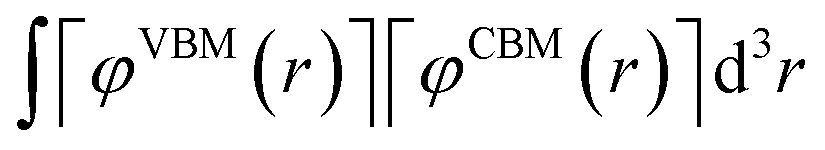DOI:
10.1039/D4RA08275C
(Paper)
RSC Adv., 2025,
15, 4779-4788
Understanding the mechanism of water splitting on (111) and (001) surfaces of CsPbI2Br: time-domain ab initio analysis and DFT study†
Received
21st November 2024
, Accepted 13th January 2025
First published on 13th February 2025
Abstract
Photochemical splitting of water is a promising source of clean and sustainable energy. Perovskites are increasingly being used as photocatalysts. In this paper, we have presented nonadiabatic quantum dynamics simulations (NAMD) and ab initio simulation studies of photocatalytic splitting of water on the (111) and (001) surfaces of CsPbI2Br. The simulations not only helped identify the surface on which splitting occurred but also provided atomistic insights into this behavior. We proposed a three-step reaction mechanism, comprising photogeneration of charge carriers, followed by hole transfer from the iodine atom to water and splitting of water at the interface. Subsequent to water splitting, a hydrogen bond was formed between H and I. The splitting occurred due to the shifting of p-orbitals of the oxygen atom in the presence of light. We have computed the charge carrier lifetime on the (111) and (001) surfaces. The overlap integral between the conduction band minima (CBM) and valence band maxima (VBM) was suppressed on the (111) surface compared to that on the (001) surface. As a result, charge carriers remained separated for a longer time on the (111) surface and could participate in the water splitting process.
1 Introduction
We are currently facing two major challenges: environmental pollution and the energy crisis.1 A number of studies have stressed the efficacious utilization of solar energy to overcome these limitations.2,3 Among the various methods, photocatalytic technique has shown remarkable potential. Photocatalytic splitting of water is the process in which decomposition of water is driven by energy of sunlight. An appropriate catalytic material is required for its technological applications. In the last few years, perovskites have drawn the attention of the scientific community for photochemical water splitting. A number of photocatalysts have been studied in the past years, such as metal oxides (e.g. TiO2, ZnO, WO3, and CeO2),4–7 phosphides (InP, CoP and Ni2P),8–10 nitrides (GaN and C3N4)11,12 and metal sulphides (MoS2 and WS2).13,14 Photocatalysts, such as TiO2, ZnO and MnS, have limitations that their band gap is >3 eV and they can be activated only by UV light. Photocatalysts such as MoS2 and C3N4 have band gaps <3 eV, but the lifetime of charge carriers generated in the presence of light is very less, thus hampering their technological applications.
Metal halide perovskites exhibit outstanding optoelectronic and photocatalytic properties due to their high absorption coefficient, ease of fabrication, and extended diffusion length of charge carriers. These properties enable their use in a variety of applications, such as photovoltaics, LEDs, solar fuels and scintillators. To commercially utilize perovskite materials across diverse applications, the key requisites are high shelf life, efficiency and low cost of fabrication. Hybrid organic–inorganic halide perovskites have drawn the attention of the scientific community on account of their spectacular performance in solar cell devices. The power conversion efficiency of solar cells has exceeded 23%. One of the major drawbacks of these materials is their poor stability at elevated temperatures and under humid conditions. Wang et al. concluded that methylammonium lead iodide (MAPbI3) decomposes into MAI and PbI2, with MAI further transforming into CH3NH2 and HI.15 Uncapped MAPbI3 decomposes within a few minutes. The breakdown is due to the fact that MA+ has a large dipole moment, leading to a resonance coupling effect between MA+ and water molecules, resulting in the formation of hydrogen bonds.
It has been observed that replacing organic elements with inorganic elements improves their stability against thermal decomposition.15 Pristine cesium lead iodide (CsPbI3) has a band gap (Eg = 1.73 eV) but it has limited stability and it transforms into an orthorhombic phase, which is non-perovskite at ambient temperature.16 CsPbBr3 is considered to be the most stable cesium-based perovskite material. However, it is characterized by a large band gap making it not an optimum material for solar harvesting applications.16 Mixed halide perovskite CsPbI2Br does not suffer from stability issues compared to CsPbI3 and is characterized by an optimum value of band gap, which makes it suitable for optical and photocatalytic applications. Wang et al. synthesized CsPbBr3/Pt–TiO2 composites and achieved hydrogen production from hydrolysis.17 Song et al. synthesized CsPbBr3/polyaniline materials with a photocatalytic hydrogen production having the efficiency of 4.81 mmol h−1 g−1.18 There are only a few studies in the literature, where cesium lead halide perovskites have been explored as photocatalysts. Most of the studies have focussed only on bromine-based halides. A detailed atomistic study is necessary for the development of effective photocatalysts to be employed for water splitting.
In this paper, we used ab initio-time dependent DFT molecular dynamics simulations to study the fundamental mechanism for photocatalytic splitting of water at the (001) and (111) CsPbI2Br/water interface. NAMD simulations helped us to find the important electron excitations induced by photons. We conclude that the hydrogen transfer reaction in CsPbI2Br is driven by holes and have also established a relationship between the re-forming of the bond and the transfer of a charge during the simulation. We also studied the properties of CsPbI2Br (001), (111) surfaces and the mechanism of their degradation in the presence of H2O and OH− species. We performed the first principles calculations to study the origin of the electronic structure of H2O and OH− on the (111) and (001) surfaces of CsPbI2Br.
2 Computational details
Vienna ab initio simulation package developed by the Fakultat fur Physik of the Universitat Wien was used to perform density functional theory (DFT) simulations.19 Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was used with generalized gradient approximation (GGA)20,21 to relax the bulk crystalline structure of CsPbI2Br. We chose 400 eV to be the energy cut-off for the plane wave basis set. Monkhorst k point mesh 8 × 8 × 1 was used to perform the DFT simulations. Geometrical optimization was carried out until the convergence criteria of residual force on the atom was equal to 0.05 eV per angstrom. We employed slab models for the (001) and (111) planes. The interaction between the periodic images can be avoided by using a vacuum of 15 Å.
Ab initio NAMD simulations were performed with the help of Ab Initio Dynamics (PYXAID) code.22,23 The decoherence-induced surface hopping (DISH) approach was used in NAMD simulations.24 NAMD simulations have been used previously in literature to study the mechanism of water splitting25,26 and for estimation of the lifetime of charge carriers.27,28 A 100 fs ab initio MD simulation trajectory was generated with the help of 10 attoseconds timestep. We used ab initio atomistic thermodynamics to compute the Gibbs free energy (ΔG) according to the equation.
| | |
ΔGads = ΔEDFT + ΔEZPE − TΔS
| (1) |
In this equation, Δ
EDFT is the electronic energy obtained from DFT calculations, Δ
EZPE and Δ
S are the difference in the zero-point energy and entropy, respectively, and
T is the temperature (298.15 K).
3 Results and discussion
3.1 NAMD calculations
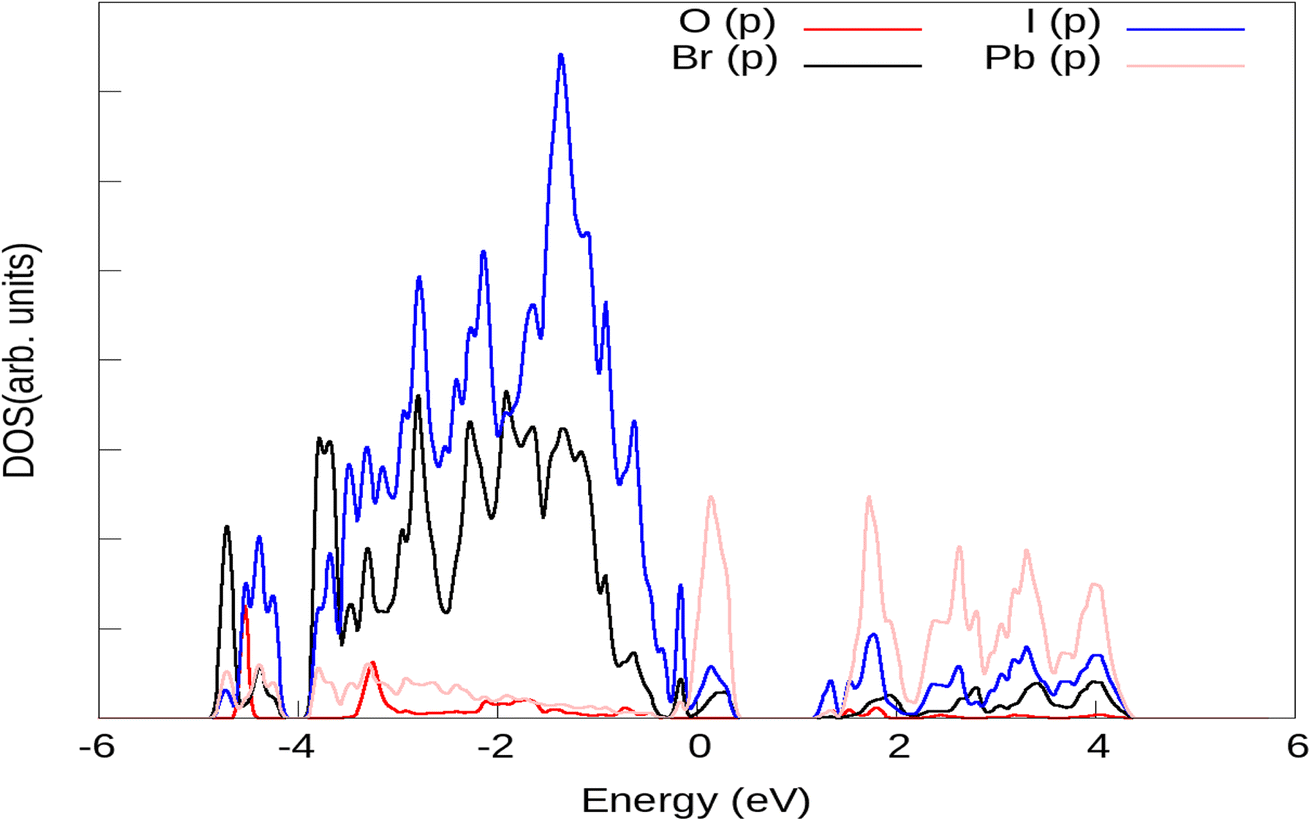
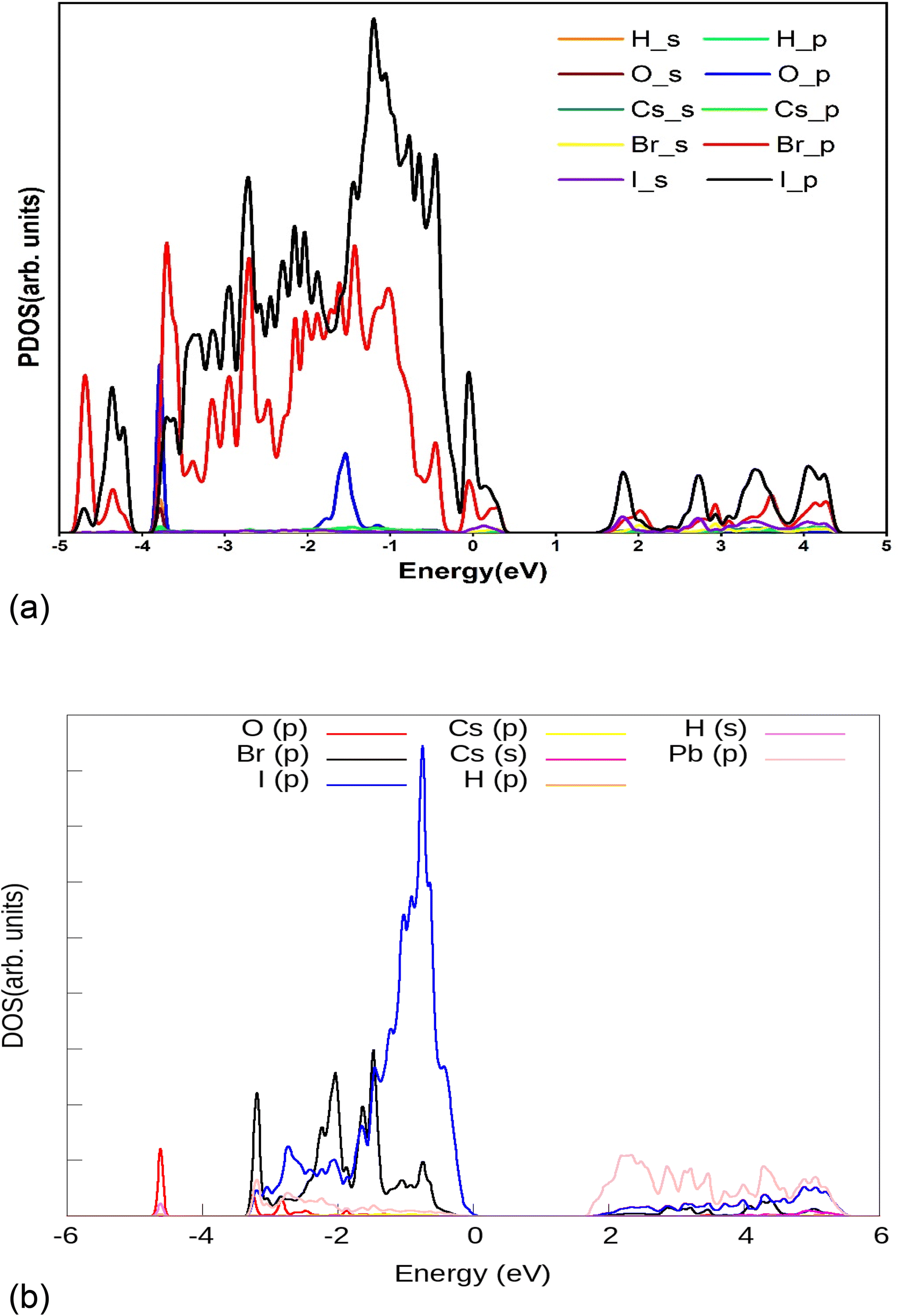
Results of the electronic structure calculation, as presented in Fig. 1 for simulation of H2O on 111 surfaces of CsPbI2Br show that the valence band of (111) surface of CsPbI2Br/water interface is due to the p orbitals of Br and I species. The major contribution is from the p orbitals of the iodine atom. The contribution of s orbitals of I and Br are insignificant. The energy of 2p orbitals of O species is lower than the energy of 2p orbitals localized on iodine atoms. The energy of the hole would be substantially smaller if it is localized on the I species than on the O atom. The hole energy increases as one moves away from the edge of the valence band. If the hole is placed deep in the valence band edge, i.e. at energies −3 eV, where the contribution of O atoms is significant, then the hole will finally migrate to I atoms, where energy is minimum. Back-transfer of the hole from the I orbitals to the orbitals of oxygen is inhibited by the Boltzmann factor because the energy difference between the p orbitals of the I atom and the p orbitals of the O atom is comparatively larger of the order of about 2 eV. As a result, the oxidation of water is not possible. In order to carry out this process, the hole must be localized on the adsorbed water molecules. Under the influence of sunlight, there is a migration of the hole at (111) surface of the CsPbI2Br/water interface. There is a significant change in atomic orbitals near the valence band edge. The valence band edge is still dominated by I atoms, however, the orbitals of O atom are shifted near the edge of the valence band as shown in Fig. 2. Thus, non-adiabatic thermally controlled transitions are possible from low-lying I states to the O-states in the presence of light. Thus, there is an increased possibility of hole localization on O atoms, making oxidation of water on the CsPbI2Br surface feasible.
 |
| | Fig. 1 Projected density of states (PDOS) of the (111) surface of the CsPbI2Br/H2O interface, plotted from NAMD simulations. | |
 |
| | Fig. 2 Projected density of states (PDOS) of the (111) surface of the CsPbI2Br/H2O interface in the presence of light, from NAMD simulations. | |
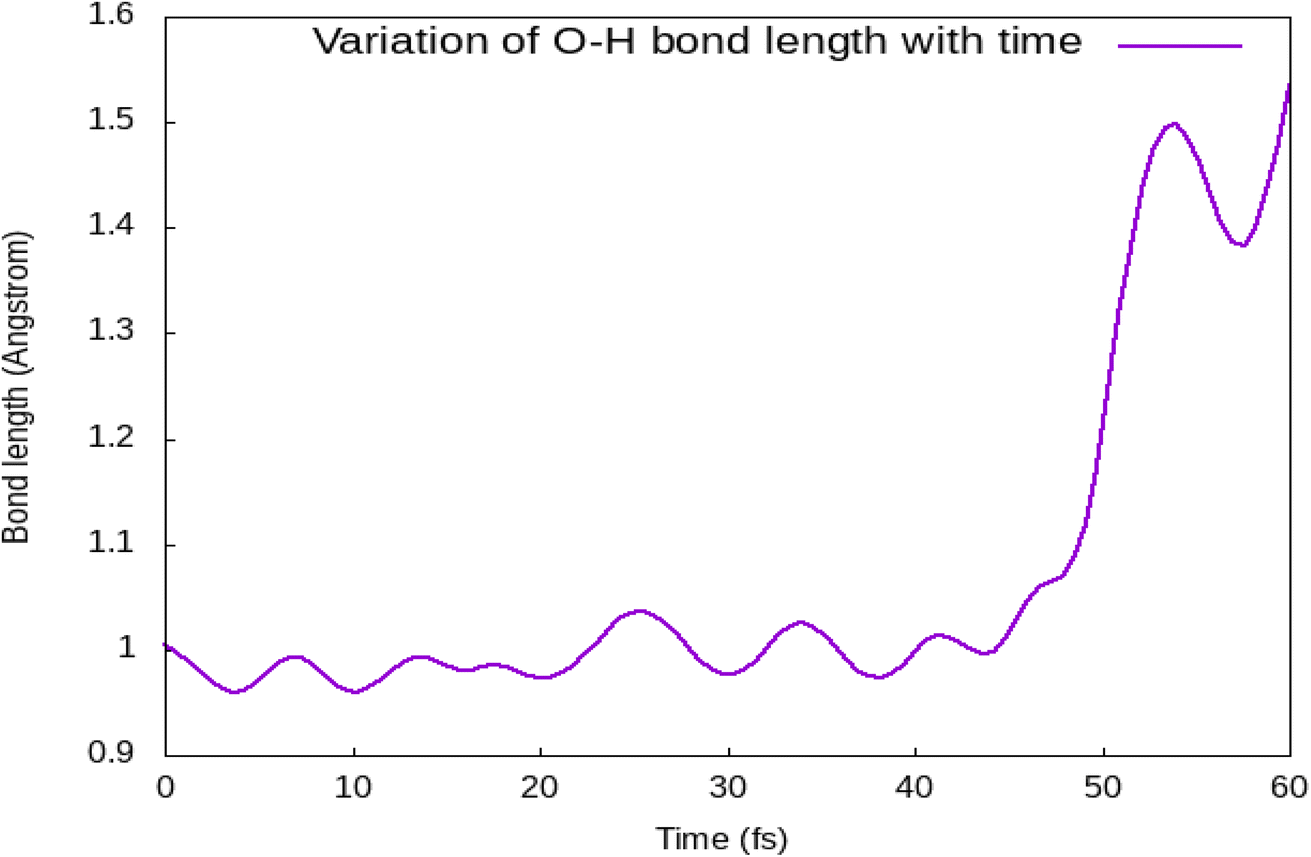
Fig. 3 shows a change in O–H bond length as a function of time for a water molecule that splits during the simulation. We have observed that during the first 20 fs, the water molecule comes near the iodine atom forming the hydrogen bond with it, and splits between 40–60 fs after transforming the hydrogen atom to the I atom. In order to identify the driving force for such photocatalytic splitting, we calculated the electron density of each atom by Hirshfeld charge analysis. During the period 0–20 fs, after photoexcitation, there is a high charge transfer from the I to the Br atom (Fig. 4). There is a hole transfer from the Br to the I atom. During the time span of 30–40 fs, we find a hole transfer from I to the water molecule. This has been possible due to the shifting of p-orbitals of the O atom in the presence of light. This leads to a decrease in O–H bond strength, resulting in vibration excitation. After this, between 40–60 fs, we discovered a reverse hole transfer from splitting water molecule to the I atom leading to a reduction charge transfer of water (Fig. 5). After the transfer of the hole from water to I, there is formation of hydrogen bond between H and I. Most studies have focused on bromine-based halides for photocatalytic splitting of water.17,18 We have considered a mixed halide (Iodine and Bromine) perovskite. The bond between hydrogen and iodine is weaker in comparison to the bond between hydrogen and bromine. A weak bond has a long bond length, which makes it easier for iodine to release hydrogen thus making hydrogen evolution feasible on (111) surface.
 |
| | Fig. 3 Time evolution of the O–H distance of the splitting water molecule on the (111) CsPbI2Br/H2O interface. | |
 |
| | Fig. 4 Time evolution of electron populations for (a) iodine atoms and (b) bromine atoms. | |
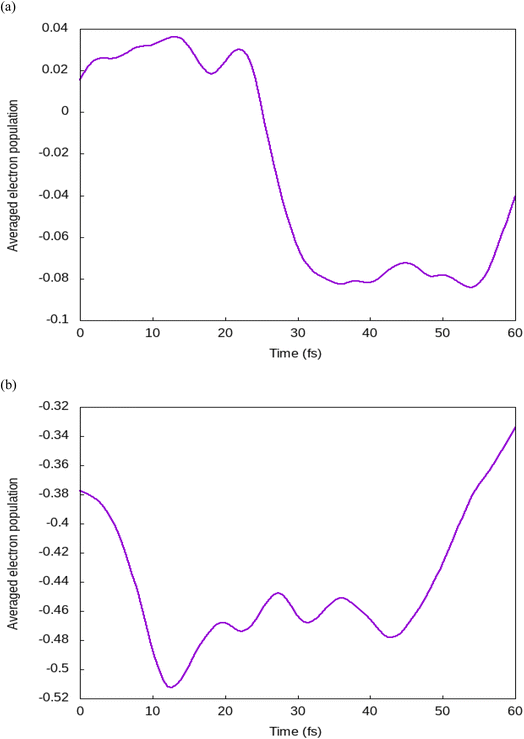 |
| | Fig. 5 Time evolution of electron populations for (a) hydrogen atoms and (b) oxygen atoms. | |
We have carried out the NAMD simulations of H2O on the (001) CsPbI2Br surface. There is no change in the O–H bond length during the simulation on the (001) surface as shown in Fig. S1.† We have also calculated the overlap between wavefunctions of valence band maxima (VBM) and conduction band minima (CBM) according to the following equation.
| |
 | (2) |
We have computed the charge carrier lifetime. Fig. 6 depicts the decay of the first excited state population due to the non-radiative recombination of charge carriers along with the lifetime of the charge carriers. Charge carriers (holes and electrons on the (111) interface show a lifetime of 71 fs, in comparison (001) shows a lifetime of 29 fs). A higher lifetime of charge carriers on the (111) plane is due to the less overlap integral between VBM and CBM, resulting in their reduced recombination on (111).
 |
| | Fig. 6 Population decay and electron–hole recombination dynamics on the (111) CsPbI2Br/H2O interface and the (001) CsPbI2Br/H2O interface. | |
After an electron–hole pair is created, there is reorganization in the electronic structure at the interface. The hole relaxes to the edge of CsPbI2Br by losing energy and coupling to phonons. During the relaxation process, the hole can move into the interfacial water molecules. After photoexcitation, the hole migrates to the CsPbI2Br water interface. The region of the semiconductor around the hole becomes positively charged. The conduction band edge is contributed by I atoms and the orbitals of the oxygen atoms also contribute to the CB edge. Thus, non-adiabatic thermally activated transitions from low-lying I states to O states become realistic.
Chauhan et al. have carried out the NAMD simulations to study the photocatalytic water-splitting properties of the PtSSe/ζ-phosphorene heterostructure.29 They have concluded that PtSSe/ζ-phosphorene possesses a high carrier mobility and suitable band alignment. You et al. have studied the photocatalytic splitting of water in the presence of light.30 They have suggested that water molecule, which forms two hydrogen bonds with nitrogen atoms lead to the photocatalytic reaction under irradiation.
After the formation of hydroxyl species, these species can participate in molecular transformation involving the oxygen evolution reaction (OER).
| | |
S-OH− + h+ → S-O*− + H+
| (3) |
| | |
S-O*− + H2O + h+ → S-OOH− + H+
| (4) |
| |
 | (5) |
| |
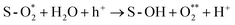 | (6) |
Here, S represents the surface.
We have carried out the NAMD simulations for OH− species on (001) and (111) surfaces of CsPbI2Br. We have found that hydroxyl species form a strong bond with both surfaces but there is no splitting of the O–H bond. One can conclude from NAMD simulations that OER is not feasible on CsPbI2Br surfaces whereas there is a splitting of water on the (111) surface.
3.2 Ab initio calculations
DFT simulations were carried out using the VASP software to support the NAMD simulations. In order to elucidate the interaction between molecule and surface, we present the electronic structure calculation between the (111) and (001) surfaces of CsPbI2Br and various species such as H2O and OH−.
We have calculated minimum energy structures, relaxed adsorption geometry, and binding energy, and plotted the electron localization function (ELF), Charge density difference graphs (CDD) and partial density of states (PDOS). We have taken several initial geometries of H2O, OH− on (001) and (111) surfaces. The (001) and (111) surfaces of CsPbI2Br were selected as absorptive surfaces. We have cleaved the (001) and (111) surfaces from the bulk CsPbI2Br and built a periodic slab.
The adsorption energy formula is
| | |
Eads = Esystem − (Eslab + Emolecule)
| (7) |
when
Eads is negative, then the process occurs spontaneously and the reaction is considered to be exothermic. The adsorption energies of H
2O on (111) and (001) surfaces are 0.38 eV and 0.11 eV, respectively (Fig. S2a and b
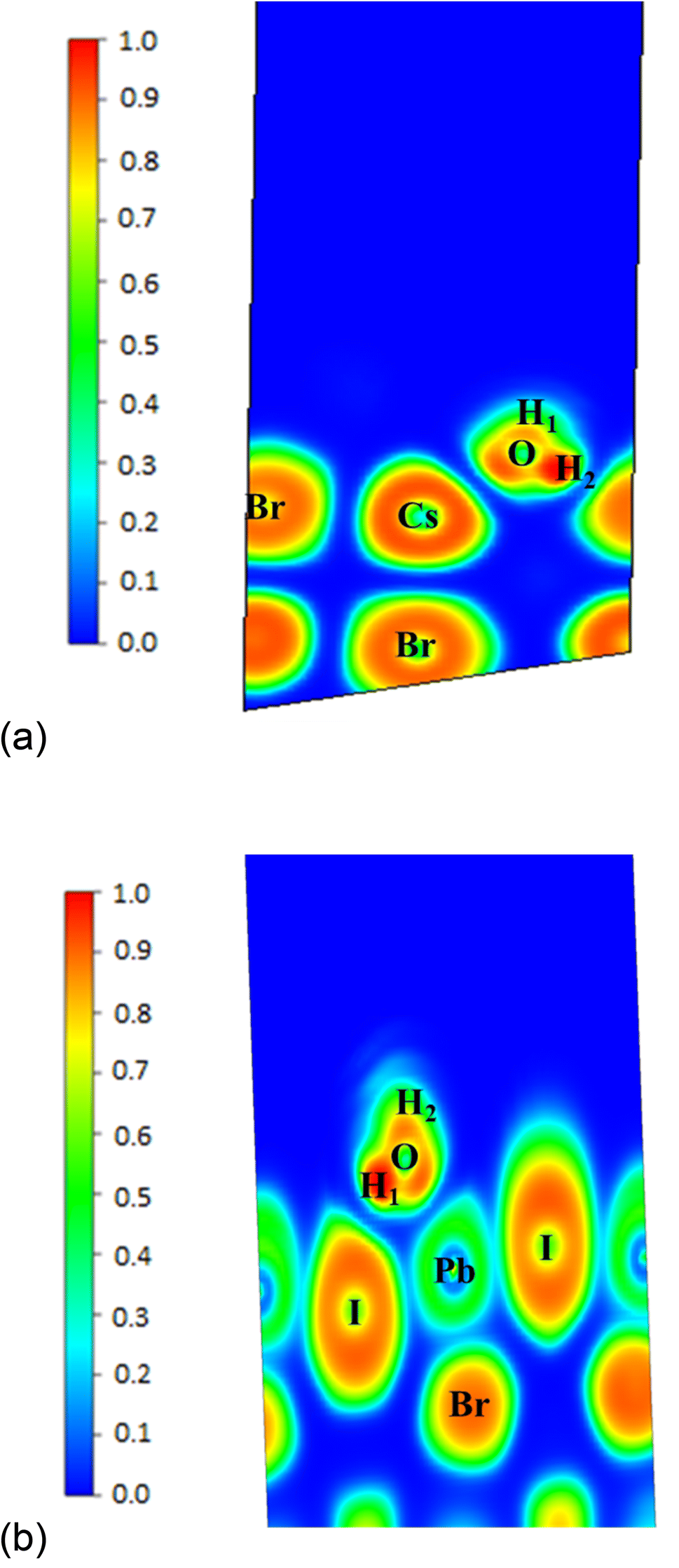
†). The values confirmed that the water is physically adsorbed on the (111) surface. We plotted the ELF graphs. ELF is a dimensionless quantity. Its value lies between 0 and 1, when ELF = 1, it corresponds to perfect localization. The ELF graphs of the adsorption of H
2O on the (111) surface of CsPbI
2Br confirmed that there are covalent interactions between H
2O and the atoms of the (111) surface whereas the corresponding graphs for the surface (001) indicated the absence of both covalent and ionic interactions between H
2O and the (001) surface (
Fig. 7a and b). This is the reason that we have observed the photocatalytic splitting of the water on the (111) surface but not on the (001) surface. The covalent interactions of H
2O on (111) surface are further verified from PDOS graphs. The graph shows the overlapping of the p-orbital of oxygen and the p-orbital of the I atom (
Fig. 8a). In the case of (001) CsPbI
2Br, we have not observed the overlapping of orbitals (
Fig. 8b).
 |
| | Fig. 7 (a) Electron localization function (ELF) graph of H2O on the (111) surface of CsPbI2Br. (b) Electron localization function (ELF) graph of H2O on the (001) surface of CsPbI2Br. | |
 |
| | Fig. 8 (a) Projected density of states (PDOS) of the (111) surface of the CsPbI2Br/H2O interface, plotted from ab initio simulations. (b) Projected density of states (PDOS) of the (001) surface of the CsPbI2Br/H2O interface, plotted from ab initio simulations. | |
Adsorption energies of OH− on (111) and (001) are 0.82 eV and 0.78 eV, respectively. The values confirmed that the adsorption of OH− species is strong on both surfaces. The relaxed configuration is shown in Fig. S3a and b.† The oxygen atom is adsorbed vertically on the (001) surface and horizontally on the (111) surface. The O–Pb bond order is of the order 2.15 angstrom and 2.21 angstrom for (111) and (001), respectively. CDDs are plotted to illustrate the electronic charge distribution on adsorption of the OH− species (Fig. S4a and b†). Cyan and yellow represent the depletion and accumulation of the electronic charge, respectively. Plots have shown that there is a transfer of charge from the OH− to the surface atoms. It has been described by Sabatier that in order for optimal catalyst, the interaction between substrate and catalyst must not be too weak nor too strong. It is because of this reason that we have observed the splitting of hydrogen from H2O on (111), where adsorption is neither strong nor weak. However, the binding energy of H2O on (001) is weak and OH− radical on both the surfaces (111) and (001) is quite strong.
3.3 Gibb's free energy calculation for the dissociation of water
Fig. 9 shows Gibb's free energy for the dissociation of water on (111) and (001) surfaces of CsPbI2Br. Vibrational analysis was used to determine the zero-point energy of all adsorbates. The vibrational modes are derived from the eigenvalues of the system's mass-weighted Hessian matrix. The Hessian matrix is computed using the finite difference approximation with a step size of 0.015 Å. Vibrational contributions to substrate entropies are regarded insignificant and counted as zero in TΔS. The vibrational contributions to the entropies of free molecules in the gas phase are taken from the NIST database31 at 298.15 K and 1 atm. All these values for different species are listed in Table 1. Water dissociation on perovskite surfaces is influenced by the crystallographic orientation of the surface. This is because the orientation of the perovskite crystals might affect the energetics and density of active sites for the dissociation of water. Here, we studied the Gibbs free energy change of water dissociation reaction on the (001) and (111) surfaces of CsPbI2Br. The water dissociation process involves two steps: initial water adsorption and subsequent dissociation into hydroxide (OH−) and proton (H+) ions. The Gibbs free energies change for the steps involved in water dissociation are shown in Table 2 and the reaction path is shown in Fig. 9. The initial adsorption of water on the CsPbI2Br (111) plane results in a negative Gibbs free energy (ΔG < 0), indicating a thermodynamically spontaneous process. This shows a significant contact between the water molecules and the surface sites of the (111) plane, resulting in effective adsorption. After adsorption, water molecules separate into OH− and H+ ions. This dissociation step has also a negative Gibbs free energy of −0.3 eV, proving its spontaneous character on the (111) plane. The favourable thermodynamics of both steps underline the (111) plane's ability to facilitate water dissociation, making it very relevant for catalytic and energy applications utilising perovskite materials.
 |
| | Fig. 9 Gibbs free energy reaction path for the dissociation of water on the [001] and [111] surfaces of CsPbI2Br. | |
Table 1 Zero-point energy corrections and entropic contributions to free energies (eV), where * represents the active sites
| |
EDFT (eV) |
ZPE (eV) |
TS (eV) |
| H2 |
−6.76 |
0.27 |
0.40 |
| H2O |
−14.22 |
0.56 |
0.67 |
| *H2O [001] vdW |
−175.24303 |
0.638591 |
0.183844 |
| *H2O [111] vdW |
−197.98968 |
0.641159 |
0.162666 |
| *OH− [001] vdW |
−171.42240 |
0.319702 |
0.152852 |
| *OH− [111] vdW |
−194.62107 |
0.344538 |
0.100174 |
Table 2 Gibbs free energy changes of water dissociation steps on the [001] and [111] planes of CsPbI2Br, where * represents the active sites
| |
Reaction |
ΔEreact = ΔEDFT + ΔZPE (eV) |
ΔG (eV) |
| 001 vdW |
Step 1: * + H2O → *H2O |
−0.486869 |
−0.01071 |
| Step 2: *H2O → *OH− + H+ |
0.256741 |
0.08773 |
| 111 vdW |
Step 1: * + H2O → *H2O |
−0.549911 |
−0.052577 |
| Step 2: *H2O → *OH− + H+ |
−0.173011 |
−0.31052 |
The situation for the (001) plane of CsPbI2Br is quite different. Water adsorption is not thermodynamically favourable. The dissociation of the water molecule on the (001) surface may require additional energy as the Gibbs free energy in this shows a positive value.
4 Conclusion
We report nonadiabatic quantum dynamics simulation of the splitting of water driven by light on (111) and (001) surfaces of CsPbI2Br. We captured the quantum motion of charge carriers (electrons and holes) on CsPbI2Br surfaces. We observed the transfer of the hole from I to water and the subsequent proton transfer event. After the relaxation of energy, the hole is localized on oxygen species, thus making the splitting of water possible from both thermodynamics and kinetic points of view on the (111) surface. First principles calculation has shown that splitting of water on (111) is possible due to the fact that adsorption energy values of water are not too large or too small whereas the value is negligible on (001) surface. Ab initio calculations indicated the covalent interaction between water and the (111) surface of CsPbI2Br. OH− species are strongly adsorbed on (111) and the (001) plane. Ionic interactions drive the adsorption of OH− on CsPbI2Br planes.
Data availability
All data supporting the findings of this study are included within the manuscript and as part of the ESI.†
Author contributions
Aman Kaura: conceptualization, methodology, software, writing – original draft. Harjot Sidhu: methodology, software, writing, Neelam Minhas: methodology, software, Gh Mustafa: data curation, methodology Gurinder Singh: validation, writing – review & editing. JK Goswamy: supervision.
Conflicts of interest
There are no conflicts to declare.
References
- L. Wang, S. Li, I. M. Ahmad, G. Zhang, Y. Sun, Y. Wang, C. Sun, C. Jiang, P. Cui and D. Li, Sci. Total Environ., 2023, 887, 164055 CrossRef CAS PubMed.
- S. Kuskaya, F. Bilgili, E. Mugaloglu, K. Khan, M. E. Hoque and N. Toguc, Renew. Energy, 2023, 206, 858–871 CrossRef CAS.
- J. Zheng and B. Zeng, Sustain. Energy Technol. Assess., 2023, 57, 103303 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. Ghosh, P. S. Das, D. Sarkar, S. Pal, M. K. Naskar, Y. S. Chaudhary, S. Dey and C. Sinha, ACS Appl. Polym. Mater., 2023, 5, 9918–9930 CrossRef CAS.
- C. Xie, W. Chen, S. Du, D. Yan, Y. Zhang, J. Chen, B. Liu and S. Wang, Nano Energy, 2020, 71, 104653 CrossRef CAS.
- M. Reli, N. Ambrozova, M. Valaskova, M. Edelmannova, L. Capek, C. Schimpf, M. Motylenko, D. Rafaja and K. Koci, Energy Convers. Manage., 2021, 15, 114156 CrossRef.
- S. Yu, X. B. Fan, X. Wang, J. Li, Q. Zhang, A. Xia, S. Wei, L. Z. Wu, Y. Zhou and G. R. Patzke, Nat. Commun., 2018, 9, 4009 CrossRef PubMed.
- R. Deng, M. Guo, C. Wang and Q. Zhang, Nano Mater. Sci., 2024, 6, 139–173 CrossRef CAS.
- X. Yan, M. Xia, H. Liu, B. Zhang, C. Chang, L. Wang and G. Yang, Nat. Commun., 2023, 14, 1741 CrossRef CAS PubMed.
- N. A. A. Zulkifli, S. M. Said, M. F. M. Taib, K. Arifin, S. M. Mahmood, K. L. Woon, S. K. Patel, C. L. Tan and R. Zakaria, Mater. Today Commun., 2020, 25, 101646 CrossRef CAS.
- Y. Li, S. Zhu, Y. Liang, Z. Li, S. Wu, C. Chang, S. Luo and Z. Cui, Mater. Des., 2020, 196, 10919 Search PubMed.
- U. Gupta and C. N. R. Rao, Nano energy, 2017, 41, 49–65 CrossRef CAS.
- R. Kumar, D. Das and A. K. Singh, J. Catal., 2018, 359, 143–150 CrossRef CAS.
- Z. Wang, Z. Shi, T. Li, Y. Chen and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 1190–1212 CrossRef CAS PubMed.
- F. Bella, P. Renzi, C. Cavallo and C. Gerbaldi, Chem.–Eur. J., 2018, 24, 12183–12205 CrossRef CAS PubMed.
- H. Wang, X. H. Wang, R. T. Chen, H. F. Zhang, X. L. Wang, J. H. Wang, J. Zhang, L. C. Mu, K. F. Wu, F. T. Fan, X. Zong and C. Li, Int. J. Hydrogen Energy, 2019, 4, 40–47 CAS.
- W. Song, Y. Wang, C. Wang, B. Wang, J. Feng, W. Luo, C. Wu, Y. Yao and Z. Zou, ChemCatChem, 2021, 13, 1711–1716 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew and A. Zunger, Phys. Rev. B:Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B:Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- A. V. Akimov and O. V. Prezhdo, J. Chem. Theor. Comput., 2013, 9, 4959–4972 CrossRef CAS PubMed.
- A. V. Akimov and O. V. Prezhdo, J. Chem. Theor. Comput., 2014, 10, 789–804 CrossRef CAS PubMed.
- H. M. Jaeger, S. Fischer and O. V. Prezhdo, J. Chem. Phys., 2012, 137, 545 CrossRef PubMed.
- P. You, C. Lian, D. Chen, J. Xu, C. Zhang, S. Meng and E. Wang, Nano Lett., 2021, 21, 6449–6455 CrossRef CAS PubMed.
- A. V. Akimov, J. T. Muckerman and O. V. Prezhdo, J. Am. Chem. Soc., 2013, 23, 8682–8691 CrossRef PubMed.
- N. Minhas, G. Mustafa, K. Kaur, N. Kaur, G. Singh, A. Kaura and J. K. Goswamy, J. Phys. Chem. Solids, 2022, 170, 110905 CrossRef CAS.
- Gh. Mustafa, N. Minhas, H. Singh, J. Singh, G. Singh, A. Kaura and J. K. Goswamy, J. Solid State Chem., 2023, 322, 123981 CrossRef CAS.
- P. Chauhan and A. Kumar, Sci. Rep., 2024, 14, 21618 CrossRef CAS PubMed.
- P. You, C. Lian, D. Chen, J. Xu, C. Zhang, S. Meng and E. Wang, Nano Lett., 2021, 21, 6449 CrossRef CAS PubMed.
- Computational Chemistry Comparison and Benchmark Database, https://cccbdb.nist.gov.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
J. K. Goswamya
*b and
J. K. Goswamya