA1-13-1-0025.References
- P. De Padova and C. Quaresima, et al. , Appl. Phys. Lett., 2010, 96, 261905 CrossRef.
- A. Kara, H. Enriquez, A. P. Seitsonen, L. C. L. Y. Voon, S. Vizzini, B. Aufray and H. Oughaddou, Surf. Sci. Rep., 2012, 67, 1–18 CrossRef CAS.
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef.
- T. Morishita, K. Nishio and M. Mikami, Phys. Rev. B, 2008, 77, 081401 CrossRef.
- H. Okamoto, Y. Sugiyama and H. Nakano, Chem.–Eur. J., 2011, 17, 9864–9887 CrossRef CAS.
- G. G. Guzmán-Verri and L. C. Lew Yan Voon, Phys. Rev. B, 2007, 76, 075131 CrossRef.
- K. Takeda and K. Shiraishi, Phys. Rev. B, 1994, 50, 14916–14922 CrossRef CAS.
- E. Durgun, S. Tongay and S. Ciraci, Phys. Rev. B, 2005, 72, 075420 CrossRef.
- S. Cahangirov, M. Topsakal, E. Akturk, H. Sahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS.
- Y. Ding and J. Ni, Appl. Phys. Lett., 2009, 95, 083115 CrossRef.
- L. C. L. Y. Voon, E. Sandberg, R. S. Aga and A. A. Farajian, Appl. Phys. Lett., 2010, 97, 163114 CrossRef.
- S. Wang, Phys. Chem. Chem. Phys., 2011, 13, 11929–11938 RSC.
- C.-C. Liu, W. Feng and Y. Yao, Phys. Rev. Lett., 2011, 107, 076802 CrossRef.
- E. F. Sheka, Int. J. Quantum Chem., 2013, 113, 612–618 CrossRef CAS.
- X.-D. Wen, T. J. Cahill and R. Hoffmann, Chem.–Eur. J., 2010, 16, 6555–6566 CAS.
- R. Hoffmann, Angew. Chem., Int. Ed., 2013, 52, 93–103 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS.
- F. Guinea, M. I. Katsnelson and A. K. Geim, Nat. Phys., 2009, 6, 30–33 CrossRef.
- M. Topsakal, S. Cahangirov, E. Bekaroglu and S. Ciraci, Phys. Rev. B, 2009, 80, 235119 CrossRef.
- Y. Ma, Y. Dai, W. Wei, C. Niu, L. Yu and B. Huang, J. Phys. Chem. C, 2011, 115, 20237–20241 CAS.
- Z. H. Aitken and R. Huang, J. Appl. Phys., 2010, 107, 123531 CrossRef.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS.
- X. Wei, B. Fragneaud, C. A. Marianetti and J. W. Kysar, Phys. Rev. B, 2009, 80, 205407 CrossRef.
- Q. Peng, W. Ji and S. De, Comput. Mater. Sci., 2012, 56, 11 CrossRef CAS.
- Q. Peng, X.-J. Chen, W. Ji and S. De, Adv. Eng. Mater., 2013 DOI:10.1002/adem.201300033.
- Q. Peng, W. Ji and S. De, Phys. Chem. Chem. Phys., 2012, 14, 13385–13391 RSC.
- C. A. Marianetti and H. G. Yevick, Phys. Rev. Lett., 2010, 105, 245502 CrossRef CAS.
- G. Kresse and J. Furthuller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- R. O. Jones and O. Gunnarsson, Rev. Mod. Phys., 1989, 61, 689–746 CrossRef CAS.
- Q. Peng, C. Liang, W. Ji and S. De, Comput. Mater. Sci., 2013, 68, 320–324 CrossRef CAS.
- Q. Peng, C. Liang, W. Ji and S. De, Phys. Chem. Chem. Phys., 2013, 15, 2003–2011 RSC.
- Q. Peng, C. Liang, W. Ji and S. De, Appl. Phys. A: Mater. Sci. Process., 2013 DOI:10.1007/s00339-013-7551-4.
- Q. Peng, X.-J. Chen, S. Liu and S. De, RSC Adv., 2013, 3, 7083–7092 RSC.
- H. Şahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R. T. Senger and S. Ciraci, Phys. Rev. B, 2009, 80, 155453 CrossRef.
- N. D. Drummond, V. Zólyomi and V. I. Fal'ko, Phys. Rev. B, 2012, 85, 075423 CrossRef.
- Z. Ni, Q. Liu, K. Tang, J. Zheng, J. Zhou, R. Qin, Z. Gao, D. Yu and J. Lu, Nano Lett., 2012, 12, 113–118 CrossRef CAS.
- P. D. Padova, C. Quaresima, C. Ottaviani, P. M. Sheverdyaeva, P. Moras, C. Carbone, D. Topwal, B. Olivieri, A. Kara, H. Oughaddou, B. Aufray and G. L. Lay, Appl. Phys. Lett., 2010, 96, 261905 CrossRef.
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Appl. Phys. Lett., 2010, 97, 223109 CrossRef.
- B. Feng, Z. Ding, S. Meng, Y. Yao, X. He, P. Cheng, L. Chen and K. Wu, Nano Lett., 2012, 12, 3507–3511 CrossRef CAS.
- E. Cerda and L. Mahadevan, Phys. Rev. Lett., 2003, 90, 074302 CrossRef CAS.
- W. Bao, F. Miao, Z. Chen, H. Zhang, W. Jang, C. Dames and C. N. Lau, Nat. Nanotechnol., 2009, 4, 562–566 CrossRef CAS.
- Y. Zhang and F. Liu, Appl. Phys. Lett., 2011, 99, 241908 CrossRef.
- G. Tsoukleri, J. Parthenios, K. Papagelis, R. Jalil, A. C. Ferrari, A. K. Geim, K. S. Novoselov and C. Galiotis, Small, 2009, 5, 2397–2402 CrossRef CAS.
- D. Yoon, Y.-W. Son and H. Cheong, Nano Lett., 2011, 11, 3227–3231 CrossRef CAS.
- W. Pan, J. Xiao, J. Zhu, C. Yu, G. Zhang, Z. Ni, K. Watanabe, T. Taniguchi, Y. Shi and X. Wang, Sci. Reports, 2012, 2, 893 Search PubMed.
- R. Quhe, J. Zheng, G. Luo, Q. Liu, R. Qin, J. Zhou, D. Yu, S. Nagase, W.-N. Mei, Z. Gao and J. Lu, NPG Asia Mater., 2012, 4, e6 CrossRef.
- M. Topsakal, S. Cahangirov and S. Ciraci, Appl. Phys. Lett., 2010, 96, 091912 CrossRef.
- Q. Peng and S. De, Phys. E., 2012, 44, 1662–1666 CrossRef CAS.
- Q. Peng, W. Ji and S. De, Nanoscale, 2013, 5, 695–703 RSC.
- R. T. Sanderson, Chemical bonds and bond energy, Academic Press, New York, 1976, vol. 1d Search PubMed.
- Y. Hiki, Annu. Rev. Mater. Sci., 1981, 11, 51 CrossRef CAS.
- K. Brugger, J. Appl. Phys., 1965, 36, 768 CrossRef.
- Q. Peng, A. R. Zamiri, W. Ji and S. De, Acta Mech., 2012, 223, 2591–2596 CrossRef.
- M. Topsakal, E. Aktürk and S. Ciraci, Phys. Rev. B, 2009, 79, 115442 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |