
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cross-coupling of organic fluorides with allenes: a silyl-radical-relay pathway for the construction of α-alkynyl-substituted all-carbon quaternary centres†
Jun
Zhou
 a,
Zhengyu
Zhao
a,
Soichiro
Mori
b,
Katsuhiro
Yamamoto
b and
Norio
Shibata
*ab
a,
Zhengyu
Zhao
a,
Soichiro
Mori
b,
Katsuhiro
Yamamoto
b and
Norio
Shibata
*ab
aDepartment of Nanopharmaceutical Sciences, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan. E-mail: nozshiba@nitech.ac.jp
bDepartment of Life Science and Applied Chemistry, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan
First published on 1st March 2024
Abstract
Controlling the transformation of versatile and reactive allenes is a considerable challenge. Herein, we report an efficient silylboronate-mediated cross-coupling reaction of organic fluorides with allenes to construct a series of sterically demanding α-ethynyl-containing all-carbon quaternary centers (ACQCs), using catalyst-free silyl-radical-relay reactions to selectively functionalize highly inert C–F bonds in organic fluorides. The key to the success of this transformation lies in the radical rearrangement of an in situ-generated allenyl radical to form a bulky tertiary propargyl radical; however, the transformation does not show efficiency when using the propargyl isomer directly. This unique reaction enables the cross-coupling of a tertiary carbon radical center with a C(sp2)–F bond or a benzylic C(sp3)–F bond. α-Ethynyl-containing ACQCs with (hetero)aromatic substituents and benzyl were efficiently synthesized in a single step using electronically and sterically diverse organic fluorides and allenes. The practical utility of this protocol is showcased by the late-stage functionalization of bioactive molecules and the modification of a liquid crystalline material.
Introduction
All-carbon quaternary centers (ACQCs) exhibit rigidity and structural diversity and are key structural units that occur frequently in many natural products, pharmaceuticals, and bioactive molecules.1 Moreover, at least 12% of the 200 top-selling prescription drugs in the US since 2011 contain a stereo-quaternary carbon center.2 Therefore, the construction of ACQCs presents a quite attractive challenge for organic synthetic chemists.3 In particular, ACQCs that contain an alkyne moiety serve as versatile intermediates and basic functionalized groups in organic transformations.4 Generally, the synthetic methods for alkyne-containing ACQCs involve either the direct introduction of alkynyl moieties into target molecules5 or the transformation from halogenated allenes in the presence of Knochel reagents.6 However, the aforementioned methodologies are associated with major drawbacks, including the use of transition-metal (TM) catalysts, dimerization of terminal alkynes, β-H elimination of branched tertiary alkyl units, and/or the reliance on special functionalized precursors. Therefore, the development of efficient strategies to overcome these limitations and the extremely strong steric effect to realize alkynyl-substituted ACQCs through C–C bond coupling reactions remains challenging.C–C bond formation is a perpetual subject of interest in organic chemistry and represents one of the most important transformations for the manufacture of products used in daily life.7 The majority of C–C bond coupling reactions require TM catalysts and/or organic (pseudo)halides (Ar/alkyl–X; X = e.g., I, Br, Cl, OTf, OMs).8 However, organic fluorides are rarely employed as coupling partners because the C–F bond is rather inert and has a higher bond dissociation energy (BDE) (e.g., fluorobenzene: 126 kcal mol−1; 1-fluoropropane: 114 kcal mol−1) compared with the corresponding C–I/Br/Cl bonds.9 Additionally, with the rapid development of synthetic methodologies,10 the abundance and ready accessibility of organofluorine compounds11 make them attractive functional moieties as well as building blocks for further organic transformations.
Yet, the activation of robust C–F bonds remains a major challenge in contemporary chemistry. In this context, TM catalysis has proven a promising strategy for the direct functionalization of otherwise unreactive C–F bonds via the oxidative addition of C–F bonds to TMs followed by selective functionalization, providing access to the desired defluorinated molecules.12 However, the selective activation of C–F bonds usually either suffers from high oxidative-addition barriers, thus the employment of highly elaborate TM catalysts13 and/or forcing conditions14 is often indispensable, or confined to multi/poly-fluorinated arenes15 (Fig. 1A). Therefore, an alternative method that efficiently bypasses the high barriers required for the progress of the oxidative addition and that promotes the smooth coupling of C–F and C–H bonds under mild conditions would make a great contribution to this field.
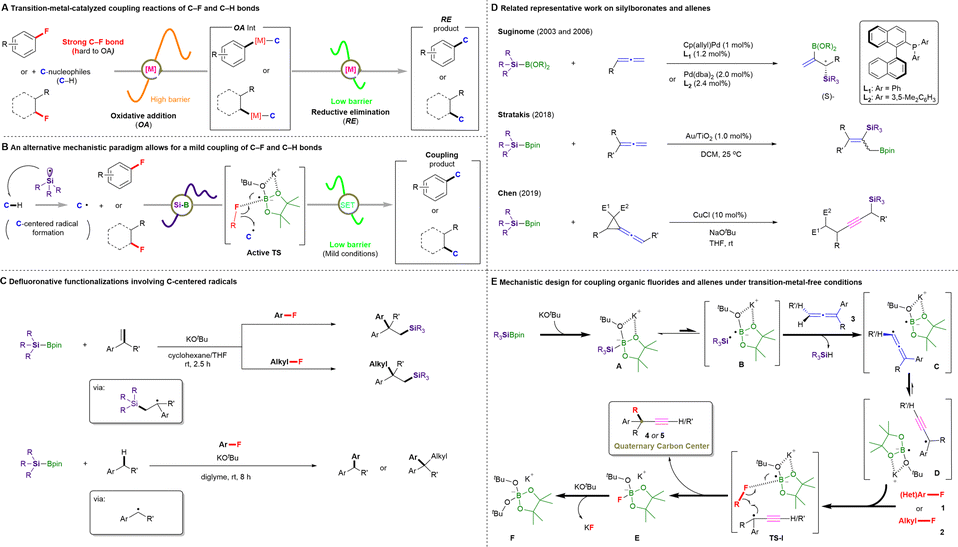 | ||
| Fig. 1 Cross-couplings of C–F and C–H bonds. (A) Transition-metal-catalyzed coupling reactions of C–F and C–H bonds. (B) A low-barrier cross-coupling of C–F and C–H bonds enabled by silylboronate and KOtBu. (C) Our previous defluoronative functionalization work involving C-centered radicals using silylboronates and organic fluorides. (D) Related representative work on silylboronates and allenes. (E) Mechanistic design for the cross-coupling of organic fluorides and allenes under transition-metal-free conditions (this work). | ||
Specifically, we envisioned that in situ-generated silyl radicals would quickly abstract a proton from targeted C–H reactants to form the corresponding C-centered radicals, thus enabling subsequent catalyst-free transformations with organic fluorides via the activation of an inert C–F bond to realize C–C coupling products (Fig. 1B). Such silyl-radical-relay reactions have already been demonstrated by our recently reported TM-free silylboronate (R3SiBpin)-mediated cross-couplings of organic fluorides with alkenes/arylmethanes (Fig. 1C).16
Recently, the unique structural features and versatile reactivity of allenes enabled them to be not only a versatile functional group that is incorporated in natural products, pharmaceuticals, and organic materials,17 but also serve as an ideal platform for the development of new methodologies in synthetic transformations, chiral ligands or even catalysts.18 In this context, reactions between silylboronates and allenes have been pioneered by the Suginome group19 and further explored by Stratakis;20 however, most hitherto reported protocols require TM catalysts and many provide silaboration products with one double bond retained. To date, only the Chen group21 has reported the successful transformation of vinylidene cyclopropanes into propargylic silanes in the presence of copper(I) chloride and NaOtBu (Fig. 1D). Nevertheless, the silyl moiety is involved in the final products.
Based on these previous results, we designed a new silylboronate-mediated cross-coupling of organic fluorides with allenes to access ACQCs that feature an α-ethynyl group via C–F bond activation and radical rearrangement at room temperature (Fig. 1E).16,22 Initially, R3SiBpin and potassium tert-butoxide (KOtBu) in an ether-based solvent smoothly generate intermediate A. Owing to the radical-initiation properties of KOtBu23 and the steric demand of intermediate A, A splits into the bulky frustrated radical pair B,24 which consists of a trialkylsilyl radical (˙SiR3) and a boron-radical species (B˙), via homolytic cleavage of the Si–B bond. The silyl radical in B then directly abstracts a hydrogen atom from the allene (3) to form allenyl radical-containing frustrated radical pair C, which could easily isomerize to the sterically highly demanding propargylic radical-containing frustrated radical pair D.25 Subsequently, D would attract organic fluoride 1 or 2 by preferential interaction between the F atom and the B center to afford TS-I. Finally, the desired ethynyl-containing product with a quaternary carbon center (4 or 5) would be obtained via C–C bond coupling, accompanied by the release of E ([Bpin(OtBu)F]K), which would promptly react with another equivalent of KOtBu to provide a stable [Bpin(OtBu)2] species and KF.
Results and discussion
Silylboronate-mediated cross-coupling reactions of aryl fluorides and aryl allenes
As depicted in our mechanistic hypothesis for the proposed C–C coupling (Fig. 1E), we expected that a silyl radical generated from the silylboronate and KOtBu could abstract a hydrogen atom from the allene terminal. Therefore, we initiated the cross-coupling reactions by using 4-fluorobiphenyl (1a) and penta-1,2-dien-3-ylbenzene (3a) as model substrates. The desired product, 4-(3-phenylpent-1-yn-3-yl)biphenyl (4aa), which possesses a quaternary carbon center with an α-ethynyl moiety, was obtained in 34% yield under the standard reaction conditions [Et3SiBpin (2.0 equiv.), KOtBu (4.0 equiv.), THF, room temperature; entry 1, Table 1]. Furthermore, among the silylboronates tested under the same conditions, the Suginome reagent (PhMe2SiBpin) improved the yield of 4aa to 41% (entry 2). Subsequent optimization focused on screening the number of reactant equivalents, solvent, and reaction time in the presence of PhMe2SiBpin and finally afforded 4aa in 94% yield under the optimized conditions (entry 3; for details, see the ESI; Tables S1–S4†). Control experiments showed that the reaction did not proceed in the absence of silylboronate or KOtBu (entries 4). Moreover, the desired product was not obtained using other bases such as KOMe, NaOtBu, LiOtBu, or KHMDS (entries 5 and 6). This indicated that the countercations in MOtBu exhibited superior ability for K+ over Na+ or Li+ in facilitating this transformation may lie in its established capacity to function as a single-electron reductant.23 Decreasing the amount of PhMe2SiBpin or KOtBu resulted in lower yields of 4aa (86% and 51%, respectively; entries 7 and 8). Replacing PhMe2SiBpin with Et3SiBpin under otherwise identical optimal conditions gave 4aa in only 75% yield (71% isolated; entry 9). Additionally, the use of an inadequate amount of 3a (2.0 equiv.) had a negative effect on the reaction yield (entry 10).|
|
||
|---|---|---|
| Entry | Variation from the “standard conditions” | 4aa (%) |
| a Unless otherwise noted, the standard reactions refer to: 1a (17.2 mg, 0.1 mmol), 3a (43.2 mg, 0.3 mmol), PhMe2SiBpin (78.7 mg, 0.3 mmol), KOtBu (67.2 mg, 0.6 mmol), and diglyme (1.0 mL); room temperature; 12 h. b Determined by 19F NMR and 1H NMR spectroscopy using 3-fluoropyridine as the internal standard. c The isolated yield is shown in parentheses. | ||
| 1 | Et3SiBpin (2.0 equiv.), KOtBu (4.0 equiv.), THF | 34 |
| 2 | PhMe2SiBpin (2.0 equiv.), KOtBu (4.0 equiv.), THF | 41 |
| 3 | None | 94 |
| 4 | Without PhMe2SiBpin or KOtBu | 0 |
| 5 | KOMe instead of KOtBu | 0 |
| 6 | NaOtBu, LiOtBu or KHMDS instead of KOtBu | Trace |
| 7 | PhMe2SiBpin (2.5 equiv.) and KOtBu (5.5 equiv.) instead | 86 |
| 8 | PhMe2SiBpin (2.0 equiv.) and KOtBu (4.0 equiv.) instead | 51 |
| 9c | Et3SiBpin instead of PhMe2SiBpin | 75(71) |
| 10 | 3a (2.0 equiv.) instead | 78 |
| 11 | PhMe2SiBpin (1.5 equiv.) and KOtBu (3.0 equiv.) and 3a (1.5 equiv.) at 50 °C | 79 |
| 12 | Et3SiBpin (1.5 equiv.) and KOtBu (3.0 equiv.) and 3a (1.5 equiv.) at 50 °C | 38 |

It should also be noted here that the Suginome reagent usually generates the undesired side product 1,2-di-tert-butoxy-1,1,2,2-tetramethyldisilane26 (same polarity as 4aa), which is formed by the dimerization of the tert-butoxydimethylsilyl radical (tBuOMe2Si˙), which renders the purification of 4aa difficult via column chromatography on silica gel. Instead, the use of Et3SiBpin afforded pure 4aa, albeit in a lower yield. Furthermore, the slightly elevated reaction temperature (50 °C) could significantly reduce the consumption of both the Suginome reagent, KOtBu, and allene 3a to half the amount, with 79% yield (entry 11). However, only a 38% yield was observed when PhMe2SiBpin was replaced by Et3SiBpin under the same conditions as in entry 11 (entry 12).
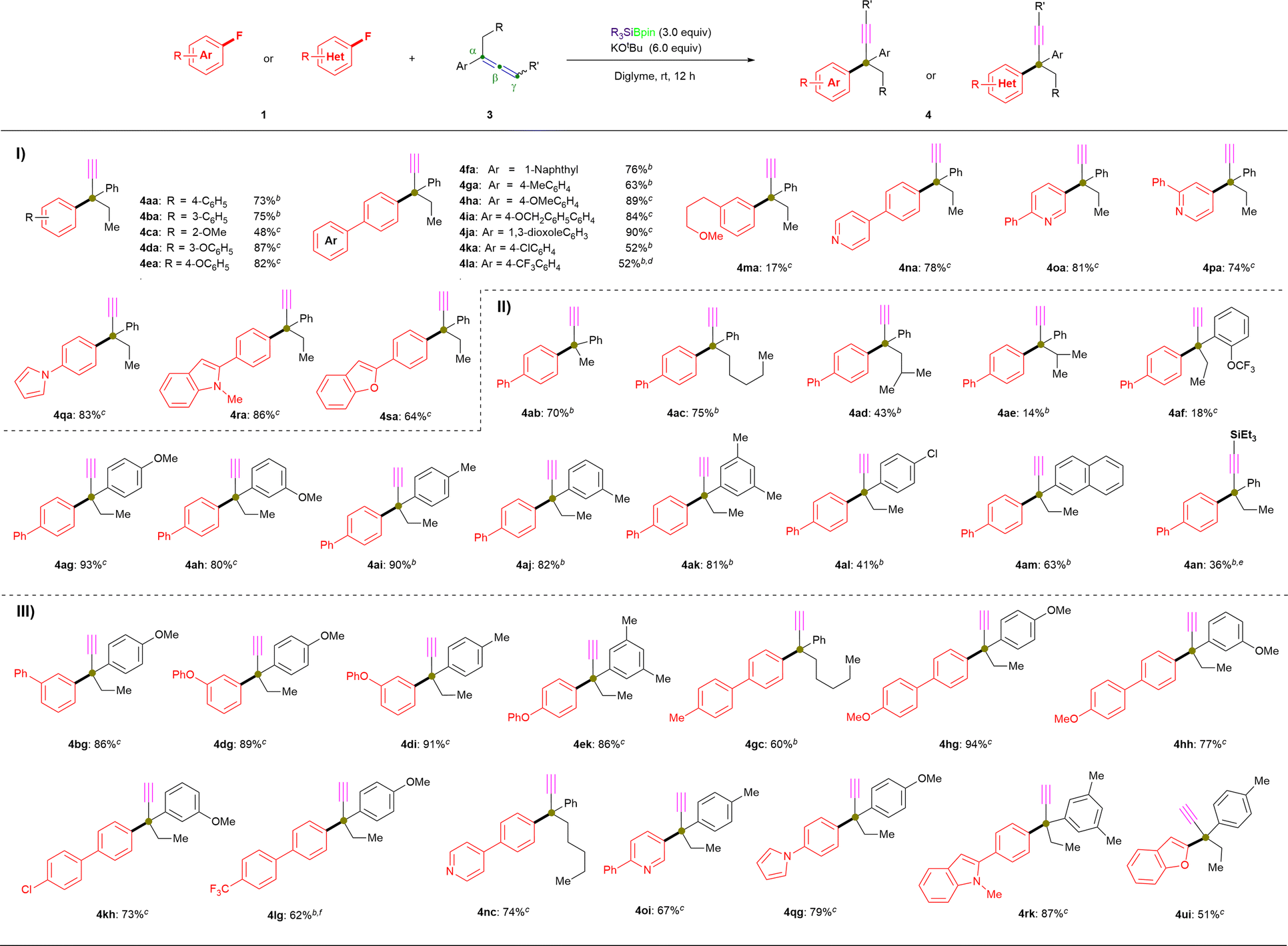 | ||
| Fig. 2 Substrate scope of 1 and 3. a Unless otherwise noted, all reactions were conducted using 1 (0.2 mmol), 3 (3.0 equiv.), R3SiBpin (3.0 equiv.), KOtBu (135 mg, 6.0 equiv.), and diglyme (2.0 mL) at room temperature for 12 h, and isolated yields are shown. b Reactions were performed using Et3SiBpin. c Reactions were performed using PhMe2SiBpin. d Reaction was performed for 4 h. e4aa was obtained in 61% yield. f Reaction was performed for 2 h. | ||
Next, the scope of allenes (3) was examined via coupling with 1a under the standard conditions (Fig. 2-II). Initially, we evaluated α-position-substituted allenes (methyl: 3b; n-pentyl: 3c; iso-butyl: 3d; iso-propyl: 3e) with 1a using Et3SiBpin as the silylboronate reagent, which provided the coupling products (4ab: 70%; 4ac: 75%) in similar yield, while the yields of the sterically hindered products (4ad: 43%; 4ae: 14%) were significantly decreased. Similarly, ortho-OCF3-substituted allene 3f also successfully afforded defluoronative product 4af, albeit only in 18% yield. Moreover, allenes that bear electron-donating (4-OMe: 3g; 3-OMe: 3h; 4-Me: 3i; 3-Me: 3j; 3,5-di-Me: 3k), electron-withdrawing (4-Cl: 3l) or 2-naphthyl (3m) groups underwent defluoroamination to afford the desired products in good-to-high yield (4ag: 93%; 4ah: 80%; 4ai: 90%; 4aj: 82%; 4ak: 81%; 4al: 41%; 4am: 63%). Moreover, triethylsilyl-substituted allene at the γ-position (3n) was evaluated with 1a under the standard reaction conditions (entry 8, Table 1); however, 3n afforded a mixture of 4an (36% yield) and 4aa (61% yield). Other γ-position-substituted allenes (n-heptyl, 3p; phenyl, 3r; dimethyl, 3s) and purely aliphatic allene 3-ethylhepta-1,2-diene (3w) failed to afford any desired products, which should be attributed to the deprotonation of γ-C(sp2)–H in allenes 3 which is a crucial step in this transformation (for more details, see the ESI†). These above results strongly support our hypothesis of a radical-relay reaction, as shown in Fig. 1E, because the aryl moiety in allenes 3 increases the stability of the generated radical, which is indispensable for a successful transformation.
After evaluating the ranges of 1 and 3 in this coupling reaction, we then further demonstrated the scope of this feasible coupling reaction using various combinations of 1 and 3 (Fig. 2-III). Fluoroarenes with phenyl (1b), electron-donating (1d, 1e, 1g, and 1h), and electron-withdrawing (1k and 1l) substituents were coupled with various allenes to generate the desired α-ethynyl-substituted quaternary center products 4 in good yields (4bg: 86%; 4dg: 89%; 4di: 91%; 4ek: 86%; 4gc: 60%; 4hg: 94%; 4hh: 77%; 4kh: 73%; 4lg: 62%), and even the complete transformation of 1l into 4lg was achieved within 2 hours with the potentially cleavable C–F bonds of the trifluoromethyl group remaining intact. The reactions of N-heterocycle-containing aryl fluorides (1n, 1o, 1q, and 1r) with substituted-allenes (3c, 3g, 3i, and 3k) under standard conditions mediated by PhMe2SiBpin resulted in pyridine derivatives and 1H-pyrrole derivatives in good-to-high yields (4nc: 74%; 4oi: 67%; 4qg: 79%; 4rk: 87%). Additionally, 2-fluorobenzofuran (1u) is also an acceptable coupling partner, giving 4ui in a moderate yield of 51%.
Furthermore, we repeated the defluorinative cross-coupling reactions under the heating reaction conditions (entry 11, Table 1) by using representative aryl fluorides 1 with allene 3a (Fig. 3). First, as shown in Fig. 3-I, four types of π-extended aryl fluorides substituted with electron-donating (1a, 1h and 1j) and withdrawing (1k) groups were evaluated, and the desired α-ethynyl-containing ACQCs were observed in 1H NMR yields of ≤82% (4aa: 79%; 4ha: 75%; 4ja: 82%; 4ka: 73%) and with 77% isolated yield for 4ja. Notably, N-containing heteroaromatic fluorides (1n–1t) were successfully coupled with 3a under the same conditions (entry 11, Table 1) to afford the corresponding coupling products with an ACQC in moderate to high 1H NMR yields (4na: 62%; 4oa: 32%; 4qa: 46%; 4ra: 89%), whereas desired product 4ta could be obtained in 68% isolate yield by coupling active 2-fluoro-5-phenylpyridine (1t) and allene 3a. Similarly, 2-fluorobenzofuran (1u) and the simplest polycyclic aromatic fluoride (1v) were efficiently coupled with allene 3a to afford 4ua and 4va in 66% and 59% yield, respectively. Additionally, fluorobenzene substituted with triethylsilyl (1w), trifluoromethyl (1x), bromo (1y), methyl (1z), methoxyl (1aa), and N,N-dimethyl (1ab) were also coupled with allene 3a, albeit in low yields (4wa: 40% isolated yield; 4xa: 41% isolated yield; 4ya: 12%; 4za: 23%; 4aaa: 13%; 4aba: 0%). Thus, the lower yield regarding the substituted fluorobenzenes (especially for 1z, 1aa, and 1ab) could be explained by their C(sp2)–F bond possessing higher BDE than that of substituted biaryl fluorides. In this case, a complex mixture (containing 12 or 15) was obtained rather than the desired products with an ACQC. However, carbonyl containing fluorobenzenes, such as carboxylate (1ac), resulted in no desired product under the aforementioned conditions with or without heating (Fig. 3-I).
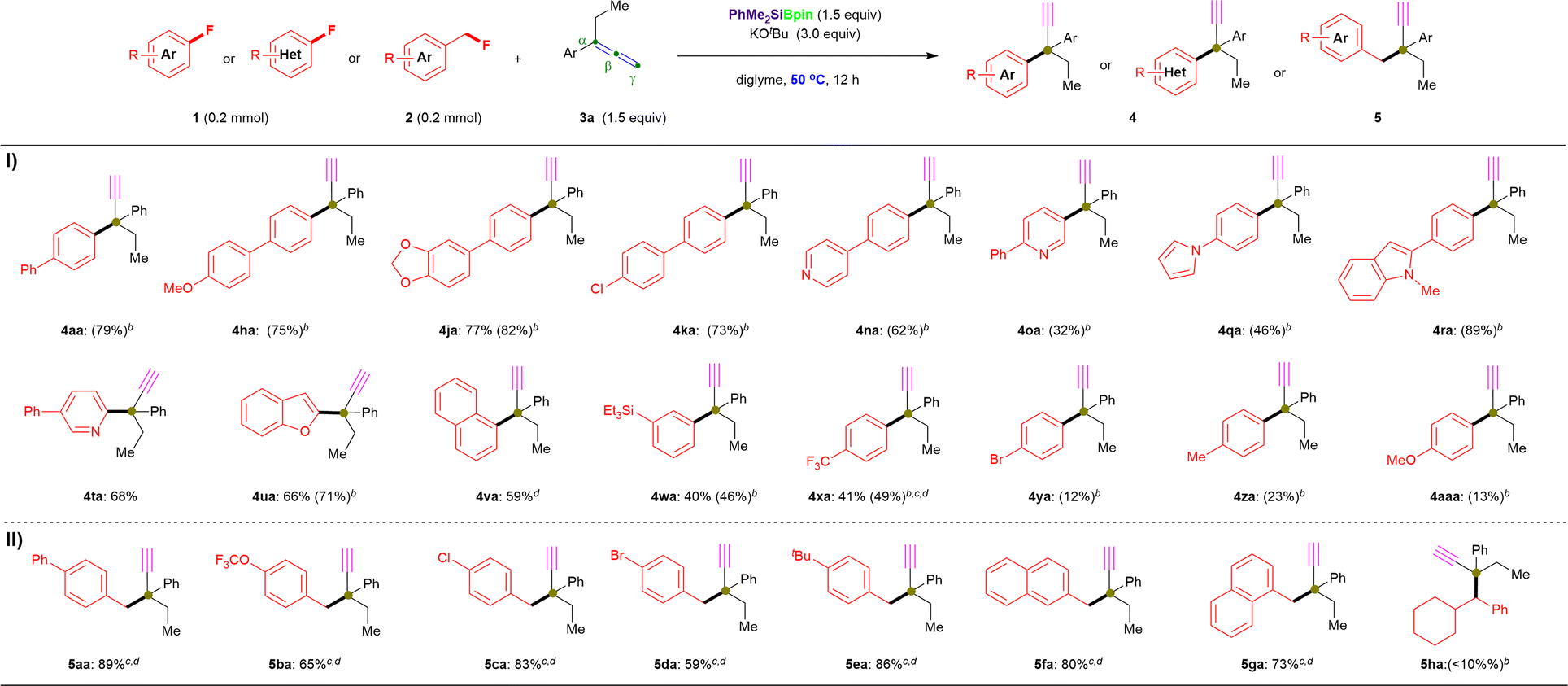 | ||
| Fig. 3 Further scopes and limitations. a Unless otherwise noted, all reactions were conducted using 1 or 2 (0.2 mmol), 3a (43.2 mg, 1.5 equiv.), PhMe2SiBpin (78.7 mg, 1.5 equiv.), KOtBu (67.5 mg, 3.0 equiv.), and diglyme (1.5 mL) at 50 °C for 12 h. b The 1H NMR yields are shown in the parentheses. c Reactions were performed at room temperature. d Reactions were performed using Et3SiBpin. | ||
To our delight, the cross-coupling of benzyl fluorides 2 with allene 3a under similar reaction conditions while at room temperature were also successful in affording desired products with ACQCs (Fig. 3-II). Primary benzyl fluorides bearing electron-donating substituents (phenyl: 2a; tert-butyl: 2e) and electron-withdrawing substituents (trifluoromethoxy: 2b; chloro: 2c; bromo: 2d) reacted efficiently with allene to afford the corresponding products in up to 89% yield (5aa: 89%; 5ba: 65%; 5ca: 83%; 5da: 59%; 5ea: 86%). Additionally, the naphthyl-containing primary fluorides (2f and 2g) successfully yielded the desired products (5fa: 80%; 5ga: 73%) in good yields regardless of the α/β-substituted position on the naphthalene ring. However, secondary alkyl fluorides 2h furnished the desired coupling product 5ha in <10% 1H NMR yield, even when the reaction was performed at 50 °C. Additionally, the primary aliphatic alkyl fluorides (2i and 2j) did not afford the corresponding products even under heating.
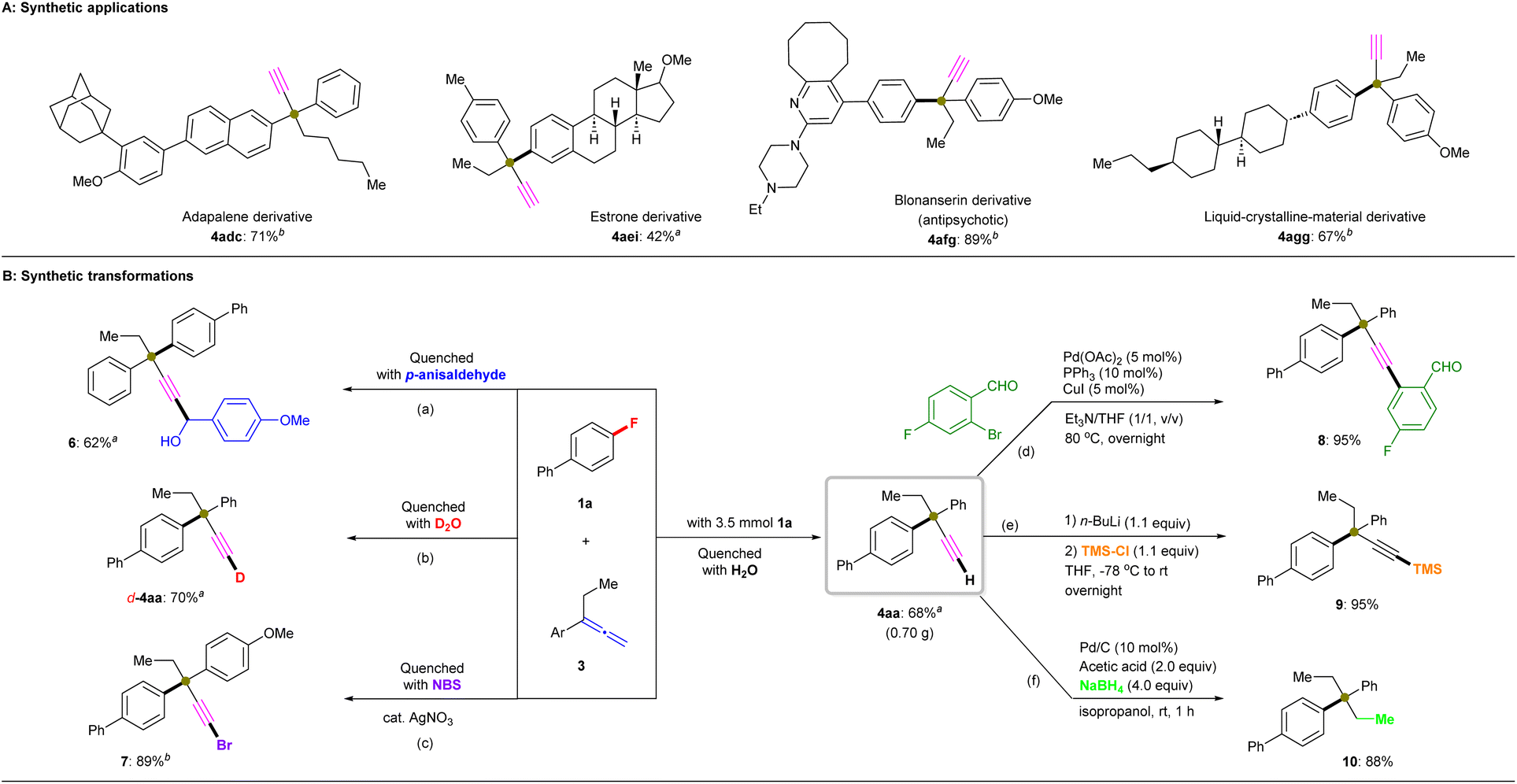 | ||
| Fig. 4 Synthetic utility. (A) Synthetic applications. Unless otherwise noted, all reactions were conducted using 1 (0.2 mmol), 3 (3.0 equiv.), R3SiBpin (3.0 equiv.), KOtBu (135 mg, 6.0 equiv.), and diglyme (2.0 mL) at room temperature for 12 h (isolated yields are shown). (B) Synthetic transformations. a Reaction performed using Et3SiBpin. b Reaction performed using PhMe2SiBpin. (a) 1a (0.2 mmol), 3a (0.6 mmol), p-anisaldehyde (0.3 mmol); (b) 1a (0.2 mmol), 3a (0.6 mmol), D2O (2.0 mL); (c) 1a (0.2 mmol), 3g (0.6 mmol), NBS (0.4 mmol), AgNO3 (0.1 mmol); (d) 4aa (0.2 mmol), Pd(OAc)2 (0.01 mmol), PPh3 (0.02 mmol), CuI (0.01 mmol), aryl bromide (0.24 mmol), Et3N/THF (1.0 mL, 1/1, v/v), 80 °C, overnight; (e) 4aa (0.2 mmol), n-BuLi (0.24 mmol), TMS-Cl (0.22 mmol), THF (1.0 mL), −78 °C to rt, overnight; (f) 4aa (0.2 mmol), Pd/C (0.02 mmol), acetic acid (0.04 mmol), NaBH4 (0.8 mmol), rt, 1 h. | ||
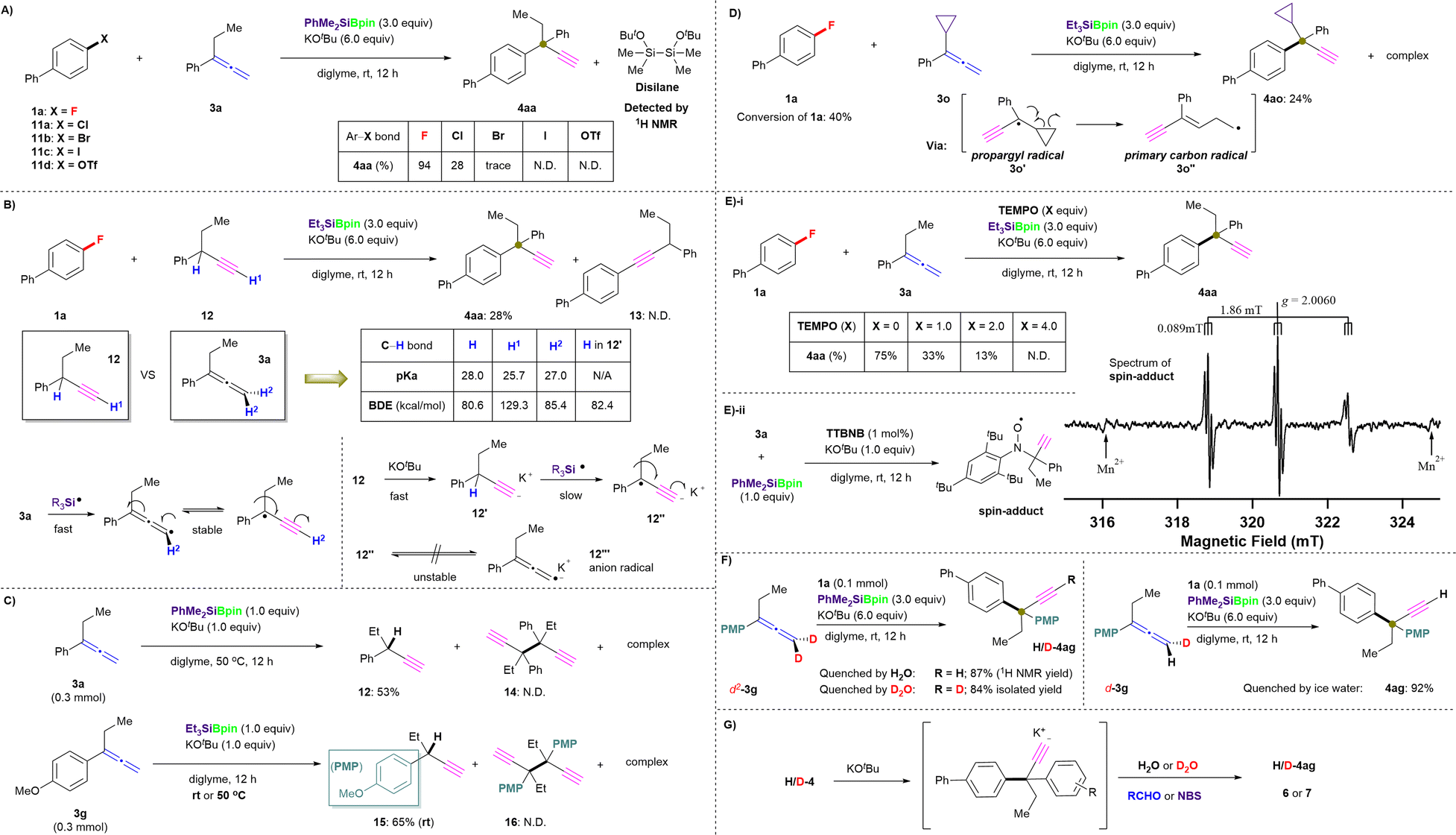 | ||
| Fig. 5 Mechanistic study. (A) Chemoselectivites of organic (pseudo)halides Ar–X. (B) An attempt to use an alternative process. (C) Homocoupling attempt of generated propargyl radicals. (D) Radical ring-opening reaction. (E) Effect of TEMPO on this silylboronate-mediated coupling reaction (i) and ESR experiment in the presence of spin trapping reagent TTBNB (ii). (F) Kinetic-isotope-effect experiments. (G) Possible mechanism for the quenching process. | ||
The kinetic-isotope effect of the C–H/C–D cleavage under ionic conditions is more substantially observed than that by the radical reactions.32 Thus, we evaluated 1a in combination with deuterated allenes (d2-3g and d-3g) in several parallel reactions under the standard conditions in the presence of PhMe2SiBpin (Fig. 5F). However, independent of the existence of deuterium in allene 3g, the formation of D- or H-4ag depends on the quenching method, i.e., on using H2O or D2O, even when quenching with ice water. The formed products 4/5 should exist as potassium acetylides in the reaction mixture, as the excess of tBuOK could easily result in a further deprotonation process. Therefore, the acetylide can be captured by D+, H+, Br+, RCHO, etc. (Fig. 5G). Besides, due to the high acidity of the C(sp)–H/D moiety in the terminal alkynyl position of 4ag, the H/D exchange occurs easily during the workup steps.32,33 Interestingly, the reaction time and yield were almost identical independent of the use of 3g, d2-3g, or d-3g. Therefore, we concluded that the C(sp2)–H/D bond cleavage should occur prior to the C–F bond cleavage, and thus the C(sp2)–H/D bond cleavage should not be the rate-limiting step. All observations in the mechanistic study led us to conclude that this defluorinative cross-coupling reaction proceeds via a single-electron transfer (SET)/radical process, in accordance with our mechanistic hypothesis shown in Fig. 1E.
Conclusions
In summary, we have realized the first cross-coupling reaction of organic fluorides with allenes to construct a library of α-ethynyl-containing all-carbon quaternary centers via C–F bond and C(sp2)–H bond radical cross-couplings using a silyl-radical-relay strategy. The C–F bond cleavage occurs concomitant with the formation of an isomerized propargylic radical, which takes place through cleavage of a C(sp2)–H bond, to cooperatively form a new C–C(sp3) bond. A notable feature of this cross-coupling reaction is that the in situ-generated silyl radical is able to directly abstract a proton from a C(sp2)–H bond of the allene to form an allenyl radical, which then easily isomerizes to form a propargylic radical that exerts a more profound steric influence. Significantly, in this transformation it is not possible to use the corresponding propargyl isomers directly instead of the allenes. This method proceeds under very mild conditions and efficiently obviates the use of TM catalysis or light irradiation to allow a range of para-, meta-, and even ortho-substituted (hetero)aryl fluoride, benzyl fluoride and allene substrates to undergo the normally challenging defluorinative coupling process to afford all-carbon quaternary centers in moderate-to-excellent yield, with good functional-group compatibility and C–F bond selectivity. This radical-coupling system was further extended to the late-stage functionalization of several biologically active molecules.Data availability
The data that support the fndings of this study are available within the article and the ESI.† Details about materials and methods, experimental procedures, characterization data,mechanistic studies, ESR study and NMR spectral are included. All relevant data are also available from the authors.Author contributions
JZ optimized the reaction conditions. JZ and ZZ surveyed the substrate scope, analyzed the data, and then discussed the results with NS. JZ, ZZ, and SM prepared the starting materials. ZZ and KY performed the ESR experiment and analyzed the data. JZ and NS wrote the manuscript. NS supervised the project. All authors contributed to the manuscript and have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was supported by the Japan Science and Technology Agency's (JST) CREST program entitled “Precise Material Science for Degradation and Stability” (JPMJCR21L1) Japan.Notes and references
- (a) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS PubMed; (b) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed; (c) Y. Liu, S. J. Han, W. B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (d) X. P. Zeng, Z. Y. Cao, Y. H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS; (e) T. Ling and F. Rivas, Tetrahedron, 2016, 72, 6729–6777 CrossRef CAS; (f) W. Chen and H. Zhang, Sci. China: Chem., 2016, 59, 1065–1078 CrossRef CAS; (g) C. Li, S. S. Ragab, G. Liu and W. Tang, Nat. Prod. Rep., 2020, 37, 276–292 RSC; (h) Z. Xin, H. Wang, H. He and S. Gao, Tetrahedron Lett., 2021, 71, 153029 CrossRef CAS; (i) W. Xue, X. Jia, X. Wang, X. Tao, Z. Yin and H. Gong, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC.
- (a) M. Bartholow, Pharmatimes, 2012, 78, 48–51 Search PubMed; (b) Z. X. Wang and B. J. Li, J. Am. Chem. Soc., 2019, 141, 9312–9320 CrossRef CAS PubMed.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) B. Wang and Y. Q. Tu, Acc. Chem. Res., 2011, 44, 1207–1222 CrossRef CAS PubMed; (c) L. Süsse and B. M. Stoltz, Chem. Rev., 2021, 121, 4084–4099 CrossRef.
- (a) X. Huang, S. Wu, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2016, 7, 12382 CrossRef PubMed; (b) S. L. Zhang, W. W. Zhang and B. J. Li, J. Am. Chem. Soc., 2021, 143, 9639–9647 CrossRef CAS PubMed; (c) T. B. Poulsen, L. Bernardi, J. Alemán, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2007, 129, 441–449 CrossRef CAS; (d) M. B. Haughey, K. E. Christensen, D. L. Poole and T. J. Donohoe, Chem. Sci., 2021, 12, 13392–13397 RSC; (e) J. A. Dabrowski, F. Gao and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 4778–4781 CrossRef CAS; (f) Q. Q. Zhang, S. Y. Chen, E. Lin, H. Wang and Q. Li, Org. Lett., 2019, 21, 3123–3126 CrossRef CAS.
- (a) C. H. Ding and X. L. Hou, Chem. Rev., 2011, 111, 1914–1937 CrossRef CAS; (b) W. Liu, L. Li and C. J. Li, Nat. Commun., 2015, 6, 6526–6531 CrossRef CAS; (c) K. Tsuchida, Y. Senda, K. Nakajima and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 9728–9732 CrossRef CAS PubMed; (d) B. V. M Teodoro and L. F. Silva, J. Org. Chem., 2017, 82, 11787–11791 CrossRef; (e) Y. Xia, S. Feng, Z. Liu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 7891–7894 CrossRef CAS; (f) M. Q. Tian, Z. Y. Shen, X. Zhao, P. J. Walsh and X. H. Hu, Angew. Chem., Int. Ed., 2021, 60, 9706–9711 CrossRef CAS.
- (a) A. M. Caporusso, S. Filippi, F. Barontini and P. Salvadori, Tetrahedron Lett., 2000, 41, 1227–1230 CrossRef CAS; (b) A. M. Caporusso, A. Zampieri, L. A. Aronica and D. Banti, J. Org. Chem., 2006, 71, 1902–1910 CrossRef CAS PubMed.
- (a) E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, Wiley, New York, 1989 Search PubMed; (b) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS; (c) N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS; (d) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (e) G. Brahmachari, RSC Adv., 2016, 6, 64676–64725 RSC.
- (a) J. Tsuji, Transition Metal Reagents and Catalysts: Innovations in Organic Synthesis, Wiley, Chichester, UK, 2000 Search PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (d) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- (a) J. A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119–6146 CrossRef CAS; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS; (c) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed; (d) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed; (e) Y. Zhu, J. Han, J. Wang, N. Shibata, M. Sodeoka, V. A. Soloshonok, J. A. S. Coelho and F. D. Toste, Chem. Rev., 2018, 118, 3887–3964 CrossRef CAS PubMed; (f) S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452–1455 CrossRef CAS PubMed; (g) B. Xing, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2018, 57, 9896–9900 CrossRef CAS.
- (a) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed; (b) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- (a) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (c) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (d) L. Fu, Q. Chen and Y. Nishihara, Chem. Rec., 2021, 21, 3394–3410 CrossRef CAS PubMed; (e) B. Zhao, T. Rogge, L. Ackermann and Z. Shi, Chem. Soc. Rev., 2021, 50, 8903–8953 RSC; (f) R. Ma, H. Hu, X. Li, G. Mao, Y. Song and S. Xin, Catalysis, 2022, 12, 1665 CAS.
- (a) V. P. W. Böhm, C. W. K. Gstöttmayr, T. Weskamp and W. A. Herrmann, Angew. Chem., Int. Ed., 2001, 40, 3387–3389 CrossRef; (b) P. Tian, C. Feng and T. P. Loh, Nat. Commun., 2015, 6, 7472–7478 CrossRef PubMed; (c) D. Zell, U. Dhawa, V. Müller, M. Bursch, S. Grimme and L. Ackermann, ACS Catal., 2017, 7, 4209–4213 CrossRef CAS.
- (a) M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505–19511 CrossRef CAS; (b) D. Yu, L. Lu and Q. Shen, Org. Lett., 2013, 15, 940–943 CrossRef CAS PubMed; (c) J. Li, C. Wu, B. Zhou and P. J. Walsh, J. Org. Chem., 2018, 83, 2993–2999 CrossRef CAS PubMed; (d) Y. Yin, X. Yue, Q. Zhong, H. Jiang, R. Bai, Y. Lan and H. Zhang, Adv. Synth. Catal., 2018, 360, 1639–1643 CrossRef CAS; (e) J. He, K. Yang, J. Zhao and S. Cao, Org. Lett., 2019, 21, 9714–9718 CrossRef CAS PubMed.
- (a) T. Schaub, P. Fischer, A. Steffen, T. Braun, U. Radius and A. Mix, J. Am. Chem. Soc., 2008, 130, 9304–9317 CrossRef CAS PubMed; (b) S. Senaweera and J. D. Weaver, J. Am. Chem. Soc., 2016, 138, 2520–2523 CrossRef CAS PubMed; (c) Z. J. Luo, H. Y. Zhao and X. Zhang, Org. Lett., 2018, 20, 2543–2546 CrossRef CAS PubMed.
- (a) J. Zhou, B. Jiang, Y. Fujihira, Z. Zhao, T. Imai and N. Shibata, Nat. Commun., 2021, 12, 3749–3757 CrossRef CAS PubMed; (b) J. Zhou, Z. Zhao, B. Jiang, K. Yamamoto, Y. Sumii and N. Shibata, Chem. Sci., 2023, 14, 4248–4256 RSC.
- (a) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed; (b) S. Song, J. Zhou, C. Fu and S. Ma, Nat. Commun., 2019, 10, 507–515 CrossRef PubMed; (c) C. Y. He, Y. X. Tan, X. Wang, R. Ding, Y. F. Wang, F. Wang, D. Gao, P. Tian and G. Q. Lin, Nat. Commun., 2020, 11, 4293–4300 CrossRef CAS PubMed.
- (a) D. R. Taylor, Chem. Rev., 1967, 67, 317–359 CrossRef CAS; (b) Modern Allene Chemistry Vol. 1, ed., N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004, DOI:10.1002/9783527619573; (c) S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef PubMed; (d) X. Pu, X. Qi and J. M. Ready, J. Am. Chem. Soc., 2009, 131, 10364–10365 CrossRef CAS PubMed; (e) F. Cai, X. Pu, X. Qi, V. Lynch, A. Radha and J. M. Ready, J. Am. Chem. Soc., 2011, 133, 18066–18069 CrossRef CAS PubMed; (f) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed; (g) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989–1000 CrossRef CAS PubMed; (h) B. S. Schreib, M. Son, F. A. Aouane, M. H. Baik and E. M. Carreira, J. Am. Chem. Soc., 2021, 143, 21705–21712 CrossRef CAS PubMed; (i) Y. Wang, S. G. Scrivener, X. D. Zuo, R. Wang, P. N. Palermo, E. Murphy, A. C. Durham and Y. M. Wang, J. Am. Chem. Soc., 2021, 143, 14998–15004 CrossRef CAS PubMed; (j) W. Xu, X. Cong, K. An, S. Lou, Z. Li, M. Nishiura, T. Murahashi and Z. Hou, Angew. Chem., Int. Ed., 2022, 61, e202210624 CrossRef CAS PubMed.
- (a) M. Suginome, T. Ohmura, Y. Miyake, S. Mitani, Y. Ito and M. Murakami, J. Am. Chem. Soc., 2003, 125, 11174–11175 CrossRef CAS PubMed; (b) T. Ohmura, H. Taniguchi and M. Suginome, J. Am. Chem. Soc., 2006, 128, 13682–13683 CrossRef CAS PubMed.
- M. Kidonakis and M. Stratakis, ACS Catal., 2018, 8, 1227–1230 CrossRef CAS.
- J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 8800–8804 CrossRef CAS PubMed.
- J. Zhou, Z. Zhao and N. Shibata, Nat. Commun., 2023, 14, 1847–1855 CrossRef CAS PubMed.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, E. Leonard, A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
- (a) D. W. Stephan, J. Am. Chem., Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (b) D. Schilter, Nat. Rev. Chem., 2018, 2, 255 CrossRef CAS; (c) L. L. Liu and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3454–3463 RSC; (d) F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. Leest and J. C. Slootweg, Angew. Chem., Int. Ed., 2020, 59, 22210–22216 CrossRef CAS PubMed; (e) A. Dasgupta, E. Richards and R. L. Melen, Angew. Chem., Int. Ed., 2021, 60, 53–65 CrossRef CAS PubMed.
- (a) Y. Yu and J. Zhang, Eur. J. Org Chem., 2022, 2022, e202201017 CrossRef; (b) L. Liu, R. M. Ward and J. M. Schomaker, Chem. Rev., 2019, 119, 12422–12490 CrossRef CAS PubMed; (c) Q. Liu, J. Zheng, X. Zhang and S. Ma, Nat. Commun., 2022, 13, 3302–3311 CrossRef CAS PubMed.
- H. Saito, K. Nogi and H. Yorimitsu, Angew. Chem., Int. Ed., 2018, 57, 11030–11034 CrossRef CAS PubMed.
- P. Thangsan, T. Rukkijakan, B. Thanaussavadate, K. Yiamsawat, J. Sirijaraensre and P. Gable, Org. Biomol. Chem., 2023, 21, 1501–1513 RSC.
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280–2283 CrossRef CAS PubMed.
- A. T. Tran, V. A. Huynh, E. M. Friz, S. K. Whitney and D. B. Cordes, Tetrahedron Lett., 2009, 50, 1817–1819 CrossRef CAS.
- Q. Yang, Y. Li, J. Yang, Y. Liu, L. Zhang, S. Luo and J. Cheng, Angew. Chem., Int. Ed., 2020, 59, 19282–19291 CrossRef CAS PubMed.
- J. Maury, S. Jammi, F. Vibert, S. R. A. Marque, D. Siri, L. Feray and M. Bertrand, J. Org. Chem., 2012, 77, 9081–9086 CrossRef CAS PubMed.
- (a) K. B. Wiberg, Chem. Rev., 1955, 55, 713–743 CrossRef CAS; (b) S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS.
- (a) S. P. Bew, G. D. Hiatt-Gipson, J. A. Lovell and C. Poullain, Org. Lett., 2012, 14, 456–459 CrossRef CAS PubMed; (b) B. Chatterjee and C. Gunanathan, Chem. Commun., 2016, 52, 4509–4512 RSC; (c) W. Xiong, F. Shi, R. Cheng, B. Zhu, L. Wang, P. Chen, H. Lou, W. Wu, C. Qi, M. Lei and H. Jiang, ACS Catal., 2020, 10, 7968–7978 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06617g |
| This journal is © The Royal Society of Chemistry 2024 |