
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stretched or wrinkled? Looking into the polymer conformation within polymersome membranes†
Christiane
Effenberg
 and
Jens
Gaitzsch
*
and
Jens
Gaitzsch
*
Leibniz-Institut für Polymerforschung Dresden e. V., Germany. E-mail: gaitzsch@ipfdd.de
First published on 30th April 2024
Abstract
Self-assembly of amphiphilic block-copolymers into polymersomes is a well-established concept. In this membrane, the hydrophilic part is considered to be loosely assembled towards the solvent, and the hydrophobic part on the inside of the membrane is considered to be more densely packed. Within the membrane, this hydrophobic part could now have a stretched conformation or be a random coil, depending on the available space and also on the chemical nature of the polymer. We now analysed the literature for works on polymersomes that determined the membrane thickness via cryo-TEM and analysed the hydrophobic part of their polymers for their conformation. Over all available block-copolymers, a variety of trends became obvious: the longer a hydrophobic block, the more coiled the conformation and the bulkier the side chains, the more stretched the polymer became. Polymers with less conformational freedom like semi-crystalline ones were present in a more stretched conformation. Both trends could be exemplified on various occasions in this cross-literature meta-study. This overview hence provides additional insight into the physical chemistry of block-copolymer membranes.
Introduction
Since their discovery in 1999, vesicles of amphiphilic block-copolymers have quickly found their way into modern research.1–3 Such vesicles are also called polymersomes and are a hollow sphere that is surrounded by a bilayer of the polymeric amphiphile. One great advantage of polymersomes is their ability to carry a variety of bioactive payloads like enzymes, DNA or RNA. For this purpose, polymersomes are branded as stable compartments that transport their payload safely to the target, where it is delivered upon an external trigger.4–7 Most of these promises rely on a stable hydrophobic part as the chemical versatile building material for the membrane that prohibits premature leakage.7–10 Their lipid counterparts, the liposomes, usually cannot be held to the same standard following decreased mechanical stability.11One major argument for the superior mechanical stability of polymersomes over liposomes is the ability of polymers to coil up, effectively supporting the membrane better than lipids in a fully stretched conformation. A typical lipid membrane stretches 4–5 nm12,13 and considering an average hydrophobic lipid of 18 carbon–carbon bonds, this means that 36 carbon–carbon bonds are aligned within the membrane. Extending this thought to polymersomes, one realises that things are little different there. The membrane formed by PG14-b-PBO27, for example, spans 11 nm (2.5 times the size of a lipid membrane), but the polymer contains 81 bonds in the hydrophobic block (4.5 times the amount of bonds found in a lipid).14 This underpins the aforementioned assumption that polymers are present in a coiled state, which then contributes to their mechanical stability. It has also been shown that polymers can change their conformation if an inserted protein, for example, demands it.15
As they can change the conformation, this raises the question of the equilibrium conformation of a hydrophobic polymer within a native polymersome membrane. Understanding the conformation, what drives it and how it could be altered within a given block-copolymer system, or how changing the polymer affects the polymer conformation, is hence key to design mechanically robust polymersomes. Within a typical depiction, the hydrophobic parts of the polymers (red in Fig. 1) meet each other in the middle of the membrane and then a wobbled line is drawn towards the outside of the membrane, where the hydrophilic part of the polymer (blue in Fig. 1) takes over. This line can be shown in a stretched conformation (Fig. 1A) or in a coiled conformation (Fig. 1B), depending on the original artist. Behind such sketches lies the scientific question on whether the polymer is present in a stretched conformation or in a perfectly random coil. A standard assumption could be that the actual conformation is “in between”. In previous studies with the group of late Wolfgang Meier, we have had a look at this and calculated the theoretical maximum length of the polymer as well as the dimensions of a perfectly random coil.16 To the best of our knowledge, no other group has looked into the polymer conformation within the polymersome membrane so far. Initially, we only looked into the boundaries of a stretched molecule and random coil and noted that the actual conformation was in between both extremes for all noted self-assemblies (micelles, multi-compartment-micelles, vesicles and similar). In the follow-up work on PEG–PEHOx (all polymer acronyms are explained in Table 1), we noted that polymer conformations tend to become less stretched with increasing block length, going from 25% stretched (48 repeating units) to 17% stretched (138 repeating units).17 The less hydrophobic PG–PBO block-copolymers even went up to being 51% stretched for 27 repeating units of PBO.14 However, all of these were isolated measurements and calculations and no further comparison was investigated.
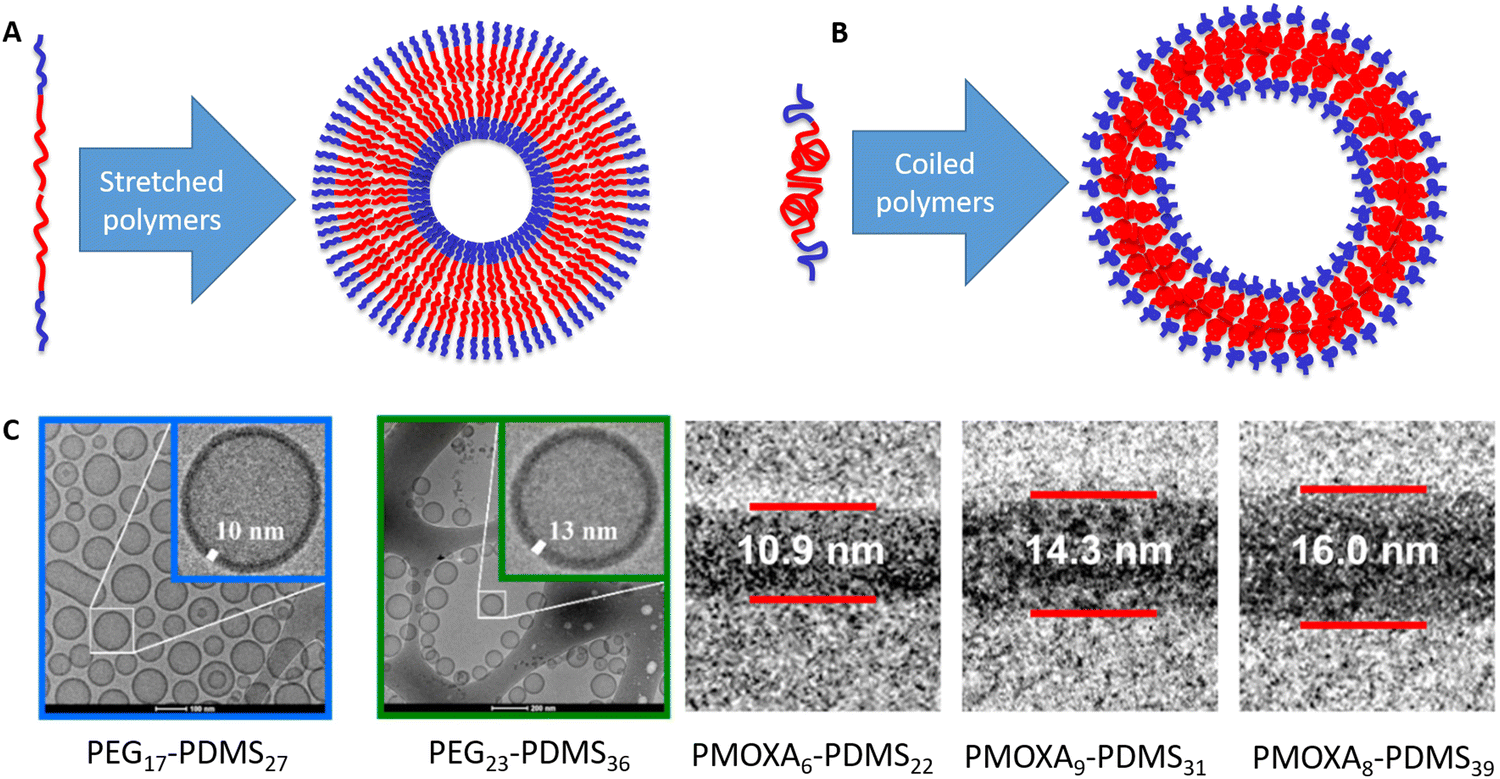 | ||
| Fig. 1 Amphiphilic block-copolymers (blue = hydrophilic, red = hydrophobic) can self-assemble into polymersomes. (A) Amphiphilic block-copolymer and polymersome with a stretched polymer conformation. (B) Amphiphilic block-copolymer and polymersome with a coiled polymer conformation. (C) Examples for cryo-TEM images that the respective authors used to measure the membrane thickness. The examples for PEG–PDMS are from the study by Fauquignon et al.18 reproduced using the creative commons licence CC BY 4.0. The examples for PMOXA–PDMS were adapted with permission from Itel et al.19 Copyright {2019} American Chemical Society. | ||
| Polymer acronym | Long name | Bonds per repeating unit + notable deviations |
|---|---|---|
| PA444 | Poly((400-acryloxybutyl) 2,5-di(40-butyloxybenzoyloxy) benzoate) | 2 |
| PA6ester1 | Poly(4′-methoxyphenyl 4-(6′′-(acryloyloxy)hexyloxy) benzoate) | 2 |
| PAA | Poly(acrylic acid) | 2 |
| PAGE | Poly(allyl glycidyl ether) | 3 |
| PBD | Poly(butadiene) | 2 |
| PBO | Poly(butylene oxide) | 3 |
| PCL | Poly(ε-caprolactone) | 7 |
| PCMA | Poly(coumarin methacrylate) | 2 |
| PDEAEMA | Poly(diethylaminoethyl methacrylate) | 2 |
| PDEAMA | Poly(diethylaminoethyl methacrylate) | 2 |
| PDMAEMA | Poly(dimethylaminoethyl methacrylate) | 2 |
| PDMIBMA | Poly(dimethylmaleimidobutyl methacrylate) | 2 |
| PDMIHMA | Poly(6-(3,4-dimethylmaleimidio)hexyl methacrylate) | 2 |
| PDMS | Poly(dimethylsiloxane) | 2 (SI–O bond: 164 pm, bond angle: 126.5°)20 |
| PDPA | Poly(diisopropylaminoethyl methacrylate) | 2 |
| PDPAEMA | Poly(2-(N,N′-diisopropylamino)ethyl methacrylate) | 2 |
| PDPAMA | Poly(diisopropylamino ethyl methacrylate) | 2 |
| PEE | Poly(ethylethylene) | 2 |
| PEG (=PEO) | Poly(ethylene glycol) | 3 |
| PEHOx | Poly(2-ethylhexyl oxazoline) | 3 |
| PEO | Poly(ethylene oxide) → always noted as PEG throughout the study for consistency | 3 |
| PEtOz | Poly(2-ethyl-2-oxazoline) | 3 |
| PFcMA | Poly(2-(methylacryloyloxy)ethyl ferrocene carboxylate) | 2 |
| PG | Poly(glycidol) | 3 |
| PGlyMA | Poly(glycidyl methacrylate) | 2 |
| PGMA | Poly(glycerol monomethacrylate) | 2 |
| PHPMA | Poly(2-hydroxypropyl methacrylate) | 2 |
| PMA | Poly(methyl acrylate) | 2 |
| PMAzo444 | Poly(4-butyloxy-20-(400-methacryloyloxybutyloxy)-4-(4-butyloxybenzoyloxy)azobenzene) | 2 |
| PMeSPG | Poly(N-3-(methylthio)propyl glycine) | 3 |
| PMOXA | Poly(methyl oxazoline) | 3 |
| PNIPAM | Poly(N-isopropylacrylamide) | 2 |
| PNAM | Poly(N-acryloylmorpholine) | 2 |
| PNAT | Poly(N-acryloylthiomorpholine) | 2 |
| PPDMI | Poly(perylene diester monoimide) | 2 |
| PPS | Poly(propylene sulphide) | 3 |
| PS | Polystyrene | 2 |
| PSS | Poly(styrene sulfonate) | 2 |
| PtBGE | Poly(tert-butyl glycidyl ether) | 3 |
| PTMC | Poly(trimethylene carbonate) | 6 |
| PTPEMA | Poly(tetraphenylethene methacrylate) | 2 |
| PVCL | Poly(N-vinylcaprolactam) | 2 |
In this work, we thus compared polymer conformations within polymersomes obtained from amphiphilic AB-diblock-copolymers across the literature. Polymersomes of ABA or ABC triblock-copolymers were looked at separately as they impose a conformation restriction on the hydrophobic block (polymer spans through the membrane) and also because there is much less data available on that. If the self-assembly of these di-and triblock-copolymers into polymersomes was confirmed by cryo-TEM (examples shown in Fig. 1C) and the membrane thickness was thus determined, the degree of stretching within the hydrophobic block could be calculated. The approach allowed for a meta-study across the literature of the conformation of a polymer within a membrane. Effects of the degree of polymerisation (same polymer), different hydrophilic blocks (same hydrophobic block), and the influence of polymeric properties like melting temperature amongst others, could now be looked into. Our evaluation of almost 90 block-copolymers promised insights into how polymers actually look within a membrane, what determines their conformation and hence be a viable basis to improve vesicle models in the future.
Results & discussion
Theoretical considerations
In order to qualify this meta-study, the original research had to meet the following criteria: (i) published in a peer-review journal, (ii) vesicles were proven by cryo-TEM (examples shown in Fig. 1C), (iii) the membrane thickness was determined via cryo-TEM, (iv) the chemical composition of the hydrophobic block had to be retrievable in terms of chemical structure and repeating unit. Since dispersity values are not always reported and are only relevant for the hydrophobic block here, they have been left out of the discussion but were mentioned when they could notably contribute to measurement errors. It was also required for the analytical data of the block-copolymers to be available for verification purposes. This excluded all commercially sourced amphiphilic block-copolymers, where the authors did not validate the chemical composition after purchase. Focussing on membrane thicknesses determined via cryo-TEM allowed to assume a comparatively similar approach by different authors to determine the membrane thickness, as it is a directly measurable read-out from a recorded image. Small-angle X-ray scattering (SAXS), for example, does require a specially trained co-worker to record and interpret relative data and may hence be subjected to a larger measurement and evaluation error across different publications. Small subjective differences by ±1 nm cannot be ruled out for cryo-TEM as well but were considered to be smaller than for other methods like SAXS. If all data were present, the hydrophobic block of the amphiphilic block-copolymers could be analysed as follows. At first, the contour length of the polymer was calculated using the following eqn (1):14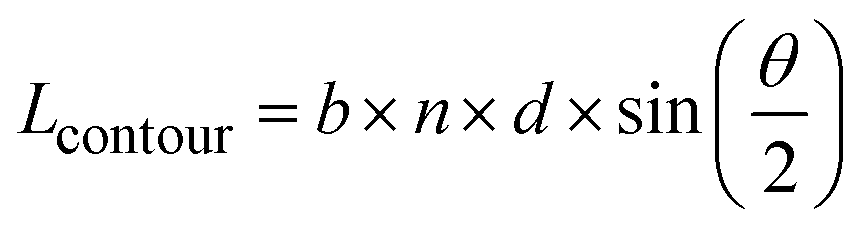 | (1) |
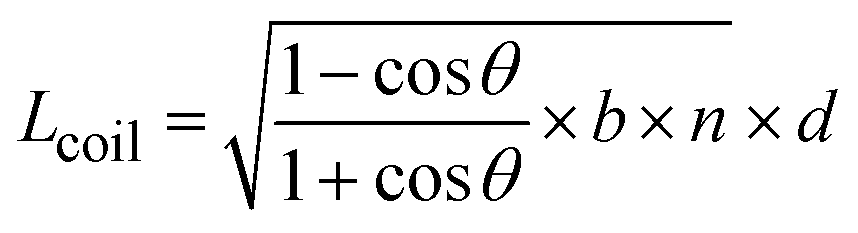 | (2) |
| Leff = x × Lcontour + (1 − x) × Lcoil | (3) |
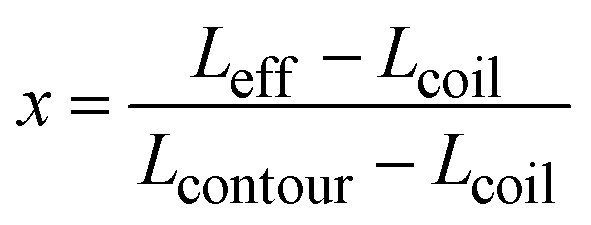 | (4) |
| Ref. | Polymer by type of hydrophobic blocka | Bonds in hydrophobic part | L eff/nmb | % stretchedc | Self-assembly techniqued |
|---|---|---|---|---|---|
| a All polymers were prepared using controlled radical polymerisation or a living polymerisation method such as ring-opening polymerisation. b As per cryo-TEM reported in the noted reference. c Calculated using the formula mentioned in the main text. d Cosolvent = cosolvent technique = solvent switch, electro = electroformation, emulsification = emulsification and solvent diffusion method, emulsion = inverted emulsion, film = film rehydration, nanoprec = nanoprecipitation, PISA = polymerisation induced self-assembly, pH switch = pH switch from acidic to basic, rehydration = rehydration without film formation. All details can be found in the respective publications. e These degrees of stretching are physically impossible and likely originate from the large side chains that contribute to the membrane thickness as discussed in the main text. | |||||
| AB diblock copolymers | |||||
| PDMS | |||||
| 18 | PEG8-b-PDMS14 | 28 | 3.6 | 79 | Film |
| 18 | PEG13-b-PDMS23 | 46 | 4.3 | 47 | Film |
| 18 | PEG17-b-PDMS27 | 54 | 5.0 | 48 | Film |
| 18 | PEG23-b-PDMS36 | 72 | 6.6 | 49 | Film |
| 19 | PMOXA6–PDMS22 | 44 | 5.5 | 78 | Electro |
| 19 | PMOXA9–PDMS31 | 62 | 7.2 | 72 | Electro |
| 19 | PMOXA8–PDMS39 | 78 | 8.0 | 61 | Electro |
| 19 | PMOXA14–PDMS65 | 130 | 10.7 | 46 | Electro |
| 21 | PMOXA11-b-PDMS68 | 136 | 8.0 | 27 | Film |
| No heteroatoms | |||||
| 3, 22 and 23 | PEG40–PEE37 | 74 | 4.0 | 32 | Electro, film |
| 23 | PEG26–PBD46 | 92 | 4.8 | 32 | Film |
| 22 and 23 | PEG50–PBD55 | 110 | 5.3 | 29 | Electro, film |
| 22 and 23 | PEG80–PBD125 | 250 | 7.4 | 16 | Electro, film |
| 23 | PEG150–PBD250 | 500 | 10.5 | 11 | Film |
| PEG derivatives | |||||
| 14 | (R/S)-PG14-b-(R/S)-PBO26 | 78 | 5.6 | 50 | Cosolvent |
| 14 | (R)-PG14-(R)-PBO26 | 78 | 5.8 | 54 | Cosolvent |
| 14 | (S)-PG14-b-(S)-PBO27 | 81 | 5.5 | 47 | Cosolvent |
| 24 | PEG17-b-PPS30 | 90 | 4.5 | 24 | Film |
| 16 | PEG45-b-PEHOx95 | 285 | 9.0 | 15 | Film, cosolvent |
| 16 | PEG45-b-PEHOx128 | 384 | 12.1 | 17 | Film, cosolvent |
| Bulky side chain in hydrophobic block | |||||
| 25 | PEG45-b-PA4447 | 14 | 3.0 | 251e | Emulsion |
| 26 and 27 | PEG45-b-PA4447 | 14 | 5.3 | 504e | Emulsion, nanoprec. |
| 26 and 27 | PEG45-b-PMAazo44412 | 24 | 7.3 | 340e | Emulsion, nanoprec. |
| 26 | PEG45-b-PA6ester120 | 40 | 5.0 | 101 | Emulsion |
| 26 | PEG91-b-(PB33-g-Chol) | 66 | 6.8 | 83 | Emulsion |
| Semi-crystalline or high Tg hydrophobic polymers | |||||
| 28 | PEG45-b-PS206 | 412 | 11.0 | 15 | Cosolvent |
| 29 | PEG45-b-PS230 | 460 | 13.0 | 17 | Cosolvent |
| 30 | PEG44-b-PS292 | 584 | 13.0 | 13 | Cosolvent |
| 31 | PEG45–PCL44 | 308 | 8.8 | 16 | Rehydration |
| 32 | PEG45-b-PTMC96 | 576 | 7.3 | 4 | Cosolvent |
| 32 | PEG45-b-PTMC144 | 864 | 8.8 | 3 | Cosolvent |
| 32 | PEG45-b-PTMC170 | 1020 | 9.6 | 3 | Cosolvent |
| Non-bulky (meth)acrylates | |||||
| 33 | PEG43-b-P(NIPAM21-co-PDMI9) | 60 | 4.0 | 44 | Cosolvent |
| 33 | PEG43-b-P(NIPAM21-co-PDMI9) | 60 | 4.8 | 58 | Cosolvent |
| 33 | PEG43-b-P(NIPAM21-co-PDMI9) | 60 | 5.5 | 70 | Cosolvent |
| 33 | PEG43-b-P(NIPAM21-co-PDMI9) | 60 | 7.2 | 101 | Cosolvent |
| 34 | PEG43-b-P(NIPAM23-co-PDMI19) | 84 | 5.0 | 39 | Cosolvent |
| 35 | PNAM25-b-PNAT25 | 50 | 6.5 | 112 | PISA |
| 35 | PNAM25-b-PNAT50 | 100 | 8.6 | 67 | PISA |
| 35 | PNAM25-b-PNAT70 | 140 | 9.7 | 51 | PISA |
| 36 | PEG45-b-PMeSPG17 | 51 | 4.5 | 66 | Nanoprec. |
| 36 | PEG45-b-PMeSPG71 | 213 | 6.5 | 16 | Nanoprec. |
| 37 | PEG16-b-PMA70 | 140 | 6.2 | 27 | Rehydration |
| 37 | PEG45-b-PMA70 | 140 | 5.6 | 22 | Rehydration |
| 37 | PAA10-b-PMA70 | 140 | 5.5 | 21 | Rehydration |
| Photo cross-linked membranes | |||||
| 38 | PEG45-b-P(DEAEMA36-co-TPEMA6) | 84 | 7.4 | 68 | Nanoprec. |
| 39 | PEG45-b-P(FcMA17-co-DEAEMA48-co-DMIHMA16) | 162 | 7.0 | 26 | Emulsification |
| 40 | PEG45-b-P(DPAEMA59-co-DMIHMA24) | 166 | 9.8 | 42 | pH switch |
| 41 | PEG45-b-P(DPAEMA57-co-DMIHMA27) | 168 | 13.5 | 63 | pH switch |
| 41 | PEG45-b-P(DEAEMA70–DMIBMA20) | 180 | 8.1 | 29 | pH switch |
| 42 | PEG45-b-P(DEAEMA73-s-DMIBMA19) | 184 | 8.8 | 32 | pH switch |
| 43 | PEG45-b-P(DEAEMA77-s-DMIBMA18) | 190 | 9.5 | 34 | pH switch |
| 44 | PEG45-b-(PDEAEMA49-co-PDMAEMA27-co-PDMIBMA24) | 200 | 5.3 | 11 | pH switch |
| 45 | PEG45-b-P(DEAEMA78-s-DMIBMA23) | 202 | 8.0 | 24 | pH switch |
| 46 | PEG45-b-P(DEAEMA82-s-DMIBMA20) | 204 | 7.3 | 20 | pH switch |
| 47 | PEG45-b-P(DEAEMA81-co-DMIBMA23) | 208 | 7.0 | 19 | pH switch |
| 48 | PEG45-b-P(DEAEMA83–DMIBMA23) | 212 | 7.0 | 18 | pH switch |
| 44 | PEG45-b-(PDEAEMA49-co-PDMAEMA31-co-PDMIBMA29) | 218 | 5.7 | 12 | pH switch |
| 40 | PEG45-b-P(DEAMA83-co-DMIBMA28) | 222 | 8.6 | 24 | pH switch |
| 49 | PEG45-b-P(DEAEMA89-s-DMIBMA24) | 226 | 7.5 | 19 | pH switch |
| 49 | PEG45-b-P(DMEAEMA45–DEAEMA45–DMIBMA24) | 228 | 7 | 16 | pH switch |
| 39 | PEG45-b-P(FcMA19-co-DEAEMA83-co-DMIBMA33) | 270 | 6.5 | 11 | Emulsification |
| 50 | PEG77.5N3-b-P(DEAEMA130-co-DMIBMA32) | 324 | 13.0 | 27 | pH switch |
| Polymers from PISA | |||||
| 51 | PEG113-b-P(HPMA320-co-GlyMA80) | 800 | 14.0 | 9 | PISA |
| 52 | PEG113-b-PHPMA400 | 800 | 12.5 | 8 | PISA |
| 53 | PGMA59–PHPMA400 | 800 | 14.0 | 9 | PISA |
| 54 | PGMA62–PHPMA600 | 1200 | 21.4 | 11 | PISA |
| 54 | PGMA62–PHPMA700 | 1400 | 25.0 | 11 | PISA |
| 54 | PGMA62–PHPMA800 | 1600 | 26.7 | 10 | PISA |
| 54 | PGMA62–PHPMA900 | 1800 | 29.9 | 10 | PISA |
| 54 | PGMA62–PHPMA1000 | 2000 | 35.1 | 12 | PISA |
| Triblock-copolymers | |||||
| ABA triblock-copolymers | |||||
| 55 | PEG22-b-P(S-stat-CMA)118-b-PEG22 | 236 | 14.0 | 44 | Cosolvent |
| 55 | PEG45-b-P(S-stat-CMA)206-b-PEG45 | 412 | 21.0 | 38 | Cosolvent |
| 19 | PMOXA3–PDMS19–PMOXA3 | 38 | 6.0 | 114 | Electro |
| 19 | PMOXA6–PDMS34–PMOXA6 | 68 | 9.2 | 91 | Electro |
| 19 | PMOXA6–PDMS44–PMOXA6 | 88 | 10.7 | 79 | Electro |
| 19 | PMOXA7–PDMS49–PMOXA7 | 98 | 12.1 | 81 | Electro |
| 19 | PMOXA12–PDMS63–PMOXA12 | 126 | 13.4 | 67 | Film |
| 56 | PMOXA17–PDMS67–PMOXA17 | 134 | 11.7 | 51 | Film |
| 19 | PMOXA12–PDMS87–PMOXA12 | 174 | 16.2 | 57 | Electro |
| 56 | PVCL10–PDMS65–PVCL10 | 130 | 14.6 | 72 | Film |
| 57 | PEG16PPS50PEG16 | 150 | 8.0 | 30 | Film |
| ABC triblock copolymers | |||||
| 58 | PEG45–PDPA85–PSS22 | 170 | 13.9 | 64 | pH switch |
| 17 | PEG45-b-PEHOx48-b-PEtOz10 | 144 | 6.3 | 26 | Cosolvent |
| 17 | PEG45-b-PEHOx62-b-PEtOz35 | 186 | 8.2 | 28 | Cosolvent |
| 17 | PEG45-b-PEHOx65-b-PEtOz19 | 195 | 7.8 | 24 | Cosolvent |
| 17 | PEG45-b-PEHOx87-b-PEtOz10 | 261 | 9.9 | 21 | Film |
| 17 | PEG45-b-PEHOx139-b-PEtOz10 | 417 | 12.9 | 19 | Film |
| 59 | PEG42-b-PAGECOOH12-b-PtBGE22 | 36 | 4.1 | 95 | Cosolvent |
This discussion will now be grouped into AB-diblock-copolymers and ABA/ABC triblock-copolymers and it will focus on general trends that can be derived from the obtained data for almost 90 block-copolymers. Similar to Table 2, this discussion will follow polymers with similar or comparable hydrophobic blocks in order to make general trends more easily visible.
AB diblock-copolymers
PDMS is one of the most common hydrophobic polymers in self-assembly and was also one of the first ones used. Consequently, a number of block-copolymers combining PDMS either with PEG or PMOXA have been reported. Generally speaking, short PDMS blocks are in a much more stretched conformation than long ones. In PEG8-b-PDMS14,18 for example, PDMS is 79% stretched and in PMOXA6–PDMS2219 it is quite similar with 78% stretching. In longer ones, this number drops to 50% stretching for PEG23-b-PDMS3618 and notably further to 27% for PMOXA11-b-PDMS68.19 Even though there are no big outliers within the PEG and PMOXA series, the PDMS bits notably exhibit greater stretching when combined with PMOXA. They appear to reach a plateau of about 48% stretching for 23–36 repeating units, when combined with PEG, and drop from 79% to 61% stretching for 22 and 39 repeating units, respectively, when combined with PMOXA (Fig. 2A). As no other hydrophobic block exhibited the transition from PEG to PMOXA, this trend of more stretching with PMOXA could not be generalised.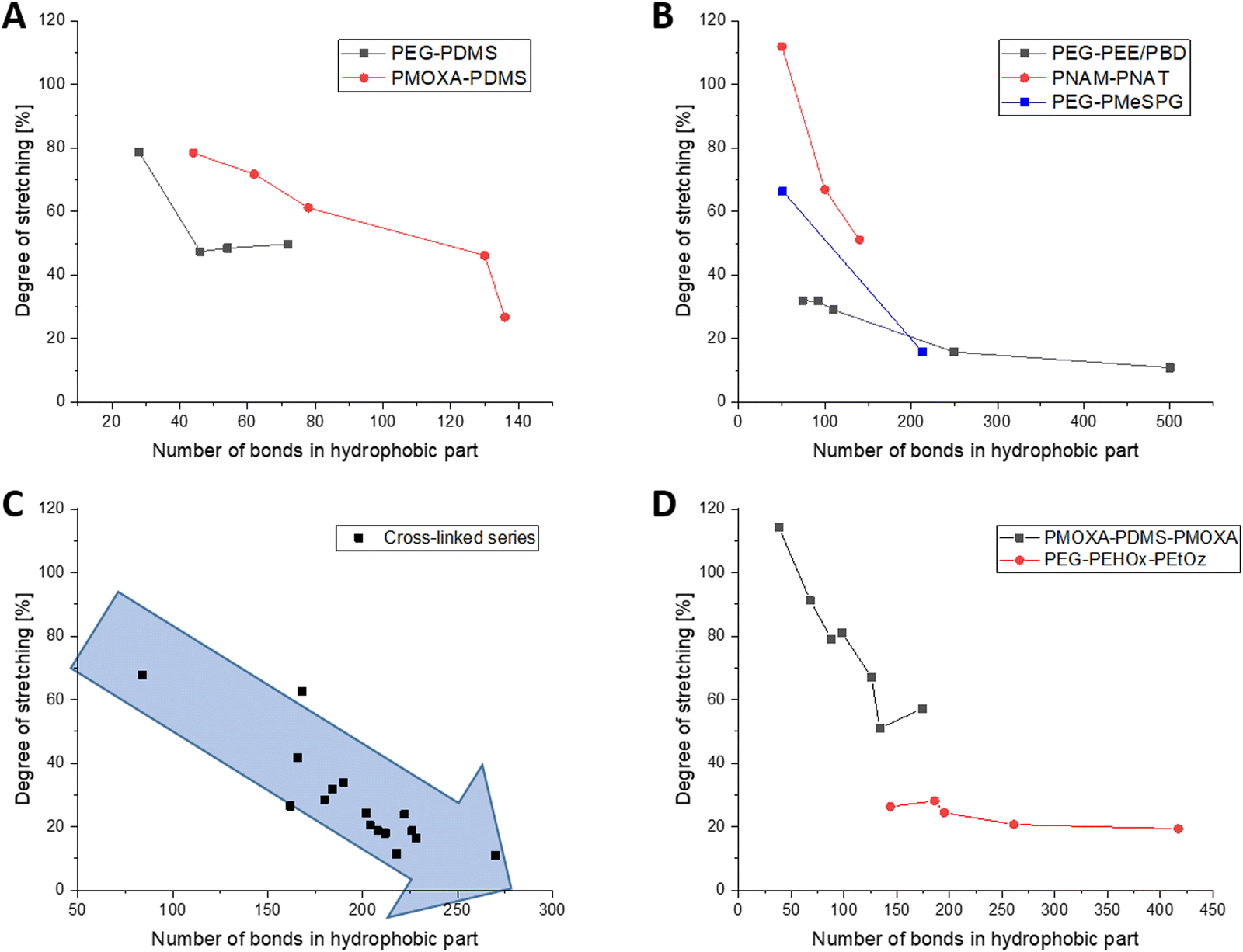 | ||
| Fig. 2 Development of the degree of stretching for the various groups of polymers, always dependent on the amount of chemical bonds in the backbone of the hydrophobic part. (A) Diblock-copolymers with PDMS as the hydrophobic part. (B) Series of AB diblock copolymers, where more than one polymer have been reported, see the inserted caption for polymer names. (C) All photo cross-linked and pH sensitive polymersomes. The blue arrow has been added to highlight the decreasing degree of stretching with increasing length of the hydrophobic block. (D) ABA and ABC triblock copolymers, where a series of polymers have been reported. | ||
The series of PBD and PEE as saturated counterparts then extends this series of polymers with a relatively simple structure in their repeating unit. Here, the trend of polymers that become less stretched with increasing length becomes once again very much apparent. The series starts at 32% stretching for PEG40–PEE373,22,23 and goes down to 11% stretching for the considerably longer PEG150–PBD250,23 hence strongly underpinning the previously observed trend (Fig. 2B).
As for PEG derivatives as a hydrophobic block, only a limited number of polymers with PBO (3 examples)14 and PPS (1 example)24 were available. Within these four datasets, all hydrophobic blocks were of similar length (26–30 repeating units), making them comparable between each other. While the PBO blocks were around 50% stretched, the PPS chain was only 24% stretched. The ethyl side chain present in PBO, but not in PPS, could be a reason for this as a side chain can prevent polymer folding for sterical reasons and consequently lead to a more stretched polymer conformation. Both previously reported block-copolymers PEG45-b-PEHOx95 and PEG45-b-PEHOx12816 technically also fall into this category with 3 atoms per repeating in their main chain. Likely owing to their long hydrophobic parts, the degrees of stretching are very similar with both 15% and 17% being relatively low.
Testing the argument for the side chain, polymers with rather bulky or very long side chains (more than 10 C or O atoms) were examined next. With repeating units as low as seven in PEG45-b-PA4447,25 the linker moiety between the hydrophilic and hydrophobic block and most crucially, the dispersity of the polymer now became relevant as well and can explain the calculated yet impossible stretching of over 250%. However, a notable measurement error seems to be apparent with this kind of polymers as the same group of authors reported 6 nm and 11 nm of membrane thickness in different publications.26,27 It is reasonable to assume that for shorter numbers of repeating units, bend side chains partially present longer chains (dispersity) and the linker moieties extend the hydrophobic part of the membrane. As a consequence, the calculated degree of stretching becomes formally too high, which explains the calculated numbers of over 400% degrees of stretching. For an increasing number of repeating units like for PEG45-b-PA6ester120,26 a realistic number of 100% stretching could be calculated. Both examples, however, strongly suggest that the trend stated above is correct and polymer side chains do prevent dense coiling and support a stretched conformation.
Semi-crystalline polymers or those with a high glass-transition temperature behaved in the exact opposite way. These polymers either have a high incentive for close packing (building crystalline domains) or lack the mobility to leave their energetically preferred coiled state (high glass transition temperature). All polymers with PCL,31 PTMC32 or PS28–30 in their hydrophobic blocks preferred coiled conformations, ranging between 3% and 17% stretching. Having 300 and more atoms in the main chain of their hydrophobic block made all of them long polymers, giving another incentive for low degrees of stretching. It is hence not entirely clear if the lack of mobility or the high degree of polymerisation caused the low degree of stretching. For the polymers with a comparable amount of atoms in their main chain, PEG44-b-PS292 (584 atoms)30 and PEG45-b-PTMC96 (576 atoms),32 the PS chain is more stretched (13% stretching) than the PTMC chain (3–4% stretching), again strongly underpinning the argument that side chains prevent ideally coiled structures.
Several other methacrylic derivatives have been synthesised as well but are difficult to evaluate for a series, but this opened the opportunity to look into different trends. For example, PEG43-b-P(NIPAM21-co-PDMI9)33 has 4 reported values, ranging from 44% to 100% of stretching when altering the amount of tetrahydrofuran (THF) during self-assembly. Taking our method, the most amount of THF leads to the most stretched polymers, most likely because of high chain mobility in the good solvent THF. While this is an interesting observation, it cannot be verified further as more data from different polymer systems are missing. Of some interest is also the mini-series of PEG10-45-b-PMA7037 as it is the only one with an altering length of the hydrophilic polymer, while maintaining a constant length of the hydrophobic polymer. With 21–27% of stretching for all polymers and no apparent trend, this influence seems to be negligible. Albeit from a low sample size, the mini-series in PNAM25-b-PNAT25–70 (from about 100% to 50% of stretching)35 and PEG45-b-PMeSPG17–71 (66% to 16% stretching)36 follow the general trend that the polymers with a low degree of polymerisation prefer a more stretched conformation (Fig. 2B).
The photo cross-linked polymersome membranes studied by Appelhans and Voit et al. have been studied widely over the past 15 years and thankfully provided the largest cohesive data set for this analysis. To keep everything comparable, only block-copolymers with PEG45 were taken into consideration. As it was the longest block-copolymers in this series, an exception was made for PEG77.5N3-b-P(DEAEMA130-co-DMIBMA32)50 to extend the series as much as possible. Plotting all of them into one graph revealed the same tendency as previously observed that stretching decreased notably with increasing degree of polymerisation with the hydrophobic part of the membrane. Neither the alkyl residue on the pH responsive part (methyl, ethyl, iso-propyl) nor the spacer in the photo cross-linker (butyl or hexyl) appeared to have notable impact on the degree of stretching. It decreased from 68% stretching for PEG45-b-P(DEAEMA36-co-TPEMA6; 42 RU)38 over 42% for PEG45-b-P(DPAMA59-co-DMIHMA24; 83 RU)40 and 24% of PEG45-b-P(DEAMA83-co-DMIBMA28; 111 RU)40 to 11% for PEG45-b-P(FcMA19-co-DEAEMA83-co-DMIBMA33; 135 RU).39 The latter is especially notable as even the ferrocene residue did not alter the overall trend in the degree of stretching for high degrees of polymerisation (Fig. 2C).
A similar approach can be used to assess the conformation in polymers obtained from the polymerisation-induced self-assembly (PISA). All of the ones with a measured membrane thickness in an aqueous system are from PHMPA and have a high degree of polymerisation (800–2000)52–54 and a low degree of stretching with 8–11% stretched polymer chains. Following the argument of previously mentioned polymers, this follows the trend of polymers with a high degree of polymerisation exhibiting a low degree of stretching. While this could be expected, the argument should be treated with caution with PISA as the PISA process within the membrane may not necessarily result in alignment along the cross-section of the membrane. For the same reason, these polymers are not in an energetically relaxed state because tensions due to the polymerisation were never released from the system. The real degree of stretching of polymers from PISA may hence be determined using the polymerisation method and not by the degree of polymerisation. Owing to the generally high degrees of polymerisation, however, the exact effect of PISA as a simultaneous polymerisation and self-assembly method cannot be determined from the available data.
ABA and ABC triblock-copolymers
There are notably less publications on the self-assembly of ABA or ABC triblock-copolymers (A and C hydrophilic), let alone ones that have all information to be considered for this meta-study. Generally speaking, an ABA or ABC triblock copolymer can be in an I-shape (A and C (second A) on opposite sides) or in a U-shape (A and C (second A) on the same side).60 For the sake of simplicity, an I-shape will be assumed for this overview as the general trends would remain the same for the U-shape, and only the degrees of stretching would be cut by half. There are two interesting series of publications from the group of the late Wolfgang Meier, which cover PDMS as well as PEHOx in such triblock-Copolymers. Within both series, the trend of more stretched polymers at a lower amount of repeating units does extend. For PDMS, this decreases from about 100% stretching for 19 repeating units to about 50% stretching for 67 repeating units (Fig. 2D, chemical bonds shown).19,56 These are generally higher than the ones for AB-diblock copolymers of PDMS, which saw 78% stretching for 22 repeating units and 27% for 68 repeating units. Because the hydrophilic parts are now on the opposite side of the membrane, this understandably pulls the hydrophobic part further apart.The trend for decreasing stretching with increasing chain length also holds true for the PMOXA–PEHOx–PEtOz system, although not as pronounced. Stretching here decreased from 26% for 48 repeating units to 19% for 139 repeating units (Fig. 2D).17 Compared to their AB-diblock counterparts with 95 and 128 repeating units of PEHOx and 15% and 17% of stretching, respectively,16 the triblock-copolymers with 87 and 139 repeating units of PEHOx also showed a larger degree of stretching (20% and 19%, respectively). Although notably less different than for PDMS, the triblock copolymers are still more stretched. Following the relatively high degree of polymerisation for PEHOx, the generally less stretched chains can be expected to show a lower difference in absolute terms.
Conclusion
Generally spoken, the longer a polymer became, the more coiled it became. This held true for all polymers in this series, regardless of if they had additional functional groups, were cross-linked or involved diblock- or triblock copolymers. Polymers with a longer side chain had the tendency to be less coiled following the steric restrictions of the side chain. Conversely, polymers with a high glass transition temperature or semi-crystalline polymers showed a low level of stretching as the chains lack the mobility to rearrange into a stretched conformation. There was also an indication that PDMS-containing polymers were more stretched in ABA triblock-copolymers than in AB diblock-copolymers, although this could not be verified with a second polymer system.It can hence be hypothesised that a polymer is more stretched towards the hydrophilic part of the membrane and begins to coil up once it penetrates deeper into the hydrophobic block. This is reasonable, considering that a hydrophobic polymer would always minimise the contact area with the hydrophilic surroundings of the solvent. A direct or stretched pathway to the hydrophobic part of the membrane would serve this purpose. Shorter hydrophobic blocks hardly reach this stage and are hence more stretched.
With these results, it is now better explainable, why polymers with entirely different packing parameters, i.e. different hydrophilic-to-hydrophobic balances like PEG40–PEE373,22,23 and PEG45-b-PTMC170,32 can both form polymersomes. While the first example has a mass ratio of 0.85 (1800 g mol−1 to 2100 g mol−1), the latter has a ratio of 0.12 (2000 g mol−1 to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), and they exhibit decisively different degrees of stretching with 32% and 3%, respectively. Polymer conformation is hence a factor to consider when designing polymersomes.
000 g mol−1), and they exhibit decisively different degrees of stretching with 32% and 3%, respectively. Polymer conformation is hence a factor to consider when designing polymersomes.
We hope that our study motivates more researchers to take a closer look into the conformation of their polymers and it is certain that this meta-study already provides a valuable insight into polymer conformations within the membrane of polymersomes.
Author contributions
Christiane Effenberg: methodology, formal analysis, Investigation, data curation, writing – review and editing. Jens Gaitzsch: conceptualisation, Methodology, Resources, writing – original draft, writing – review and editing, Supervision, project administration.Conflicts of interest
The authors have no competing financial interests to declare.Acknowledgements
The authors thank Dr. Riccardo Wehr and Dr. Davy Daubian, whose discussions helped setting up this train of thought in the first place and also thank Prof. Giuseppe Battaglia and Prof. Nico Bruns for helpful discussions.References
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- D. E. Discher, B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef PubMed.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS PubMed.
- H. De Oliveira, J. Thevenot and S. Lecommandoux, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 525–546 CAS.
- J. Leong, J. Y. Teo, V. K. Aakalu, Y. Y. Yang and H. Kong, Adv. Healthcare Mater., 2018, 7, e1701276 CrossRef PubMed.
- S. Moreno, B. Voit and J. Gaitzsch, Colloid Polym. Sci., 2021, 299, 309–324 CrossRef CAS.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- A. Najer, D. L. Wu, D. Vasquez, C. G. Palivan and W. Meier, Nanomedicine, 2013, 8, 425–447 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- L. Messager, J. Gaitzsch, L. Chierico and G. Battaglia, Curr. Opin. Pharmacol., 2014, 18, 104–111 CrossRef CAS PubMed.
- S. Ohki, J. Theor. Biol., 1970, 26, 277–287 CrossRef CAS PubMed.
- N. Ritzmann, J. Thoma, S. Hirschi, D. Kalbermatter, D. Fotiadis and D. J. Müller, Biophys. J., 2017, 113, 1181–1186 CrossRef CAS PubMed.
- R. Wehr, E. C. dos Santos, M. S. Muthwill, V. Chimisso, J. Gaitzsch and W. Meier, Polym. Chem., 2021, 12, 5377–5389 RSC.
- F. Itel, A. Najer, C. G. Palivan and W. Meier, Nano Lett., 2015, 15, 3871–3878 CrossRef CAS PubMed.
- D. Daubian, J. Gaitzsch and W. Meier, Polym. Chem., 2020, 11, 1237–1248 RSC.
- D. Daubian, A. Fillion, J. Gaitzsch and W. Meier, Macromolecules, 2020, 53, 11040–11050 CrossRef CAS.
- M. Fauquignon, E. Ibarboure, S. Carlotti, A. Brûlet, M. Schmutz and J.-F. Le Meins, Polymers, 2019, 11, 2013 CrossRef CAS PubMed.
- F. Itel, M. Chami, A. Najer, S. Lorcher, D. L. Wu, I. A. Dinu and W. Meier, Macromolecules, 2014, 47, 7588–7596 CrossRef CAS.
- J. A. González Calderón, D. Contreras López, E. Pérez and J. Vallejo Montesinos, Polym. Bull., 2020, 77, 2749–2817 CrossRef.
- K. Jaskiewicz, M. Makowski, M. Kappl, K. Landfester and A. Kroeger, Langmuir, 2012, 28, 12629–12636 CrossRef CAS PubMed.
- H. Aranda-Espinoza, H. Bermudez, F. S. Bates and D. E. Discher, Phys. Rev. Lett., 2001, 87, 208301 CrossRef CAS PubMed.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 8203–8208 CrossRef CAS.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS PubMed.
- E. Mabrouk, D. Cuvelier, L.-L. Pontani, B. Xu, D. Lévy, P. Keller, F. Brochard-Wyart, P. Nassoy and M.-H. Li, Soft Matter, 2009, 5, 1870–1878 RSC.
- L. Jia and M.-H. Li, Liq. Cryst., 2014, 41, 368–384 CrossRef CAS.
- S. Hocine, A. Brûlet, L. Jia, J. Yang, A. Di Cicco, L. Bouteiller and M.-H. Li, Soft Matter, 2011, 7, 2613–2623 RSC.
- Y. Men, F. Peng, Y. Tu, J. C. M. van Hest and D. A. Wilson, Polym. Chem., 2016, 7, 3977–3982 RSC.
- K. T. Kim, J. H. Zhu, S. A. Meeuwissen, J. J. L. M. Cornelissen, D. J. Pochan, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2010, 132, 12522–12524 CrossRef CAS PubMed.
- S. A. Meeuwissen, K. T. Kim, Y. Chen, D. J. Pochan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 7070–7073 CrossRef CAS PubMed.
- R. Górecki, F. Antenucci, K. Norinkevicius, L. Elmstrøm Christiansen, S. T. Myers, K. Trzaskuś and C. Hélix-Nielsen, Langmuir, 2021, 37, 2079–2090 CrossRef PubMed.
- C. Lebleu, L. Rodrigues, J.-M. Guigner, A. Brûlet, E. Garanger and S. Lecommandoux, Langmuir, 2019, 35, 13364–13374 CrossRef CAS PubMed.
- C. K. Wong, A. F. Mason, M. H. Stenzel and P. Thordarson, Nat. Commun., 2017, 8, 1240 CrossRef PubMed.
- C. K. Wong, A. D. Martin, M. Floetenmeyer, R. G. Parton, M. H. Stenzel and P. Thordarson, Chem. Sci., 2019, 10, 2725–2731 RSC.
- F. H. Sobotta, M. T. Kuchenbrod, F. V. Gruschwitz, G. Festag, P. Bellstedt, S. Hoeppener and J. C. Brendel, Angew. Chem., Int. Ed., 2021, 60, 24716–24723 CrossRef CAS PubMed.
- Y. Deng, H. Chen, X. Tao, S. Trépout, J. Ling and M.-H. Li, Chin. Chem. Lett., 2020, 31, 1931–1935 CrossRef CAS.
- A. F. Mason and P. Thordarson, ACS Macro Lett., 2016, 5, 1172–1175 CrossRef CAS PubMed.
- D. Zhang, Y. Fan, H. Chen, S. Trépout and M.-H. Li, Angew. Chem., Int. Ed., 2019, 58, 10260–10265 CrossRef CAS PubMed.
- S. Moreno, H. Hübner, C. Effenberg, S. Boye, A. Ramuglia, D. Schmitt, B. Voit, I. M. Weidinger, M. Gallei and D. Appelhans, Biomacromolecules, 2022, 23, 4655–4667 CrossRef CAS PubMed.
- K. Zhang, S. Moreno, X. Wang, Y. Zhou, S. Boye, D. Voigt, B. Voit and D. Appelhans, Biomacromolecules, 2023, 24, 2489–2500 CrossRef CAS PubMed.
- X. Wang, S. Moreno, S. Boye, P. Wang, X. Liu, A. Lederer, B. Voit and D. Appelhans, Adv. Sci., 2021, 8, 2004263 CrossRef CAS PubMed.
- R. Ccorahua, S. Moreno, H. Gumz, K. Sahre, B. Voit and D. Appelhans, RSC Adv., 2018, 8, 25436–25443 RSC.
- B. Iyisan, A. C. Siedel, H. Gumz, M. Yassin, J. Kluge, J. Gaitzsch, P. Formanek, S. Moreno, B. Voit and D. Appelhans, Macromol. Rapid Commun., 2017, 38, 1700486 CrossRef PubMed.
- M. Palinske, U. L. Muza, S. Moreno, D. Appelhans, S. Boye, R. Schweins and A. Lederer, Macromol. Chem. Phys., 2023, 224, 2200300 CrossRef CAS.
- S. Moreno, S. Boye, A. Lederer, A. Falanga, S. Galdiero, S. Lecommandoux, B. Voit and D. Appelhans, Biomacromolecules, 2020, 21, 5162–5172 CrossRef CAS PubMed.
- S. Moreno, P. Sharan, J. Engelke, H. Gumz, S. Boye, U. Oertel, P. Wang, S. Banerjee, R. Klajn, B. Voit, A. Lederer and D. Appelhans, Small, 2020, 16, 2002135 CrossRef CAS PubMed.
- F. Rajabasadi, S. Moreno, K. Fichna, A. Aziz, D. Appelhans, O. G. Schmidt and M. Medina-Sánchez, Adv. Mater., 2022, 34, 2204257 CrossRef CAS PubMed.
- D. Wang, S. Moreno, S. Boye, B. Voit and D. Appelhans, Chem. Commun., 2021, 57, 8019–8022 RSC.
- X. Xu, S. Moreno, S. Boye, P. Wang, B. Voit and D. Appelhans, Adv. Sci., 2023, 10, 2207214 CrossRef CAS PubMed.
- P. Wang, S. Moreno, A. Janke, S. Boye, D. Wang, S. Schwarz, B. Voit and D. Appelhans, Biomacromolecules, 2022, 23, 3648–3662 CrossRef CAS PubMed.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O’Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS PubMed.
- S. Varlas, T. J. Neal and S. P. Armes, Chem. Sci., 2022, 13, 7295–7303 RSC.
- Q. Zhang, R. Zeng, Y. Zhang, Y. Chen, L. Zhang and J. Tan, Macromolecules, 2020, 53, 8982–8991 CrossRef CAS.
- T. Chidanguro, E. Ghimire and Y. C. Simon, J. Mater. Chem. B, 2020, 8, 8914–8924 RSC.
- Y. Yang, A. Alford, V. Kozlovskaya, S. Zhao, H. Joshi, E. Kim, S. Qian, V. Urban, D. Cropek, A. Aksimentiev and E. Kharlampieva, ACS Appl. Polym. Mater., 2019, 1, 722–736 CrossRef CAS PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Mueller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- J. Gaitzsch, S. Hirschi, S. Freimann, D. Fotiadis and W. Meier, Nano Lett., 2019, 19, 2503–2508 CrossRef CAS PubMed.
- S. Miwa, R. Takahashi, C. Rössel, S. Matsumoto, S. Fujii, J. H. Lee, F. H. Schacher and K. Sakurai, Langmuir, 2018, 34, 7813–7820 CrossRef CAS PubMed.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00239c |
| This journal is © The Royal Society of Chemistry 2024 |