
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA scalable synthesis of adjuvanting antigen depots based on metal–organic frameworks†
Ryanne N.
Ehrman‡
a,
Olivia R.
Brohlin‡
a,
Yalini H.
Wijesundara
a,
Sneha
Kumari
a,
Orikeda
Trashi
a,
Thomas S.
Howlett
a,
Ikeda
Trashi
a,
Fabian C.
Herbert
a,
Arun
Raja
a,
Shailendra
Koirala
a,
Nancy
Tran
a,
Noora M.
Al-Kharji
a,
Wendy
Tang
a,
Milinda C.
Senarathna
a,
Laurel M.
Hagge
a,
Ronald A.
Smaldone
 a and
Jeremiah J.
Gassensmith
*ab
a and
Jeremiah J.
Gassensmith
*ab
aDepartment of Chemistry and Biochemistry, The University of Texas at Dallas, 800 West Campbell Rd., Richardson, TX 75080, USA. E-mail: gassensmith@utdallas.edu
bDepartment of Biomedical Engineering, The University of Texas at Dallas, 800 West Campbell Rd., Richardson, TX 75080, USA
First published on 24th January 2024
Abstract
Vaccines have saved countless lives by preventing and even irradicating infectious diseases. Commonly used subunit vaccines comprising one or multiple recombinant proteins isolated from a pathogen demonstrate a better safety profile than live or attenuated vaccines. However, the immunogenicity of these vaccines is weak, and therefore, subunit vaccines require a series of doses to achieve sufficient immunity against the pathogen. Here, we show that the biomimetic mineralization of the inert model antigen, ovalbumin (OVA), in zeolitic imidazolate framework-8 (ZIF-8) significantly improves the humoral immune response over three bolus doses of OVA (OVA 3×). Encapsulation of OVA in ZIF-8 (OVA@ZIF) demonstrated higher serum antibody titers against OVA than OVA 3×. OVA@ZIF vaccinated mice displayed higher populations of germinal center (GC) B cells and IgG1+ GC B cells as opposed to OVA 3×, indicative of class-switching recombination. We show that the mechanism of this phenomenon is at least partly owed to the metalloimmunological effects of the zinc metal as well as the sustained release of OVA from the ZIF-8 composite. The system acts as an antigen reservoir for antigen-presenting cells to traffic into the draining lymph node, enhancing the humoral response. Lastly, our model system OVA@ZIF is produced quickly at the gram scale in a laboratory setting, sufficient for up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 vaccine doses.
000 vaccine doses.
Introduction
Edward Jenner discovered that the administration of cowpox blister fluid, which contained live cowpox virus, effectively prevented smallpox; these efforts established the modern field of vaccination and significantly reduced infectious disease-related mortality.1–3 Over time, vaccine formulations have evolved, with subunit vaccines emerging as a preferred option, owing to their enhanced safety profile compared to early formulations such as inactivated and live vaccines.4,5 Subunit vaccines comprise one or more proteins called antigens (Ag) isolated from a specific pathogen.6 However, these proteinaceous subunit vaccines—like most biomacromolecule-based drugs—are delicate and easily destroyed by environmental factors, requiring cold-chain transportation.7–9 Additionally, they are often weakly immunogenic and need several doses—typically called boosters—to achieve and maintain complete immunity against the pathogen.10–13 To achieve high antibody titers and immune memory, many of today's modern vaccines require a prime-boost regimen, where one or more additional doses of vaccine are needed, even in formulations that use adjuvants.12,14 However, patient compliance decreases with the number of doses required.15 The distaste for needles and vaccine side effects deters patients from receiving additional doses, taking a significant toll on the idea of herd immunity.16–20 Recent work has shown that sustained release of an antigen from an injected “depot” over several days provokes a more robust immune response than repeat injections.21–23 This suggests that a continuous release of Ag from an injection should outperform several injections, thereby removing the need for follow-up doses.The emergence of biomimetic mineralization of delicate biomaterials in a metal–organic framework (MOF) has allowed for the development of thermally and enzymatically protected biological material from numerous stressors.24–30 Recent work has shown that the biofriendly zeolitic imidazolate framework-8 (ZIF-8)-encapsulated vaccines promote a more robust immune response against viral nanoparticles or whole-cell bacteria when injected subcutaneously.24,25,31–33 These results are striking, but in all cases, the vaccines used have been “self-adjuvanting” as the RNA or DNA within a virus or the lipopolysaccharides on the inactivated bacteria can induce their own strong immune response. Further, it is not clear what happens to the ZIF inside the skin, nor has size dependence on the toxicity of the particles in vivo been assessed as they reside for so long within the tissue.
This work details the MOF encapsulation of a model antigen, ovalbumin (OVA), in ZIF-8 (OVA@ZIF). By adjusting the metal-to-ligand ratio, we can adjust the size of our vaccine composites. We formulated nano- and micro-sized OVA@ZIF (n-OVA@ZIF) and (μ-OVA@ZIF), respectively (Scheme 1A). We show the process of MOF shell degradation within tissue over several days, which provides a constant supply of antigens to the immune system.34–36 This provokes a stronger immune response over bolus vaccine.37 Constant supply of antigen through the depot effect promotes the formation of more developed germinal centers (GCs—Scheme 1B) within follicular lymph nodes38–44 GCs are the location of B cell development, which is crucial for a strong and protective antibody response. Additionally, we show that, as the ZIF degrades, the draining lymph node has increased zinc concentrations. Zinc is known to promote immune activation, particularly in combination with an antigen, and so its presence in conjunction with the resulting robust immune response suggests the metal may be helping adjuvant the immune response. The resulting Ag release and ZIF dissolution from the ZIF depot promote GC development, resulting in a single injection of OVA@ZIF producing more antibodies than three bolus injections of OVA (OVA 3×). Finally, we show that the synthesis of OVA@ZIF is easily scaled in the laboratory to multi-gram quantities.
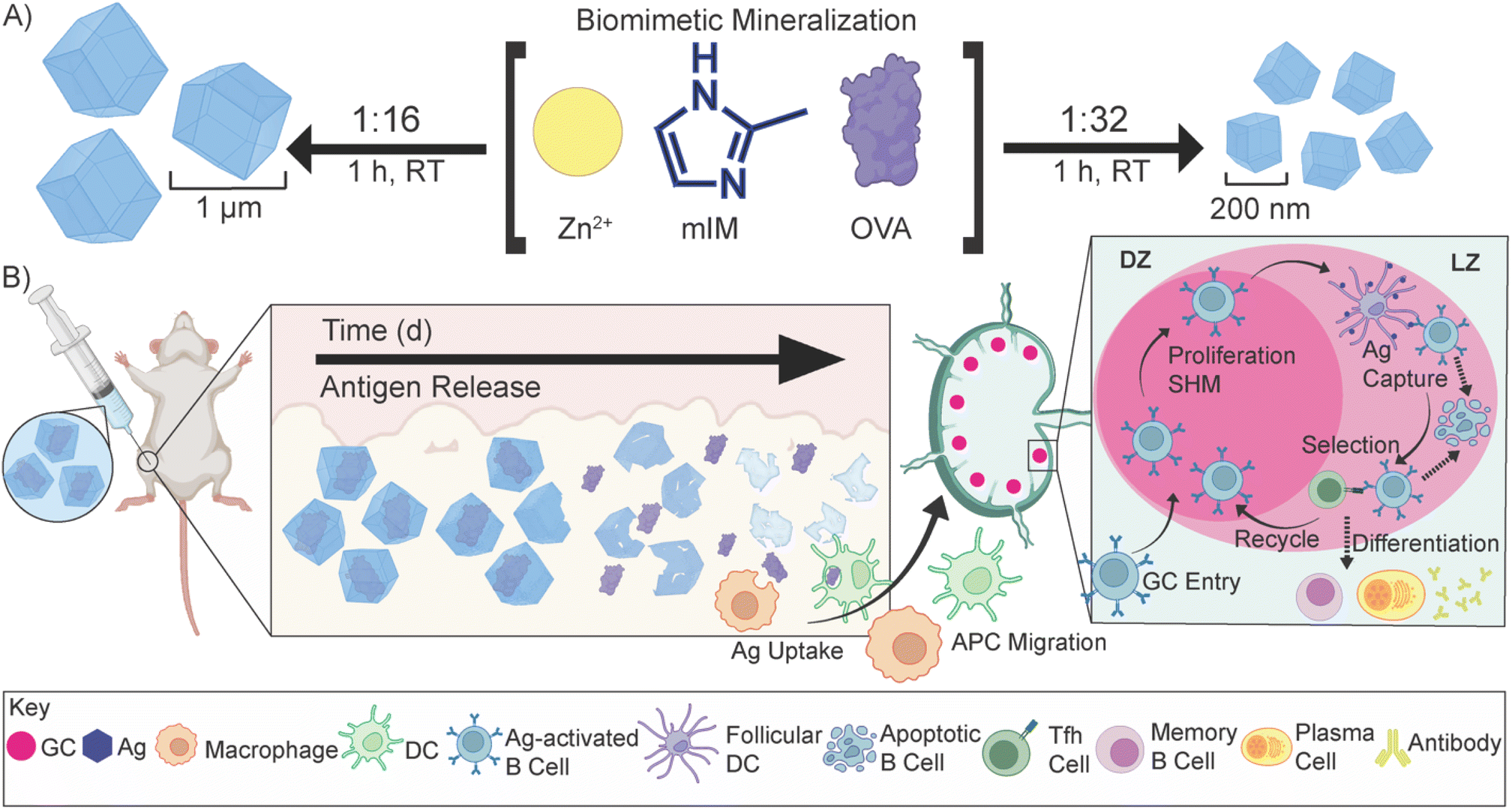 | ||
| Scheme 1 Biomimetic mineralization of OVA@ZIF and sustained release of antigens. (A) Synthetic scheme of μ-OVA@ZIF and n-OVA@ZIF. In a “one pot” reaction, Zn2+, mIM, and ovalbumin are suspended in water and react for 1 h at room temperature. By adjusting the ligand-to-metal ratio, we can tune the size of these ZIFs. We formulated a 1:16, micron-sized (∼1 μm) and 1:32 nano-sized (∼200 nm) μ-OVA@ZIF and n-OVA@ZIF, respectively. (B) Sustained release of antigens following vaccination, allowing for the formation of GCs. The ZIF-8 crystals slowly degrade in the body, exposing the antigen to the immune system and allowing for a constant supply of antigen to the GC. | ||
Results and discussion
Synthesis and cytotoxicity
In a one-pot aqueous synthesis, OVA, 2-methyl imidazole (mIM), and Zn2+ react at room temperature for 1 h to form the OVA@ZIF composite through biomimetic mineralization—an in situ process where the protein templates nucleate the MOF crystal growth yielding total encapsulation of the protein within the ZIF-8 framework.45–47 By adjusting the metal-to-ligand ratios, we can tune the size of these composites; higher metal-to-ligand ratios tend to yield larger particles.48 To investigate if size plays an important role in the compatibility of our system, we synthesized two formulations of OVA@ZIF. A 1:32 metal-to-ligand ratio yielded nano-sized particles approximately 200 nm in diameter n-OVA@ZIF, and a 1:16 metal-to-ligand ratio yielded micron-sized, approximately 1 μm μ-OVA@ZIF. Scanning electron microscopy (SEM) micrographs of both μ-OVA@ZIF and n-OVA@ZIF show approximately 1 μm and 200 nm crystals, respectively (Fig. 1A and B). Powder X-ray diffraction (PXRD) diffractograms confirm the crystallinity of both ZIF-8 formulations and the vaccine formulations μ-OVA@ZIF and n-OVA@ZIF (Fig. 1C). To help visualize release kinetics and study uptake via flow cytometry, cyanine-7 (Cy7) labeled OVA (OVA[Cy7]) were produced and encapsulated in both micro-sized (μ-OVA[Cy7]@ZIF) and nano-sized (n-OVA[Cy7]@ZIF) formulations using the previously mentioned formula. We used the fluorescence of the resulting supernatant to determine encapsulation efficiency (Fig. 1D). The encapsulation efficiency of both formulations was greater than 98%. μ-OVA[Cy7]@ZIF and n-OVA[Cy7]@ZIF crystals were imaged through confocal laser scanning microscopy (CLSM) to confirm that the fluorescence is due to the presence of OVA[Cy7]. CLSM images of non-labeled μ- and n-OVA@ZIF further confirm that the fluorescent intensity is from fluorophore-conjugated protein (Fig. S1 and S2†). The n-OVA@ZIF formulation has a lower drug loading ratio of 1:37 w/w ovalbumin to ZIF composite, as compared to μ-OVA@ZIF, which has a 1:28 w/w drug loading ratio. The MOF in these ZIF composites maintains porosity and surface area in all ZIF formulations, as demonstrated in Fig. S3† by N2 gas adsorption isotherms. As expected, the OVA@ZIF formulations have slightly reduced surface area as a portion of the mass is now made up of protein. Additional characterization, including X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FT-IR) spectroscopy of all the ZIF formulations, can be found in Fig. S4 and S5,† respectively. These results further prove we have produced ZIF-8.
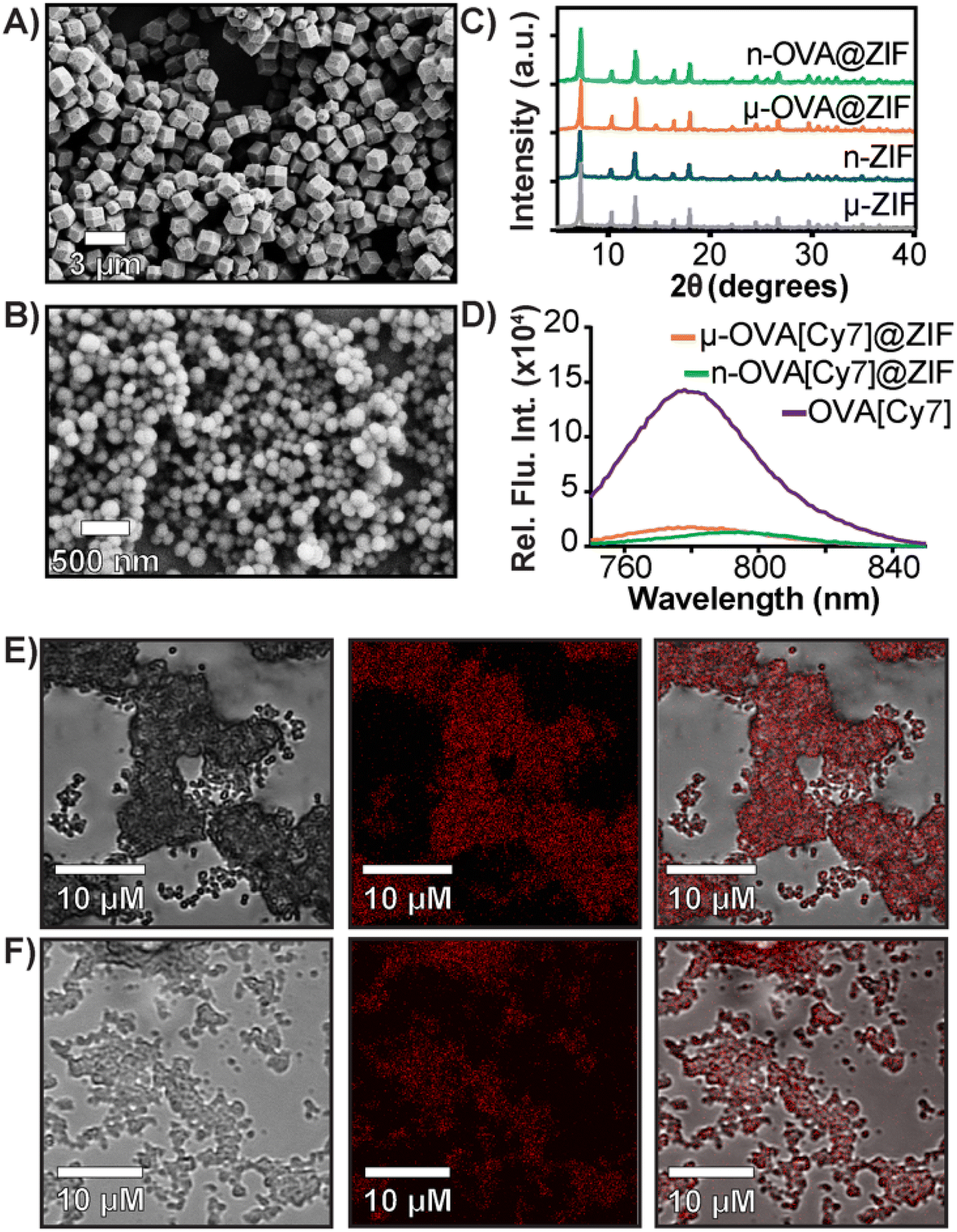 | ||
| Fig. 1 Characterization of OVA@ZIF. (A) SEM images of μ-OVA@ZIF and (B) n-OVA@ZIF. (C) PXRD diffractogram of OVA@ZIF composites. (D) Encapsulation efficiency of μ-OVA[Cy7]@ZIF and n-OVA[Cy7]@ZIF as measured by Cy7 fluorescence in the supernatant (λex = 756 nm and λem = 779 nm). (E) CLSM images of μ-OVA[Cy7]@ZIF and (F) n-OVA[Cy7]@ZIF. Brightfield images are shown on the left, fluorescent in the middle, and merged channels on the right. | ||
The safety and toxicology of ZIF-8 have been an area of substantial research;49–51 however, to the best of our knowledge, it has yet to be benchmarked against current FDA-approved therapeutics. To that end, we conducted a study to compare ZIF-8 formulations against the adjuvant alum, which is nearing a century of use in vaccine formulations.52 The alum formulations that are typically used to adjuvant an immune response in humans consist of either aluminum hydroxide (alum(OH)) or aluminum phosphate (alum(PO4)).53 We conducted in vitro cytotoxicity comparisons between alum salts and our ZIF-8 formulations using lactate dehydrogenase (LDH) assay to assess the safety profiles. We utilized immortalized murine RAW 264.7 macrophages (Fig. 2A) as a standard lab-strain and primary bone marrow-derived macrophages (BMDMs) to represent more realistic toxicities in fresh primary cells. Remarkably, alum(OH) exhibited significant toxicity in both cell assays, while alum(PO4) demonstrated higher toxicity in the BMDM assay than in the RAW 264.7 assay. Conversely, the μ-OVA@ZIF formulation showed no toxicity in either cell assay at tested concentrations, while n-OVA@ZIF displayed reduced cell viability in the RAW 264.7 assay without significant cytotoxicity in primary BMDMs. Overall, our results indicate that alum salts have a stronger toxic profile than our μ-OVA@ZIF formulation and are at least comparable with n-OVA@ZIF. This aligns with existing literature,54,55 and our study provides evidence that ZIF-8 is a less toxic adjuvant than the FDA-approved alum. Thus, our findings establish that μ-OVA@ZIF is the least toxic among all tested materials. In addition, we assessed cellular uptake using flow cytometry in RAW 264.7 macrophages and found uptake of μ-OVA[Cy7]@ZIF to be statistically insignificant (p = 0.09) compared to n-OVA[Cy7]@ZIF, yet superior to naked OVA[Cy7] (Fig. 2B).
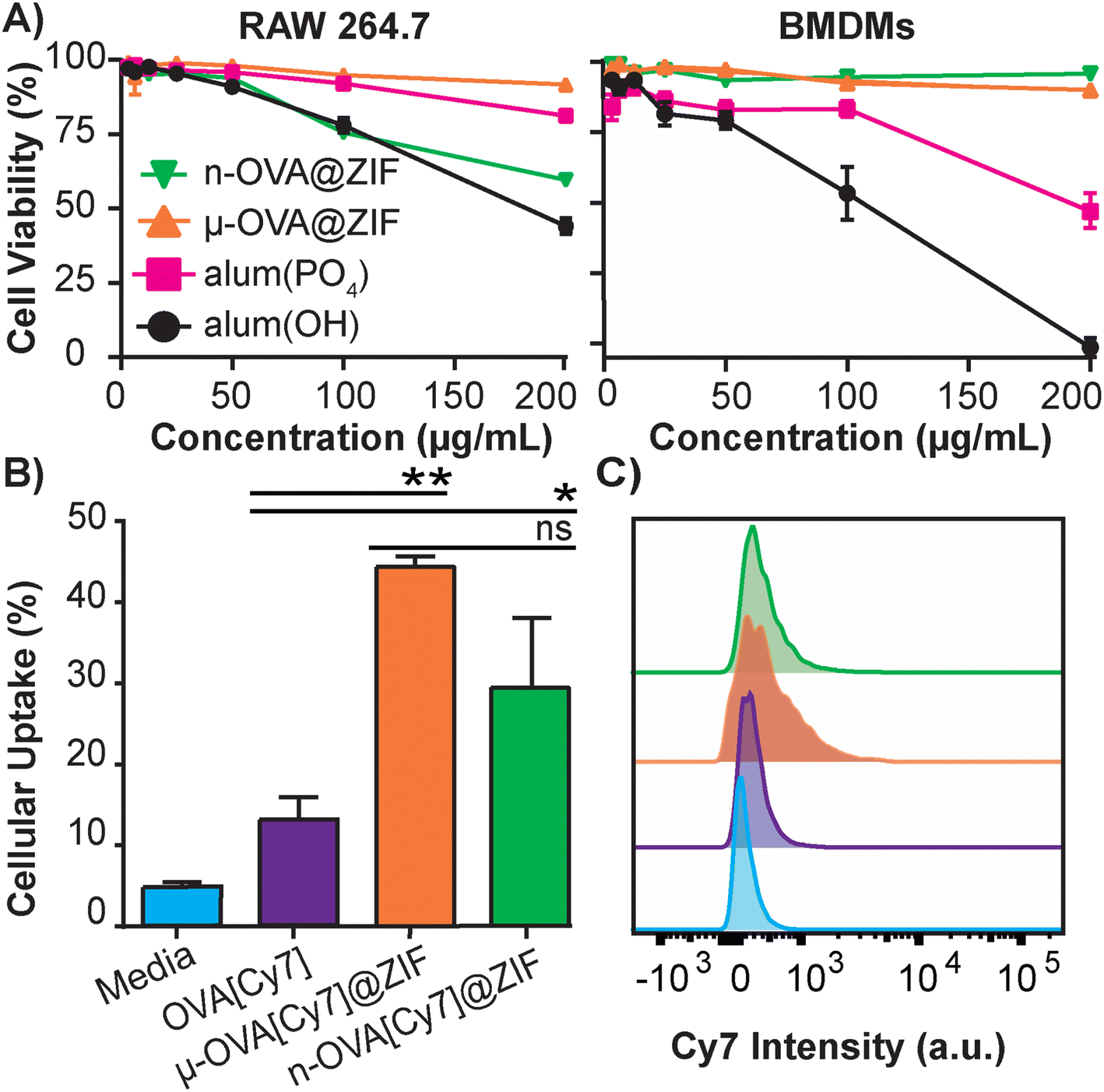 | ||
| Fig. 2 In vitro studies of OVA@ZIF. (A) Cell viability after 4 h incubation of ZIFs and alum in immortalized RAW 264.7 cells (left) and primary BMDMs (right) using LDH assay. (B) Flow cytometry analysis of the cellular uptake of μ-OVA[Cy7]@ZIF and n-OVA[Cy7]@ZIF by RAW 264.7 macrophage following 4 h incubation. (C) Representative histogram overlay of flow cytometry plot illustrating the Cy7 intensity of RAW 264.7 macrophages following uptake experiment. Statistical significance was determined by an unpaired Student's t-test (P > 0.05 is ns, P ≤ 0.05 is *, P ≤ 0.01 is **, P ≤ 0.001 is ***, and P ≤ 0.0001 is ****). | ||
Tissue residency and time-dependent dissolution
To assess the tissue residency of our formulations, μ-OVA[Cy7]@ZIF, n-OVA[Cy7]@ZIF, and OVA[Cy7] (10 μg of OVA[Cy7]) were subcutaneously injected into the right flank of female BALB/c mice (n = 4), and Cy7 fluorescence intensity was monitored over time through fluorescent live animal imaging (Fig. 3A and B). Fluorescence intensity in the OVA[Cy7] was significantly less than the OVA[Cy7]@ZIF cohorts at 24 h (P ≤ 0.001 for both μ- and n-OVA[Cy7]@ZIF with respect to OVA[Cy7]), and the signal was completely diminished in the OVA[Cy7] cohort at 36 h. Whereas μ- and n-OVA[Cy7]@ZIF cohorts demonstrated a vastly longer tissue residency of Ag, with notable fluorescent signal until day 28 and 31, respectively. It is worth noting that there is no statistical significance between μ- and n-OVA[Cy7]@ZIF at any of the reported time points (P > 0.05). In agreement with our hypothesis, OVA@ZIF shows significant enhancement of tissue residency of the Ag. However, the n-OVA[Cy7]@ZIF injected mice developed inflammation and skin irritation at the injection site (Fig. S6†). This is likely caused by the sub-micron size of the n-OVA@ZIF particles; previous literature suggests that these smaller particle sizes can cause nanotoxicity.56,57 In contrast, the μ-OVA[Cy7]@ZIF formulation showed no signs of in vivo toxicity and demonstrated a sustained release profile similar to the n-OVA[Cy7]@ZIF. We extracted the inguinal lymph node (ILN) closest to the injection site and the contralateral inguinal lymph node (CILN) on day 14 following a single dose of OVA, n-OVA@ZIF, and μ-OVA@ZIF. Saline was used as a negative control. Both μ-OVA@ZIF and n-OVA@ZIF should predominantly exert their immunological effect within the ILN as compared to the much more distant CILN. Consequently, we expect to see morphological changes—e.g., enlargement—in the ILN as a result of the immune response compared to the CILN.58,59 Gross pathological examination found that μ-OVA@ZIF formulation produced the largest ILN, indicating that this formulation would encourage a more robust immune response (Fig. S7†). From these preliminary in vivo data, with both formulations demonstrating similar tissue residency times, we conclude that μ-OVA@ZIF is more biocompatible and appears to be more efficient at activating the immune system. Looking at both the in vitro and in vivo data, we decided that μ-OVA@ZIF was the preferred vaccine formulation and was therefore used in further experiments.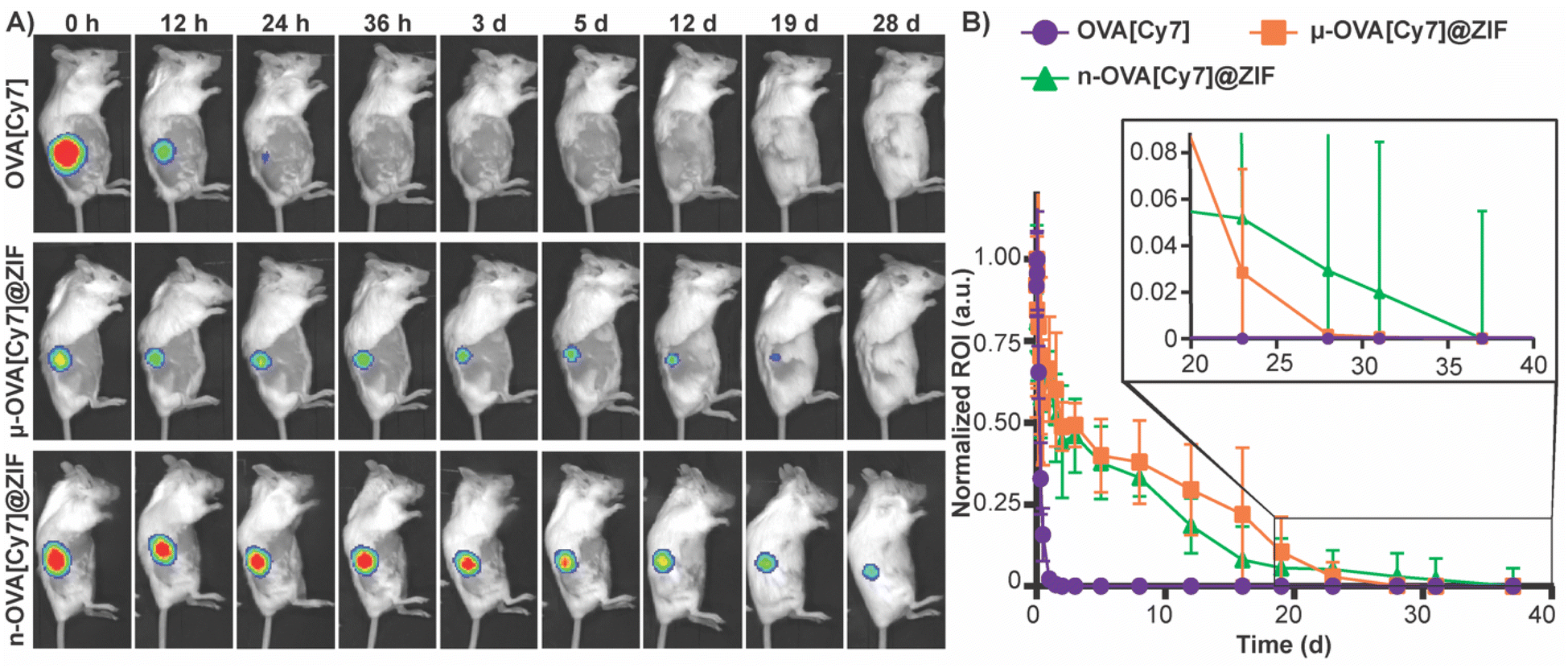 | ||
| Fig. 3 Evaluation of μ-OVA@ZIF and n-OVA@ZIF tissue residency. (A) Representative images of Cy7 fluorescence in the right flank of mice subcutaneously injected with OVA[Cy7] (top), μ-OVA[Cy7]@ZIF (middle), and n-OVA[Cy7]@ZIF (bottom) (B) normalized Cy7 fluorescence from mice subcutaneously injected with OVA[Cy7], μ-OVA[Cy7]@ZIF, and n-OVA[Cy7]@ZIF (10 μg OVA[Cy7]) over the course of 38 days (n = 4). | ||
To qualitatively assess the degradation of ZIF-8 crystals, we injected 50 μL of μ-OVA@ZIF into BALB/c mice (n = 2) (Fig. 4A). 24 h, 48 h, 72 h, and 96 h post-vaccination, the mice were euthanized, and ZIF-8 crystals were extracted from the injection site as a white bio-aggregate clump (Fig. S8†). SEM images were taken of pristine and ex vivo ZIF-8 (Fig. 4B). It is known that ZIF-8 degrades in the presence of phosphates and albumin.34 There was a definitive difference between pristine and ex vivo ZIF-8. After 24 h, some crystals maintained their rhombic dodecahedron shape, while others developed large cavitations. After 48 h, the crystals appeared to be majority amorphous in structure, and after 72 h, no obvious ZIF-8 crystals were seen in the SEM micrographs (Fig. 4B). At 96 h post-injection, we could no longer find crystals with the naked eye, though fluorescence data clearly show the presence of antigen. We thus conclude that the OVA@ZIF persists as micro or nanoparticles too small to see with the unaided eye. It is worth noting that we were able to extract less material with every successive time point.
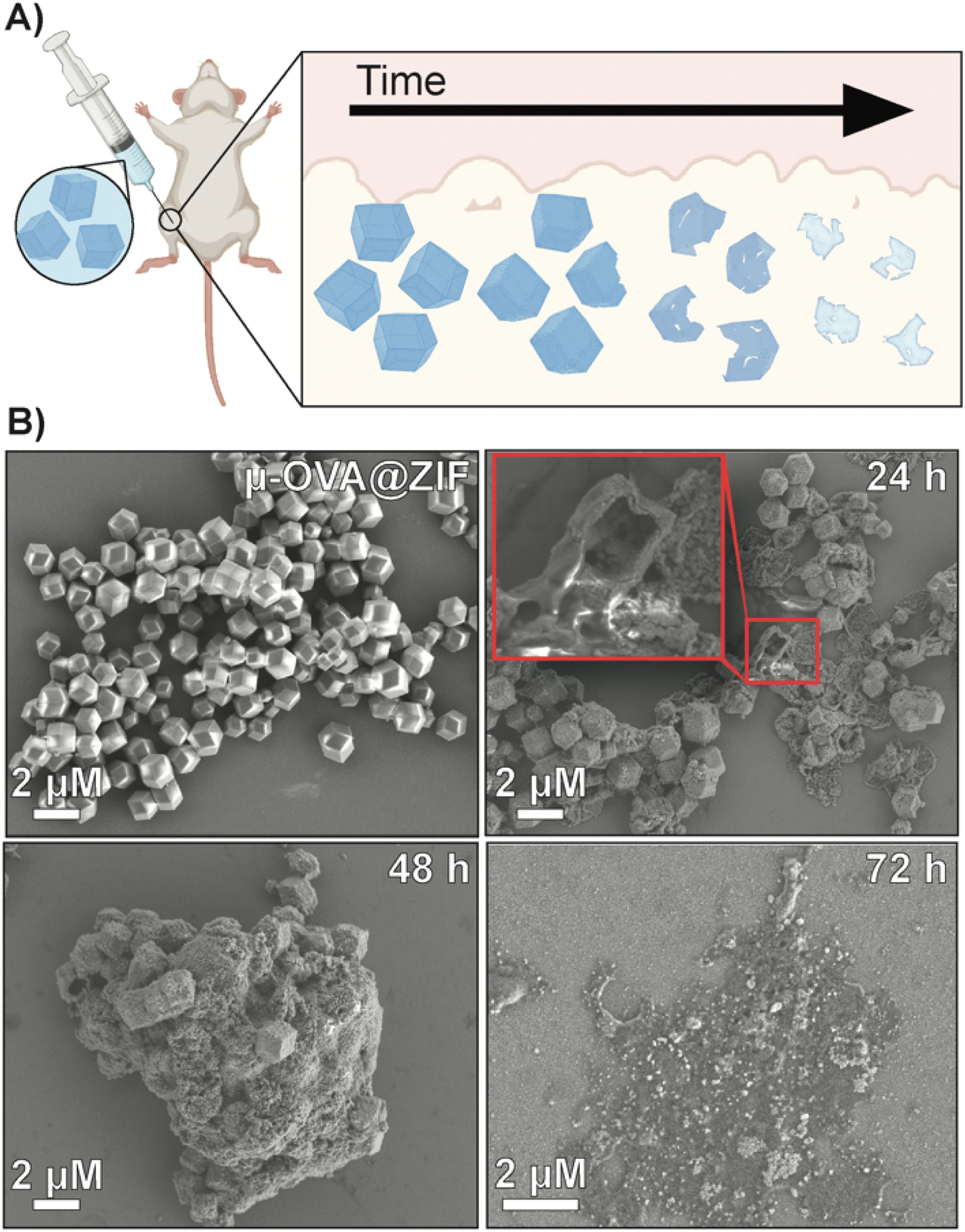 | ||
| Fig. 4 Degradation analysis of μ-OVA@ZIF post-injection. (A) Schematic representation of the degradation of ZIF-8 crystals in vivo. (B) SEM images of pristine μ-OVA@ZIF and the ex vivo extracts at 24 h, 48 h, and 72 h post-injection. | ||
The high concentration of metal in MOF carrier systems poses a challenge when translating these delivery systems to a clinical setting—thus far, only one MOF system has made it to phase I clinical trials.60 To further understand the safety and metabolism of the MOF composites, we performed an in vivo study to quantify Zn2+ distribution to major organs. We vaccinated female BALB/c mice (n = 3) with μ-OVA@ZIF and following inoculation, major organs (including the ILN, the injection site, and the flank) were extracted after 24 h, 1 w, and 2 w post-vaccination. Inductively coupled plasma mass spectrometry (ICP-MS) was used to quantify zinc concentrations in organs. Our experimental design is represented by Fig. 5A. As a negative control, naïve mice (n = 3) were used as the baseline to determine typical Zn2+ concentrations in each organ. The spleen, a crucial organ of the lymphatic system, displayed no significant changes in metal concentration (Fig. 5B). Interestingly, the ILN, contained significantly more Zn2+ 24 h post-vaccination but returned to baseline by day 7 (1 w—Figure 5C). It has been known that maintaining homeostatic zinc levels is crucial for normal immune function.61,62 However, recent studies have shown that using zinc as a therapeutic has immunogenic properties, including T cell activation and IFN-γ production (a pro-inflammatory cytokine).63,64 This warrants further investigation into zinc's role in adjuvanting the vaccine@MOF. Like the ILN, the liver demonstrates a slight yet statistically significant increase in zinc concentration at 24 h but returns to baseline by 1 w (Fig. 5D). The kidneys, lungs, and heart presented no significant change in metal content compared to the negative control (Fig. 5E, F and H). The depot site, the flank, required two weeks to return to baseline; however, this is expected as we previously demonstrated that the μ-ZIFs reside at the injection site for at least 2 w as shown in Fig. 3.
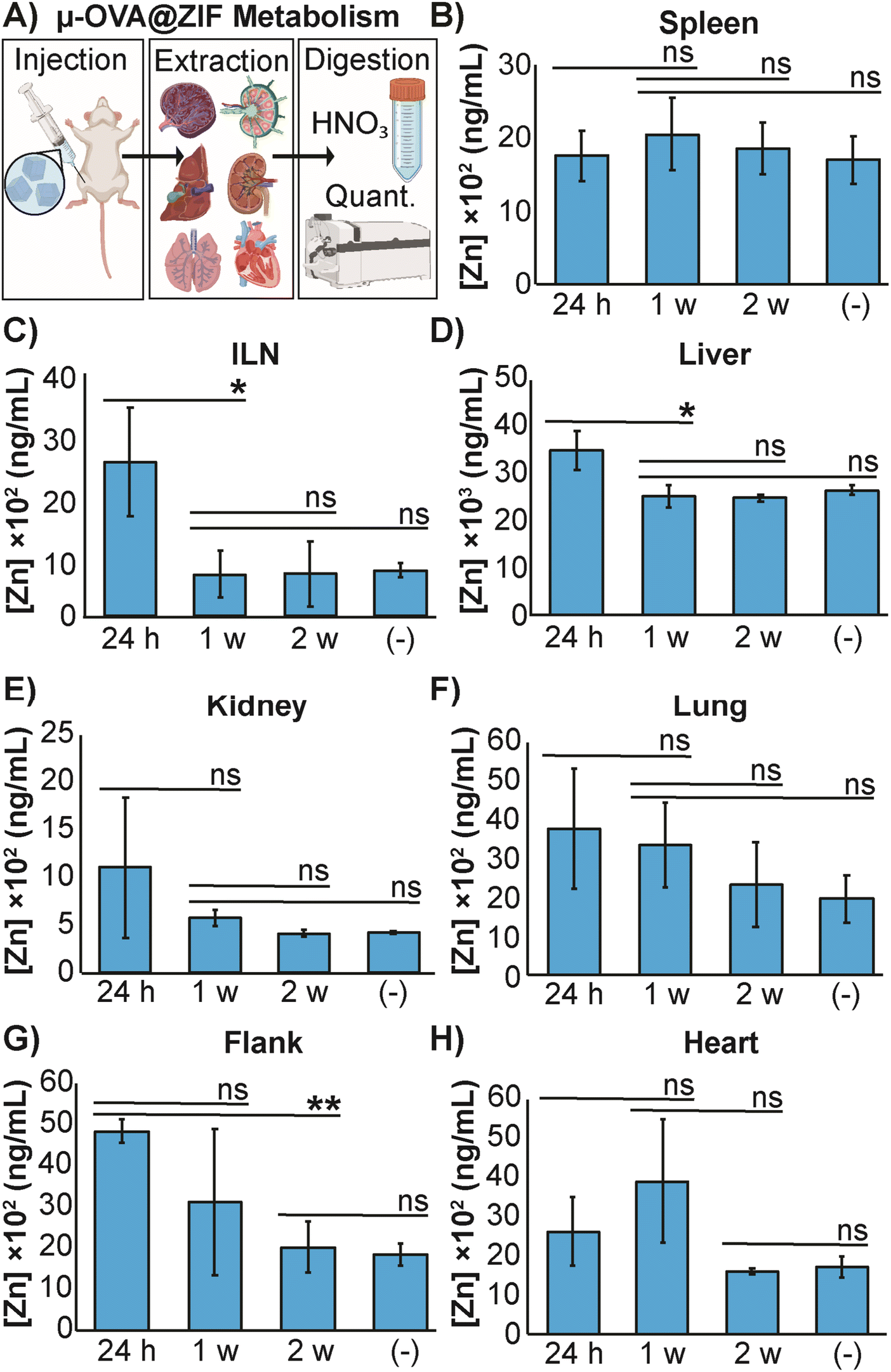 | ||
| Fig. 5 Distribution of zinc released from μ-OVA@ZIF. (A) Representative scheme illustrating the vaccination, organ extraction, sample digestion, followed by quantification of Zn2+ through ICP-MS. The concentration of Zn2+ in the (B) spleen, (C) ILN, (D) liver, (E) kidneys, (F) lungs, (G) flank and (H) heart of female BALB/c mice (n = 3) following vaccination with μ-OVA@ZIF. Organs were extracted at 24 h, 1 w, and 2 w post inoculation. (−) denotes the negative control consisting of unvaccinated mice (n = 3). Statistical significance was determined by an unpaired Student's t-test (P > 0.05 is ns, P ≤ 0.05 is *, P ≤ 0.01 is **, P ≤ 0.001 is ***, and P ≤ 0.0001 is ****). | ||
Adjuvanting of ZIF-8 and germinal center development
The ability of ZIF-8 encapsulation to enhance the immune response against the model Ag, OVA, was evaluated in vivo by looking at the humoral response. Female BALB/c mice were injected with either three bolus injections of OVA or one dose of μ-OVA@ZIF (n = 5). The single dose and two subsequent boosters delivered to each mouse contained 25 μg of OVA, so OVA 3× received thrice the amount of Ag. Mice were injected with OVA on days 0, 7, and 14 to simulate a booster dose injection schedule. μ-OVA@ZIF was injected once on day 0. To investigate the long-term humoral immunity of single dose OVA@ZIF versus OVA 3×, total IgG was investigated from days 14 to 91 following the first administration of OVA@ZIF and OVA 3× (Fig. 6A). Using ELISA, endpoint titer total IgG was determined using saline mice as the baseline. There was higher anti-OVA IgG in the serum of single dose μ-OVA@ZIF versus OVA 3× across all time points (Fig. 6B).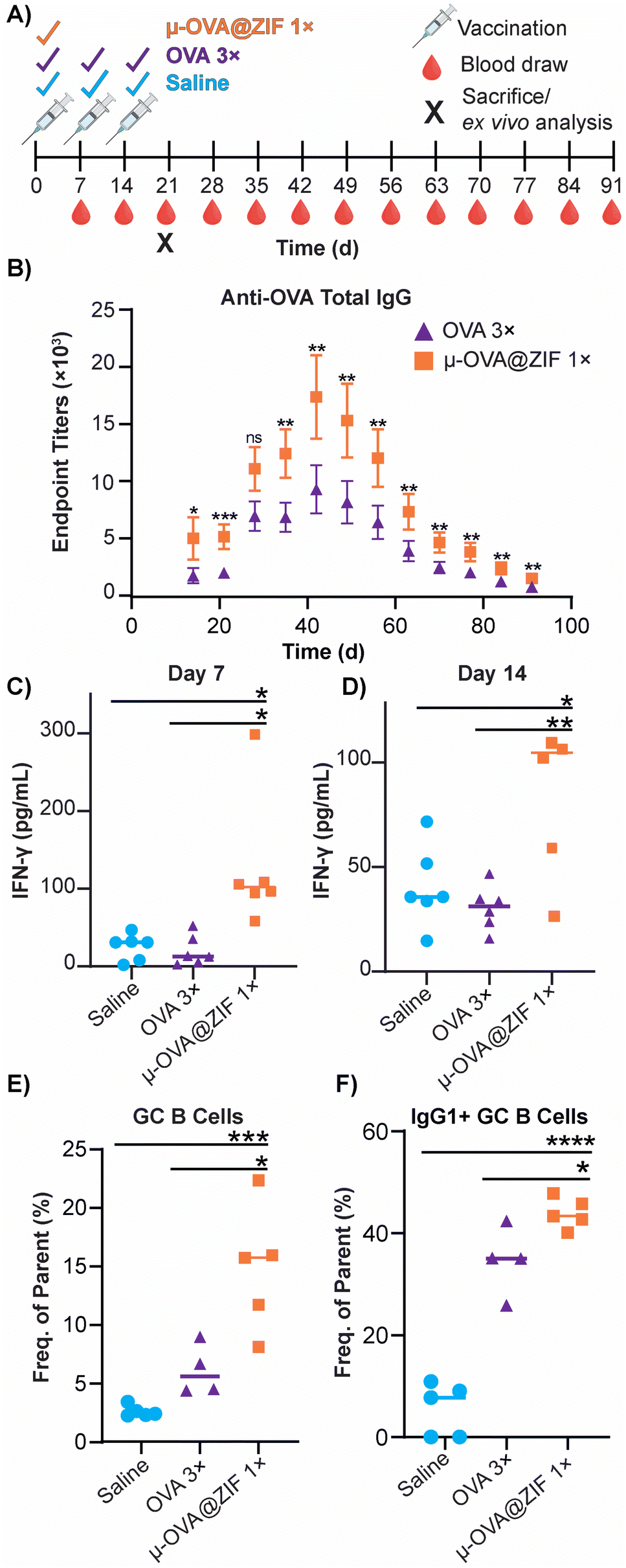 | ||
| Fig. 6 Humoral response of single dose μ-OVA@ZIF vs. OVA 3×. (A) Scheme of vaccine and blood draw schedule for determining endpoint titers of anti-OVA IgG. (B) Endpoint titers of Anti-OVA IgG from mouse serum. IFN-γ concentration (pg mL−1) in mouse serum following vaccination on (C) day 7 and (D) day 14 determined through ELISA. (E) Day 21 GC formation in the ILN of vaccinated mice. (F) Percentage of IgG1+ GC B cells in the ILN. Statistical significance was determined by an unpaired Student's t-test (P > 0.05 is ns, P ≤ 0.05 is *, P ≤ 0.01 is **, P ≤ 0.001 is ***, and P ≤ 0.0001 is ****). | ||
We observed that zinc was being trafficked into the lymph node in the previous study illustrated by Fig. 5C. This prompted us to further investigate the role zinc may have in adjuvanting our vaccine@MOF. As previously mentioned, zinc has been shown to cause IFN-γ production and T-cell activation. To check if our vaccine@MOF is inducing the production of the pro-inflammatory cytokine, IFN-γ, we determined the concentration of IFN-γ in serum through ELISA on day 7 and day 14 following vaccination of naïve BALB/c mice (n = 5) with saline (negative control), bolus OVA, and μ-OVA@ZIF. There was no significant difference between the negative control and bolus OVA. However, as we suspected, there was significantly higher concentration of IFN-γ in serum taken from the μ-OVA@ZIF cohort on both day 7 (Fig. 6C) and day 14 (Fig. 6D). This prompted us to further investigate the role ZIF alone plays in promoting the activation of T cells, specifically CD4+ T helper cells. Here, we vaccinated BALB/c mice (n = 8 saline, n = 9 μ-ZIF) with saline or μ-ZIF formulation—no antigen was present. After 72 h, the ILN was extracted and analyzed for CD4+ T effector cells through immunophenotyping via flow cytometry. CD4+ T effector cells, also known as T helper cells, are required for “helping” or activating immune cells involved in the adaptive immune system including B cells and CD8+ cytotoxic T cells. We observed significantly higher populations of CD4+ T eff cells in the ILN of μ-ZIF vaccinated mice in the ILN 72 h post-vaccination with respect to the negative control, saline (Fig. S9†). This discovery further supports our hypothesis that the presence of zinc in our vaccine@MOF is helping to promote an immune response, ultimately adjuvanting our vaccine.
GCs are crucial for successful immunization and immune memory, yielding antibody-producing plasma cells and long-lived memory B cells.65 GCs form in secondary lymphoid tissues, such as lymph nodes, typically after 14–28 days following Ag exposure.66 Ag-activated B cells enter the GC in what is known as the light zone (LZ) and undergo proliferation and rounds of somatic hypermutation (SHM), creating diverse clones of themselves. This diverse library of GC B cells will migrate to the dark zone (DZ) and begin affinity maturation where greater affinity antibodies are selected.67,68 GC B cells interact with follicular dendritic cells (DC) and T follicular helper cells (Tfh). Cells that “pass” the selection process will either differentiate into long-lived memory B cells or antibody-producing plasma cells or will return to the LZ and begin this process again. We observed higher populations of GC B cells on day 21 in the ILN of the μ-OVA@ZIF cohort compared to OVA 3× (Fig. 6E). Memory B cells and plasma B cells arise from these GC, creating long-term immune memory and increased antibody production. We observe higher antibody titers in the μ-OVA@ZIF cohort until day 91, validating our hypothesis. Additionally, we observed higher IgG1+ GC B cells in the ILN of the μ-OVA@ZIF; this is indicative of class-switching recombination (CSR), a process where B cells begin to produce higher affinity antibodies (Fig. 6F). GC formation and CSR are two crucial benchmarks for assessing a vaccine's efficacy, as immune memory, long-term antibody production, and high-affinity antibodies are needed to maintain humoral immunity. The sustained release of the Ag by encapsulation in MOF provides a constant source of Ag, better equipping the immune system than three bolus injections of Ag. Additionally, hematoxylin and eosin (H&E) staining of tissue samples collected on day 21 show no significant sign of inflammation, tissue damage, or major morphological changes compared to saline (Fig. S10†). The gating strategy used to determine cell populations for Fig. 6E andF is shown in Fig. S11.†
Scalable synthesis
Finally, to demonstrate our formulation's ease of preparation and scalability, we synthesized the μ-ZIF formulations under the same conditions previously mentioned—we increased the volumes and concentrations. We made 2 L of μ-ZIF (Fig. 7A and B) and bovine serum albumin@ZIF (μ-BSA@ZIF) (Fig. 7A and B). We encapsulated 500 mg of BSA[Cy-7] (250 μg mL−1) in the 2 L ZIF-8 reaction. The reaction was left for 1 h on the benchtop (Fig. 7B). The encapsulation efficiency was 98.1% ± 1.64, comparable to that of the 1 mL reaction of OVA@ZIF. Following a series of washes, the MOFs were dried under a vacuum, and the total weight of μ-ZIF was determined to be 15.9 g (Fig. 7C), and the drug loading ratio of albumin to ZIF is 1:32 w/w albumin to ZIF. The drug loading of the scaled reaction is similar to that of the 1 mL reaction (1:30 w/w) as we would suspect. We used BSA in this case because Ag-grade OVA is expensive, and it would be wasteful to create such a large batch of OVA@ZIF as no lab could possibly use this much material. It is worth mentioning, however, that Ag doses given to mice are typically the same as those given to humans (between 5–50 μg per dose).12,69 This is the equivalent of 20000 injections. Thus, this synthesis would prepare enough injections for a small town in an hour. PXRD diffractograms were obtained, and the sodalite crystallinity of both μ-BSA@ZIF and μ-ZIF was confirmed (Fig. 7D).
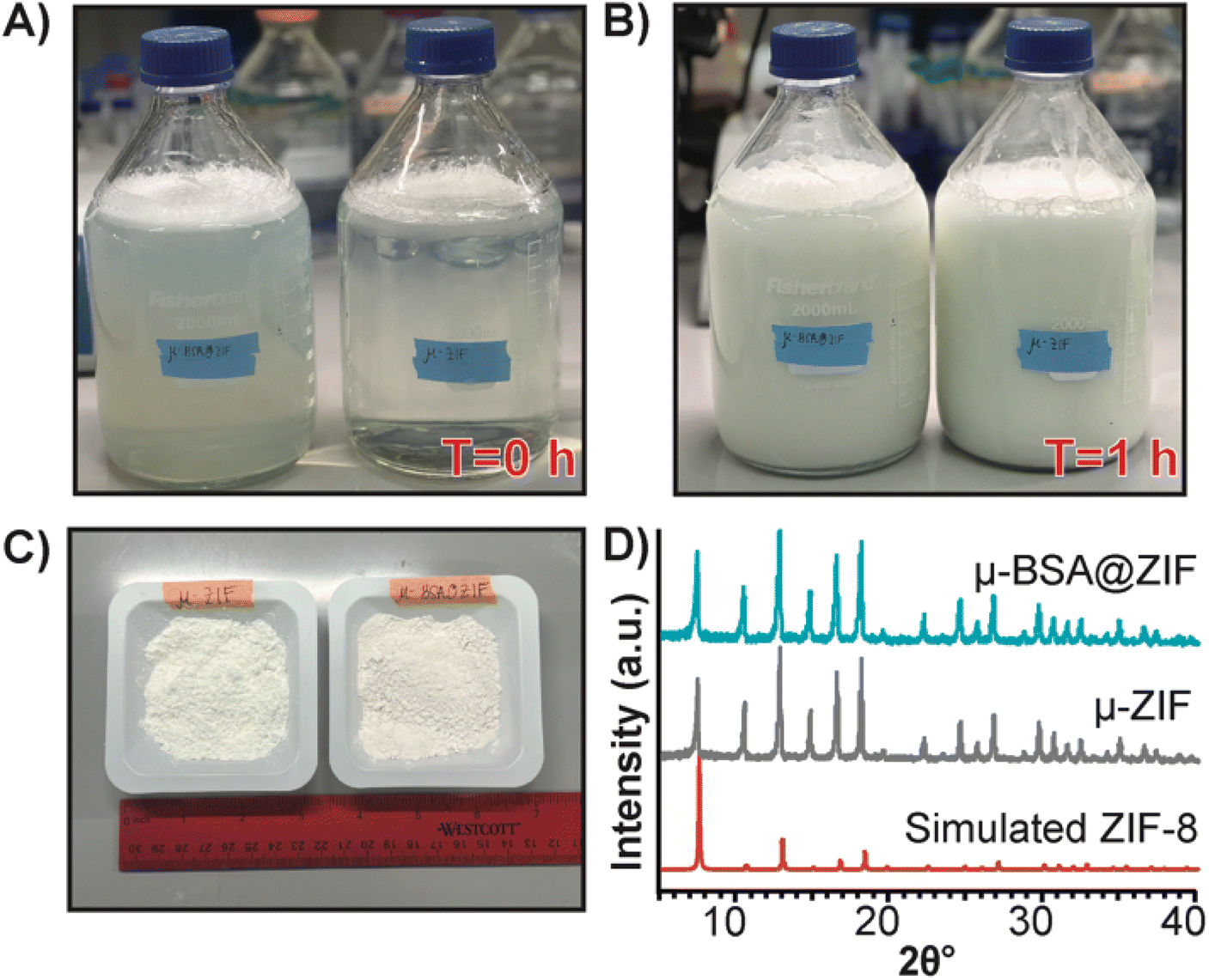 | ||
| Fig. 7 Scaled synthesis of μ-ZIF and μ-BSA[Cy7]@ZIF. (A) Image of 2 L bottles (left: μ-BSA@ZIF, right: μ-ZIF) used for scaled-up ZIF synthesis at T = 0 h and (B) at T = 1 h. (C) 15.9 g of μ-ZIF powder (left) and 16.4 g μ-BSA[Cy7]@ZIF powder (right) from 2 L synthesis. (D) PXRD diffractograms of μ-BSA[Cy7]@ZIF (top), μ-ZIF (middle), and simulated ZIF-8 (bottom) following scaled synthesis. | ||
Conclusion
ZIF-8 has been found to be highly effective in improving the stability and protection of proteinaceous vaccine formulations against thermal and enzymatic degradation. In addition, it has been demonstrated that ZIF-8 can boost the immune response generated by the vaccine, to the extent that it can overcome the need for a three-series booster regimen. In terms of the humoral immune response, μ-OVA@ZIF has shown significantly higher levels of serum antibody titers against OVA as compared to three bolus Ag doses. Not only does ZIF-8 encapsulation generate a strong immune response, but it also uses less antigen than the bolus injection. ZIF-8 encapsulation can reduce the number of needle injections and the amount of expensive, precious biomaterial. Lastly, we determined that this synthesis is easily scaled to a 2 L reaction volume while maintaining the short reaction time (1 h) and high encapsulation efficiency of protein (98%). We discovered that ZIF-8 possesses robust adjuvant properties and is less toxic than FDA-approved adjuvants like alum. Our research also indicates that zinc may play a role in promoting adjuvant activity, which is interesting and could provide considerable impetus to evaluate other MOFs as metalloimmuno-adjuvants. Zinc is known to encourage T cell maturation and the production of IFN-γ, which aligns with our findings of zinc trafficking into the lymph nodes through the metabolism of ZIF-8. These findings warrant further investigation into the role of zinc in ZIF-8's adjuvant properties to understand better and exploit this MOF-based delivery system.Methods
Chemicals
Zinc acetate (ZnOAc), 2-methyl imidazole, sodium chloride, phosphate buffer saline (PBS) pH 7.4, Dulbecco's Modified Eagle Medium (DMEM), penicillin-streptomycin, L-glutamine, BSA, Ethylenediaminetetraacetic acid (EDTA), sodium bicarbonate (NaCO3), dimethyl sulfoxide (DMSO), goat anti-mouse IgG (whole molecule)-alkaline phosphatase, magnesium chloride (MgCl2), paraformaldehyde (PFA) p-nitrophenyl phosphate (pNPP), tween-20, polyvinyl pyrrolidine (PVP), diethanolamine, potassium phosphate dibasic, potassium phosphate monobasic, hydrochloric acid, and sodium hydroxide were purchased from Sigma Aldrich (St. Louis, MO, USA) or Thermofisher Scientific (Waltham, MA, USA) and used without further modification. FBessence was purchased from VWR (Radnor, PA, USA). LDH assay, cell staining buffer (CSB), Zombie Red, Zombie UV, anti-mouse CD19 Alex Fluor 700 (AF700), anti-mouse IgG1 phyco-erythrin (PE), anti-mouse CD95 allophycocyanin (APC), and anti-mouse GL7 Alexa Fluor 488 (AF488) were purchased from Biolegend (San Diego, CA, USA). Vaccine-grade, endotoxin-free OVA was purchased from Invitrogen (Waltham, MA, USA) and Worthington Biochemical (Lakewood, NJ, USA). Bradford protein assay was purchased from BioRad Laboratories (Hercules, CA, USA). Non-fluorescent mouse diet was purchased from Lab Supply (Northlake, TX, USA). Cyanine 7-NHS (Cy7-NHS) was purchased from Lumiprobe (Cockeysville, MD, USA) and synthesized (Fig. S8–19†).Instruments
SEM micrographs were taken on Zeiss Supra 40. PXRD patterns were obtained from Rigaku SmartLab X-ray diffractometer. XPS spectra was obtained on Versaprobe II scanning XPS. FT-IR spectra was obtained on Agilent Cary 660 FT-IR. Absorbance and fluorescence spectra were obtained on Biotek Synergy H4 Hybrid microplate reader. Thermofisher Scientific Sorvall Legend Micro17, Thermofisher Scientific Sorvall Lynx 4000, and Beckman Coulter Allegra X-14R centrifuges were used for obtaining cell and ZIF-8 pellets. CLSM images were taken on Olympus FV3000 RS microscope. The concentration of Zn in the extracted organs was quantified using Agilent 7900 ICP-MS. Flow cytometry data were acquired on a BD LSRFortessa. Cell counting was carried out on a Thermo Countess II. Live animal imaging was performed on an IVIS Lumina III. Paraffin embedding was done on Histo-Core ARCADIA. Embedded tissues were processed with a Leica RM22335 microtome. H&E images were obtained on VS120 virtual slide microscope. Ultrapure water was filtered in lab with the ELGA PURELAB flex 2 system.Animals and ethics
Female BALB/c mice (4–6 weeks) were purchased from Charles Rivers Laboratories (Wilmington, MA, USA). In vivo experiments were approved by the University of Texas at Dallas Institutional Animal Care and Use Committee. This work was diligently carried out in accordance with protocol #19-06.ZIF-8 synthesis
1 M ZnOAc and 3 M mIM stocks were made in sterile water (filtered and autoclaved). The final stock solutions were filtered through a 0.22 μM syringe filter. All microscale reactions took place at a final volume of 1 mL. For the nano-sized MOFs, sterile water, ovalbumin (150 μg mL−1 in water), 2560 mM mIM, and 80 mM ZnOAc were added sequentially, vortexing after each addition. For the micro-sized MOFs, sterile water, OVA (150 μg mL−1), 640 mM mIM, and 40 mM ZnOAc were added sequentially, vortexing after each addition. The reaction vial was left for 1 h at RT. The resulting crystals were washed 3× by centrifuging at 17000 × g for 5 min. The supernatant was removed, and the pellet was resuspended in 1 mL of sterile water.
Fluorescent labeling of protein
For OVA[Cy7], 10 mg mL−1 OVA was dissolved in 1 mL of 0.1 M NaCO3 (pH 8.3). 63 μL of Cy7-NHS in DMSO dye solution was added to the OVA solution and vortexed well. The protein-dye solution was incubated overnight on a rotisserie at 4 °C. The solution was washed with water in a 10 kDa protein concentrator at 4000 × g for 10 min until the filtrate was clear. The concentration of protein was determined through Bradford protein assay.For BSA[Cy7] 1 g of BSA dissolved in 0.1 M NaCO3 (pH 8.3). 1 mg of synthesized Cy7-NHS was dissolved in 200 μL of DMSO. The Cy7-NHS was added to the protein solution and vortexed well. The protein-dye solution was incubated overnight on a rotisserie at 4 °C. The solution was washed with water in a 10 kDa protein concentrator at 4000 × g for 10 min until the filtrate was clear, indicating that the free Cy7 dye was removed. The concentration of protein was determined through Bradford Protein Assay.
Cell culture
RAW 264.7 murine macrophages were cultured in DMEM supplemented with 1% L-Glutamine, 2% penicillin-streptomycin, and 10% FBessence in a 75 cm2 culture flask. Cells were grown at 37 °C with 5% CO2. Cells were passaged at 80–95% confluency by removing media, rinsing with 0.1 M PBS, followed by an additional 0.1 M PBS, and detaching cells with a cell scraper. The cell solution was centrifuged at 300 × g for 2 min.Cytotoxicity
The night before, 1 × 105 viable RAW 264.7 cells were seeded in a 96-well plate in supplemented DMEM. Old media was aspirated. OVA, μ- and n-OVA@ZIF samples were diluted in complete DMEM and added to wells at a final volume of 100 μL. The cells were incubated with samples for 4 h at 37 °C with 5% CO2. Following the protocol from the LDH assay kit, 20 μL of lysis buffer was added to the high control and left to incubate for an additional 20 min at 37 °C with 5% CO2. Then, 100 μL of the working solution was added to each well and left to incubate in the dark for 20 min. 50 μL of stop solution was added, and the absorbance was read at 490 nm. Cell viability was determined through the following equation: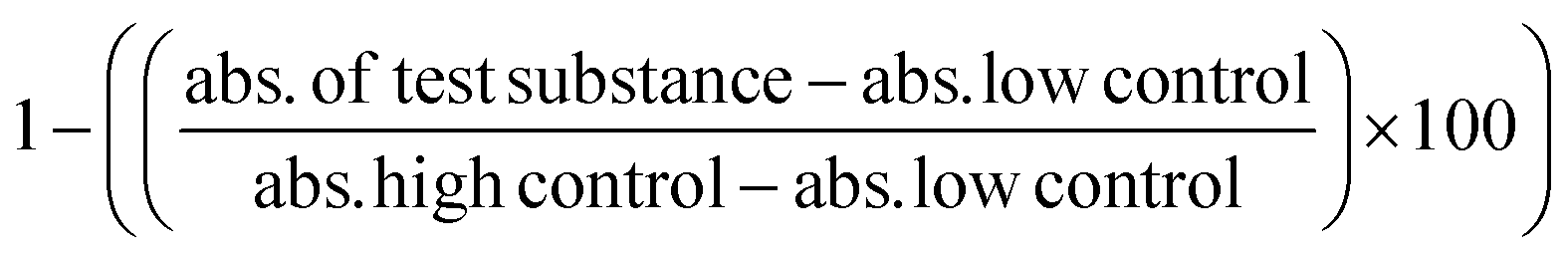 .
.
In vitro uptake
For uptake studies, 1 × 106 cells were added to a 24-well plate with 2 μg OVA[Cy7], 8 μL of μ- and n-OVA[Cy7]@ZIF (2 μg OVA). The cells were incubated with samples for 4 h at 37 °C with 5% CO2. The cells were washed 3× with 0.5 M EDTA and 3× with PBS. Cells were stained with a 1:2000 zombie red solution in PBS for 20 min on ice and in the dark. Cells were centrifuged and resuspended in CSB (0.1 M PBS, 5% FBS, 0.5 mM EDTA, and 2 mM NaN3). Flow cytometry analysis was performed on a BD LSRFortessa flow cytometer, and 100000 events were recorded. Raw data were processed and analyzed using FlowJo software. Dead cells were gated out by selecting the negative zombie red population, and Cy7 positive cells were assumed to have uptake OVA. The Histogram overlay was normalized to mode.
Tissue residency
12 female BALB/c (n = 4) mice were fed a non-fluorescent diet for at least 48 h before the start of the experiment. The day before vaccination, mice were depilated with Nair hair remover on the side of the injection. Mice were anesthetized with isoflurane before injections or imaging. Baseline images of each mouse were obtained before the injection of the sample. OVA[Cy7], μ-OVA[Cy7]@ZIF, and n-OVA[Cy7]@ZIF (10 μg OVA per dose) were injected subcutaneously with the sample into the right flank of each mouse. Mice were imaged at t = 0 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 24 h, 36 h, 2 d, 3 d, 5 d, 8 d, 12 d, 16 d, 19 d, 23 d, 28 d, 31 d, and 37 d. Data were normalized using GraphPad Prism.Ex vivo degradation
100 μL of μ-OVA@ZIF in 0.1 M saline was injected into both flanks (to increase the chance of retrieving the sample) of female BALB/c mice (n = 2). Mice were euthanized through cervical dislocation, and samples were removed from the subcutaneous layer at 24, 48, 72, and 96 h (no sample was found at 96 h). Samples were washed 3× with water.Ex vivo organ Zn concentration determination
100 μL of μ-OVA@ZIF in 0.1 M saline was subcutaneously injected into the left flank of female BALB/c mice (n = 3). At 24 h, 1 w, and 2 w. Mice were euthanized via cervical dislocation, and major organs (including flank, ILN, kidneys, lungs, liver, and spleen) was removed post-mortem. A negative control of naïve mice (n = 3) was used to determine the baseline concentration of Zn in the previously mentioned organs. Organs were first digested in 70% nitric acid. Then the digested organs were diluted to 35% nitric acid and syringe-filtered with a 0.22 μm filter. The filtered solution was finally brought up to a concentration of 2% nitric acid using ultra-pure water. Commercially available Zn standards were used for determination of the Zn concentration.T cell activation
Female BALB/c (n = 8 or n = 9) were subcutaneously vaccinated with 100 μL of 0.1 M saline or μ-ZIF in the left flank. After 72 h, the mice were sacrificed via cervical dislocation and the ILN was extracted and homogenized with a mortal and 100 μM cell strainer. Lymphocytes were stained with an antibody cocktail (anti-CD3, anti-CD4, anti-CD44, and anti-CD62L) for 1 h in the dark and on ice. Cells were washed and flow cytometry was performed to determine the percent population of T eff cells.In vivo antibody production
Female BALB/c mice (n = 5) were vaccinated subcutaneously with three doses of 100 μL 0.1 M saline and 100 μL OVA (75 μg OVA total) on days 0, 7, and 14. Additionally, one dose of μ-OVA@ZIF (25 μg OVA) on day 0. Mice were anesthetized with isoflurane, and blood was drawn weekly from day 14 until day 98. The serum was isolated by centrifuging blood samples at 1200 × g for 10 min to remove red blood cells. Serum was stored at −20 °C until further use.Endpoint titer anti-OVA IgG ELISA
96-well Nunc titer plate was coated the night before with 100 μL 1 μg mL−1 OVA in coating buffer (0.05 M NaCO3 buffer pH 9.6). The plate was left to incubate overnight at 4 °C. Plate was washed 4× with 300 μL of wash buffer (0.1 M PBS 0.05% w/v tween-20 pH 7.4). The plate was blocked with 200 μL of assay diluent (1% BSA in wash buffer) for 1 h at 37 °C. The plate was washed 5× with wash buffer, and 100 μL of serially diluted serum (100×–3200×) in assay diluent was added to each well. The plate was incubated at 37 °C for 1.5 h. The plate was subsequently washed 4× with wash buffer, followed by the addition of 100 μL of (1:2000) alkaline phosphatase-conjugated goat anti-mouse IgG in conjugate buffer (0.02 g of PVP mL−1 assay diluent). The plate was incubated at 37 °C for 1 h. The plate was incubated at 37 °C for 1 h. The plate was washed 4× with wash buffer. 100 μL of 1 mg mL−1 pNPP in substrate buffer (1 M diethanolamine buffer, 0.5 mM MgCl2 pH 9.8) was added to the plate and left in the dark to incubate for 15 min or until color developed. Absorbance was read at 450 nm at 15 and 30 min. The endpoint titer was determined by plotting the dilution factor against 450 nm absorbance. The logarithmic curve was fit, and the endpoint titer was determined by the intersection of logarithmic fit to the average baseline (saline).
In vivo GC study
Female BALB/c mice (n = 5) were vaccinated subcutaneously with three doses of 100 μL 0.1 M saline and 100 μL OVA (75 μg OVA total) on days 0, 7, and 14. Additionally, one dose of μ-OVA@ZIF (25 μg OVA) on day 0. Mice were anesthetized before vaccination. On day 21, mice were euthanized via cervical dislocation, and the ILN was extracted along with other major organs (heart, lung, liver, and kidney) for H&E staining. A single-cell suspension was obtained by carefully passing cells through a 100 μM cell strainer. Cells were washed 1× with 0.1 M PBS, resuspended in 1 mL of 0.1 M PBS, and counted using trypan blue to determine live/dead cells. Cells were stained with Zombie UV (1:2000) in 0.1 M PBS for 30 min on ice in the dark. Subsequently, the antibody cocktail (anti-mouse CD19 AF700, anti-mouse IgG1 PE, anti-mouse CD95 APC, and anti-mouse GL7 AF 488 in CSB) was added to the cells. The cells were left to stain for 30 minutes on ice in the dark. Cells were washed 3× with FACS buffer. Flow cytometry analysis was carried out on a BD LSRFortessa flow cytometer. Raw data were processed and analyzed using FlowJo software.
H&E
Organs were incubated in 4% PFA for 48 h at ambient T on a shaker. Tissues were washed 3× with 0.1 M PBS, placed into tissue cassettes, and then into 70% ethanol. The organs were embedded in paraffin wax. Each organ was sectioned into 5 μm section using a rotary microtome. The sections were collected and stained with H&E for pathological analysis.Scaled ZIF-8 synthesis
2 L of μ-ZIF was synthesized by adding 640 mM mIM, 40 mM ZnOAc, bringing the final volume to 2 L with MilliQ water. 2 L of μ-BSA[Cy7]@ZIF was synthesized by 640 mM mIM, 40 mM ZnOAc, 500 mg of BSA[Cy7]. The reaction bottles were vigorously shaken after adding the precursors and left to incubate for 1 h at ambient T. Solutions were transferred to 1 L centrifuge bottles and centrifuged for 30 min at 17000 × g. The supernatant was discarded, and ZIF was resuspended in 200 mL of water, transferred to 50 mL disposable centrifuge tubes, and washed 2× with water. Tubes were then placed under a vacuum for drying.
Data availability
Raw and processed data used to create figures and displayed in the ESI† can be accessed at OSFHome at the following: https://doi.org/10.17605/OSF.IO/7MHQU.Author contributions
Conceptualization: J. J. G. Methodology: R. N. E., O. R. B., Y. H. W., S. Kumari, F. C. H., investigation: R. N. E., O. R. B., Y. H. W., S. Kumari, I. T., O. T., T. S. H., F. C. H., A. R., S. Koirala, N. T., N. M. A., W. T., M. C. S., L. M. H. Manuscript preparation: R. N. E. and J. J. G.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Dr Rockford Draper (University of Texas at Dallas) for the gift of RAW 264.7 cells. Also, we thank the University of Texas at Dallas Lab Animal Resource Center and its staff for their assistance in the care and maintenance of animals. J.J.G. would like to thank the National Science Foundation (DMR- 2003534) and the Welch Foundation (AT-198920190330) for their support.References
- D. A. Henderson, Public Health Rep., 1997, 112, 116–121 CAS.
- S. A. Plotkin, Nat. Med., 2005, 11, S5–S11 CrossRef CAS PubMed.
- P. Pezzotti, S. Bellino, F. Prestinaci, S. Iacchini, F. Lucaroni, L. Camoni, M. M. Barbieri, W. Ricciardi, P. Stefanelli and G. Rezza, Vaccine, 2018, 36, 1435–1443 CrossRef PubMed.
- P. M. Moyle and I. Toth, ChemMedChem, 2013, 8, 360–376 CrossRef CAS PubMed.
- A. Vartak and S. J. Sucheck, Vaccines, 2016, 4, 12 CrossRef PubMed.
- M. Hansson, P.-A. k. Nygren and S. Ståhl, Biotechnol. Appl. Biochem., 2000, 32, 95–107 CrossRef CAS PubMed.
- R. P. Welch, H. Lee, M. A. Luzuriaga, O. R. Brohlin and J. J. Gassensmith, Bioconjugate Chem., 2018, 29, 2867–2883 CrossRef CAS PubMed.
- R. Riccò, W. Liang, S. Li, J. J. Gassensmith, F. Caruso, C. Doonan and P. Falcaro, ACS Nano, 2018, 12, 13–23 CrossRef PubMed.
- D. M. Matthias, J. Robertson, M. M. Garrison, S. Newland and C. Nelson, Vaccine, 2007, 25, 3980–3986 CrossRef CAS PubMed.
- M. Heidary, V. H. Kaviar, M. Shirani, R. Ghanavati, M. Motahar, M. Sholeh, H. Ghahramanpour and S. Khoshnood, Front. Microbiol., 2022, 13, 927306 CrossRef PubMed.
- J. Poorolajal, M. Mahmoodi, R. Majdzadeh, S. Nasseri-Moghaddam, A. Haghdoost and A. Fotouhi, Vaccine, 2010, 28, 623–631 CrossRef CAS PubMed.
- S. B. Manoff, S. L. George, A. J. Bett, M. L. Yelmene, G. Dhanasekaran, L. Eggemeyer, M. L. Sausser, S. A. Dubey, D. R. Casimiro, D. E. Clements, T. Martyak, V. Pai, D. E. Parks and B.-A. G. Coller, Vaccine, 2015, 33, 7126–7134 CrossRef CAS PubMed.
- Y. Liao, Y. Chen, B. Chen, Z. Liang, X. Hu, B. Xing, J. Yang, Q. Zheng, Q. Hua and C. Yan, Front. Immunol., 2022, 13, 1017590 CrossRef CAS PubMed.
- É. Racine, V. Gilca, R. Amini, M. Tunis, S. Ismail and C. Sauvageau, Vaccine, 2020, 38, 6205–6214 CrossRef PubMed.
- C. B. Bridges, T. L. Watson, N. P. Nelson, M. Chavez-Torres, P. Fineis, B. Ntiri-Reid, E. Wake, J. M. Leahy, A. K. Kurian, M. A. K. Hall and E. D. Kennedy, Vaccine, 2019, 37, 5111–5120 CrossRef PubMed.
- A. Taddio, M. Ipp, S. Thivakaran, A. Jamal, C. Parikh, S. Smart, J. Sovran, D. Stephens and J. Katz, Vaccine, 2012, 30, 4807–4812 CrossRef PubMed.
- J. C. Nelson, R. C. L. Bittner, L. Bounds, S. Zhao, J. Baggs, J. G. Donahue, S. J. Hambidge, S. J. Jacobsen, N. P. Klein, A. L. Naleway, K. M. Zangwill and L. A. Jackson, Am. J. Public Health, 2009, 99, S389–S397 CrossRef PubMed.
- C. L. Ventola, Pharmacol. Ther., 2016, 41, 426–436 Search PubMed.
- S. K. Kurosky, K. L. Davis and G. Krishnarajah, Vaccine, 2016, 34, 387–394 CrossRef PubMed.
- A. Taddio, C. M. McMurtry, C. Logeman, V. Gudzak, A. de Boer, K. Constantin, S. Lee, R. Moline, E. Uleryk, T. Chera, N. E. MacDonald and B. Pham, Vaccine, 2022, 40, 7526–7537 CrossRef PubMed.
- H. Wang, L. Cui, Y. Luo, X. Zhou, R. Liu, Q. Chen, Y. Guan and Y. Zhang, Biomater. Adv., 2022, 137, 212812 CrossRef CAS PubMed.
- K. J. McHugh, R. Guarecuco, R. Langer and A. Jaklenec, J. Controlled Release, 2015, 219, 596–609 CrossRef CAS PubMed.
- E. C. Gale, A. E. Powell, G. A. Roth, E. L. Meany, J. Yan, B. S. Ou, A. K. Grosskopf, J. Adamska, V. C. T. M. Picece, A. I. d'Aquino, B. Pulendran, P. S. Kim and E. A. Appel, Adv. Mater., 2021, 33, 2104362 CrossRef CAS PubMed.
- O. R. Brohlin, R. N. Ehrman, F. C. Herbert, Y. H. Wijesundara, A. Raja, A. Shahrivarkevishahi, S. D. Diwakara, R. A. Smaldone and J. J. Gassensmith, ACS Appl. Nano Mater., 2022, 5, 13697–13704 CrossRef CAS.
- F. C. Herbert, S. S. Abeyrathna, N. S. Abeyrathna, Y. H. Wijesundara, O. R. Brohlin, F. Carraro, H. Amenitsch, P. Falcaro, M. A. Luzuriaga, A. Durand-Silva, S. D. Diwakara, R. A. Smaldone, G. Meloni and J. J. Gassensmith, Nat. Commun., 2021, 12, 2202 CrossRef CAS PubMed.
- S. Kumari, Y. H. Wijesundara, T. S. Howlett, M. Waliullah, F. C. Herbert, A. Raja, I. Trashi, R. A. Bernal and J. J. Gassensmith, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218247120 CrossRef CAS PubMed.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- J. Yang and Y.-W. Yang, Small, 2020, 16, 1906846 CrossRef CAS PubMed.
- S. Kumari, R. N. Ehrman and J. J. Gassensmith, Matter, 2023, 6, 2570–2573 CrossRef CAS.
- M. A. Luzuriaga, F. C. Herbert, O. R. Brohlin, J. Gadhvi, T. Howlett, A. Shahrivarkevishahi, Y. H. Wijesundara, S. Venkitapathi, K. Veera, R. Ehrman, C. E. Benjamin, S. Popal, M. D. Burton, M. A. Ingersoll, N. J. De Nisco and J. J. Gassensmith, ACS Nano, 2021, 15, 17426–17438 CrossRef CAS PubMed.
- M. A. Luzuriaga, R. P. Welch, M. Dharmarwardana, C. E. Benjamin, S. Li, A. Shahrivarkevishahi, S. Popal, L. H. Tuong, C. T. Creswell and J. J. Gassensmith, ACS Appl. Mater. Interfaces, 2019, 11, 9740–9746 CrossRef CAS PubMed.
- Y. H. Wijesundara, F. C. Herbert, O. Trashi, I. Trashi, O. R. Brohlin, S. Kumari, T. Howlett, C. E. Benjamin, A. Shahrivarkevishahi, S. D. Diwakara, S. D. Perera, S. A. Cornelius, J. P. Vizuet, K. J. Balkus, R. A. Smaldone, N. J. De Nisco and J. J. Gassensmith, Chem. Sci., 2022, 13, 13803–13814 RSC.
- M. A. Luzuriaga, C. E. Benjamin, M. W. Gaertner, H. Lee, F. C. Herbert, S. Mallick and J. J. Gassensmith, Supramol. Chem., 2019, 31, 485–490 CrossRef CAS PubMed.
- C. Cheng, C. Li, X. Zhu, W. Han, J. Li and Y. Lv, J. Biomater. Appl., 2019, 33, 1373–1381 CrossRef CAS PubMed.
- Z. Teng, F. Hou, M. Bai, J. Li, J. Wang, J. Wu, J. Ru, M. Ren, S. Sun and H. Guo, J. Mater. Chem. B, 2022, 10, 2853–2864 RSC.
- M. A. Luzuriaga, A. Shahrivarkevishahi, F. C. Herbert, Y. H. Wijesundara and J. J. Gassensmith, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnology, 2021, 13, e1735 CAS.
- H. H. Tam, M. B. Melo, M. Kang, J. M. Pelet, V. M. Ruda, M. H. Foley, J. K. Hu, S. Kumari, J. Crampton, A. D. Baldeon, R. W. Sanders, J. P. Moore, S. Crotty, R. Langer, D. G. Anderson, A. K. Chakraborty and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6639–E6648 CrossRef CAS PubMed.
- K. M. Cirelli and S. Crotty, Curr. Opin. Immunol., 2017, 47, 64–69 CrossRef CAS PubMed.
- A. V. Boopathy, A. Mandal, D. W. Kulp, S. Menis, N. R. Bennett, H. C. Watkins, W. Wang, J. T. Martin, N. T. Thai, Y. He, W. R. Schief, P. T. Hammond and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16473–16478 CrossRef CAS PubMed.
- G. A. Roth, E. C. Gale, M. Alcántara-Hernández, W. Luo, E. Axpe, R. Verma, Q. Yin, A. C. Yu, H. Lopez Hernandez, C. L. Maikawa, A. A. A. Smith, M. M. Davis, B. Pulendran, J. Idoyaga and E. A. Appel, ACS Cent. Sci., 2020, 6, 1800–1812 CrossRef CAS PubMed.
- K. M. Cirelli, D. G. Carnathan, B. Nogal, J. T. Martin, O. L. Rodriguez, A. A. Upadhyay, C. A. Enemuo, E. H. Gebru, Y. Choe, F. Viviano, C. Nakao, M. G. Pauthner, S. Reiss, C. A. Cottrell, M. L. Smith, R. Bastidas, W. Gibson, A. N. Wolabaugh, M. B. Melo, B. Cossette, V. Kumar, N. B. Patel, T. Tokatlian, S. Menis, D. W. Kulp, D. R. Burton, B. Murrell, W. R. Schief, S. E. Bosinger, A. B. Ward, C. T. Watson, G. Silvestri, D. J. Irvine and S. Crotty, Cell, 2019, 177, 1153–1171.e28 CrossRef CAS PubMed.
- B. S. Ou, O. M. Saouaf, J. Baillet and E. A. Appel, Adv. Drug Delivery Rev., 2022, 187, 114401 CrossRef CAS PubMed.
- N. J. Kräutler, D. Suan, D. Butt, K. Bourne, J. R. Hermes, T. D. Chan, C. Sundling, W. Kaplan, P. Schofield, J. Jackson, A. Basten, D. Christ and R. Brink, J. Exp. Med., 2017, 214, 1259–1267 CrossRef PubMed.
- N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 2018, 9, 4217–4223 RSC.
- L. Tong, S. Huang, Y. Shen, S. Liu, X. Ma, F. Zhu, G. Chen and G. Ouyang, Nat. Commun., 2022, 13, 951 CrossRef CAS PubMed.
- F. Carraro, J. D. Williams, M. Linares-Moreau, C. Parise, W. Liang, H. Amenitsch, C. Doonan, C. O. Kappe and P. Falcaro, Angew. Chem., Int. Ed., 2020, 59, 8123–8127 CrossRef CAS PubMed.
- M. D. R. Weber, T. L. Baker, B. Dao, C. Kwon and F. Tian, Cryst. Growth Des., 2020, 20, 2305–2312 CrossRef CAS.
- S. Kumari, T. S. Howlett, R. N. Ehrman, S. Koirala, O. Trashi, I. Trashi, Y. H. Wijesundara and J. J. Gassensmith, Chem. Sci., 2023, 14, 5774–5782 RSC.
- M. Hoop, C. F. Walde, R. Riccò, F. Mushtaq, A. Terzopoulou, X.-Z. Chen, A. J. deMello, C. J. Doonan, P. Falcaro, B. J. Nelson, J. Puigmartí-Luis and S. Pané, Appl. Mater. Today, 2018, 11, 13–21 CrossRef.
- J. Lin, L. Huang, H. Ou, A. Chen, R. Xiang and Z. Liu, RSC Adv., 2021, 11, 21414–21425 RSC.
- M. Kool, K. Fierens and B. N. Lambrecht, J. Med. Microbiol., 2012, 61, 927–934 CrossRef CAS PubMed.
- H. HogenEsch, Front. Immunol., 2013, 3 Search PubMed.
- D. L. Portuondo, A. Batista-Duharte, L. S. Ferreira, C. R. de Andrade, C. Quinello, D. Téllez-Martínez, M. L. de Aguiar Loesch and I. Z. Carlos, Vaccine, 2017, 35, 4430–4436 CrossRef CAS PubMed.
- N. Goto, H. Kato, J.-i. Maeyama, K. Eto and S. Yoshihara, Vaccine, 1993, 11, 914–918 CrossRef CAS PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- K. Donaldson and C. A. Poland, Curr. Opin. Biotechnol., 2013, 24, 724–734 CrossRef CAS PubMed.
- J. I. Andorko, K. L. Hess and C. M. Jewell, Arch Aesthetic Plast Surg, 2015, 17, 323–338 CAS.
- T. J. Moyer, A. C. Zmolek and D. J. Irvine, J. Clin. Investig., 2016, 126, 799–808 CrossRef PubMed.
- N. Tyagi, Y. H. Wijesundara, J. J. Gassensmith and A. Popat, Nat. Rev. Mater., 2023, 8, 701–703 CrossRef.
- I. Wessels, M. Maywald and L. Rink, Nutrients, 2017, 9 CAS.
- A. S. Prasad, Mol. Med., 2008, 14, 353–357 CAS.
- M.-K. Ko, H. W. Kim, S. H. Park, J.-H. Park, S.-M. Kim and M. J. Lee, Virus Res., 2023, 335, 199189 CrossRef CAS PubMed.
- A. Collins, P. Møller, G. Gajski, S. Vodenková, A. Abdulwahed, D. Anderson, E. E. Bankoglu, S. Bonassi, E. Boutet-Robinet, G. Brunborg, C. Chao, M. S. Cooke, C. Costa, S. Costa, A. Dhawan, J. de Lapuente, C. D. Bo’, J. Dubus, M. Dusinska, S. J. Duthie, N. E. Yamani, B. Engelward, I. Gaivão, L. Giovannelli, R. Godschalk, S. Guilherme, K. B. Gutzkow, K. Habas, A. Hernández, O. Herrero, M. Isidori, A. N. Jha, S. Knasmüller, I. M. Kooter, G. Koppen, M. Kruszewski, C. Ladeira, B. Laffon, M. Larramendy, L. L. Hégarat, A. Lewies, A. Lewinska, G. E. Liwszyc, A. L. de Cerain, M. Manjanatha, R. Marcos, M. Milić, V. M. de Andrade, M. Moretti, D. Muruzabal, M. Novak, R. Oliveira, A.-K. Olsen, N. Owiti, M. Pacheco, A. K. Pandey, S. Pfuhler, B. Pourrut, K. Reisinger, E. Rojas, E. Rundén-Pran, J. Sanz-Serrano, S. Shaposhnikov, V. Sipinen, K. Smeets, H. Stopper, J. P. Teixeira, V. Valdiglesias, M. Valverde, F. van Acker, F.-J. van Schooten, M. Vasquez, J. F. Wentzel, M. Wnuk, A. Wouters, B. Žegura, T. Zikmund, S. A. S. Langie and A. Azqueta, Nat. Protoc., 2023, 18, 929–989 CrossRef CAS PubMed.
- T. Hägglöf, M. Cipolla, M. Loewe, S. T. Chen, L. Mesin, H. Hartweger, M. A. ElTanbouly, A. Cho, A. Gazumyan, V. Ramos, L. Stamatatos, T. Y. Oliveira, M. C. Nussenzweig and C. Viant, Cell, 2023, 186, 147–161 CrossRef PubMed.
- J. H. Lee, H. J. Sutton, C. A. Cottrell, I. Phung, G. Ozorowski, L. M. Sewall, R. Nedellec, C. Nakao, M. Silva, S. T. Richey, J. L. Torre, W.-H. Lee, E. Georgeson, M. Kubitz, S. Hodges, T.-M. Mullen, Y. Adachi, K. M. Cirelli, A. Kaur, C. Allers, M. Fahlberg, B. F. Grasperge, J. P. Dufour, F. Schiro, P. P. Aye, O. Kalyuzhniy, A. Liguori, D. G. Carnathan, G. Silvestri, X. Shen, D. C. Montefiori, R. S. Veazey, A. B. Ward, L. Hangartner, D. R. Burton, D. J. Irvine, W. R. Schief and S. Crotty, Nature, 2022, 609, 998–1004 CrossRef CAS PubMed.
- N. S. De Silva and U. Klein, Nat. Rev. Immunol., 2015, 15, 137–148 CrossRef CAS PubMed.
- G. D. Victora and M. C. Nussenzweig, Annu. Rev. Immunol., 2012, 30, 429–457 CrossRef CAS PubMed.
- M. Heidary, V. H. Kaviar, M. Shirani, R. Ghanavati, M. Motahar, M. Sholeh, H. Ghahramanpour and S. Khoshnood, Front. Microbiol., 2022, 13, 927306 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06734c |
| ‡ These authors contributed equally to this work and should be considered as co-first authors for this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |