
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorescent acyclic cucurbituril solid supramolecular multicolour delayed fluorescence behaviour†
Man
Huo
,
Shuang-Qi
Song
,
Xian-Yin
Dai
,
Fan-Fan
Li
,
Yu-Yang
Hu
and
Yu
Liu
 *
*
College of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: yuliu@nankai.edu.cn
First published on 26th February 2024
Abstract
Organic photoluminescent macrocyclic hosts have been widely advanced in many fields. Phosphorescent hosts with the ability to bind organic guests have rarely been reported. Herein, acyclic cucurbituril modified with four carboxylic acids (ACB-COOH) is mined to present uncommon purely organic room-temperature phosphorescence (RTP) at 510 nm with a lifetime of 1.86 μs. Its RTP properties are significantly promoted with an extended lifetime up to 2.12 s and considerable quantum yield of 6.29% after assembly with a polyvinyl alcohol (PVA) matrix. By virtue of the intrinsic self-crimping configuration of ACB-COOH, organic guests, including fluorescence dyes (Rhodamine B (RhB) and Pyronin Y (PyY)) and a drug molecule (morphine (Mor)), could be fully encapsulated by ACB-COOH to attain energy transfer involving phosphorescent acyclic cucurbituril. Ultimately, as-prepared systems are successfully exploited to establish multicolor afterglow materials and visible sensing of morphine. As an expansion of phosphorescent acyclic cucurbituril, the host afterglow color can be readily regulated by attaching different aromatic sidewalls. This study develops the fabrication strategies and application scope of a supramolecular phosphorescent host and opens up a new direction for the manufacture of intelligent long-lived luminescent materials.
Cucurbit[n]urils (CB[n])-based supramolecular organic luminescent emission has attracted a considerable amount of attention and has been broadly used in many fields, including biological imaging,1 organic light-emitting diodes,2 sensing and detection.3 The macrocyclic-confinement derived from the rigid pumpkin-shaped structure can apparently adjust the guest aggregation performance, and thus various intelligent organic light-emitting materials in aqueous media and in the solid state have been developed.4 Recently, it has been documented that CB[n] (n = 6, 7, 8, or 10) can induce high-efficiency RTP by forming stable complexes with phosphorescent guests in the solid state and in an aqueous medium.5 The stiff cavity of CB[n] effectively enhances intersystem crossing (ISC) and significantly inhibits the vibration of chromophore molecules, thus reducing nonradiative transitions.6 Besides, the hydrophobicity of the CB[n] cavity effectively suppresses the quenching of the triplet state of the phosphor by water molecules or oxygen. Corresponding reports have focused on the macrocyclic or assembly confinement effect towards phosphorescent guests, and the macrocycle itself cannot produce emissive characteristics. Additionally, it is a formidable challenge to achieve the chemical modification of CB[n], which greatly hinders the application scope of functional luminescent materials. In advanced studies, acyclic cucurbiturils derived from a glycoluril tetramer (ACB) have been explored as a class of novel macrocycle,7 consisting of ACB to which aromatic terminals have been attached. Acyclic cucurbiturils possess good water-solubility and impart an overall C-shaped configuration, which shows unique molecular recognition properties and self-assembly performance, in particular revealing a high affinity for various drugs and some organic dye molecules.8 By connecting different aromatic sidewalls, diverse acyclic cucurbituril derivatives with luminescent performance have been successfully designed and synthesized,9 which endows them with externally adjustable structures and revolutionary properties, thus greatly expanding their application potential in smart luminescent materials.
With the development of supramolecular photoluminescent macrocyclic hosts,10 the host–guest interaction provides a desirable platform for potentially regulating photoluminescence performance.11 In contrast to fluorescent macrocyclic hosts, phosphorescent ones promise prominent advantages, such as long lifetime and large Stokes shifts, which can mitigate interference from background fluorescence.12 To achieve efficient phosphorescence emission, three basic conditions need to be considered: ensuring effective ISC, minimizing nonradiative relaxation and reducing quenching loss of the triple-excited state.13 Apart from CB macrocyclic confinement phosphorescence emission, structural immobilization of macrocyclic hosts can also realize triplet emission for the host itself. Some feasible strategies have been proposed, such as macrocyclization of an organic luminophore,14 doping,15 crystallization16 and polymerization to build phosphorescent macrocyclic materials.17 For instance, Tang et al. described a series of crown-ether-based RTP materials through recrystallization strategies and achieved phosphorescent modulation after complexing with potassium.16 A cyclization-promoted RTP system has been reported by Huang and co-workers, which showed progressive enhancement of low-temperature phosphorescence from a carbazole unit to linear conjugation and thence to a cyclic conjugated molecule.18 Despite significant progress having been made in the preparation of phosphorescent systems based on CB[n]-confinement or host self-aggregation, the phosphorescence behaviour of glycoluril derivatives has so far not been reported, to the best of our knowledge. Glycoluril-based cucurbituril derivatives show improved water-solubility with considerable ability to include guests to facilitate phosphorescence performance and exhibit tuneable photophysical performance by connecting different aromatic sidewalls. In particular, phosphorescent hosts with the ability to bind organic guests for energy transfer or to influence electron transfer have not yet been developed. Unlike the traditional cascaded assembly confined triplet-to-singlet energy transfer process (TS-FRET) process, phosphorescent hosts as donors could take part directly in the TS-FRET process by encapsulating an organic dye acceptor because of the close distance between the donor and acceptor. This strategy is of cardinal significance for the promotion of increasingly sophisticated supramolecular materials.
In this study, we report a phosphorescent ACB-COOH (Scheme 1) which generated an uncommon phosphorescence emission at 510 nm. The carbonyl-rich glycoluril oligomer was introduced to boost the inefficient ISC. After embedding into the PVA matrix, it exhibited ultralong phosphorescence emission with a lifetime of 2.12 s because abundant hydrogen-bonding interactions between ACB-COOH and PVA suppressed nonradiative decay processes. Control studies in the film state revealed that the (p-phenylenebisoxy)diacetic acid (Ph-COOH)@PVA system was barely emissive, which further implied that the carbonyl-rich glycoluril oligomer with a self-crimping configuration are vital factors in producing effective afterglow emission. Benefiting from excellent host–guest recognition properties, two kinds of guest (Rhodamine B (RhB), Pyronin Y (PyY) and morphine (Mor)) were selected to be encapsulated by phosphorescent ACB-COOH. The inclusion interaction with Mor led to phosphorescent quenching via a photoinduced electron transfer (PET) process, which was utilized as an effective probe for the visible detection of Mor. Interestingly, an RhB⊂ACB-COOH@PVA or a PyY⊂ACB-COOH@PVA system would show orange or red afterglow emission through TS-FRET. In view of their various lifetimes and colourful afterglow emissions, obtained films were shaped by paper cutting craft with multicolour afterglow emission and utilized for digital encryption and multi-level anti-counterfeiting.
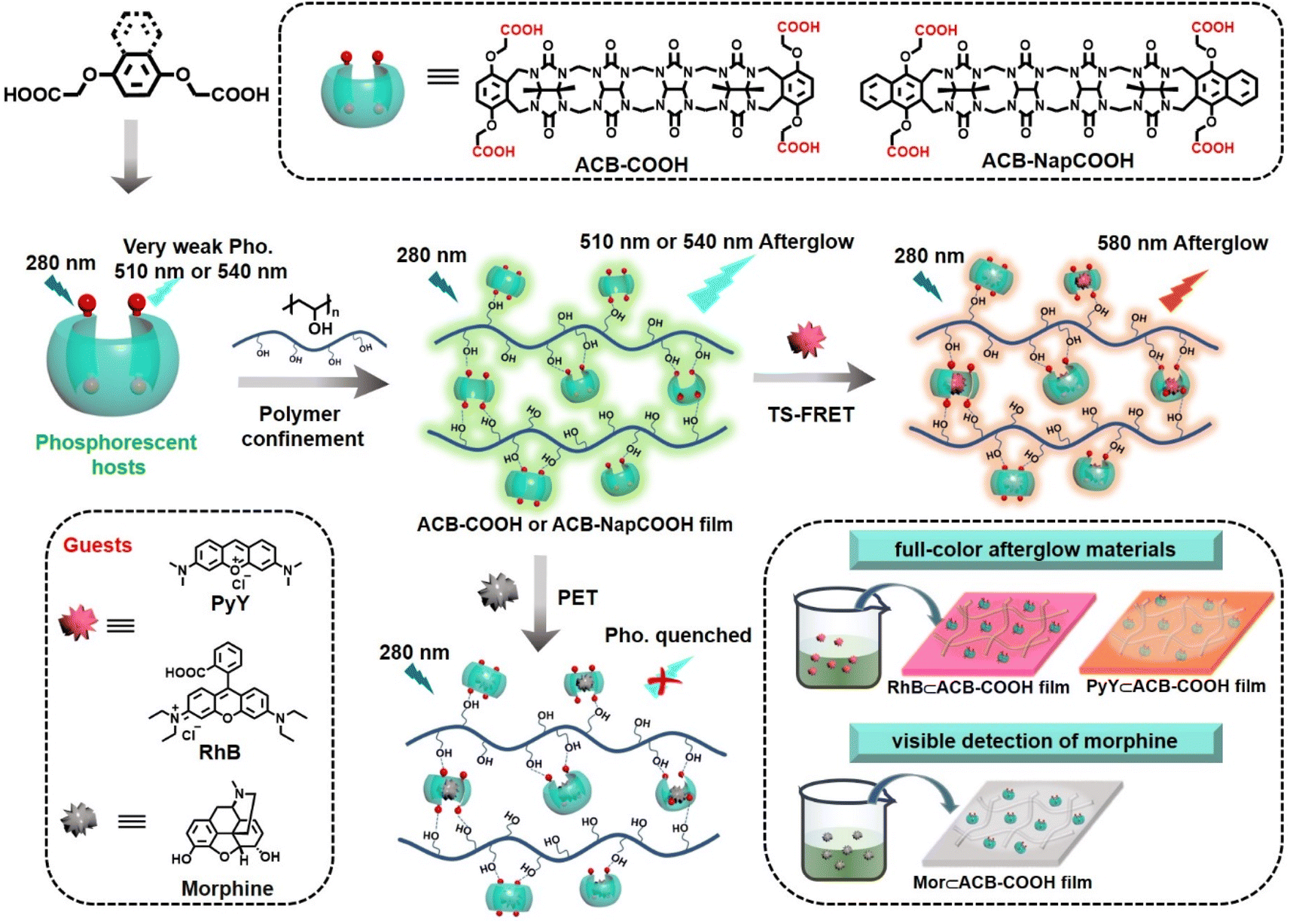 | ||
| Scheme 1 Schematic illustration of phosphorescent acyclic cucurbituril solid supramolecular multicolour delayed fluorescence. | ||
Results and discussion
Possessing a C-shaped configuration, ACB-COOH consists of ACB bridged by methylene and o-xylylene rings substituted with OCH2COOH groups as terminal walls,19 which shows unique molecular recognition properties. ACB-COOH was synthesized in accordance with previously reported procedures, and the chemical structure was characterized by nuclear magnetic resonance spectroscopy (NMR) (Fig. S1 and S2†) and high-resolution mass spectrometry (Fig. S3†). ACB-COOH defines a hydrophobic cavity and shows high affinity for organic ammonium ions through electrostatic interactions. The UV-vis absorption spectrum of ACB-COOH in aqueous solution was measured, showing maximum absorption at 290 nm (Fig. S7†). Considering its symmetrical structure with abundant carbonyl groups, it was expected to enable the RTP emission of ACB-COOH attributed to the attachment of potential phosphor Ph-COOH sidewalls.20 ACB-COOH powder revealed an uncommon RTP emission in the gated spectrum with a lifetime at 510 nm of 1.86 μs (Fig. S9†). Polymer confinement was confirmed as a useful method to construct amorphous materials with ultralong RTP emission, because abundant inter-/intramolecular interactions greatly restrict molecular motion.21 It is well documented that PVA has a considerable oxygen barrier, which plays an extremely important role in the preparation of polymer-based RTP film materials, especially with hydrophilic molecules owing to its effective rigidification effect.22 Encouragingly, ACB-COOH was doped into the rigid PVA matrix to fabricate the amorphous ACB-COOH@PVA film. An aqueous solution of ACB-COOH@PVA was prepared by heating and ultrasonic treatment, and then, the above solution was drop-casted onto quartz flakes followed by drying thoroughly to remove water to promote the generation of hydrogen-bonding interactions between ACB-COOH and the PVA matrix. As shown in Fig. 1a, the obtained ACB-COOH@PVA film exhibited a main emission peak at 320 nm with a lifetime of 2.04 ns (Fig. 1f), which was attributed to fluorescence emission. In the time-resolved (gated) spectrum of the ACB-COOH film (Fig. 1a), there was a prominent emission peak at 510 nm. The time-resolved emission decay curve described a phosphorescence lifetime of the ACB-COOH@PVA film at 510 nm of up to 2.12 s (Fig. 1e), while the corresponding quantum yield was 6.29% (Fig. S8†). In contrast, the gated spectrum of the neat PVA film was silent (Fig. S10†). The above results suggested that the phosphorescence emission originated from ACB-COOH and the ACB-COOH@PVA film possessing abundant hydrogen-bonding networks efficiently limited the phosphor vibration to inhibit the nonradiative relaxation channels of triplet excitons. We further measured the excitation-dependent phosphorescence emission maps of the ACB-COOH@PVA film (Fig. 1b). When the excitation wavelength changed from 260 nm to 360 nm, the phosphorescence emission was dominated by emission centered at around 510 nm, with the optimal excitation being 280 nm. It is highly significant that the amorphous host with long-lived supramolecular phosphorescence was obtained under ambient conditions. To investigate the effect of doping concentration on RTP performance, different concentrations of ACB-COOH-doped PVA films were prepared (Fig. S11†). Upon increasing the weights of ACB-COOH, the RTP emission intensity and lifetime gradually increased (Fig. S12†). We determined the optimal doping concentration to be 1.7 wt% in subsequent research into the photophysical properties of the ACB-COOH@PVA system. Moreover, when the doping percentage of ACB-COOH (1.7 wt%) was fixed, the phosphorescence performance of ACB-COOH was studied with different concentrations of PVA. The optimal concentration was determined to be 60 mg mL−1 (Fig. S13†). When the ACB-COOH@PVA film absorbed water vapor from the atmosphere, its RTP intensity was gradually decreased (Fig. S14†). Other polymer matrices, such as sodium polyacrylate (PAAS) and polyacrylamide (PAM), were employed to conduct control experiments (Fig. S15†). ACB-COOH/PAAS and ACB-COOH/PAM systems showed almost identical profiles of steady-state and gated spectra to the ACB-COOH@PVA system. For the phosphorescence lifetime at 510 nm, that of the ACB-COOH/PAAS system was measured to be 129.2 ms and that of the ACB-COOH/PAM system was 272.2 ms, greatly inferior to the ACB-COOH@PVA system.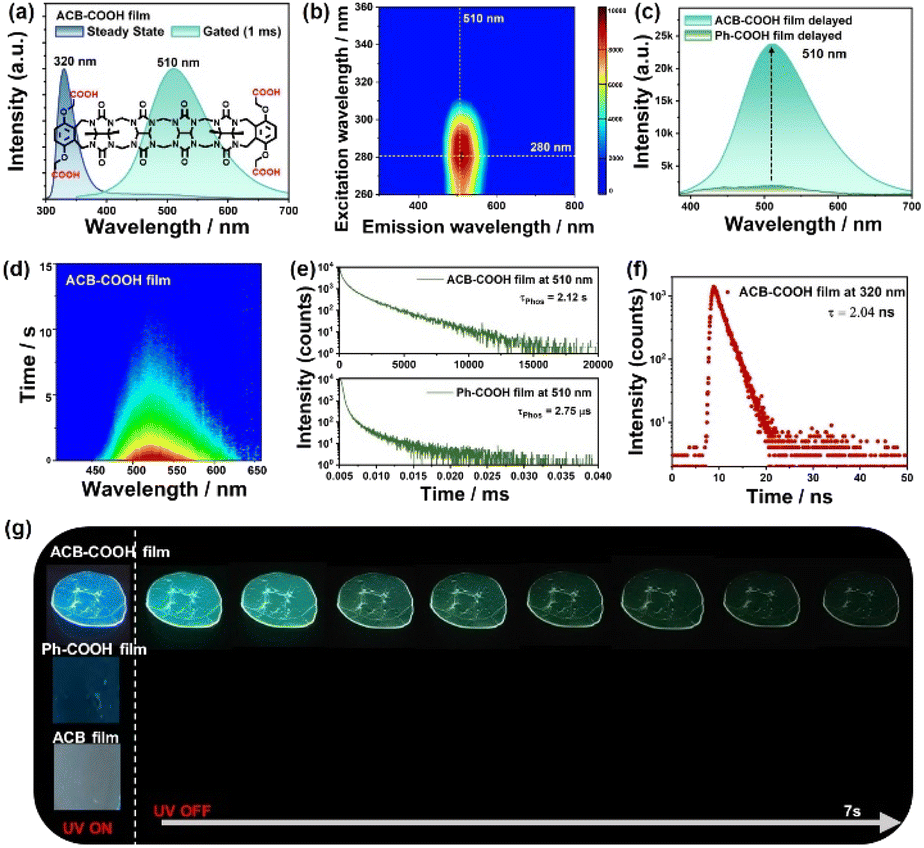 | ||
| Fig. 1 (a) Steady-state and gated (delay time: 1 ms) spectra of the ACB-COOH film. (b) Phosphorescence excitation mapping of the ACB-COOH film. (c) Delayed spectra of the ACB-COOH film and Ph-COOH film. (d) Transient emission spectrum of the ACB-COOH film, excited at 280 nm. (e) Phosphorescence lifetime decay curves of the ACB-COOH film and Ph-COOH film at 510 nm. (f) Fluorescence lifetime of the ACB-COOH film at 320 nm. (g) Afterglow performance of ACB-COOH, Ph-COOH and ACB films under a 254 nm UV lamp as well as removal of the excitation. | ||
To gain deeper insight into the phosphorescence emission of the ACB-COOH@PVA system, some control experiments were conducted, taking advantage of Ph-COOH, ACB and diethyl 2,2′-(phen-1,4-ylenedioxy)bis(acetate) (Ph-COOEt). First, the phosphorescence properties of Ph-COOH-, ACB-, and ACB-COOH-doped PVA films were observed before and after 280 nm UV excitation under ambient conditions. A clear green afterglow lasting 10 s was captured in the ACB-COOH film, but no afterglow was noticed in the other films after turning off the UV lamp (Fig. 1g and Movie S1†). Furthermore, the detailed photophysical performances of the above films were described with delayed spectra and time-resolved emission decay curves. Ph-COOH-doped film showed a very weak emission peak at 510 nm in the delayed spectra which was assigned to π–π stacking interaction between two Ph-COOH from its aggregation state in the PVA film.23 Compared with that, the RTP intensity of the ACB-COOH@PVA film presented dramatic strengthening under same experimental conditions (Fig. 1c and S16†). Significantly, the lifetimes of Ph-COOH- and Ph-COOEt-doped films at 510 nm were measured to be only 2.75 μs and 2.80 μs, respectively (Fig. 1e and S17†), further manifesting fantastic RTP emission features for the ACB-COOH@PVA film with a second-level lifetime. This phenomenon was mainly caused by multiple carbonyl and nitrogen atoms from ACB greatly boosting spin orbit coupling and triggering effective ISC processes, resulting in uncommon long-lived phosphorescence behaviour in the acyclic cucurbituril. At the same time, the carbonyl and nitrogen atoms synergistically participated in the formation of hydrogen-bonding networks with the PVA matrix to restrain the nonradiative transitions of the triplet-state excitons.24 In addition, the ACB-doped PVA film showed phosphorescence emission at 480 nm, and the phosphorescence lifetime was 7.50 ms (Fig. S18†), implying that the phosphorescence emission of ACB-COOH@PVA stemmed mainly from the aromatic sidewall Ph-COOH rather than the ACB moiety. Furthermore, to investigate the close-range interaction between ACB and phosphors, a physical mixture of ACB and Ph-COOH was incorporated into the PVA matrix, which displayed much weaker phosphorescence emission at 425 nm with a lifetime of 16.06 ms (Fig. S19†). Additionally, cucurbituril, with a defined rigid pumpkin-shaped structure and characterized by a perfect cyclized conformation, was exploited to create a macrocyclic-confined phosphorescence assembly. CB[7] was employed to interact with Ph-COOH to explore the comparison effect of the cyclization conformation on phosphorescence properties. As demonstrated in Fig. S20†, CB[7]@Ph-COOH-doped PVA films revealed RTP emission at 400 nm, and the corresponding lifetime was 0.112 ms. The above results jointly demonstrated that the covalent attachment and self-crimping configuration greatly limited the vibration of phosphor and enhanced ISC processes to achieve effective phosphorescence behaviour in the acyclic cucurbituril.
To further prove the phosphorescence property of ACB-COOH, temperature-dependent emission spectra as well as time-resolved emission decay curves were obtained. With the temperature increasing from 77 K to 298 K, the phosphorescence emission intensity and lifetime of ACB-COOH powder apparently decreased (Fig. 2b, c and S21†). Remarkably, the phosphorescence emission peak was located at 425 nm at 77 K, which would redshift to 510 nm as the temperature rose to 298 K. The green long-lived phosphorescence emission around 510 nm was ascribed to a π–π stacking interaction between two Ph-COOH in ACB-COOH. Furthermore, we speculated that the emission at 425 nm at 77 K was assigned to the isolated Ph-COOH chromophore.25 To confirm this hypothesis, the phosphorescence spectrum of Ph-COOH in N,N-dimethylformamide (DMF) at extremely low concentration was measured at 77 K (Fig. 2d). It showed a characteristic blue emission at around 420 nm, representing the phosphorescence emission of isolated Ph-COOH, which suggested that the phosphorescence spectrum of ACB-COOH at 77 K at around 420 nm belonged to isolated sidewalls. We also performed the temperature-dependent emission spectra of the ACB-COOH@PVA film and tested corresponding lifetimes (Fig. 2a and S22–S24†). As shown in Fig. 2d, the photograph of the ACB-COOH film at 77 K displayed bright blue afterglow emission after removing the excitation light source. Temperature-related spectral experiments have confirmed its RTP emission properties, excluding thermal activation delayed fluorescence (TADF).
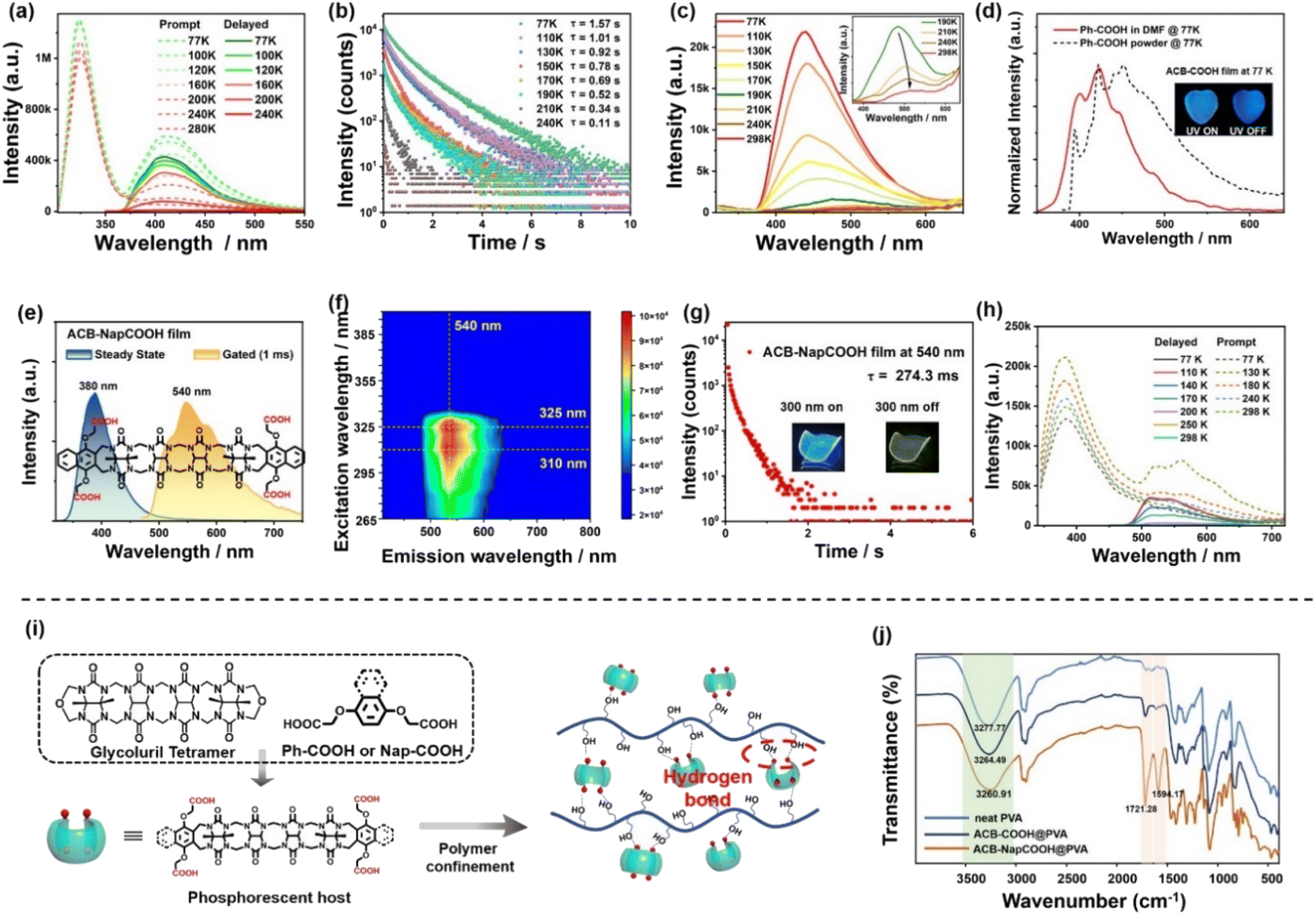 | ||
| Fig. 2 (a) Temperature-dependent prompt spectra and delayed spectral curves of the ACB-COOH film. (b) Temperature-dependent lifetime decay curves of the ACB-COOH powder from 77 to 298 K. (c) Temperature-dependent delayed spectra of the ACB-COOH powder from 77 to 298 K. Inset: enlarged view of temperature-dependent delayed spectra of the ACB-COOH powder from 190 to 298 K. (d) Normalized phosphorescence spectra of Ph-COOH in DMF and the Ph-COOH powder at 77 K. Inset: photograph of ACB-the COOH film at 77 K under a 254 nm UV lamp as well as removal of the excitation. (e) Steady-state and gated (delay time: 1 ms) spectra of the ACB-NapCOOH film. (f) Phosphorescence excitation mapping of the ACB-NapCOOH film. (g) Phosphorescence lifetime decay curves of the ACB-NapCOOH film at 540 nm. Inset: a photograph of the ACB-NapCOOH film before and after turning off the UV lamp (300 nm) under an ambient atmosphere. (h) Temperature-dependent prompt spectra and delayed spectral curves of the ACB-NapCOOH powder. (i) Schematic illustration of phosphorescent acyclic cucurbituril doped into a PVA matrix. (j) FT-IR spectra of neat PVA, ACB-COOH@PVA, ACB-NapCOOH@PVA films. | ||
Further, 4-(carboxymethoxynaphthalen-1-yloxy) acetic acid (Nap-COOH) modified ACB (ACB-NapCOOH) was synthesized to develop the ultralong phosphorescence behaviour of solid supramolecular acyclic cucurbituril (Fig. S4–S6†).26 ACB-NapCOOH possessing an extended aromatic conjugated structure exhibited good hydrophilicity owing to its solubilizing propoxycarboxylate side-chains. The steady-state and delayed spectra of the ACB-NapCOOH powder exhibited fluorescence emission at 380 nm and RTP emission at 540 nm, and the lifetime of RTP emission was 1.72 μs (Fig. S25†). Finally, the ACB-NapCOOH@PVA film was prepared using the aforementioned method. As depicted in Fig. 2e, the ACB-NapCOOH@PVA film displayed a fluorescence emission band at 380 nm according to the steady-state photoluminescence spectrum, and the fluorescence lifetime was 6.69 ns (Fig. S26†). The ACB-NapCOOH@PVA film presented a dominant redshifted emission band at around 540 nm compared with the ACB-COOH@PVA system in the gated spectrum, attributed to the extension of the π-conjugate structure of aromatic capping groups. Moreover, an obvious yellow afterglow of ACB-NapCOOH@PVA film could be seen by the naked eye under ambient conditions (Fig. 2g (inset)). According to the time-resolved photoluminescent decay curve of the ACB-NapCOOH@PVA film, the phosphorescence lifetime was determined to be 274.3 ms (Fig. 2g). The difference in phosphorescent lifetime between the ACB-COOH film and ACB-NapCOOH film was due to the aggregation-induced quenching of naphthalene.27 The excitation-dependent phosphorescence emission maps of the ACB-NapCOOH@PVA film demonstrated that it had a certain emission centered at around 540 nm with a change of excitation wavelength between 265 nm and 400 nm (excitation: 310 nm and emission: 540 nm) (Fig. 2f). Moreover, the temperature-dependent emission spectra were recorded for the ACB-NapCOOH powder (Fig. 2h). With increasing temperature, the phosphorescence intensity and lifetime revealed a decreasing trend, which eliminated the possibility of TADF for the emission band at 540 nm. The phosphorescence lifetime decay curves showed that the lifetime of ACB-NapCOOH powder at 77 K was 578.3 ms (Fig. S27†). The delay spectrum of Nap-COOH in a low concentration of DMF at 77 K was measured, which represented the delayed emission from a locally (monomer) excited state. In comparison, the Nap-COOH powder and ACB-NapCOOH@PVA exhibited broader redshifted emission spectra, which was attributed to J-type aggregate formation (Fig. S28†). Additionally, the phosphorescence quantum yield of the ACB-NapCOOH@PVA film was measured to be 4.87% (Fig. S29†). As a control, the photophysical performance of Nap-COOH-doped PVA films was also studied. The delayed spectrum of the Nap-COOH-doped PVA film displayed a similar profile to the ACB-NapCOOH-doped PVA film (Fig. S30†), while the phosphorescence emission intensity was lower than that of ACB-NapCOOH@PVA. The phosphorescence lifetime of the Nap-COOH film at 540 nm was 69.0 ms. From above combined results, it was concluded that the phosphorescence emission of ACB-NapCOOH@PVA could stem from the aromatic capping groups, and the abundant carbonyl groups and PVA-based hydrogen bonding network synergistically enhanced the inherent phosphorescence efficiency.
The uncommon phosphorescence behaviour of acyclic cucurbituril encouraged us to investigate the emission mechanism. The unhydrolyzed compounds, ACB-COOEt and ACB-NapCOOEt, were employed for embedding into the PVA matrix. The profiles of the phosphorescence spectrum of the ACB-COOEt-doped film showed little change from ACB-COOH@PVA, but the phosphorescence performance was unsatisfactory. The lifetime of the ACB-COOEt-doped film at 510 nm was 71.9 ms (Fig. S31†). Similarly, the delayed spectrum of the ACB-NapCOOEt-doped PVA film presented much weaker phosphorescence emission than the ACB-NapCOOH-doped PVA film (Fig. S32†). The lifetime of the ACB-NapCOOEt film was measured to be only 30.6 μs. This was because ACB-COOEt and ACB-NapCOOEt have poor compatibility with PVA, and hydrogen-bonding interactions within unhydrolyzed ACB-COOEt- or ACB-NapCOOEt-doped PVA films were not sufficient to create a rigid environment. Thus, the outstanding phosphorescence emission of the ACB-COOH@PVA film should be related to the rigidification effect involving PVA. Delay-time-dependent experiments for ACB-COOH, Ph-COOH, ACB-NapCOOH and Nap-COOH at 298 K and at 77 K were conducted, and there was no significant change in the emission intensity at different delay times (Fig. S33 and S34†). The amorphous morphology of the obtained films was confirmed via powder X-ray diffraction (Fig. S35†). Furthermore, Fourier transform infrared spectroscopy was used to study the formation of multiple hydrogen-bonds within the acyclic cucurbituril derivative-doped PVA matrix (Fig. 2j). Compared with neat PVA, the ACB-COOH@PVA system showed two emerging peaks at 1721.28 cm−1 and 1594.17 cm−1, attributed to the carbonyl of ACB. The peak position of the hydroxy group (–OH) on PVA shifted from 3277.77 cm−1 for neat PVA to 3260.91 cm−1 for the ACB-COOH@PVA film and to 3264.49 cm−1 for the ACB-NapCOOH@PVA film, indicating the establishment of a hydrogen-bonding network between the chromophores and PVA chains (Fig. 2i).
Possessing a self-crimping C-shaped conformation, the acyclic cucurbituril derivatives can adapt their flexible cavity to accommodate organic guests such as some fluorescent dyes and drug molecules. RhB, PyY and Mor were chosen for the host–guest study. First, inclusion behaviours were characterized through UV-vis spectrophotometric titration and 1H NMR. The Job's plot suggested a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio between ACB-COOH and RhB or PyY (Fig. S36 and S37†). The association constant (Ks) was 3.23 × 105 M−1 from nonlinear least-squares analysis according to absorbance intensity changes for RhB at 570 nm in aqueous solution (Fig. S38†). Fluorescence titration experiments also demonstrated that an RhB⊂ACB-COOH complex with a stoichiometric ratio of 1:1 was formed (Fig. S41†). The association constant (Ks) was determined to be 1.00 × 105 M−1 from absorbance intensity changes for PyY at 550 nm (Fig. S39†). Subsequently, we performed 1H NMR spectroscopy to research the host–guest interaction and recorded the 1H NMR spectra of RhB and PyY alone and of a 1:4 mixture of RhB or PyY and ACB-COOH. The 1H NMR spectrum of ACB-COOH with RhB or PyY showed an upfield shift for aromatic resonance, which suggested that complexation had occurred with the formation of RhB⊂ACB-COOH or PyY⊂ACB-COOH complexes (Fig. S40 and S42†). Mor is a well-known narcotic drug belonging to opioids.28 Complexes of ACB-COOH and Mor displayed an upfield shift of protons on the morphinan ring, indicating that they fit into the cavity of ACB-COOH (Fig. 3e). The binding constant (Ks) was determined to be 4.63 × 105 M−1 (Fig. S43†). As illustrated in Fig. 3a, a gradual quenching trend could be seen from the phosphorescence emission spectra of ACB-COOH at 510 nm with an increase in the concentration of Mor, and a standard curve could be described using the equation: y = 21.87 × x + 0.10 with a correlation coefficient (R2) of 0.9774 (Fig. 3d). The RTP lifetime declined from 1513.0 ms to 402.0 ms after the introduction of Mor (Fig. 3b). In comparison, heroin, another narcotic drug structurally similar to Mor, was employed to validate the effectiveness and generalizability of the proposed method, which revealed a mild quenching effect on the phosphorescence emission intensity of the ACB-COOH film (Fig. S45†and 3f). This was mainly due to the lower affinity of heroin with ACB-COOH than Mor with ACB-COOH. The XRD results demonstrated that the addition of Mor would not destroy the ACB-COOH-doped PVA film (Fig. S44†). Therefore, it could be deduced that the quenching mechanism should follow either the PET mechanism or the Förster resonance energy transfer (FRET) mechanism.29 The UV/vis absorption spectrum of Mor had almost no spectral overlap with the emission spectrum of ACB-COOH, reflecting that the PET mechanism can be rationalized as the control factor over the quenching process (Fig. S46† and 3c). As displayed in Fig. 3g, no obvious afterglow phenomenon was observed after removal of the excitation, which could be used as a visible phosphorescence probe for the detection of Mor.
:1 stoichiometric ratio between ACB-COOH and RhB or PyY (Fig. S36 and S37†). The association constant (Ks) was 3.23 × 105 M−1 from nonlinear least-squares analysis according to absorbance intensity changes for RhB at 570 nm in aqueous solution (Fig. S38†). Fluorescence titration experiments also demonstrated that an RhB⊂ACB-COOH complex with a stoichiometric ratio of 1:1 was formed (Fig. S41†). The association constant (Ks) was determined to be 1.00 × 105 M−1 from absorbance intensity changes for PyY at 550 nm (Fig. S39†). Subsequently, we performed 1H NMR spectroscopy to research the host–guest interaction and recorded the 1H NMR spectra of RhB and PyY alone and of a 1:4 mixture of RhB or PyY and ACB-COOH. The 1H NMR spectrum of ACB-COOH with RhB or PyY showed an upfield shift for aromatic resonance, which suggested that complexation had occurred with the formation of RhB⊂ACB-COOH or PyY⊂ACB-COOH complexes (Fig. S40 and S42†). Mor is a well-known narcotic drug belonging to opioids.28 Complexes of ACB-COOH and Mor displayed an upfield shift of protons on the morphinan ring, indicating that they fit into the cavity of ACB-COOH (Fig. 3e). The binding constant (Ks) was determined to be 4.63 × 105 M−1 (Fig. S43†). As illustrated in Fig. 3a, a gradual quenching trend could be seen from the phosphorescence emission spectra of ACB-COOH at 510 nm with an increase in the concentration of Mor, and a standard curve could be described using the equation: y = 21.87 × x + 0.10 with a correlation coefficient (R2) of 0.9774 (Fig. 3d). The RTP lifetime declined from 1513.0 ms to 402.0 ms after the introduction of Mor (Fig. 3b). In comparison, heroin, another narcotic drug structurally similar to Mor, was employed to validate the effectiveness and generalizability of the proposed method, which revealed a mild quenching effect on the phosphorescence emission intensity of the ACB-COOH film (Fig. S45†and 3f). This was mainly due to the lower affinity of heroin with ACB-COOH than Mor with ACB-COOH. The XRD results demonstrated that the addition of Mor would not destroy the ACB-COOH-doped PVA film (Fig. S44†). Therefore, it could be deduced that the quenching mechanism should follow either the PET mechanism or the Förster resonance energy transfer (FRET) mechanism.29 The UV/vis absorption spectrum of Mor had almost no spectral overlap with the emission spectrum of ACB-COOH, reflecting that the PET mechanism can be rationalized as the control factor over the quenching process (Fig. S46† and 3c). As displayed in Fig. 3g, no obvious afterglow phenomenon was observed after removal of the excitation, which could be used as a visible phosphorescence probe for the detection of Mor.
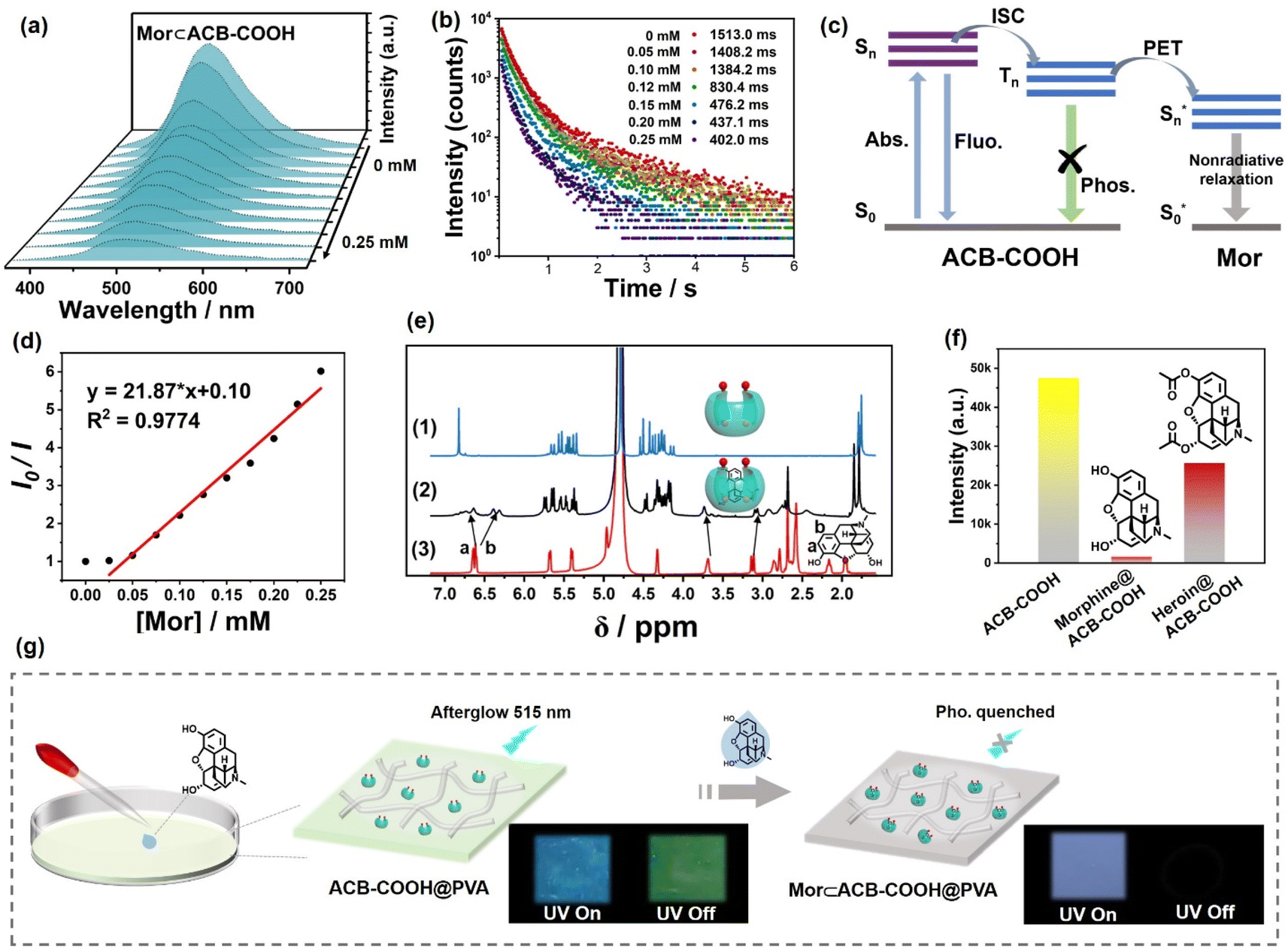 | ||
| Fig. 3 (a) Phosphorescence emission spectra of ACB-COOH upon incremental addition of Mor (0–0.25 mM). (b) Phosphorescence lifetime of the Mor⊂ACB-COOH film excited at 280 nm upon addition of Mor (0–0.25 mM). (c) Proposed diagram of the PET process between ACB-COOH and Mor. (d) Calibration curves of ACB-COOH toward Mor in the concentration range of 0–0.25 mM at room temperature. (e) 1H NMR spectra recorded (600 MHz, RT, D2O) for (1) ACB-COOH (2 mM), (2) an equimolar mixture of Mor and ACB-COOH, and (3) Mor (2 mM). (f) Phosphorescence intensity of the ACB-COOH film, morphine@ACB-COOH film and heroin@ACB-COOH film. (g) Schematic illustration of the construction of the Mor⊂ACB-COOH film and afterglow performance of the ACB-COOH film and Mor⊂ACB-COOH film under a 254 nm UV lamp as well as removal of the excitation. | ||
FRET takes place between singlet states of the donor and acceptor, and the spin angular momentum is conserved in dipole–dipole interactions at an appropriate distance.30 TS-FRET is a process in which energy is transferred from the triplet state of the phosphorescence donor to the singlet state of the acceptor.31 Different ratios of host–guest complexes were embedded into the PVA matrix to obtain a series of RhB⊂ACB-COOH@PVA films. The considerable overlap between the absorption spectra of RhB and PyY and RTP spectrum of ACB-COOH was in accord with the basic theory of FRET (Fig. 4c). Moreover, the host–guest interaction ensured a close distance between the donor and acceptor. As illustrated in Fig. 4a, as the ratio of ACB-COOH/RhB gradually increased from 50:1 to 50:8, the emission intensity of ACB-COOH decreased, while the emission peak at 580 nm attributed to RhB increased, indicating the existence of TS-FRET from ACB-COOH and RhB. Notably, the donor emission at around 510 nm was negligible when the donor/acceptor ratio was 50:8, implying a highly efficient TS-FRET process owing to the remarkable host–guest interaction between ACB-COOH and RhB. Meanwhile, the lifetime for the RhB⊂ACB-COOH@PVA film at 510 nm declined from the original 2.03 s to 32.1 ms (Fig. 4d), further confirming the occurrence of the TS-FRET process. The PRET efficiency was calculated to be 98.4% according to the decrease in donor lifetime. The lifetime at 580 nm for the RhB⊂ACB-COOH@PVA film was measured to be 43.8 ms (Fig. 4e), representing persistent luminescence based on the delayed sensitization of the singlets of RhB resulting from energy transfer involving triplets. In particular, the delayed fluorescence intensity at 580 nm was gradually intensified instead of being quenched even under a higher ratio of ACB-COOH/RhB, because the RhB molecule was encapsulated by the cavity of ACB-COOH to suppress fluorescence quenching caused by its self-aggregation. Additionally, a red afterglow could be seen by the naked eye after turning off the excitation source, demonstrating long-lived fluorescence from commercially available dyes (Fig. 4g and Movie S2†). PyY, another commercially available fluorescent dye molecule, was chosen as the guest donor in this study. As depicted in Fig. 4c, the absorption spectra of PyY had an appropriate spectral overlap with the RTP spectrum of ACB-COOH. A series of PyY⊂ACB-COOH@PVA films were prepared by doping PyY⊂ACB-COOH inclusion complexes into the PVA matrix. As seen from Fig. 4b, there was an increased emission peak located at 580 nm from PyY accompanied by a decline in the phosphorescence emission intensity at 510 nm, implying effective TS-FRET from ACB-COOH to PyY. The emission lifetime at 510 nm for PyY⊂ACB-COOH@PVA was 254.3 ms (Fig. 4d), and the TS-FRET efficiency could be calculated as 87.4%. At the same time, it was found that the degree of phosphorescence quenching of the PyY⊂ACB-COOH@PVA system was less than that of the RhB⊂ACB-COOH@PVA system, consistent with the phenomenon of the weaker inclusion interaction between PyY and ACB-COOH. In addition, the PyY⊂ACB-COOH@PVA film exhibited an orange afterglow with a lifetime of 108.8 ms at 580 nm (Fig. 4e and g). Notably, no delayed emission was detected for PyY⊂ACB-COOH@PVA and RhB⊂ACB-COOH@PVA under optimal excitation wavelengths for PyY or RhB (Fig. S47 and S48†) which excluded the persistent luminescence originating from the phosphorescence of the dyes. Triplet–triplet energy transfer (TTET) is a spin-enabled photophysical process used for direct electron exchange between molecules or intra-molecular fragments, and it can be used to construct efficient phosphorescent materials.32 The normalized delayed emission of RhB⊂ACB-COOH at 590 nm was identical to the fluorescence emission of pure RhB, indicating the typical delayed-fluorescence (DF) character of RhB emission rather than phosphorescent emission, which could exclude the TTET process (Fig. S49†). As the temperature increased from 77 K to 330 K, the delayed emission intensity at 580 nm gradually decreased. The lifetime of RhB⊂ACB-COOH@PVA at 580 nm was 0.73 s at 77 K (Fig. S50†). Unlike ACB-COOH encapsulating Mor to cause an electron transfer process, ACB-COOH encapsulated organic dyes (RhB or PyY) so that the TS-FRET process took place between the triplet state of the phosphorescence donor and singlet state of the acceptor. Benefiting from the specific cavity-encapsulation superiority of ACB-COOH, guest-dependent multicolour afterglow adjustment was achieved.
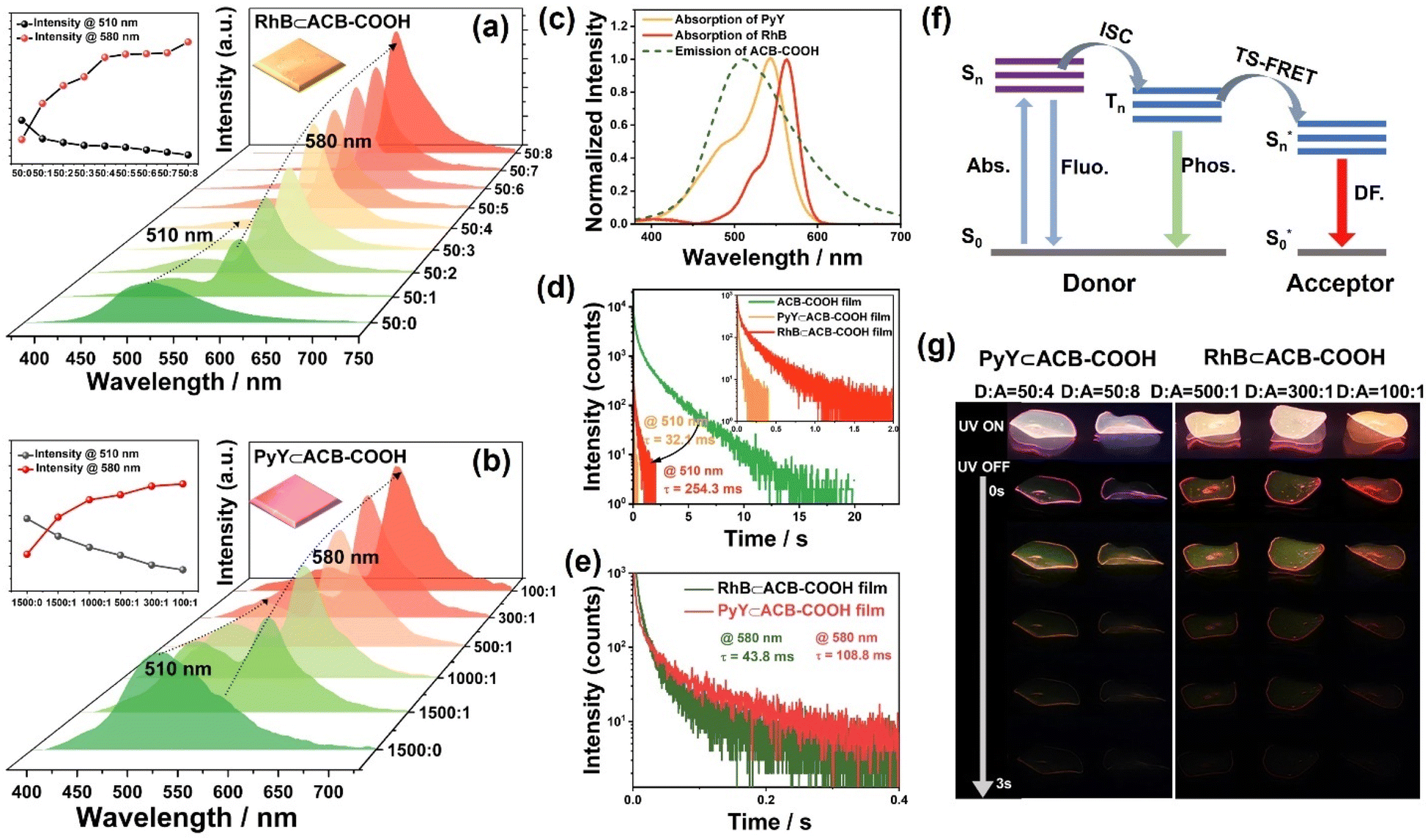 | ||
| Fig. 4 (a) Phosphorescence emission spectra of ACB-COOH upon incremental addition of RhB. Inset: changes in phosphorescence intensity at 510 nm and 580 nm. (b) Phosphorescence emission spectra of ACB-COOH upon incremental addition of PyY. Inset: changes in phosphorescence intensity at 510 nm and 580 nm. (c) Absorption spectrum of RhB and PyY and phosphorescence emission spectrum of ACB-COOH films. (d) Phosphorescence lifetime of the ACB-COOH film, RhB⊂ACB-COOH film and PyY⊂ACB-COOH film at 510 nm. (e) Phosphorescence lifetime of RhB⊂ACB-COOH and PyY⊂ACB-COOH films at 580 nm. (f) Proposed diagram of the TS-FRET process between ACB-COOH and acceptors. (g) Photograph of the RhB⊂ACB-COOH film and PyY⊂ACB-COOH film under different donor/acceptor ratios. | ||
Taking advantage of the features of processability and flexibility of the PVA film, a transparent rabbit pattern was tailored with ACB-COOH@PVA film (Fig. 5a). The rabbit crafted from the ACB-COOH@PVA film presented a green afterglow after removal of the excitation (Fig. 5b and Movie S3†). Interestingly, a red-emitting rabbit could be obtained by taking advantage of the RhB⊂ACB-COOH@PVA film under a 254 nm UV lamp, and an obvious red afterglow could be seen after turning off the UV lamp (Fig. 5c and Movie S4†). Notably, ACB-COOH@PVA and Ph-COOH@PVA were used as anti-counterfeiting ink for digital encryption (Fig. 5d). Exposed to a 254 nm UV lamp, the number “8888” was highlighted. After removing the UV lamp, the digital information “2023” was then displayed. Given various lifetimes and colourful afterglow emissions, a colourful flower was designed. The petals were coloured with RhB⊂ACB-COOH, the leaves were coloured with ACB-COOH, and the stem was coloured with ACB-NapCOOH. Under a 254 nm UV lamp radiation, the petals, leaves and stem of the flower emitted red, green and yellow, respectively (Fig. 5e). As time passed, the red petals disappeared, and the yellow stem and green leaves could be captured. Finally, only the green leaves could be seen by the naked eye. This unique phosphorescent acyclic cucurbituril has developed a new method for designing exquisite work and encrypting key information.
 | ||
| Fig. 5 (a) Transparent crafted rabbits obtained by a paper-cutting method based on the flexible ACB-COOH@PVA film and photographs of the crafted rabbit taken under daylight. (b) Photographs of the crafted rabbit of the ACB-COOH@PVA film under a 254 nm UV lamp as well as removal of the excitation. (c) Photographs of the rabbits crafted with the RhB⊂ACB-COOH@PVA film under a 254 nm UV lamp as well as removal of the excitation. (d) Schematic illustration of digital encryption fabricated with ACB-COOH@PVA and Ph-COOH@PVA. (e) Schematic illustration of a flower coloured by RhB⊂ACB-COOH, ACB-COOH and ACB-NapCOOH before and after excitation by a 254 nm ultraviolet lamp. | ||
Conclusions
In conclusion, a non-phosphorescent Ph-COOH sidewall was modified onto ACB to form a phosphorescent host ACB-COOH, which revealed an ultralong solid supramolecular purely organic phosphorescence with a lifetime of 2.12 s via doping ACB-COOH into a PVA matrix within a rigid molecular environment. In view of the host–guest interaction based on ACB-COOH endowed with a self-crimping C-shaped configuration, the phosphorescence behaviour could be modulated by encapsulating organic dyes or drug molecules. A promising probe for detecting Mor could be established depending on the inclusion interaction between Mor and ACB-COOH following the PET process. Moreover, the afterglow colour of the film was regulated from green to orange after including RhB or PyY in the cavity of ACB-COOH through a TS-FRET process. The two-dimensional film based on a supramolecular phosphorescent host not only presents a potential application in multicolour afterglow emission, phosphorescence sensing and drug detection but also expends the scope of intelligent long-lived luminescent materials.Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
M. Huo and Y. Liu conceived and directed this study. M. Huo conducted the synthesis, characterization, and analysis of all materials described with assistance from S.-Q Song, X.-Y. Dai, F.-F. Li and Y.-Y Hu. M. Huo wrote this manuscript. Y. Liu supervised the work and edited the manuscript. All authors discussed and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant nos. 22131008 and 21971127).References
- (a) A. T. Bockus, L. C. Smith, A. G. Grice, O. A. Ali, C. C. Young, W. Mobley, A. Leek, J. L. Roberts, B. Vinciguerra, L. Isaacs and A. R. Urbach, J. Am. Chem. Soc., 2016, 138, 16549–16552 CrossRef CAS PubMed; (b) H. Bai, Z. Liu, T. Zhang, J. Du, C. Zhou, W. He, J. H. C. Chau, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, ACS Nano, 2020, 14, 7552–7563 CrossRef CAS; (c) Y. Li, Y. Dong, X. Miao, Y. Ren, B. Zhang, P. Wang, Y. Yu, B. Li, L. Isaacs and L. Cao, Angew. Chem., Int. Ed., 2018, 57, 729–733 CrossRef CAS PubMed; (d) Z. Li, S. Sun, Z. Yang, S. Zhang, H. Zhang, M. Hu, J. Cao, J. Wang, F. Liu, F. Song, J. Fan and X. Peng, Biomaterials, 2013, 34, 6473–6481 CrossRef CAS PubMed.
- (a) Y. Xia, S. Chen and X.-L. Ni, ACS Appl. Mater. Interfaces, 2018, 10, 13048–13052 CrossRef CAS PubMed; (b) W. Zhang, Y. Luo, X.-L. Ni, Z. Tao and X. Xiao, Chem. Eng. J., 2022, 446, 136954 CrossRef CAS; (c) M. X. Yang, Y. Luo, W. Zhang, W. H. Lin, J. He, P. H. Shan, Z. Tao and X. Xiao, Chem.–Asian J., 2022, 17, e202200378 CrossRef CAS PubMed.
- (a) Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS PubMed; (b) G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528–7535 CrossRef CAS PubMed; (c) S. Sonzini, J. A. McCune, P. Ravn, O. A. Scherman and C. F. van der Walle, Chem. Commun., 2017, 53, 8842–8845 RSC.
- (a) H.-J. Kim, P. C. Nandajan, J. Gierschner and S. Y. Park, Adv. Funct. Mater., 2018, 28, 1705141 CrossRef; (b) G. Wu, I. Szabó, E. Rosta and O. A. Scherman, Chem. Commun., 2019, 55, 13227–13230 RSC; (c) G. Wu, Y. J. Bae, M. Olesińska, D. Antón-García, I. Szabó, E. Rosta, M. R. Wasielewski and O. A. Scherman, Chem. Sci., 2020, 11, 812–825 RSC; (d) X.-L. Ni, S. Chen, Y. Yang and Z. Tao, J. Am. Chem. Soc., 2016, 138, 6177–6183 CrossRef CAS PubMed; (e) X. Shi, X. Zhang, X.-L. Ni, H. Zhang, P. Wei, J. Liu, H. Xing, H.-Q. Peng, J. W. Y. Lam, P. Zhang, Z. Wang, H. Hao and B. Z. Tang, Macromolecules, 2019, 52, 8814–8825 CrossRef CAS; (f) G. Wu, Z. Huang and O. A. Scherman, Angew. Chem., Int. Ed., 2020, 59, 15963–15967 CrossRef CAS PubMed; (g) X. Yang, R. Wang, A. Kermagoret and D. Bardelang, Angew. Chem., Int. Ed., 2020, 59, 21280–21292 CrossRef CAS PubMed.
- (a) Z. Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed; (b) S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS; (c) J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS; (d) F. Liu, H. Yang, D. Sun, F. Gao, X. Zhang, Z. Zhao, X. Han and S. Liu, Chem. Sci., 2022, 13, 7247–7255 RSC.
- (a) X. Y. Dai, M. Huo, X. Dong, Y. Y. Hu and Y. Liu, Adv. Mater., 2022, 34, e2203534 CrossRef; (b) W. L. Zhou, Y. Chen, Q. Yu, H. Zhang, Z. X. Liu, X. Y. Dai, J. J. Li and Y. Liu, Nat. Commun., 2020, 11, 4655 CrossRef CAS PubMed; (c) D.-A. Xu, Q.-Y. Zhou, X. Dai, X.-K. Ma, Y.-M. Zhang, X. Xu and Y. Liu, Chin. Chem. Lett., 2022, 33, 851–854 CrossRef CAS; (d) X. K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, e2007476 CrossRef PubMed; (e) H. Sun and L. Zhu, Aggregate, 2023, 4, e253 CrossRef CAS; (f) L. Zhan, Y. Xu, T. Chen, Y. Tang, C. Zhong, Q. Lin, C. Yang and S. Gong, Aggregate, 2024, e485 CrossRef.
- (a) X. Lu and L. Isaacs, Angew. Chem., Int. Ed., 2016, 55, 8076–8080 CrossRef CAS; (b) D. Mao, Y. Liang, Y. Liu, X. Zhou, J. Ma, B. Jiang, J. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 12614–12618 CrossRef CAS PubMed; (c) C. Shen, D. Ma, B. Meany, L. Isaacs and Y. Wang, J. Am. Chem. Soc., 2012, 134, 7254–7257 CrossRef CAS PubMed; (d) D. Ma, G. Hettiarachchi, D. Nguyen, B. Zhang, J. B. Wittenberg, P. Y. Zavalij, V. Briken and L. Isaacs, Nat. Chem., 2012, 4, 503–510 CrossRef CAS.
- (a) A. T. Brockett, W. Xue, D. King, C.-L. Deng, C. Zhai, M. Shuster, S. Rastogi, V. Briken, M. R. Roesch and L. Isaacs, Chem, 2023, 9, 881–900 CrossRef CAS PubMed; (b) B. Zhang and L. Isaacs, J. Med. Chem., 2014, 57, 9554–9563 CrossRef CAS PubMed; (c) S. Jiang, J. Yang, L. Ling, S. Wang and D. Ma, Anal. Chem., 2022, 94, 5634–5641 CrossRef CAS PubMed; (d) D. Ma, B. Zhang, U. Hoffmann, M. G. Sundrup, M. Eikermann and L. Isaacs, Angew. Chem., Int. Ed., 2012, 51, 11358–11362 CrossRef CAS PubMed.
- (a) T. Minami, N. A. Esipenko, B. Zhang, M. E. Kozelkova, L. Isaacs, R. Nishiyabu, Y. Kubo and P. Anzenbacher Jr, J. Am. Chem. Soc., 2012, 134, 20021–20024 CrossRef CAS PubMed; (b) S. Murkli, J. Klemm, D. King, P. Y. Zavalij and L. Isaacs, Chemistry, 2020, 26, 15249–15258 CrossRef CAS PubMed; (c) E. G. Shcherbakova, B. Zhang, S. Gozem, T. Minami, P. Y. Zavalij, M. Pushina, L. D. Isaacs and P. Anzenbacher Jr, J. Am. Chem. Soc., 2017, 139, 14954–14960 CrossRef CAS PubMed.
- (a) Q. Li, Y. Wu, J. Cao, Y. Liu, Z. Wang, H. Zhu, H. Zhang and F. Huang, Angew. Chem., Int. Ed., 2022, 61, e202202381 CrossRef CAS PubMed; (b) X. Y. Lou and Y. W. Yang, J. Am. Chem. Soc., 2021, 143, 11976–11981 CrossRef CAS PubMed; (c) H. Wu, Y. Wang, L. O. Jones, W. Liu, B. Song, Y. Cui, K. Cai, L. Zhang, D. Shen, X. Y. Chen, Y. Jiao, C. L. Stern, X. Li, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 16849–16860 CrossRef CAS PubMed; (d) Z. Li, Z. Yang, Y. Zhang, B. Yang and Y. W. Yang, Angew. Chem., Int. Ed., 2022, 61, e202206144 CrossRef CAS PubMed; (e) S. Li, K. Liu, X. C. Feng, Z. X. Li, Z. Y. Zhang, B. Wang, M. Li, Y. L. Bai, L. Cui and C. Li, Nat. Commun., 2022, 13, 2850 CrossRef CAS PubMed.
- (a) E. Li, K. Jie, Y. Fang, P. Cai and F. Huang, J. Am. Chem. Soc., 2020, 142, 15560–15568 CrossRef CAS PubMed; (b) H. Y. Zhou, D. W. Zhang, M. Li and C. F. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117872 CrossRef CAS PubMed; (c) H. Duan, Y. Li, Q. Li, P. Wang, X. Liu, L. Cheng, Y. Yu and L. Cao, Angew. Chem., Int. Ed., 2020, 59, 10101–10110 CrossRef CAS PubMed.
- (a) M. Huo, X. Y. Dai and Y. Liu, Adv. Sci., 2022, 9, e2201523 CrossRef PubMed; (b) F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Nat. Commun., 2022, 13, 186 CrossRef CAS; (c) X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed; (d) X.-F. Wang, W.-J. Guo, H. Xiao, Q.-Z. Yang, B. Chen, Y.-Z. Chen, C.-H. Tung and L.-Z. Wu, Adv. Funct. Mater., 2020, 30, 1907282 CrossRef CAS; (e) Y. Fan, S. Liu, M. Wu, L. Xiao, Y. Fan, M. Han, K. Chang, Y. Zhang, X. Zhen, Q. Li and Z. Li, Adv. Mater., 2022, 34, e2201280 CrossRef PubMed.
- (a) H. Shi, W. Yao, W. Ye, H. Ma, W. Huang and Z. An, Acc. Chem. Res., 2022, 55, 3445–3459 CrossRef CAS PubMed; (b) S. Garain, S. M. Wagalgave, A. A. Kongasseri, B. C. Garain, S. N. Ansari, G. Sardar, D. Kabra, S. K. Pati and S. J. George, J. Am. Chem. Soc., 2022, 144, 10854–10861 CrossRef CAS PubMed; (c) Y.-J. Ma, X. Fang, G. Xiao and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202114100 CrossRef CAS PubMed; (d) B. Zhou, G. Xiao and D. Yan, Adv. Mater., 2021, 33, 2007571 CrossRef CAS PubMed; (e) H. Sun, S. Shen and L. Zhu, ACS Mater. Lett., 2022, 4, 1599–1615 CrossRef CAS.
- S. Li, Z.-Y. Zhang, J.-F. Lv, L. Li, J. Li and C. Li, J. Mater. Chem. A, 2023, 11, 4957–4962 RSC.
- H. J. Wang, W. W. Xing, Z. H. Yu, H. Y. Zhang, W. W. Xu and Y. Liu, Adv. Opt. Mater., 2022, 10, 2201903 CrossRef CAS.
- P. Wei, X. Zhang, J. Liu, G. G. Shan, H. Zhang, J. Qi, W. Zhao, H. H. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9293–9298 CrossRef CAS PubMed.
- W. Zhu, H. Xing, E. Li, H. Zhu and F. Huang, Macromolecules, 2022, 55, 9802–9809 CrossRef CAS.
- H. Zhu, I. Badia-Dominguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. Ruiz Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164–2169 CrossRef CAS PubMed.
- D. Ma, P. Y. Zavalij and L. Isaacs, J. Org. Chem., 2010, 75, 4786–4795 CrossRef CAS PubMed.
- Z. Wang, A. Li, Z. Zhao, T. Zhu, Q. Zhang, Y. Zhang, Y. Tan and W. Z. Yuan, Adv. Mater., 2022, 34, 2202182 CrossRef CAS PubMed.
- (a) J. Guo, C. Yang and Y. Zhao, Acc. Chem. Res., 2022, 55, 1160–1170 CrossRef CAS PubMed; (b) L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed; (c) H. Li, X. Xue, Y. Cao, H. Cheng, A. Luo, N. Guo, H. Li, G. Xie, Y. Tao, R. Chen and W. Huang, J. Am. Chem. Soc., 2023, 145, 7343–7351 CrossRef CAS PubMed; (d) X. Zhang, M. Zeng, Y. Zhang, C. Zhang, Z. Gao, F. He, X. Xue, H. Li, P. Li, G. Xie, H. Li, X. Zhang, N. Guo, H. Cheng, A. Luo, W. Zhao, Y. Zhang, Y. Tao, R. Chen and W. Huang, Nat. Commun., 2023, 14, 475 CrossRef CAS; (e) A. Cheng, H. Su, X. Gu, W. Zhang, B. Zhang, M. Zhou, J. Jiang, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202312627 CrossRef CAS PubMed; (f) H. Su, K. Hu, W. Huang, T. Wang, X. Zhang, B. Chen, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218712 CrossRef CAS PubMed; (g) L. Gao, J. Huang, L. Qu, X. Chen, Y. Zhu, C. Li, Q. Tian, Y. Zhao and C. Yang, Nat. Commun., 2023, 14, 7252 CrossRef CAS PubMed; (h) Z. Li, S. Cao, Y. Zheng, L. Song, H. Zhang and Y. Zhao, Adv. Funct. Mater., 2024, 34, 2306956 CrossRef CAS.
- (a) Y. Zhang, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li, Z. Cai, Y. Zhao and C. Yang, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef CAS PubMed; (b) Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou, Y. Yang, X. Chen, C. Yang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS PubMed; (c) H. Wu, W. Chi, Z. Chen, G. Liu, L. Gu, A. K. Bindra, G. Yang, X. Liu and Y. Zhao, Adv. Funct. Mater., 2019, 29, 1807243 CrossRef; (d) Y. Yang, Y. Liang, Y. Zheng, J.-A. Li, S. Wu, H. Zhang, T. Huang, S. Luo, C. Liu, G. Shi, F. Sun, Z. Chi and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201820 CrossRef CAS PubMed; (e) X. Yao, H. Ma, X. Wang, H. Wang, Q. Wang, X. Zou, Z. Song, W. Jia, Y. Li, Y. Mao, M. Singh, W. Ye, J. Liang, Y. Zhang, Z. Liu, Y. He, J. Li, Z. Zhou, Z. Zhao, Y. Zhang, G. Niu, C. Yin, S. Zhang, H. Shi, W. Huang and Z. An, Nat. Commun., 2022, 13, 4890 CrossRef CAS PubMed.
- Y. Deng, P. Li, J. Li, D. Sun and H. Li, ACS Appl. Mater. Interfaces, 2021, 13, 14407–14416 CrossRef CAS PubMed.
- (a) Y. Wang, S. Tang, Y. Wen, S. Zheng, B. Yang and W. Z. Yuan, Mater. Horiz., 2020, 7, 2105–2112 RSC; (b) S. Zheng, T. Zhu, Y. Wang, T. Yang and W. Z. Yuan, Angew. Chem., Int. Ed., 2020, 59, 10018–10022 CrossRef CAS PubMed.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed.
- S. Jiang, S. Lan, D. Mao, X. Yang, K. Shi and D. Ma, Chem. Commun., 2018, 54, 9486–9489 RSC.
- Y. Chen, J. Chen, T. Yu, Y. Zeng and Y. Li, Acta Chim. Sin., 2023, 81, 450–455 CrossRef CAS.
- S. Murkli, J. Klemm, A. T. Brockett, M. Shuster, V. Briken, M. R. Roesch and L. Isaacs, Chemistry, 2021, 27, 3098–3105 CrossRef CAS PubMed.
- M. Huo, X. Y. Dai and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 27171–27177 CrossRef CAS PubMed.
- H.-Q. Peng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS PubMed.
- (a) D. Wasserberg, S. C. J. Meskers and R. A. J. Janssen, J. Phys. Chem. A, 2007, 111, 1381–1388 CrossRef CAS PubMed; (b) D. Volyniuk, V. Cherpak, P. Stakhira, B. Minaev, G. Baryshnikov, M. Chapran, A. Tomkeviciene, J. Keruckas and J. V. Grazulevicius, J. Phys. Chem. C, 2013, 117, 22538–22544 CrossRef CAS; (c) A. Kirch, M. Gmelch and S. Reineke, J. Phys. Chem. Lett., 2019, 10, 310–315 CrossRef CAS PubMed; (d) S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed; (e) X. K. Ma, Q. Cheng, X. Zhou and Y. Liu, JACS Au, 2023, 3, 2036–2043 CrossRef CAS PubMed; (f) F.-F. Shen, Y. Chen, X. Dai, H.-Y. Zhang, B. Zhang and Y. Liu, Chem. Sci., 2021, 12, 1851–1857 RSC; (g) A. Cravcenco, M. Hertzog, C. Ye, M. N. Iqbal, U. Mueller, L. Eriksson and K. Börjesson, Sci. Adv., 2019, 5, eaaw5978 CrossRef CAS PubMed.
- (a) L. Wei, F. Gao, C. He, Q. He, P. Jin, Y. Rong, T. Zhao, C. Yang and W. Wu, Sci. China: Chem., 2023, 66, 3546–3554 CrossRef CAS; (b) X. Y. Dai, M. Huo and Y. Liu, Nat. Rev. Chem, 2023, 7, 854–874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00160e |
| This journal is © The Royal Society of Chemistry 2024 |