DOI:
10.1039/D4SC00093E
(Edge Article)
Chem. Sci., 2024,
15, 7072-7078
A tin analogue of propadiene with cumulated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) Sn double bonds†
Sn double bonds†
Received
5th January 2024
, Accepted 25th March 2024
First published on 2nd April 2024
Abstract
The synthesis, structure, and properties of a stable, linear 2-stannapropadiene are reported. The identical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Sn bonds in this 2-stannapropadiene are the shortest hitherto reported C–Sn bonds. This 2-stannapropadiene features a 119Sn NMR signal at 507 ppm for the central tin atom, indicative of an unsaturated Sn4+ oxidation state. Due to the inert-pair effect, the tin atom displays a pronounced preference for the +2 oxidation state over the +4 oxidation state. Nevertheless, by employing silyl substituents, it is possible to disrupt the inert-pair effect, leading to the formation of an isolable 2-stannapropadiene with a linear structure centered on a Sn4+ atom. Treatment of this 2-stannapropadiene with SnBr2·dioxane resulted in the formation of a novel four-membered cyclic 1,1-dibromo-1,3-distannetane, which was subsequently reduced to afford the corresponding stable four-membered cyclic bis(stannylene).
Sn bonds in this 2-stannapropadiene are the shortest hitherto reported C–Sn bonds. This 2-stannapropadiene features a 119Sn NMR signal at 507 ppm for the central tin atom, indicative of an unsaturated Sn4+ oxidation state. Due to the inert-pair effect, the tin atom displays a pronounced preference for the +2 oxidation state over the +4 oxidation state. Nevertheless, by employing silyl substituents, it is possible to disrupt the inert-pair effect, leading to the formation of an isolable 2-stannapropadiene with a linear structure centered on a Sn4+ atom. Treatment of this 2-stannapropadiene with SnBr2·dioxane resulted in the formation of a novel four-membered cyclic 1,1-dibromo-1,3-distannetane, which was subsequently reduced to afford the corresponding stable four-membered cyclic bis(stannylene).
Introduction
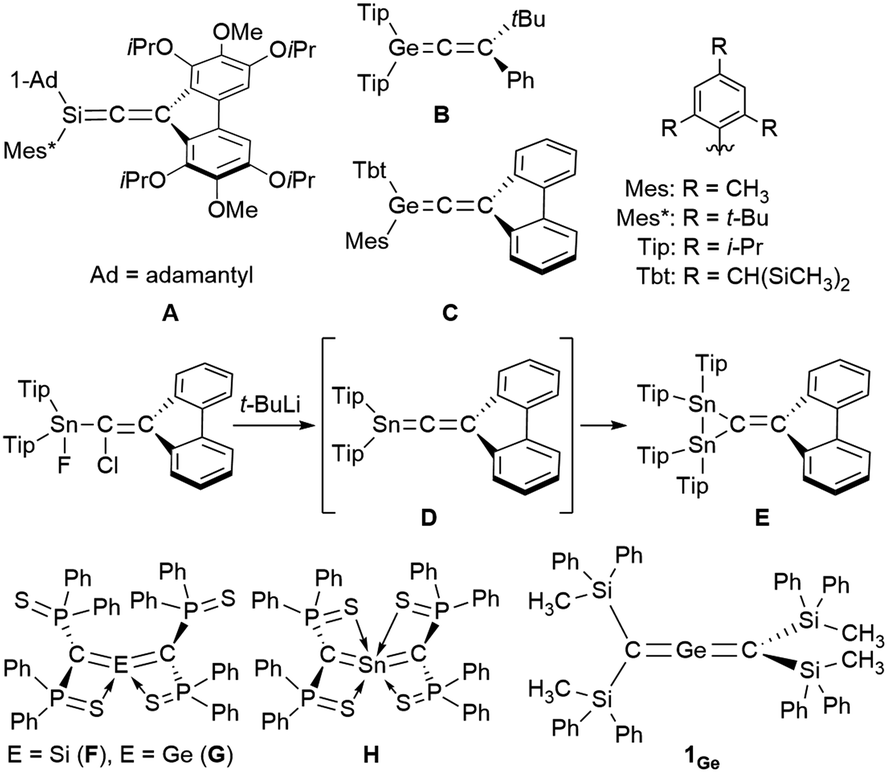
Zero-valent, di-coordinated group-14-element compounds, the so-called called ylidones, have been extensively studied over the last decade, and C, Si, Ge, Sn, and Pb analogues have already been reported.1–10 Ylidones represent one of the isomers of the heavier analogues of allenes; however most ylidones have been characterized as zero-valent compounds (E0) rather than allenes (E4+). The first 1-metallaallenes reported were the Si analogues (A in Fig. 1) synthesized by West et al. in 1993 and the Ge analogues (B and C) independently synthesized in 1998 by West as well as Tokitoh and Okazaki.11–13 Subsequently, the synthesis, reactivity, and unique properties of their analogues were investigated.14–18 Interestingly, an attempted synthesis of 1-stannapropadiene (D) along a similar synthetic route was reported by Escudié in 2004; however this led to an unprecedented stable distannirane (E) via a [2 + 1] cycloaddition between a transient stannylene and a 1-stannapropadiene (D).19 This was the first tangible piece of evidence for the transient formation of a 1-stannapropadiene. In contrast, there is only one example each for the Si, Ge and Sn 2-metallaallene analogues (F, G, and H) using the same diphenythiophosphinoyl groups (Ph2(S)P–) on the terminal carbon atoms of their allene moieties.20–22 Importantly, the structural features of these compounds are not consistent with classical allene character due to the coordination of the sulfur atom in the substituents to the central metal atoms. Accordingly, tin analogues of allenes with cumulative CSn π-bonds have remained elusive thus far. Earlier studies have reported on the isolation and characterization of a linear 2-germapropadiene (1Ge) by using bulky silyl substituents.23 A single-crystal X-ray electron-density-distribution (EDD) analysis of 1Ge allowed us to obtain the differential electron-density map of 1Ge, which suggested two orthogonal π-bonds for the CGeC moiety. Thus, 1Ge represents the first example of a stable, structurally characterized germanium-centered heteroallene with a linear structure. Furthermore, the reactivity of 1Ge is consistent with that of an allene rather than a tetrylone. To further investigate the properties of the corresponding 2-stannapropadiene, 119Sn NMR spectroscopy should offer a useful diagnostic tool for the determination of the electronic structure of the target compound. Herein, we report the synthesis and properties of a 2-stannapropadiene (1Sn). The combined results of X-ray crystallographic and NMR spectroscopic analyses suggest that 1Sn exists as an allene-type structure in the solid state and in solution. The linear structure of 1Sn with cumulative CSn double bonds was confirmed by X-ray crystallography for the first time.
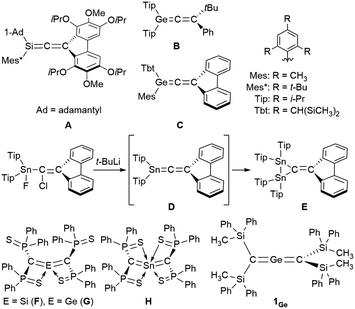 |
| | Fig. 1 Heteroallenes that contain heavier group-14 atoms and related compounds. | |
Results and discussion
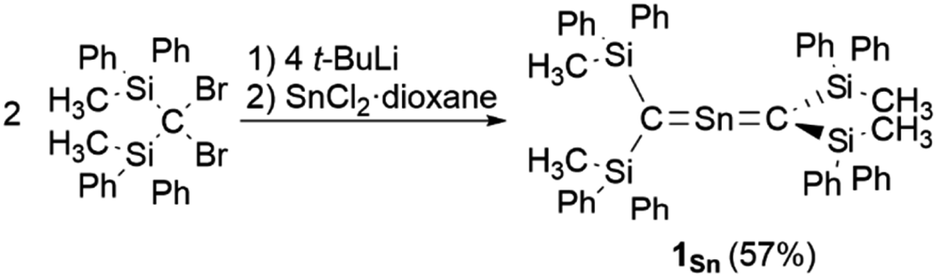
As previously reported, 1Ge can be obtained from the reaction between bis(silyl)carbenoid RSi2CLiBr, which is generated in situ from the reaction of RSi2CBr2 with t-BuLi (2 eq.) in THF at −95 °C, and GeCl2·dioxane (1.0 eq.) at −95 °C.24 After removal of the generated LiBr and recrystallization from hexane and benzene, 1Ge can be obtained as pale-yellow crystals in 50% yield. The tin analogue (1Sn) was prepared in a similar manner, i.e., the bis(silyl)carbenoid was generated using the previously outlined procedure, and SnCl2·dioxane (0.33 eq.) was added at low temperature (Scheme 1). After removal of all inorganic salts, recrystallization from hexane and benzene afforded 2-stannapropadiene (1Sn) as yellow crystals in 57% yield. While 1Ge forms the corresponding cyclic thermal isomer quantitatively in solution after 2 h at 60 °C upon 1,3-migration of the phenyl group, 1Sn does not undergo thermal decomposition, not even in solution after 12 h at 100 °C.
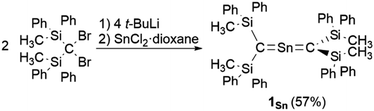 |
| | Scheme 1 Synthesis of 2-stannapropadiene 1Sn. | |
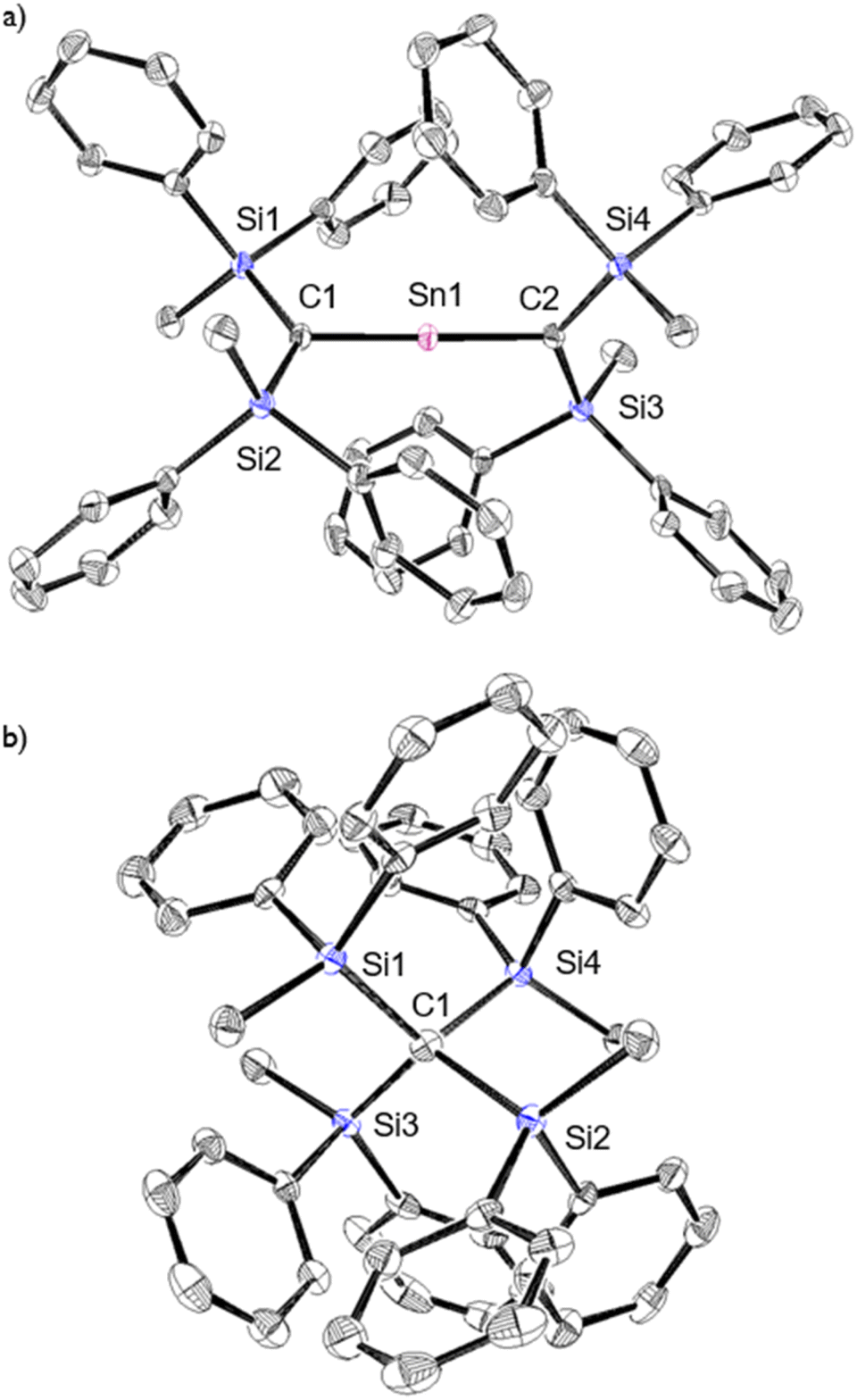
The characterization of 1Sn was accomplished using multinuclear NMR, ultraviolet-visible (UV-vis), and infrared (IR) spectroscopy as well as mass spectrometry and single-crystal X-ray diffraction analysis (Fig. 2). The solid-state structure of 1Sn exhibits D2d symmetry with a linear C–Sn–C moiety (C–Sn–C angle: 178.06(6)°) and almost identical C–Sn bonds (C1–Sn1: 1.9787(15) Å; C2–Sn1: 1.9827(16) Å). These C–Sn bonds represent some of the shortest among the hitherto structurally characterized organotin species with CSn double bonds such as stannenes [2.003(5)–2.073(10) Å]25–29 and are significantly shorter than those of a previously reported base-stabilized 2-stannapropadiene (2.063(2) Å).20 Furthermore, the allenic moiety of the base-stabilized 2-stannapropadiene is slightly bent (171.1(1)°) with pyramidal carbon atoms (ΣCallene: 354.6°). In contrast, the two terminal carbon atoms of 1Sn are almost planar (ΣC1: 359.6°; ΣC2: 359.8°), indicating that the structural properties of 1Sn are considerably different from those of the base-stabilized 2-stannapropadiene. The C1–Si1–Si2 and C2–Si3–Si4 planes slightly deviate from a perpendicular arrangement relative to each other (79.9°), probably due to the steric repulsion among the silyl groups.
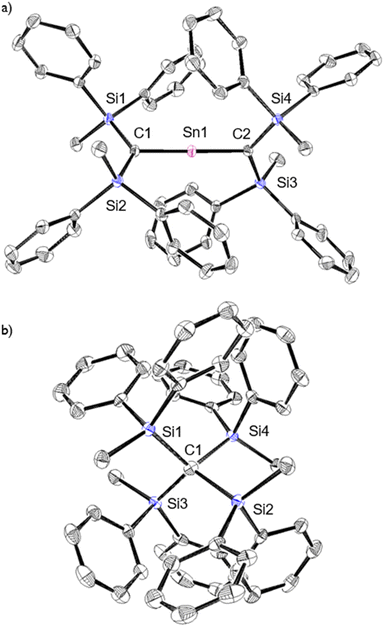 |
| | Fig. 2 (a) Top and (b) side views of the molecular structure of 1Sn in the crystalline state with thermal ellipsoids at 50% probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] for 1Sn: C1–Sn1: 1.9787(15), C2–Sn1: 1.9827(16), C1–Si1: 1.8410(16), C1–Si2: 1.8336(16), C2–Si3: 1.8423(16), C2–Si4: 1.8428(16), C1–Sn–C2: 178.06(6), Si1–C1–Si2: 130.90(9), Si1–C1–Sn1: 114.62(8), Si2–C1–Sn1: 114.07(8), Si3–C2–Si4: 133.33(9), Si3–C2–Sn1: 112.17(8), and Si4–C2–Sn1: 114.29(8). | |
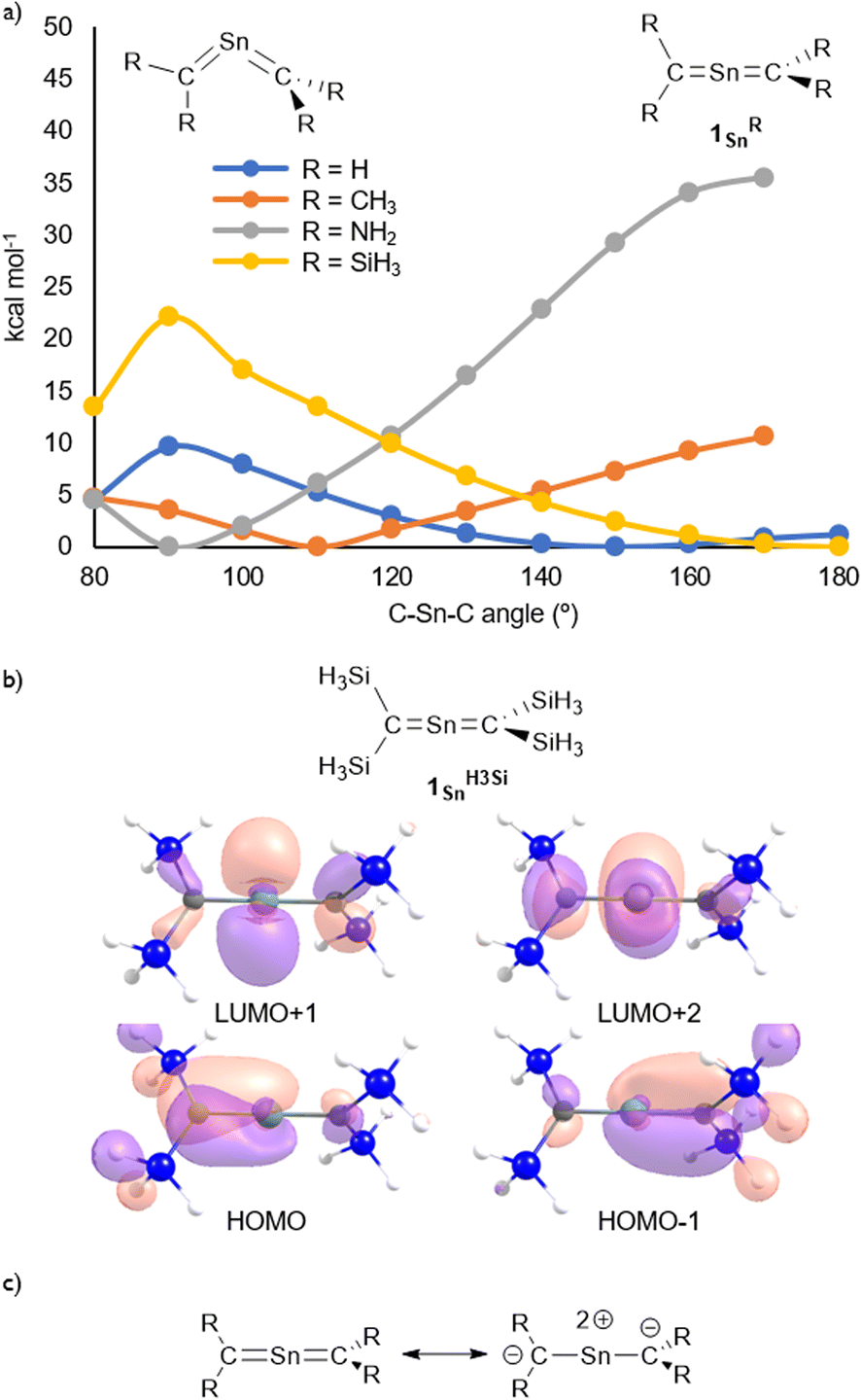
The molecular structure of 1Sn was further examined using theoretical calculations. The structural characteristics of 1Sn, which were optimized at the B3PW91-D3(bj)/Def2TZVP level for Sn and the 6-311G(2d,p) level for the rest of the atoms, are in good agreement with those obtained experimentally (Fig. S45†), i.e., the linear geometry of the CSnC moiety (C–Sn–C = 180.0°) and the two C–Sn bond lengths (1.959 Å) are close to the values obtained from the XRD analysis (C1–Sn1: 1.9787(15) Å; C2–Sn1: 1.9827(16) Å). The introduction of H3Si substituents on the 2-stannapropadiene (1SnH3Si) resulted in an optimized linear structure, whereas the H-, H3C-, and H2N-substituted 2-stannapropadienes 1SnH, 1SnH3C, and 1SnH2N exhibit bent structures, suggesting that the presence of silyl substituents on the terminal carbon affect the linear CSnC structure of 1Sn. That is, the π-electron accepting ability of the silyl groups on the terminal carbons would be crucial for the formation of the linear structure of 2-stannapropadiene. Apeloig and co-workers have reported the synthesis of a tetrasilyl-substituted distannene with a non-twisted structure that exhibits an almost perfect planar geometry due to the suitable steric effect and the silyl substituent effect.30 The frontier Kohn–Sham orbitals of 1Sn provided further information on the nature of the cumulated CSn bonds. The HOMO (π), HOMO−1 (π), LUMO+1 (π*), and LUMO+2 (π*) of 1Sn reveal features typical of compounds with a linear allene-type structure (Fig. 3b). A natural bond orbital (NBO) analysis of the optimized structure of 1Sn showed two C–Sn π-bonds that consist of almost pure 5p orbitals of the tin atom and 2p orbitals of the carbon atoms on the allene moiety.31 The calculated natural-population-analysis (NPA) charges on the tin atom (+2.06) and on each of the terminal carbon atoms (−1.95) indicate a stronger polarization in the allene moiety of 1Sn compared to that of the germanium derivative 1Ge (Ge: +1.76; C: −1.86).23
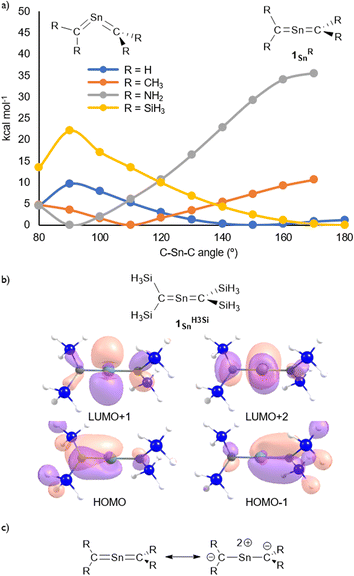 |
| | Fig. 3 (a) Optimized structures of 2-stannapropadienes calculated at the B3PW91-D3(bj)/Def2TZVP level for Sn and the 6-311G(2d,p) level for the remaining atoms. (b) Kohn–Sham orbitals of 1SnH3Si. (c) Canonical resonance structures of linear 2-stannapropadienes. | |
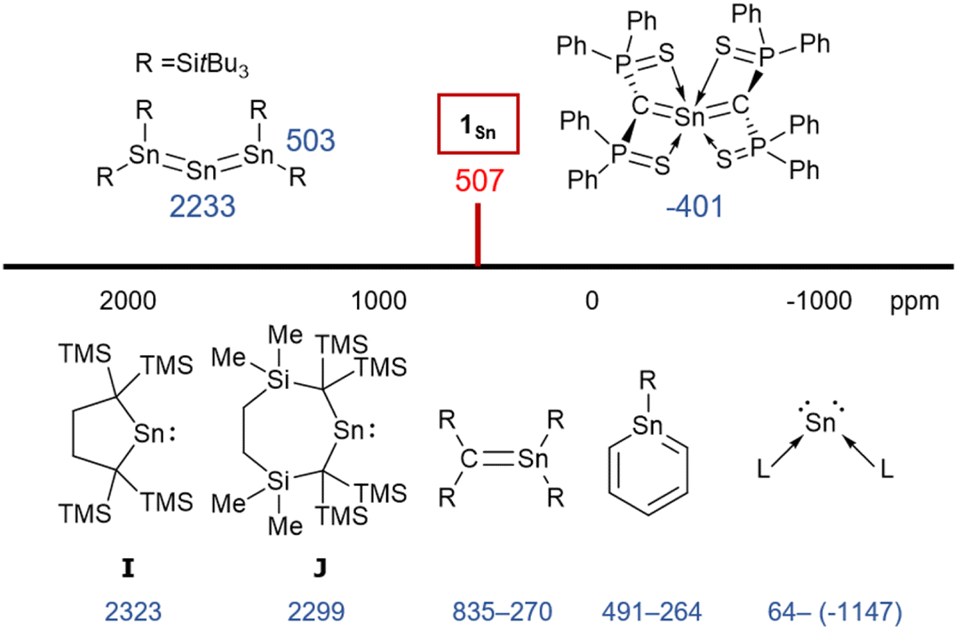
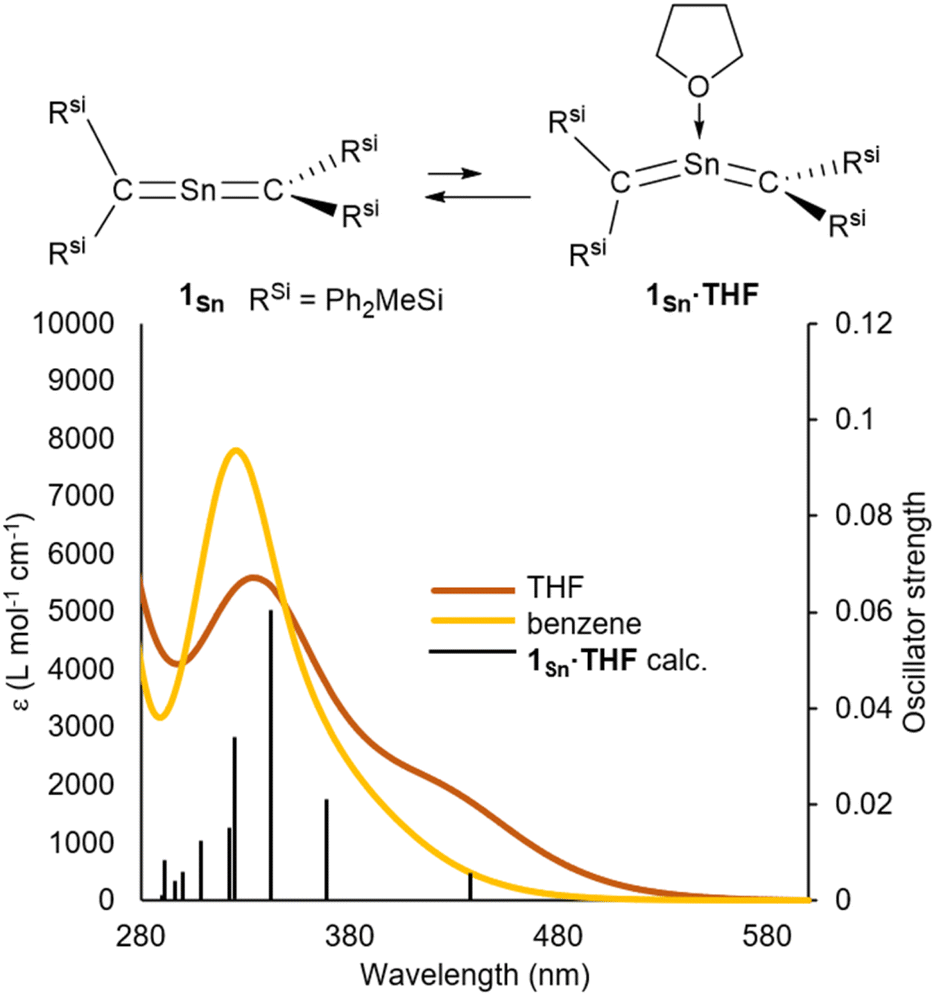
To investigate the multiple-bond character of 1Sn by vibrational spectroscopy, we recorded the IR spectrum (KBr, pellet) of 1Sn in the solid state (Fig. S53†). The CSnC asymmetric stretching frequency of 1Sn is observed at 930 cm−1 and the assignment of this IR shift was supported by DFT calculations (925 cm−1). This frequency is slightly lower than the CGeC asymmetric stretching frequency of 1Ge (973 cm−1) and significantly lower than the CCC asymmetric stretching frequency of 1,1,3,3-tetrakis(trimethylsilyl)allene (1870 cm−1).32,33 It can thus be concluded that the frequency of the stretching vibration reflects the bond strength of the allene moieties. In the 1H NMR spectrum of 1Sn in C6D6, the signal for the protons of the methyl groups was observed as a sharp singlet at 0.39 ppm, indicating a highly symmetric structure for 1Sn in solution. The resonance for the carbon nuclei of the terminal carbons in the allene moiety was observed at 107 ppm, i.e., shifted slightly down-field relative to those of previously reported 2-germapropadiene 1Ge (86 ppm) and tetrakis(trimethylsilyl)allene (64 ppm).32,33 Moreover, the 119Sn NMR signal for the tin atom was observed at 507 ppm in C6D6, indicating unsaturated Sn4+ character, similar to those of the stable stannenes (270–835 ppm) and stannaaromatic compounds (264–491 ppm) shown in Fig. 4.34–37 However, this value is significantly lower than those of previously reported stannylones ((−1147)–64 ppm), which are zero-valent tin species, and higher than those of kinetically stabilized stannylenes (2235–2323 ppm), which are Sn2+ species.38,39 The central tin atom of a tristannaallene has been observed at 2233 ppm due to the considerable unsaturated character that is similar to that in stannylenes, and a stereochemically active lone pair of electrons.40 In contrast to the resonance of the 119Sn nucleus in the base-stabilized 2-stannapropadiene (−401 ppm), the resonance of 1Sn is characteristic for an unsaturated tin compound.21 Moreover, the 119Sn NMR signal of 1Sn in THF at 20 °C was observed as a broadened peak at 497 ppm. With decreasing temperature, this signal gradually shifts to a higher field (Fig. S38†), suggesting a dynamic process. At −50 °C, the signals were observed at 475 ppm and 362 ppm. Gauge-independent atomic orbital (GIAO) 119Sn NMR calculations for the optimized structure of 1Sn afforded a value of 849 ppm. When the solvent effect for THF was considered using the SCRF method, no significant shift in the signal was observed (847 ppm). However, for 1Sn·THF, wherein one molecule of THF is coordinated to the central tin atom, a significant signal shift to 539 ppm was observed. The optimized structure of 1Sn·THF revealed a bent C–Sn–C moiety (Fig. S46†), and this distortion in the molecular geometry around the tin atom would feasibly account for the observed changes of the chemical shifts. To investigate the dynamics of 1Sn in THF, variable-temperature (VT) 1H NMR experiments were carried out. As shown in Fig. S37,† the methyl protons of 1Sn were observed as a broadened peak (0.20 ppm) at room temperature. At −50 °C, these protons were observed as two independent sharp singlet signals (0.31/0.16) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio. However, at 40 °C, all methyl protons appeared as a sharp singlet, suggesting rapid dissociation of the coordinated THF molecule from the central tin atom of 1Sn.
:3 ratio. However, at 40 °C, all methyl protons appeared as a sharp singlet, suggesting rapid dissociation of the coordinated THF molecule from the central tin atom of 1Sn.
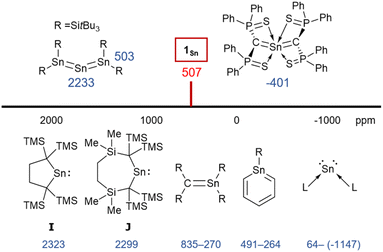 |
| | Fig. 4
119Sn NMR chemical shifts of 1Sn and related compounds. | |
The ultraviolet-visible (UV-vis) spectrum of 1Sn in benzene exhibits an absorption maximum at λmax = 328 nm (ε 8000 dm3 mol−1 cm−1), indicating the presence of π-bonding without conjugation between the two CSn double bonds (Fig. 5). This value differs significantly from those of our previously reported RSi2CChCRSi2 compounds (Ch = S: 449 nm, Se: 523 nm, and Te: 610 nm) with bent structures, suggesting strong interactions between each of the C–Ch multiple bonds due to their bent structures.41–43 And, the λmax value of 1Sn is red-shifted relative to those of 1Ge [265 nm (ε 11000 dm3 mol−1 cm−1), 272 nm (ε 12000 dm3 mol−1 cm−1), and 283 nm (ε 11000 dm3 mol−1 cm−1)] and tetrakis(trimethylsilyl)allene32,33 (273 nm, ε 36000 dm3 mol−1 cm−1), indicating a narrowing of the HOMO–LUMO gap of 1Sn relative to that of the germanium analogue 1Ge due to the insufficient orbital overlap of 5pπ–2pπ in 1Sn relative to that of 4pπ–2pπ in 1Ge (Fig. S51†). Furthermore, the absorption maxima at 265–283 nm in 2-germapropadiene are expected to overlap with the π–π* transitions of phenyl groups. In contrast, the absorption peak of 2-stannapropadiene appears at 328 nm, which does not overlap with the π–π* transitions of phenyl groups. Therefore, the epsilon value of 2-stannapropadiene is smaller than that of the germanium analogue. The UV-vis spectrum of 1Sn in THF at room temperature exhibited a pronounced shoulder peak at 430 nm, which can be attributed to the coordination of a THF molecule to the central tin atom of 1Sn (Fig. S56†). However, at 50 °C, the shoulder peak at 430 nm disappeared due to the dissociation of the coordinated THF molecule. Similarly, when donor molecules such as pyridine and 4-dimethylaminopyridine (DMAP) were added to a benzene solution of 1Sn, similar shoulder peaks were observed in the respective spectra (Fig. S55†). Time-dependent density functional theory (TD-DFT) calculations were performed for compounds 1Sn and 1Sn·donor at the B3PW91/Def2tzvp level for Sn and the 6–311++G(2d,p) level for all other atoms. The results indicate a mixture of two π–π* transitions, i.e., one at 333 nm (f = 0.0178) and another at 306 nm (f = 0.1589), which is in good agreement with the observed λmax value for 1Sn. For 1Sn·THF, the calculations suggested a mixture of two π–π* transitions associated with the bent allene moiety, whereby one occurs at 443 nm (f = 0.0067), consistent with the experimentally observed λmax value. According to the theoretical calculations, the coordination of the donor molecule to 1Sn to form 1Sn·donor (donor: THF, pyridine, or DMAP) is expected to be exothermic for all cases except for THF (for details, see Table S52†). While the coordination of the donor molecule THF to the central tin atom of 1Sn is endothermic, it is still likely that THF coordinates to the central tin atom in solution. The coordinated molecules of THF readily dissociate upon solvent distillation to regenerate 1Sn. Recrystallization in the presence of THF or DMAP leads to the formation of 1Sn, indicating that the coordination of these donors to 1Sn in the crystalline solid state is not favorable. The higher positive charge on the central tin atom in 1Sn, coupled with its lower steric demand compared to that of germanium derivative 1Ge, suggests a preference for the coordination of donor molecules to the central tin atoms.
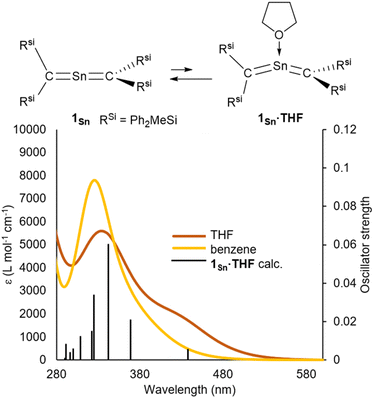 |
| | Fig. 5 (a) UV-vis spectra of 1Sn in benzene and THF at room temperature, and simulated UV-vis absorption spectrum of 1Sn·THF obtained from TD-DFT calculations, together with theoretical oscillator-strength values (black vertical lines). | |
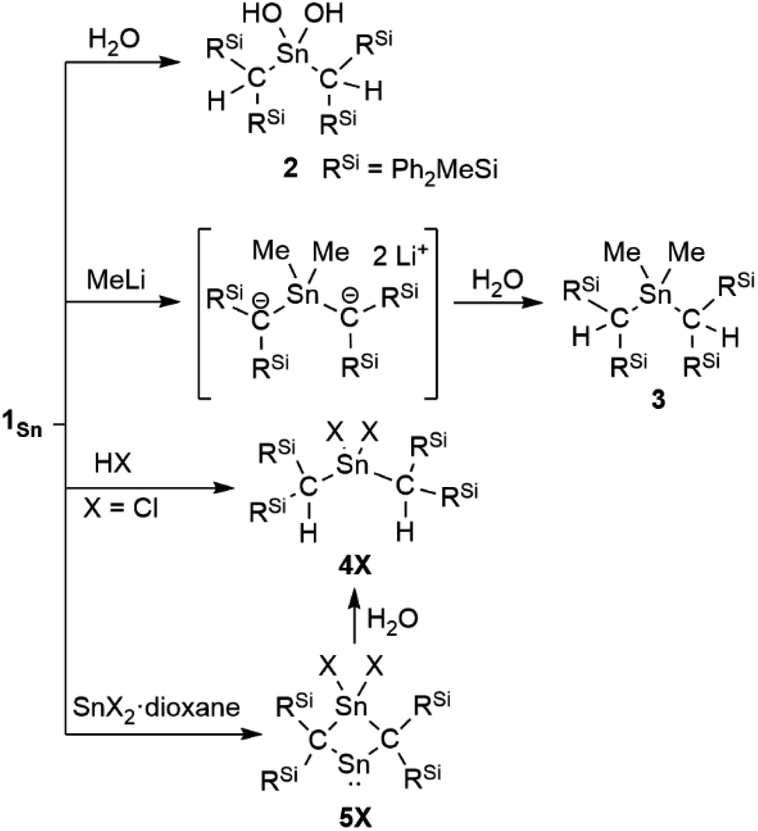
The reaction of 1Sn with H2O immediately produces stannanediol 2 (Scheme 2). Subsequently, the reaction of 1Sn with 2 eq. of MeLi followed by H2O affords the corresponding dimethylated compound 3. As expected, 1Sn readily reacts with hydrogen chloride under mild conditions to quantitatively form the corresponding dichlorostannane (4Cl). These reactivity patterns are similar to those of 2-germapropadiene (1Ge), indicating that the central tin atom can be expected to be the most electrophilic atom in the CSnC allene moiety. In order to further explore the reactivity of 1Sn, we undertook an investigation involving the reaction between 1Sn and the SnX2·dioxane complex. This reaction was motivated by our previous syntheses of four-membered cyclic compounds derived from bis(methylene)-λ4-chalcogenanes using GeX2·dioxane (X = Cl, Br).43 The reaction of 1Sn with SnX2·dioxane proceeded smoothly even at room temperature to afford the corresponding four-membered stannylenes (5X) as light brown solids, which are thermally stable but sensitive to H2O, which selectively affords decomposed 4X.
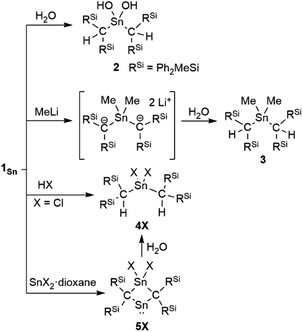 |
| | Scheme 2 Reactivity patterns of 1Sn. | |
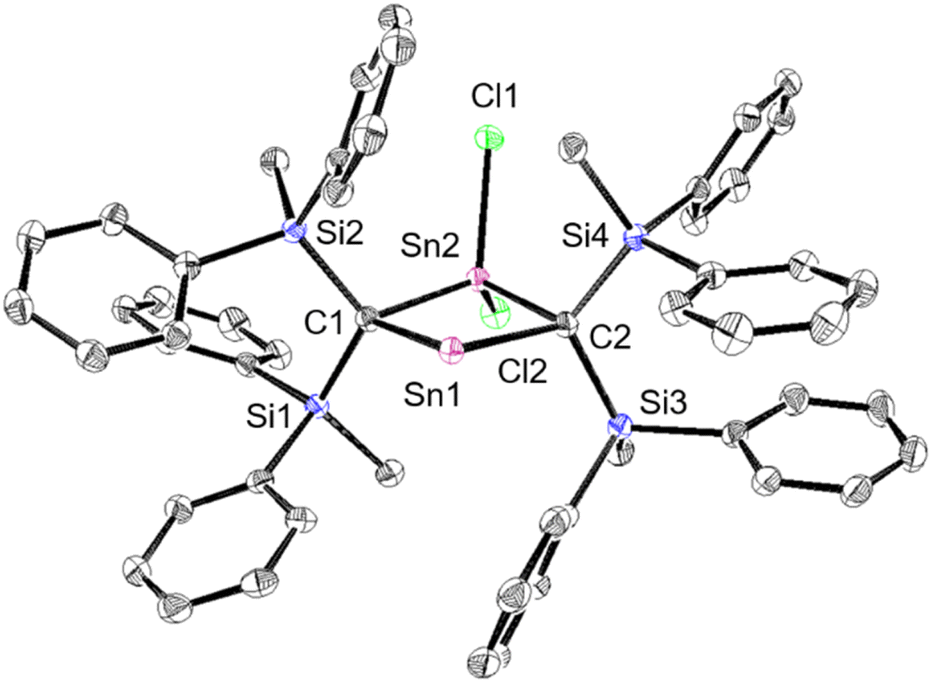
The experimental molecular structure and theoretical calculations suggest that 5Cl contains a Sn2+ and a Sn4+ atom (Fig. 6). The shortest distance between divalent tin atoms in 5Cl is 7.20 Å, which indicates that 5Cl is monomeric in the solid state. In contrast, there is a weak intermolecular interaction between Sn1⋯Sn2 in 5Cl (3.1022(5) Å), which is shorter than the sum of the van der Waals radii (4.34 Å).44 The Sn1 center adopts a typical di-coordinated Sn2+ geometry, and the Sn2 center shows a typical tetra-coordinated Sn4+ geometry. The C1–Sn1–C2 bond angle of 5Cl (87.93(5)°) is similar to that of a previously reported five-membered dialkylstannylene (86.7(2)°).38 The Sn1–C bonds in 5Cl (2.313(2) Å and 2.300(2) Å) are significantly longer than those of a hitherto reported dialkylstannylene (2.218(7) Å and 2.223(7) Å), indicating small amounts of strain due to the four-membered ring in 5Cl. The bond length of Sn2–C (2.163(2) Å and 2.173(2) Å) is identical to that of typical Sn–C bonds (2.16 Å), indicating an increased p-character of the central Sn2+ atom in the Sn1–C single bonds in 5Cl relative to that in 1Sn. The four-membered ring of 5 deviates slightly from planarity (sum of the interior angles: 358.58°). The molecular structure of 5Br is isostructural to that of 5Cl (Fig. S43†). In the 1H, 13C, and 29Si NMR spectra of 5X in C6D6 at room temperature, a signal for only one set of silyl groups was observed, suggesting a flipping of the four-membered ring of 5X. The 119Sn NMR spectrum of 5Cl in C6D6 exhibits signals at 1355 ppm for the Sn2+ nucleus and at 61 ppm for the Sn4+ nucleus. The signal for the Sn2+ nucleus of 5Cl is shifted upfield relative to those of the kinetically stabilized stannylenes (I: 2323 ppm; J: 2299 ppm; Fig. 4),38,39 indicating some kind of interaction with the Sn2+ nucleus. Unfortunately, the 119Sn NMR spectrum of 5Br could not be recorded due to prohibitively low solubility. To investigate the interactions with Sn2+, NBO calculations were conducted at the B3PW91-D3(bj)/Def2tzvp level for Sn and the 6-311+G(2d,p) level for all other atoms. The vacant p orbital of Sn2+ in 5X receives electron donation not only from the C–Si σ-bonds but also from the C–Sn4+ bonds, X–Sn4+ bonds, and π orbitals of the phenyl groups. This would increase the electron density on Sn2+, resulting in a significant upfield shift of the 119Sn signal of 5Cl. In Fig. S28,† the UV-vis spectra of 5X in benzene show absorption maxima at λmax = 466 nm for 5Cl (ε 1300 dm3 mol−1 cm−1) and λmax = 472 nm for 5Br (ε 1300 dm3 mol−1 cm−1), which are slightly blue-shifted compared to that of the five-membered cyclic stannylene I (484 nm; ε 400 dm3 mol−1 cm−1) in Fig. 4. Moreover, the absorption coefficients of 5X are significantly higher than that of I, suggesting an expansion between the phenyl moieties and the vacant p orbital of Sn2+ obtained from the DFT calculations on 5X.
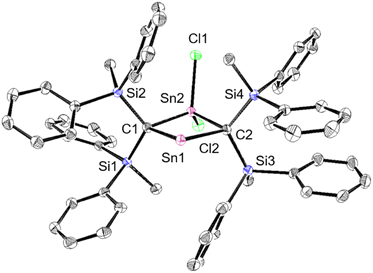 |
| | Fig. 6 Molecular structure of 5Cl in the crystalline state with thermal ellipsoids at 50% probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] for 5Cl: Sn1⋯Sn2: 3.1022(5), C1–Sn1: 2.313(2), C2–Sn1: 2.301(2), C1–Sn2: 2.163(2), C2–Sn2: 2.173(2), C1–Si1: 1.905(2), C1–Si2: 1.888(2), C2–Si3: 1.902(2), C2–Si4: 1.909(2), C1–Sn1–C2: 87.93(8), C1–Sn2–C2: 95.22(8), Sn1–C1–Sn2: 87.67(8), and Sn1–C2–Sn2: 78.76(8). | |
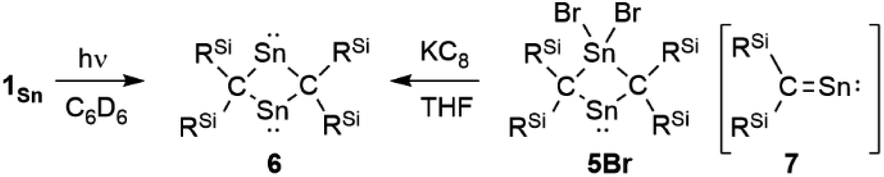
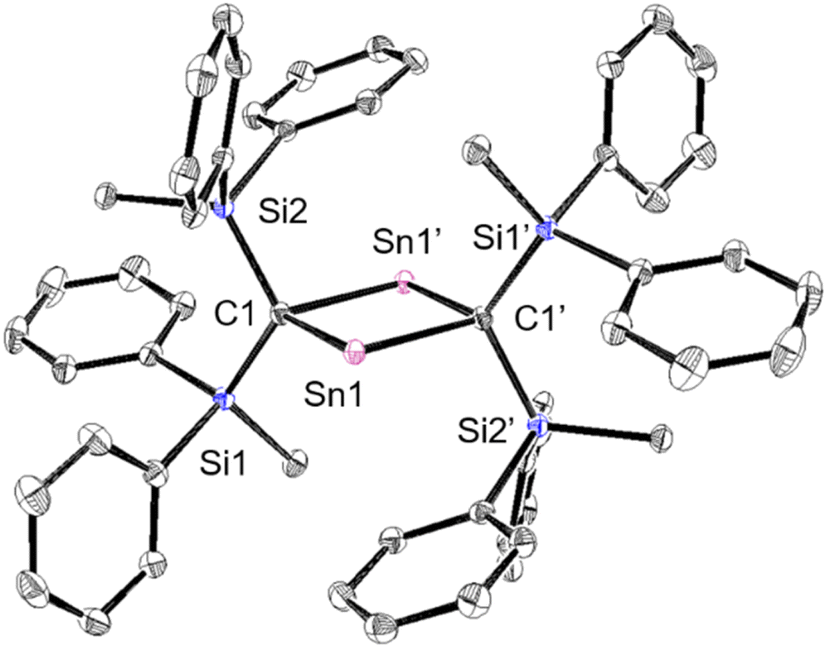
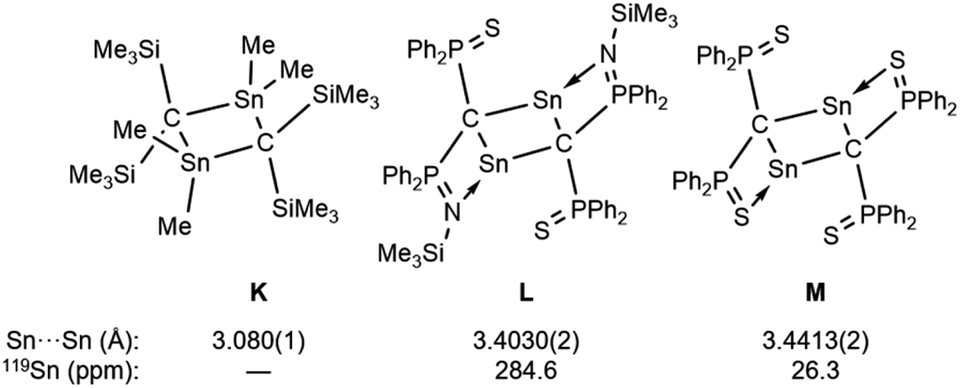
To investigate the photostability of 1Sn, we carried out photoreactions in C6D6 under irradiation from a 100 W high-pressure mercury lamp (Fig. S36†). After 3 days, one of the products obtained in the photochemical reaction was the four-membered cyclic bis(stannylene) (6). The formation of 6 implies the generation of a stannavinylidene (7) via cleavage of the CSn bond in 1Sn under irradiation. The reduction of 5Br with KC8 also afforded bis(stannylene) 6 as a purple solid in 26% yield (Scheme 3). The shortest intermolecular distance between the tin atoms in 6 (7.1223(4) Å) indicates that 6 is monomeric in the solid state (Fig. 7). The intramolecular Sn1⋯Sn1′ distance (3.311(1) Å) is similar to those of related four-membered cyclic tin analogues (3.132(2)–3.4413(2) Å) as shown in Fig. 8.45–54 The Sn1–C bonds in 6 (2.281(2) Å/2.301(1) Å) are similar to those of 5Br (2.289(5) Å/2.310(5) Å). The four-membered ring of 6 exhibits perfect planar geometry (sum of the interior angles: 360°). The NMR spectra of 6 suggested that 6 adopts a highly symmetric structure in a solution: the 1H NMR spectrum in C6D6 exhibits one set of signals due to four equivalent Ph2MeSi groups, while the 119Sn NMR spectrum of 6 shows only one signal at 2003 ppm. The chemical shift of the divalent tin nuclei in 6 is slightly upfield shifted compared to those of cyclic alkyl stannylenes I (2323 ppm)38 and J (2299 ppm)39 in Fig. 4, but significantly downfield shifted compared to that of precursor 5Cl (1355 ppm) and those of base-stabilized bis(stannylene)s (284.6–(−11) ppm).45–54
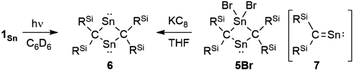 |
| | Scheme 3 Photolysis of 1Sn and reduction of 5Br. | |
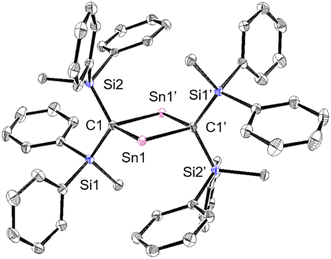 |
| | Fig. 7 Molecular structure of 6 in the crystalline state with thermal ellipsoids at 50% probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] for 6: Sn1⋯Sn1′: 3.311(1), C1–Sn1: 2.2812(13), C–Sn1′: 2.3015(14), C1–Si1: 1.8620(13), C1–Si2: 1.8707(13), C1–Sn1–C1′: 87.47(5), Sn1–C1–Sn1′: 92.53(5), and Si1–C1–Si2: 117.60(7). | |
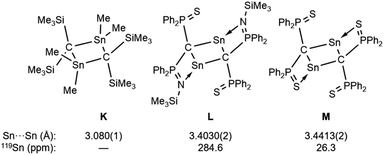 |
| | Fig. 8 Four-membered cyclic compound K and base-stabilized bis(stannylene)s L and M. | |
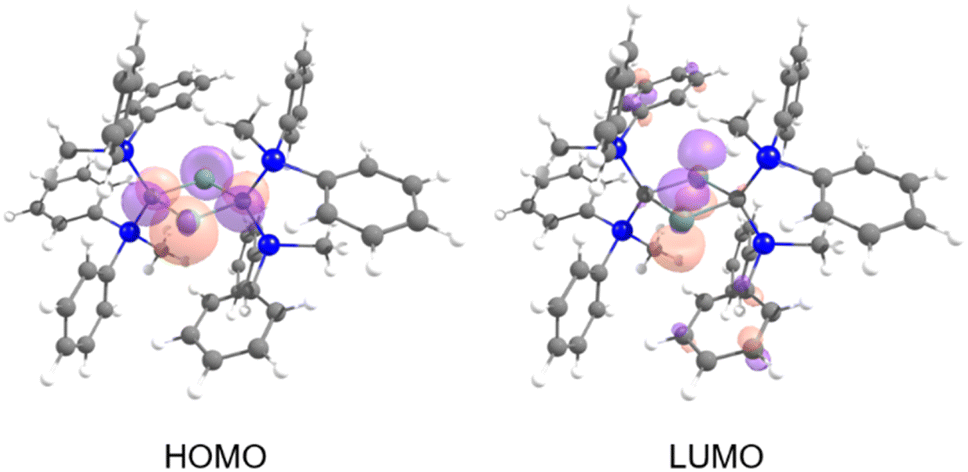
The frontier Kohn–Sham orbitals of 6 provide further information on the nature of the divalent tin moieties (Fig. 9). The HOMO represents lone pairs on two tin atoms, while the LUMO consists of the vacant p orbitals on the tin atoms, indicating the typical molecular orbitals of a stannylene. The UV-vis spectrum of 6 in benzene exhibits an absorption maximum at λmax = 558 nm (ε 700 dm3 mol−1 cm−1). Further studies on the reactivity patterns of 6 are currently in progress.
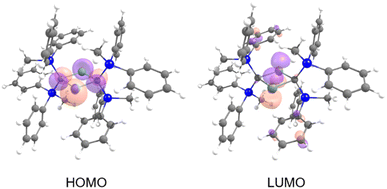 |
| | Fig. 9 Kohn–Sham orbitals of 6. | |
Conclusions
We have reported the successful synthesis of the first stable 2-stannapropadiene (1Sn) with a linear allene-type structure. The bulky silyl groups dominate the structural features and govern the stability of the 2-stannapropadiene due to the high steric demand and negative hyperconjugation by the silyl groups. A multinuclear NMR analysis of the linear heteroallene confirmed the unsaturated Sn4+ oxidation state of the central Sn atom. The reaction of 1Sn with SnX2·dioxane afforded a four-membered cyclic stannylene (5X), indicating a unique reactivity that stands in contrast to that of a previously reported 2-germapropadiene. The photolysis of 1Sn afforded bis(stannylene) 6, which is most likely obtained from the dimerization of stannavinylidene 7. Further investigations into the reactivity of 1Sn and bis(stannylene) 6 are currently in progress in our laboratories.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
The project was designed by K. S. The synthesis and characterization are conducted by T. A. All authors contributed to the writing of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by JSPS KAKENHI grants JP18K14204 and 20K05468 from MEXT (Japan). We would like to thank Dr Yuiga Nakamura, Dr Nobuhiro Yasuda, and Dr Kunihisa Sugimoto at JASRI BL02B1 and BL40XU of SPring-8, where the X-ray diffraction experiments were conducted under reference numbers 2021A1592, 2021A1578, 2021B1132, 2021B1435, 2021B1833, 2021B1798, 2022A1200, 2022A1354, 2022A1584, 2022A1626, 2022A1705, 2022B1149, 2023A1771, 2023A1925, 2023B1675, and 2023B1878.
Notes and references
- M. Wu, Y. He, L. Zhang, R. Wei, D. Wang, J. Liu, L. Leo Liu and G. Tan, Eur. J. Inorg. Chem., 2022, 2022, e202200413 CrossRef CAS.
- T. Koike, T. Nukazawa and T. Iwamoto, J. Am. Chem. Soc., 2021, 143, 14332–14341 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, A. Kaushansky, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 12916–12927 CrossRef CAS PubMed.
- T. Sugahara, T. Sasamori and N. Tokitoh, Angew. Chem., Int. Ed., 2017, 56, 9920–9923 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, S. Inoue, J. D. Epping and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 7147–7150 CrossRef CAS PubMed.
- K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, B. Niepötter, H. Wolf, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 2963–2967 CrossRef CAS PubMed.
- P. K. Majhi and T. Sasamori, Chem.–Eur. J., 2018, 24, 9441–9455 CrossRef CAS PubMed.
- J. Xu, C. Dai, S. Yao, J. Zhu and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202114073 CrossRef CAS PubMed.
- J. Xu, S. Pan, S. Yao, G. Frenking and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202209442 CrossRef CAS PubMed.
- G. E. Miracle, J. L. Ball, D. R. Powell and R. West, J. Am. Chem. Soc., 1993, 115, 11598–11599 CrossRef CAS.
- B. E. Eichler, D. R. Powell and R. West, Organometallics, 1998, 17, 2147–2148 CrossRef CAS.
- N. Tokitoh, K. Kishikawa and R. Okazaki, Chem. Lett., 1998, 27, 811–812 CrossRef.
-
B. Eichler and R. West, in Advances in Organometallic Chemistry, Academic Press, 2000, vol. 46, pp. 1–46 Search PubMed.
- B. E. Eichler, G. E. Miracle, D. R. Powell and R. West, Main Group Met. Chem., 1999, 22, 147–162 CAS.
- B. E. Eichler, D. R. Powell and R. West, Organometallics, 1999, 18, 540–545 CrossRef CAS.
- M. Trommer, G. E. Miracle, B. E. Eichler, D. R. Powell and R. West, Organometallics, 1997, 16, 5737–5747 CrossRef CAS.
- K. Sugamata and T. Sasamori, Dalton Trans., 2023, 52, 9882–9892 RSC.
- L. Baiget, H. Ranaivonjatovo, J. Escudié and H. Gornitzka, J. Am. Chem. Soc., 2004, 126, 11792–11793 CrossRef CAS.
- C. Foo, K.-C. Lau, Y.-F. Yang and C.-W. So, Chem. Commun., 2009, 6816–6818 RSC.
- W.-P. Leung, Y.-C. Chan and T. C. W. Mak, Inorg. Chem., 2011, 50, 10517–10518 CrossRef CAS PubMed.
- Y.-F. Yang, C. Foo, H.-W. Xi, Y. Li, K. H. Lim and C.-W. So, Organometallics, 2013, 32, 2267–2270 CrossRef CAS.
- K. Sugamata, T. Asakawa, D. Hashizume and M. Minoura, Angew. Chem., Int. Ed., 2023, 62, e202302836 CrossRef CAS.
- A. Inoue, J. Kondo, H. Shinokubo and K. Oshima, Chem. Lett., 2001, 30, 956–957 CrossRef.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- H. Meyer, G. Baum, W. Massa, S. Berger and A. Berndt, Angew. Chem., Int. Ed., 1987, 26, 546–548 CrossRef.
- G. Anselme, H. Ranaivonjatovo, J. Escudie, C. Couret and J. Satge, Organometallics, 1992, 11, 2748–2750 CrossRef CAS.
- Y. Mizuhata, N. Takeda, T. Sasamori and N. Tokitoh, Chem. Commun., 2005, 5876–5878 RSC.
- A. Fatah, R. El Ayoubi, H. Gornitzka, H. Ranaivonjatovo and J. Escudié, Eur. J. Inorg. Chem., 2008, 2008, 2007–2013 CrossRef.
- R. Bashkurov, N. Fridman, D. Bravo-Zhivotovskii and Y. Apeloig, Chem.–Eur. J., 2023, 29, e202302678 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- R. West and P. C. Jones, J. Am. Chem. Soc., 1969, 91, 6156–6161 CrossRef CAS.
- A. de Meijere, D. Faber, U. Heinecke, R. Walsh, T. Müller and Y. Apeloig, Eur. J. Org Chem., 2001, 2001, 663–680 CrossRef.
- C. Kaiya, K. Suzuki and M. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 7749–7752 CrossRef CAS PubMed.
- Y. Mizuhata, N. Noda and N. Tokitoh, Organometallics, 2010, 29, 4781–4784 CrossRef CAS.
- Y. Mizuhata, T. Sasamori, N. Takeda and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1050–1051 CrossRef CAS.
- Y. Mizuhata, N. Takeda, T. Sasamori and N. Tokitoh, Chem. Lett., 2005, 34, 1088–1089 CrossRef CAS.
- M. Kira, R. Yauchibara, R. Hirano, C. Kabuto and H. Sakurai, J. Am. Chem. Soc., 1991, 113, 7785–7787 CrossRef CAS.
- C. Eaborn, M. S. Hill, P. B. Hitchcock, D. Patel, J. D. Smith and S. Zhang, Organometallics, 2000, 19, 49–53 CrossRef CAS.
- N. Wiberg, H.-W. Lerner, S.-K. Vasisht, S. Wagner, K. Karaghiosoff, H. Nöth and W. Ponikwar, Eur. J. Inorg. Chem., 1999, 1211–1218 CrossRef CAS.
- K. Sugamata, T. Asakawa, Y. Urao and M. Minoura, Inorg. Chem., 2022, 61, 17641–17645 CrossRef CAS PubMed.
- K. Sugamata, Y. Urao and M. Minoura, Chem. Commun., 2019, 55, 8254–8257 RSC.
- K. Sugamata, D. Hashizume, Y. Suzuki, T. Sasamori and S. Ishii, Chem.–Eur. J., 2018, 24, 6922–6926 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- W.-P. Leung, Y.-C. Chan and T. C. W. Mak, Organometallics, 2013, 32, 2584–2592 CrossRef CAS.
- W.-P. Leung, W.-K. Chiu and T. C. W. Mak, Inorg. Chem., 2013, 52, 9479–9486 CrossRef CAS PubMed.
- W.-P. Leung, K.-W. Kan and T. C. W. Mak, Organometallics, 2010, 29, 1890–1896 CrossRef CAS.
- W.-P. Leung, C.-L. Wan, K.-W. Kan and T. C. W. Mak, Organometallics, 2010, 29, 814–820 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, M. Linnolahti, J. R. Severn, P. G. H. Uiterweerd and Z.-X. Wang, J. Organomet. Chem., 2009, 694, 3487–3499 CrossRef CAS.
- W.-P. Leung, K.-W. Wong, Z.-X. Wang and T. C. W. Mak, Organometallics, 2006, 25, 2037–2044 CrossRef CAS.
- W.-P. Leung, Q. W.-Y. Ip, S.-Y. Wong and T. C. W. Mak, Organometallics, 2003, 22, 4604–4609 CrossRef CAS.
- W. P. Leung, Z. X. Wang, H. W. Li, Q. C. Yang and T. C. Mak, J. Am. Chem. Soc., 2001, 123, 8123–8124 CrossRef CAS PubMed.
- W.-P. Leung, Z.-X. Wang, H.-W. Li and T. C. W. Mak, Angew. Chem., Int. Ed., 2001, 40, 2501–2503 CrossRef CAS PubMed.
- N. Wiberg, T. Passler, S. Wagner and K. Polborn, J. Organomet. Chem., 2000, 598, 292–303 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Teppei
Asakawa
and
Mao
Minoura
*,
Teppei
Asakawa
and
Mao
Minoura