
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the role of polymer interactions during water electrolysis under basic conditions with bifunctional cobalt corroles†
Sameeta Sahoo ,
Elizabeth K. Johnson,
Xiangru Wei,
Sen Zhang and
Charles W. Machan*
,
Elizabeth K. Johnson,
Xiangru Wei,
Sen Zhang and
Charles W. Machan*
Department of Chemistry, University of Virginia, PO Box 400319, Charlottesville, VA 22904-4319, USA. E-mail: machan@virginia.edu
First published on 31st July 2024
Abstract
With green hydrogen fuel continuing to be an important option for energy storage, studies on water-splitting reactions have attracted increasing attention. Within a multitude of parameters that have the potential to be explored to enhance water electrolysis, one of the most consequential factors is the development of an efficient electrocatalyst. The effectiveness of Co(III) corroles as electrocatalysts has largely been investigated in homogenous, non-aqueous or acidic environments. We report the use of heterogenized Co(III) corroles as bifunctional catalysts for water splitting under basic conditions, finding that the inclusion of alkyl chains on the ligand framework has a beneficial impact on electrocatalytic properties. Two new corroles have been isolated where the para positions in the fluorophenyl meso substituents of the parent cobalt(III) 5,10,15-tris(pentafluorophenyl)corrole Co(tpfpc) 1 have been modified with heptyl, [Co(ttfphc)] 2 and dodecyl [Co(ttfpdc)] 3 amines via a nucleophilic aromatic substitution reaction. The electronic structure of these new complexes and properties of the resultant catalyst inks are significantly altered relative to the parent complex by the presence of the alkyl chains, as evidenced by changes in catalytic onset potentials and Tafel behavior during water splitting at pH 14. All catalysts were found to exhibit bifunctional behavior with reasonable stability, and the interactions of the alkyl amine groups with the supporting polymer in the catalyst ink have been found to have an important role in altering corrole aggregation and therefore Co active site accessibility during deposition of the catalyst inks.
Introduction
Renewable sources of energy are an attractive answer to escalating concerns over minimizing the environmental impact1 of meeting increasing global energy demand, but still face significant barriers to wide-scale implementation. There is a need for cost-effective energy storage devices which reversibly store the electrical energy derived from renewable sources in the form of chemical energy.2 Hydrogen is attractive as a product of such reactions, because the corresponding fuel cells generate electrical energy with water as the only byproduct.3 Although the required oxygen for these fuel cell designs can be sourced directly from the atmosphere, there is a need to generate hydrogen in a carbon-neutral fashion. Water-splitting electrolyzers could serve as the source of this hydrogen fuel, if the electricity used comes from renewable sources.4The development of electrocatalysts for water splitting, despite being continuously explored, remains an ongoing challenge because of the chemical properties required for stability and activity under reaction conditions, as well as the significant scope of possible optimization parameters. Currently, there are three primary designs used for water-splitting electrolyzers. Proton-exchange membrane electrolyzers (PEMEL) offer the advantage of using a solid membrane but require the use of costly and scarce platinum group metals (PGM) to operate in strongly acidic conditions.5 Conversely, alkaline electrolyzers (AEL) can utilize non-PGM catalysts, but grapple with the challenges of low efficiency and degradation of the catalyst in harsh basic conditions, where up to 40 wt% of hydroxide electrolyte is used. Thus, there has been increasing interest in hydroxide-exchange membrane electrolyzers (HEMEL), which can use non-PGM transition metal catalysts at either pH 14 or in pure water. While AEL uses a microporous diaphragm and requires harsh basic conditions (25–40 wt% KOH) to operate, HEMEL uses a solid hydroxide exchange membrane and operates at much milder conditions, minimizing shunt currents that lead to corrosion and reduced efficiency. Furthermore, HEMEL provides a cost-effective hydrogen production route by avoiding the additional costs associated with AEL, which include compensating for the lower intrinsic hydrogen output and operational inefficiencies at varying pressures due to safety concerns related to the strong base used. If sufficient advancements are made in catalyst and device stability, HEMEL could provide a more cost-effective and practical system than PEMEL or AEL. Thus, there is a need to optimize electrocatalyst performance by understanding deactivation mechanisms and improving the stability of the catalyst/ionomer interface.6–8 While single atom catalysts (SACs) can bridge aspects of homo- and heterogeneous catalysis,9 molecular catalysts offer possible advantages in terms of characterizing the active site and chemically modulating it, which could provide a more direct route to achieving the desired properties.10–19
In particular, metallocorroles have been widely explored as electrocatalysts due to their unique electronic properties, as well as the relative ease of synthetic tuning via peripheral and core modulation.20–27 Indeed, the electrocatalytic activity of cobalt corroles and related derivatives have been studied by Kadish et al. for dioxygen reduction along with the study of the possible oxidation states under both homogeneous and heterogeneous catalytic conditions.28,29 Corroles have a contracted ring structure relative to the parent porphyrins, rendering the square planar coordination environment trianionic, which can be beneficial in stabilizing higher formal metal oxidation states, particularly those relevant to water oxidation. The use of cobalt corroles as catalysts for the hydrogen and oxygen evolution reactions (HER and OER, respectively) has been previously reported under (photo)electrochemical conditions.30,31 Relevant to the studies described here, Mondal et al. have reported the use of octa-fluoro-β-substituted Co(tpfpc) as an immobilized catalyst for the HER under acidic conditions.32 In an effort to address the higher cost for the synthesis of the octa-fluoro substituted analog, they also reported the synthesis of octa-chloro and octa-bromo derivatives, but found the fluoro derivative to have the highest catalytic activity.33 The bispyridyl analogue, Co(tpfpc)(py)2, has been reported to act as a bifunctional catalyst for HER and OER in acetonitrile.33 Replacement of two of the meso pentafluorophenyl groups with 4-aniline on the tpfpc core was also reported to result in a bifunctional water-splitting catalyst under homogeneous as well as heterogeneous conditions across pH values of 0, 7, and 14.34 The activity was proposed to benefit from both the electron-withdrawing effect of the pentafluorophenyl group enhancing the HER activity and the basicity induced by the 4-aniline group facilitating the OER activity. Finally, the immobilization of the Co(III) corrole derivatives on carbon nanotubes via covalent attachment (both short conjugated linkers and long alkane chains), as well as through π–π interactions and simple adsorption were compared.35 The hybrid electrode materials obtained from covalently linking the metallocorrole active site via a conjugated linker were found to have the highest relative catalytic activity for both the HER and the OER. The enhancement of the catalytic activity was attributed to the strong attachment of the catalysts to the carbon nanotubes stabilizing the active intermediates at all proton activities and therefore decreasing the rate of degradation.
In this study, two new Co(III) corroles with heptyl and dodecyl amine as p-substituents on the meso fluorophenyl groups are reported. These complexes have been isolated via a nucleophilic aromatic substitution reaction involving the parent cobalt(III) 5,10,15-tris(pentafluorophenyl)corrole Co(tpfpc) 1 with heptyl amine, [Co(ttfphc)] 2 and dodecyl amine, [Co(ttfpdc)] 3. All three complexes contain a single equivalent of triphenylphosphine at one of the axial coordination sites. Interestingly the Co(III) complexes of all the three corroles were found to be bifunctional, demonstrating catalytic efficacy for both the HER and the OER reactions at pH 14 (1 M KOH). Mechanistic investigations suggest that there is a benefit both to having the aminoalkyl chain attached to the corrole core, as well as in increasing its length. It is hypothesized that interactions involving the aminoalkyl groups improve their dispersion in the catalyst ink prior to deposition, improving the reaction environment by altering relative positioning with respect to other equivalents of metallocorrole, as well as the conducting carbon and polymer binder. These results include an analysis of stability and activity trends with respect to loading methods which offer insight into possible areas of future development.
Experimental results
Synthesis
Co(tpfpc) 1 was synthesized according to a reported procedure.36 Complexes 2 and 3 were synthesized by heating the free base tris-pentafluorophenyl corrole ligand in DMSO with 1-heptylamine and 1-dodecylamine, respectively,37 and subsequently metalating them by stirring with a mixture of cobalt acetate tetrahydrate, sodium bicarbonate, and triphenylphosphine in ethanol. Both substituted metallocorroles were characterized completely by NMR spectroscopy, high-resolution mass spectrometry, and UV-vis spectroscopy (Fig. S1–S13, ESI†). All characterization data are consistent with the proposed structures depicted in Fig. 1.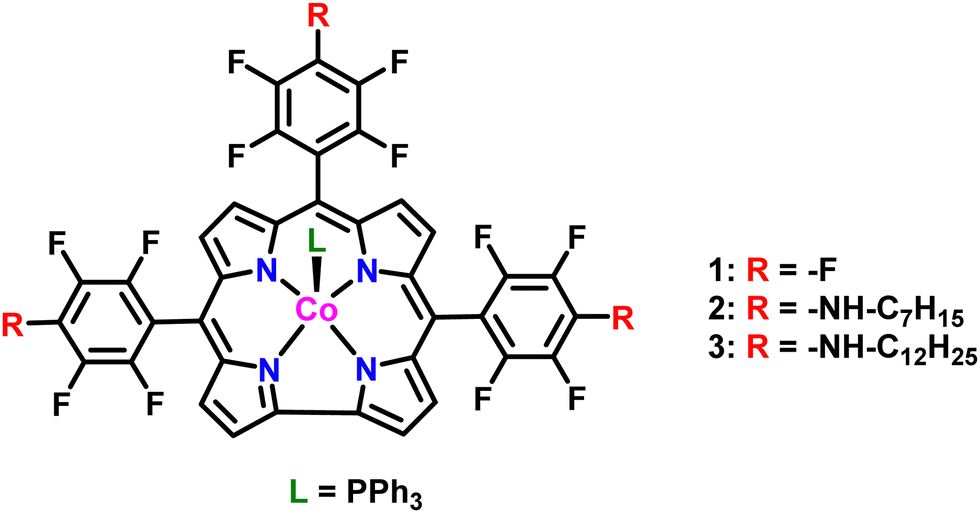 | ||
| Fig. 1 Cobalt corroles studied here as bifunctional catalysts for water splitting at pH 14. | ||
Linear sweep voltammetry (LSV) for compounds 1–3 were recorded in 1 M KOH solution with Hg/HgO as the reference electrode. A glassy carbon rod was used as counter electrode for the HER studies while a Pt wire was used as the counter electrode for the OER studies. Catalyst inks were prepared by sonicating a desired concentration of metallocorrole (1 or 3 mM, as indicated), 12.5 mg of Vulcan carbon XC-72, 0.5 mL of 5% Nafion in isopropanol, 1 mL of ethanol and 3.4 mL of dichloromethane for 30 m. For the 1 mM ink, the desired volume was deposited on EP40 carbon paper using a mechanical pipettor onto an area of 1 cm2. For the 3 mM ink, approximately 0.25 mL of ink solution was spray-coated with an airbrush on 1 cm2 of carbon paper. Representative catalyst films were characterized by solid-state UV-vis spectroscopy (Fig. S14, ESI†) and water contact angle measurements (Fig. S15, ESI†).
HER studies
Initial studies focused on the HER performance for all three complexes at pH 14 (1 M KOH) by depositing catalyst ink containing a 1 mM concentration of metallocorrole (Fig. 2). The onset potential for 1, which has three pentafluorophenyl substituents at the meso positions, was observed to be at −0.75 V vs. RHE. There is a shift to lower onset potentials with an increase in the chain lengths, with the onset potential for 2 being observed at −0.68 V while that for 3 being at −0.63 V vs. RHE. An increase in the catalytic activity is also seen with addition of the longer alkyl chain, despite the positive potential shift. The reaction overpotentials at 10 mA cm−2 upon incorporation of the amines decrease from a value of 0.96 V for 1 to 0.90 V for 2 and 0.86 V for 3. The turnover frequency (TOF) values calculated based on the amount of metal ions quantified by ICP-OES measurements (assuming all sites are active) revealed an increase in catalytic activity when compared at the same overpotential from 1 at 0.413 s−1 to 1.02 s−1 for 2 with the incorporation of the heptyl amine, a 2.5-fold increase. The further increase in aminoalkyl chain length in complex 3 achieved a TOF 2.98 s−1, which represents a 7.2-fold increase compared to 1 (Table S6, ESI†). Catalytic Tafel plots are a useful tool for comparing the catalytic activity of electrocatalysts.38,39 The Tafel slopes for complexes 1–3 were determined to be >120 mV, indicating that either the Volmer or Heyrovsky step could be rate-determining,40 although this is obscured by probable limitations in the accessibility of Co active sites in the catalyst layer. As compared to 1, which has a Tafel slope of 275 mV dec−1, there is a decrease upon the incorporation of long chain amines, with the Tafel slope for 2 dropping to 218 mV dec−1, while 3 has a comparable value of 214 mV dec−1 (Fig. S19–S21, ESI†). Solid-state UV-vis measurements suggest differences in metallocorrole aggregation between all three ink samples (Fig. S14, ESI†) compared to solution-phase measurements (Fig. S13, ESI†). Further, water contact angle measurements (Fig. S15, ESI†), suggest that a difference in the wettability of the three catalyst inks. These data are consistent with a difference in the dispersion of the metallocorrole complexes within their respective catalyst films perturbing their interactions with the working solution as well as the accessibility of Co active sites.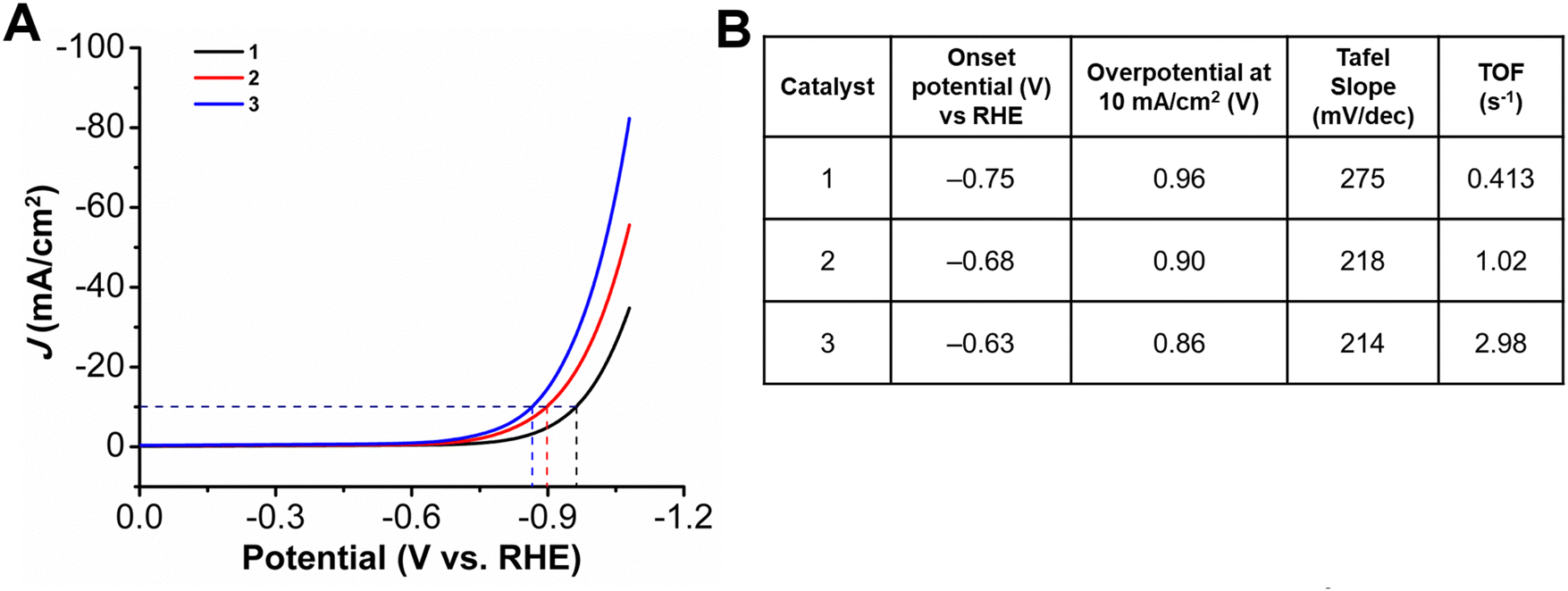 | ||
| Fig. 2 (A) LSVs and (B) summary table of HER results obtained with 1 cm2 carbon paper coated with a catalyst ink containing Vulcan carbon, Nafion, and 1.0 mM of indicated complex as the working electrode. All experiments performed under N2 saturation in 1.0 M KOH; glassy carbon counter electrode, Hg/HgO reference electrode; 100 mV s−1 scan rate. | ||
Next, the relationship between catalyst loading and HER activity was explored by preparing electrodes with a catalyst ink containing a 3 mM concentration of the metallocorrole (Fig. 3). For 1, the increase in catalyst ink concentration resulted in an improvement of catalytic activity, with the HER onset potential shifting to −0.67 V vs. RHE (80 mV more positive). On the other hand, for aminoalkyl-appended complexes 2 and 3, the increase in concentration of complexes led to a suppression of activity: onset potentials shifted to more negative potentials in both cases, accompanied by a decrease in the observed current density (Table S2, ESI†). With elevated metallocorrole concentration in the catalyst ink, there is also a decrease in the observed Tafel slope for 1, with a slope of 226 mV dec−1 (Fig. S19, ESI†), while for 2, there is approximate doubling to 404 mV dec−1 (Fig. S20, ESI†), suggestive of the Tafel slope being loading dependent.41 However, Tafel studies could not be completed for 3 at these elevated ink concentrations, due to instability under chronoamperometry conditions.
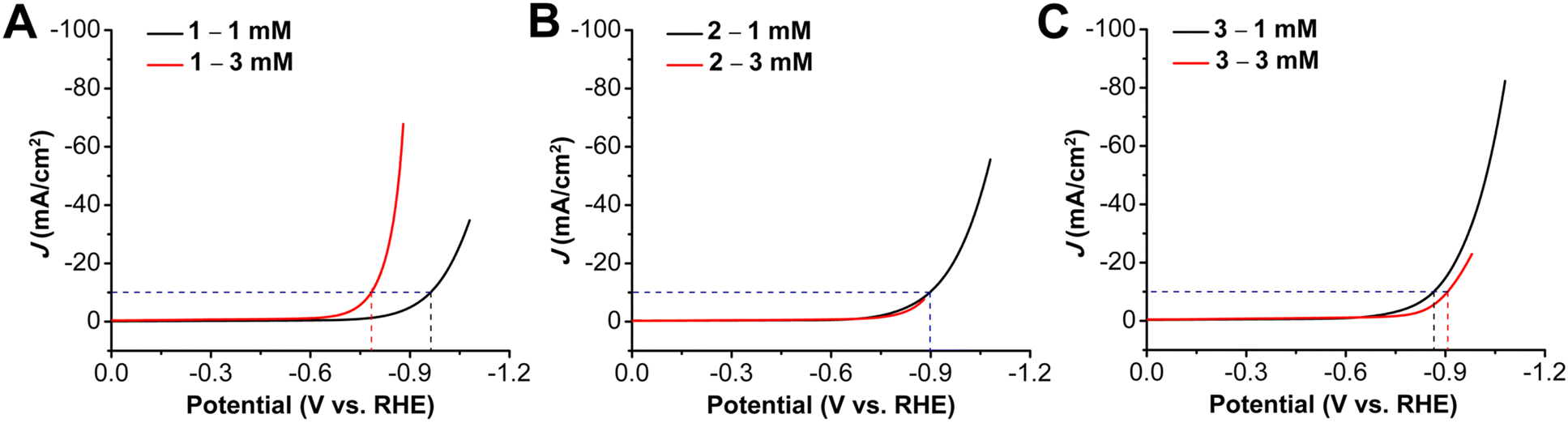 | ||
| Fig. 3 LSVs for on HER performance of 1 mM and 3 mM ink solutions of complexes (A) 1 (B) 2 and (C) 3. Obtained with 1 cm2 carbon paper coated with a catalyst ink containing Vulcan carbon, Nafion, and given concentration of indicated complex as the working electrode. All experiments performed under N2 saturation in 1.0 M KOH; glassy carbon counter electrode, Hg/HgO reference electrode; 100 mV s−1 scan rate. | ||
Stability studies conducted at constant current for the three complexes revealed distinctively different behavior for the different loadings (note that all experiments use catalyst ink on carbon paper with 1 cm2 geometric area). For complex 2, the overpotential values are almost identical at −10 mA for both 1 mM and 3 mM metallocorrole concentrations in the catalyst ink, however, there is a much larger shift of overpotential for the 3 mM ink solution of 1 (from 0.80 to 0.92 at −10 mA) (Fig. S16, ESI†). For the heptylamine-functionalized analogue 2, along with a similar increase in overpotential with the more concentrated catalyst ink, there is also a decrease in stability with a much larger difference in potential between applied currents of −10 mA and −20 mA over 30 m (Fig. S17, ESI†). For complex 3, however, despite a similar shift of overpotential from 0.90 to 0.96 for −10 mA in the 3 mM catalyst ink, there is greater stability over a longer period of time, especially under a constant applied current of −30 mA and −50 mA. Additionally, the difference in overpotential for studies at all applied currents is much less for 3 mM catalyst inks of 3 than 1 mM, confirming an increase in stability (Fig. S18, ESI†). The stability tests under chronopotentiometry conditions at −10 mA conducted for longer period of 5 h showed voltage degradations of −12.1 mV h−1, −18.1 mV h−1 and −14.1 mV h−1 for 1, 2 and 3 respectively (Fig. S28–S30, ESI†, Table S4).
Scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDS) images suggested a uniform dispersion of the metal on the electrode, before and after the stability experiments for the HER (Fig. S34–S42, ESI†). Likewise, a uniform distribution of phosphorous on the surface was also observed, consistent with the persistence of the axial ligand following the application of potential. While the SEM images did not show large regions suggestive of bulk metal structures, evidence of nanoparticulate was apparent in some of the transmission electron miscroscopy (TEM) images collected. Complex 1 did not generate nanoparticles after chronopotentiometry at −10 mA and −50 mA for 30 min, however, for 2 and 3 some nanoparticulate was observed, although there was not a consistent trend for this observation based either on chain length or applied current density (Fig. S52–S57, ESI†).
OER studies
Trends similar to those observed for the onset potentials and overpotentials at 10 mA cm−2 for the HER studies were also observed in LSVs for the OER studies (Fig. 4). While 1, with a pentafluoro substituent has an onset potential at 1.68 V vs. RHE, there is a decrease to 1.56 V for 2, with an heptylamine chain appended. With an increase in the alkyl chain length, there is a further shift in the onset potential to 1.51 V vs. RHE for 3. The effect of incorporation of alkyl chains can also be seen with the decrease of the overpotential at 10 mA cm−2 to 0.51 V and 0.46 V for 2 and 3, respectively, compared to 0.56 V for 1. The TOF values (again calculated using mass loadings obtained by ICP measurements) at the same overpotential showed a similar catalytic activity trend to that observed for the HER studies: 1 has a TOF of 0.169 s−1, 2 has a TOF of 0.301 s−1, and 3 is again the most active, with a TOF of 1.11 s−1 (Table S6, ESI†). The improved activity of 3 is a 3.6-fold increase as compared to 2 and a 6.5-fold increase as compared to 1, without the alkyl chains. There is a decrease in Tafel slope to 156 mV dec−1 for 2, as compared to 168 mV dec−1 for 1. However, for 3, with the longest aminoalkyl chain, there is an increased Tafel slope of 223 mV dec−1. The Tafel slopes for complexes 1 to 3 are again >120 mV, suggesting that while oxygen–oxygen bond formation is a possible rate-limiting step, limitations of Co site accessibility in the catalyst layer dominate the observed catalytic response.40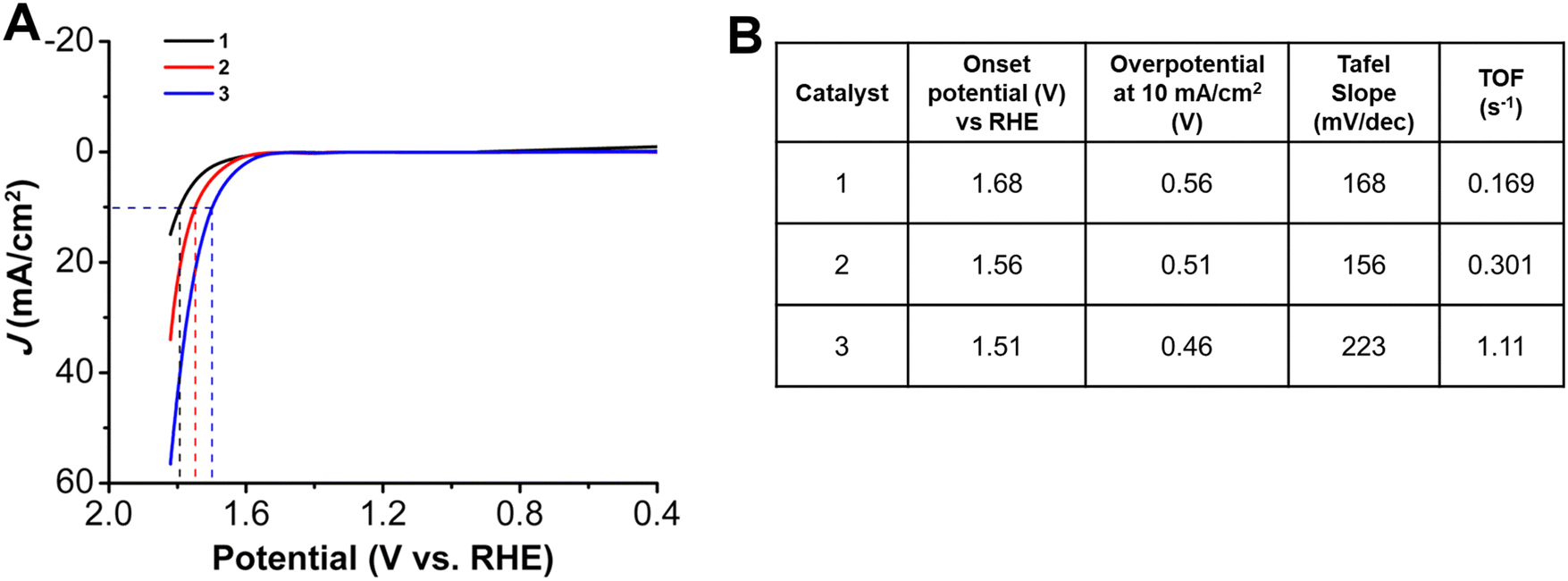 | ||
| Fig. 4 (A) LSVs and (B) summary table of the OER results obtained with 1 cm2 carbon paper coated with a catalyst ink containing Vulcan carbon, Nafion, and 1.0 mM of indicated complex as the working electrode. All experiments performed under N2 saturation in 1.0 M KOH; Pt wire counter electrode, Hg/HgO reference electrode; 100 mV s−1 scan rate. | ||
Catalyst loading studies revealed that OER performance also showed a dependence on the presence and the length of the aminoalkyl chains. With a 3 mM ink solution for 1 there was a shift in onset potential to 1.54 V vs. RHE (80 mV less positive) accompanied by a decrease in overpotential at 10 mA cm−2 to 0.42 V (Fig. 5). For 2, the OER onset potential and the overpotential at 10 mA cm−2 were almost identical to those with 1 mM ink loading. The trend to poorer performance with the higher 3 mM ink loading of the aminoalkyl-modified catalysts continues for 3, which experiences a positive voltage shift in OER onset potential to 1.63 V vs. RHE and overpotential at 10 mA cm−2 to 0.54 V. Increased catalyst loading also showed a decrease in stability for complexes 1–3 under chronopotentiometry conditions, however, the instability was more pronounced for 1 (Fig. S22–S24, ESI†). Tafel studies could not be conducted for 1 with 3 mM ink loading, due to instability of the catalyst under chronoamperometry conditions. For both 2 and 3, there was an increase in Tafel slopes to 340 mV dec−1 and 303 mV dec−1 (Fig. S26, S27 and Table S3, ESI†). The stability tests at 10 mA over a period of 5 h, showed the degradation to be as 6.04 mV h−1, 18.1 mV h−1 and 14.1 mV h−1 for 1, 2 and 3 respectively. Interestingly, the degradation rates for the alkyl amino appended catalysts 2 and 3, were almost same under both HER and OER conditions, while for 1, it was almost half under OER conditions as compared to the HER conditions (Fig. S31–S33, ESI†, Table S5).
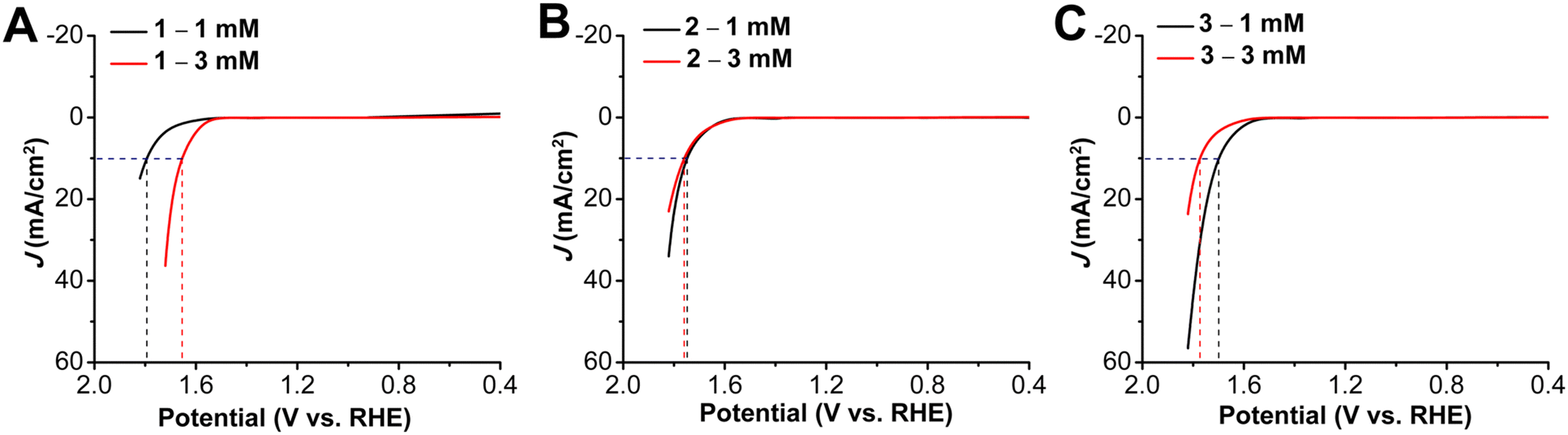 | ||
| Fig. 5 LSVs for OER performance of 1 mM and 3 mM ink solutions of complexes (A) 1 (B) 2 and (C) 3. Obtained with 1 cm2 carbon paper coated with a catalyst ink containing Vulcan carbon, Nafion, and given concentration of indicated complex as the working electrode. All experiments performed under N2 saturation in 1.0 M KOH; Pt wire electrode, Hg/HgO reference electrode; 100 mV s−1 scan rate. | ||
SEM-EDS images again confirmed uniform dispersion of Co and P, both before and after recording the LSV, as well as after the chronopotentiometry conditions of 10 mA and 50 mA for 30 mins (Fig. S43–S51, ESI†). Unlike under HER conditions, for OER neither the SEM nor the TEM images (Fig. S58–S63, ESI†) were indicative of formation of any nanoparticlate.
Discussion and conclusions
We have prepared Co(III) complexes of two new corrole ligands, obtained by nucleophilic substitution of F with aminoalkyl groups. The complexes were found to act as bifunctional catalysts for the HER and the OER reactions in 1 M KOH solution when heterogenized. In all cases, the presence of the aminoalkyl groups impacted catalytic performance. During the HER, for optimized catalyst ink loading the presence of the aminoalkyl chains lowered overpotential and improved activity. Characterization by solid-state UV-vis spectroscopy and water contact angle measurements indicated different stacking interactions between the respective metallocorroles and changes in surface wettability of the corresponding catalyst films. This correlated to the catalytic activity changes: there was a decrease in the observed Tafel slope both upon the inclusion of the aminoalkyl group as well as with an increase in the chain length of that functional group. However, under these conditions, the inconsistent appearance of nanoparticulate was detected, which could be correlated neither to chain length nor to the applied current, suggesting general instability. In all cases, ICP showed a mass loss over the course of the experiment, suggesting that the primary reason for any activity change was the loss of active sites from the electrode.Similar trends of reduced overpotential and improved TOF were noted for the OER with the aminoalkyl chains. However, in contrast to HER, the Tafel slope increased for the OER. This activity limitation is ascribed to accessibility of the Co sites in the catalyst layer and suggestive that the presence of the aliphatic chains impacts corrole aggregation. Although catalyst loss from the electrode surface was noted during the OER as well, no nanoparticulate matter was formed. For both the HER and the OER, modest decomposition rates were observed during prolonged stability tests. Based on these data, we propose that the aminoalkyl chains help to disperse the metallocorroles in the catalyst ink, decreasing the aggregation of the molecular active sites, which in turn improves the accessible number of Co active sites. This proposal is based on the activity trends noted between aminoalkyl chains of different lengths, which should not experience a substantial difference in the electronic environment at the metal center. The effect of the chain then can be to increase active site dispersity on the conducting carbon, improved accessibility of the Co active site, or some combination of the two.
The results presented here suggest that integration with the polymer binder is a possible strategy for continued improvement for the performance of molecular catalysts for water splitting. Particularly under basic conditions, this should help with catalyst dispersity and the stability of any electrolyzer configuration. Notably, even simple length changes result in quantifiable activity improvements, implying that addressing active site dispersity could address the ongoing activity limitations of heterogenizing molecular catalysts through straightforward synthetic modifications. The consequences of these conclusions are continuing to be explored in ongoing studies.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division (DE-SC00234430). The authors gratefully acknowledge Myoeum Kim and Prof. Liheng Cai for their assistance with contact angle measurements as well as Dr Poonam Rani and Prof. William Epling for their assistance with solid state UV-vis measurements.References
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, IPCC, 2021: Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, 2021 DOI:10.1017/9781009157896.
- J. S. P. R. Shukla, R. Slade, A. Al Khourdajie, R. van Diemen, D. McCollum, M. Pathak, S. Some, P. Vyas, R. Fradera, M. Belkacemi, A. Hasija, G. Lisboa, S. Luz and J. Malley, IPCC, 2022: Climate Change 2022: Mitigation of Climate Change. Contribution of Working Group iii to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, 2022 DOI:10.1017/9781009157926.
- J. Li, C. Gao, X. Lu and A. Hoseyni, Energy Sources, Part A, 2022, 44, 1173–1188 CrossRef CAS.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- R. Abbasi, B. P. Setzler, S. Lin, J. Wang, Y. Zhao, H. Xu, B. Pivovar, B. Tian, X. Chen, G. Wu and Y. Yan, Adv. Mater., 2019, 31, e1805876 CrossRef PubMed.
- A. Y. Faid, A. O. Barnett, F. Seland and S. Sunde, ACS Appl. Energy Mater., 2021, 4, 3327–3340 CrossRef CAS PubMed.
- E. K. Volk, M. E. Kreider, S. Kwon and S. M. Alia, EES Catal., 2024, 2, 109–137 RSC.
- G. A. Lindquist, J. C. Gaitor, W. L. Thompson, V. Brogden, K. J. T. Noonan and S. W. Boettcher, Energy Environ. Sci., 2023, 16, 4373–4387 RSC.
- I. Barlocco, G. Di Liberto and G. Pacchioni, Energy Adv., 2023, 2, 1022–1029 RSC.
- J. Wang, S. Dou and X. Wang, Sci. Adv., 2021, 7, eabf3989 CrossRef CAS PubMed.
- F. Zaera, Coord. Chem. Rev., 2021, 448, 214179 CrossRef CAS.
- M. Adnan Khan, S. Khan, S. Sengupta, B. Naath Mongal and S. Naskar, Coord. Chem. Rev., 2024, 504, 215679 CrossRef CAS.
- K. Muñoz-Becerra, F. J. Recio, R. Venegas and J. H. Zagal, Curr. Opin. Electrochem., 2023, 42, 101387 CrossRef.
- X. Li, H. Lei, L. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892 CrossRef CAS PubMed.
- X.-P. Zhang, H.-Y. Wang, H. Zheng, W. Zhang and R. Cao, Chin. J. Catal., 2021, 42, 1253–1268 CrossRef CAS.
- A. Kumar, V. Kumar Vashistha and D. Kumar Das, Coord. Chem. Rev., 2021, 431, 213678 CrossRef CAS.
- S. Yang, Y. Yu, X. Gao, Z. Zhang and F. Wang, Chem. Soc. Rev., 2021, 50, 12985–13011 RSC.
- J. Li, C. A. Triana, W. Wan, D. P. Adiyeri Saseendran, Y. Zhao, S. E. Balaghi, S. Heidari and G. R. Patzke, Chem. Soc. Rev., 2021, 50, 2444–2485 RSC.
- L. H. Zhang, S. Mathew, J. Hessels, J. N. H. Reek and F. Yu, ChemSusChem, 2021, 14, 234–250 CrossRef CAS PubMed.
- J. F. Barata, M. G. Neves, M. A. Faustino, A. C. Tomé and J. A. Cavaleiro, Chem. Rev., 2017, 117, 3192–3253 CrossRef CAS PubMed.
- H. Lei, X. Li, J. Meng, H. Zheng, W. Zhang and R. Cao, ACS Catal., 2019, 9, 4320–4344 CrossRef CAS.
- C. Di Natale, C. P. Gros and R. Paolesse, Chem. Soc. Rev., 2022, 51, 1277–1335 RSC.
- X. Jiang, M. L. Naitana, N. Desbois, V. Quesneau, S. Brandès, Y. Rousselin, W. Shan, W. R. Osterloh, V. Blondeau-Patissier, C. P. Gros and K. M. Kadish, Inorg. Chem., 2018, 57, 1226–1241 CrossRef CAS PubMed.
- W. R. Osterloh, V. Quesneau, N. Desbois, S. Brandès, W. Shan, V. Blondeau-Patissier, R. Paolesse, C. P. Gros and K. M. Kadish, Inorg. Chem., 2020, 59, 595–611 CrossRef CAS PubMed.
- V. Quesneau, W. Shan, N. Desbois, S. Brandès, Y. Rousselin, M. Vanotti, V. Blondeau-Patissier, M. Naitana, P. Fleurat-Lessard, E. Van Caemelbecke, K. M. Kadish and C. P. Gros, Eur. J. Inorg. Chem., 2018, 4265–4277 CrossRef CAS.
- W. Shan, N. Desbois, S. Pacquelet, B. Stéphane, Y. Rousselin, J. Conradie, A. Ghosh, C. P. Gros and K. M. Kadish, Inorg. Chem., 2019, 58, 7677–7689 CrossRef CAS PubMed.
- N. Desbois, W. R. Osterloh, D. Sabat, C. Monot, S. Brandès, M. Meyer, C. Chaar, L. Hespel, L. Lebrun, R. Baati, F. Estour and C. P. Gros, Chem. Commun., 2023, 59, 2098–2101 RSC.
- K. M. Kadish, L. Frémond, Z. Ou, J. Shao, C. Shi, F. C. Anson, F. Burdet, C. P. Gros, J.-M. Barbe and R. Guilard, J. Am. Chem. Soc., 2005, 127, 5625–5631 CrossRef CAS PubMed.
- K. M. Kadish, J. Shen, L. Frémond, P. Chen, M. El Ojaimi, M. Chkounda, C. P. Gros, J.-M. Barbe, K. Ohkubo, S. Fukuzumi and R. Guilard, Inorg. Chem., 2008, 47, 6726–6737 CrossRef CAS PubMed.
- O. V. Kharissova, Y. P. Méndez, B. I. Kharisov, A. L. Nikolaev, E. Luévano-Hipólito and L. T. González, Particuology, 2024, 90, 236–265 CrossRef CAS.
- P. D. N. Victoria, A. Vaillard, S. E. Vaillard, F. Doctorovich, B. Sarkar and N. I. Neuman, Eur. J. Inorg. Chem., 2022, e202100767 Search PubMed.
- B. Mondal, K. Sengupta, A. Rana, A. Mahammed, M. Botoshansky, S. G. Dey, Z. Gross and A. Dey, Inorg. Chem., 2013, 52, 3381–3387 CrossRef CAS PubMed.
- A. Mahammed, B. Mondal, A. Rana, A. Dey and Z. Gross, Chem. Commun., 2014, 50, 2725–2727 RSC.
- A. Kumar, S. S. P. Varshney, A. Paul and S. Jeyaraman, Dalton Trans., 2019, 48, 11345–11351 RSC.
- X. Li, H. Lei, J. Liu, X. Zhao, S. Ding, Z. Zhang, X. Tao, W. Zhang, W. Wang, X. Zheng and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 15070–15075 CrossRef CAS PubMed.
- Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427–1429 CrossRef CAS PubMed.
- T. Hori and A. Osuka, Eur. J. Org. Chem., 2010, 2379–2386 CrossRef CAS.
- C. Costentin and J.-M. Savéant, Nat. Rev. Chem., 2017, 1, 0087 CrossRef CAS.
- O. van der Heijden, S. Park, R. E. Vos, J. J. J. Eggebeen and M. T. M. Koper, ACS Energy Lett., 2024, 9, 1871–1879 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- C. Wan, Y. Ling, S. Wang, H. Pu, Y. Huang and X. Duan, ACS Cent. Sci., 2024, 10, 658–665 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional material and methods, UV-vis, NMR, and electrochemical. See DOI: https://doi.org/10.1039/d4ya00257a |
| This journal is © The Royal Society of Chemistry 2024 |