
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(IV) alkyl complexes: electronic structure contributions to Fe–C bond homolysis and migration reactions that form N–C bonds from N2†
Samuel M.
Bhutto
 ,
Reagan X.
Hooper
,
Sean F.
McWilliams
,
Brandon Q.
Mercado
and
Patrick L.
Holland
*
,
Reagan X.
Hooper
,
Sean F.
McWilliams
,
Brandon Q.
Mercado
and
Patrick L.
Holland
*
Department of Chemistry, Yale University, New Haven, Connecticut 06520, USA. E-mail: patrick.holland@yale.edu
First published on 24th January 2024
Abstract
High-valent iron alkyl complexes are rare, as they are prone to Fe–C bond homolysis. Here, we describe an unusual way to access formally iron(IV) alkyl complexes through double silylation of iron(I) alkyl dinitrogen complexes to form an NNSi2 group. Spectroscopically validated computations show that the disilylehydrazido(2−) ligand stabilizes the formal iron(IV) oxidation state through a strongly covalent Fe–N π-interaction, in which one π-bond fits an “inverted field” description. This means that the two bonding electrons are localized more on the metal than the ligand, and thus an iron(II) resonance structure is a significant contributor, similar to the previously-reported phenyl analogue. However, in contrast to the phenyl complex which has an S = 1 ground state, the ground state of the alkyl complex is S = 2, which places one electron in the π* orbital, leading to longer and weaker Fe–N bonds. The reactivity of these hydrazido(2−) complexes is dependent on the steric and electronic properties of the specific alkyl group. When the alkyl group is the bulky trimethylsilylmethyl, the formally iron(IV) species is stable at room temperature and no migration of the alkyl ligand is observed. However, the analogous complex with the smaller methyl ligand does indeed undergo migration of the carbon-based ligand to the NNSi2 group to form a new N–C bond. This migration is followed by isomerization of the hydrazido ligand, and the product exists as two isomers that have distinct η1 and η2 binding of the hydrazido group. Lastly, when the alkyl group is benzyl, the Fe–C bond homolyzes to give a three-coordinate hydrazido(2−) complex which is likely due to the greater stability of a benzyl radical compared to that for methyl or trimethylsilylmethyl. These studies demonstrate the availability of a hydrocarbyl migration pathway at formally iron(IV) centers to form new N–C bonds directly to N2, though product selectivity is highly dependent on the identity of the migrating group.
Introduction
The isolation of iron(IV) compounds has been dominated by oxo, nitrido, and imido complexes, because the π-bonds in these compounds help to stabilize this high oxidation state.1–13 In contrast, reports of organometallic iron(IV) alkyl complexes are rare.14–19 One reason for this trend is that high-valent iron alkyl species are prone to Fe–C bond homolysis to produce alkyl radicals,15,17 exemplified by the well-documented reactivity of alkyliron porphyrin and corrole complexes (Fig. 1a).20–26 In contrast, Wolczanski and coworkers reported NHC-supported alkyliron(IV) complexes that were more resistant to Fe–C homolysis, and instead underwent alkyl group migration to the imido ligand to produce the corresponding amidoiron(II) complexes (Fig. 1b).16 To our knowledge, this is the only well-characterized example of alkyl migration from a transition metal to a M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR group, though a migration step may be involved in some reactions where a metal-alkyl undergoes amination by addition of an azide.27,28 Meyer has also reported insertion of a coordinated NHC ligand into the Co–N bond of a CoNR complex,29 and there are several reports of carbenes inserting into Fe–N single bonds associated with supporting ligands.29–33
NR group, though a migration step may be involved in some reactions where a metal-alkyl undergoes amination by addition of an azide.27,28 Meyer has also reported insertion of a coordinated NHC ligand into the Co–N bond of a CoNR complex,29 and there are several reports of carbenes inserting into Fe–N single bonds associated with supporting ligands.29–33
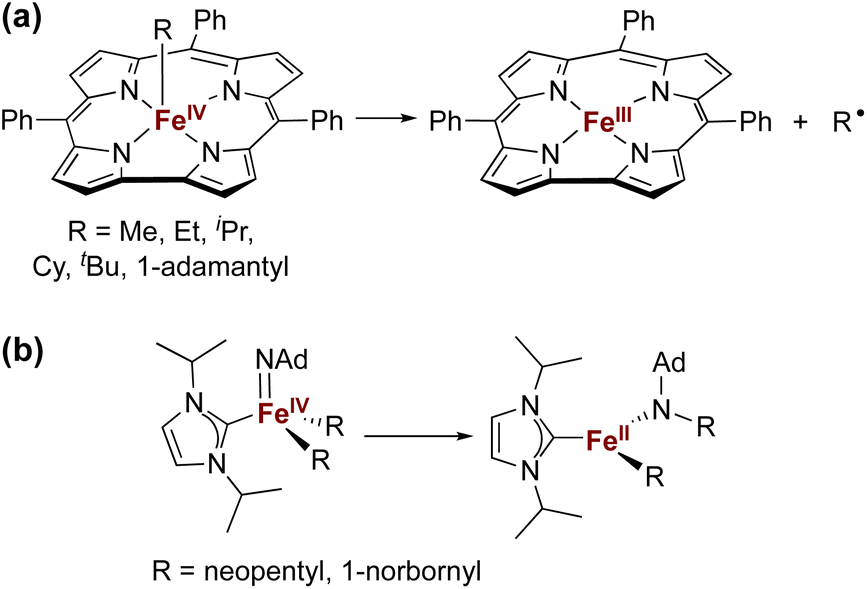 | ||
| Fig. 1 Examples of previously reported iron(IV) alkyl complexes and degradation pathways. | ||
Our research in this area emerged from the study of formally iron(IV) aryl species that undergo migration of the aryl group to the Fe-bound NNR2 group (Scheme 1).34–36 The reaction sequence of interest starts with the reaction of iron(I) aryl N2 complexes (1-N2) with two equivalents of Me3SiX (X = Br, I, OTf; OTf = trifluoromethanesulfonate) and one equivalent of reducing agent. This gives a double silylation at the distal N atom and net three-electron oxidation at the metal, resulting in a formally iron(IV) complex with aryl and hydrazido(2−) ligands (2). It is this complex that can perform the migration of the aryl group from Fe to the proximal N atom to form a new N–C bond in a hydrazido product (3).
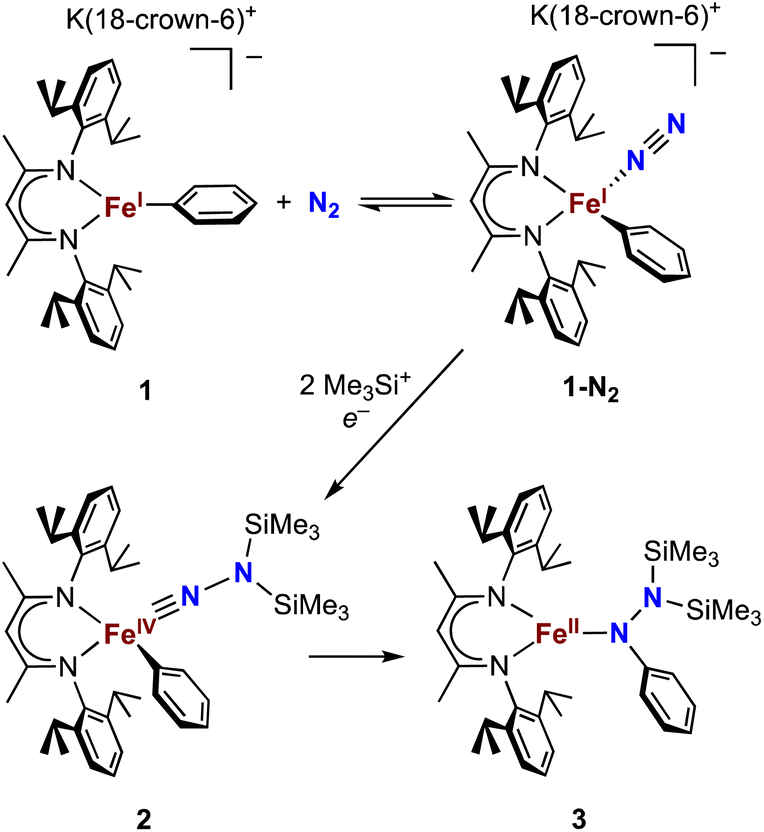 | ||
| Scheme 1 Formation of a formally iron(IV) hydrazido(2−) complex from N2, and subsequent migratory insertion of the aryl ligand. | ||
So far, the migrations of hydrocarbyl groups from Fe to a multiply-bound N ligand that we have reported have been limited to iron–aryl complexes.35,36 Since N2 binding has been reported in a β-diketiminatoiron(I) alkyl complex as well,37 we hypothesized that iron(IV) alkyl hydrazido(2−) complexes might undergo alkyl migration by analogy to the aryl migrations. Previously, Peters described hydride migration to a hydrazido(2−) ligand at a formally iron(IV) center, suggesting that migration chemistry is not limited to aryl groups.38 However, we found that alkynyl groups do not migrate in the diketiminate system.36 Here, we describe a series of iron(I) alkyl complexes that bind N2 at low temperatures and their reactivities upon N2 silylation, including the characterization of the first formally iron(IV) alkyl hydrazido(2−) complex that is stable at room temperature. In some cases, N–C bond formation occurs but in other cases homolysis causes loss of the alkyl group without N–C bond formation, and the differences give insight into the feasibility of iron(IV) in these different environments. Density functional theory (DFT) calculations elucidate the competition between Fe–C homolysis and alkyl migration pathways. A preliminary description of this research has been shared in a preprint.39
Results and discussion
Binding of N2 to iron(I) alkyl complexes
The alkyl chemistry described here starts from the known high-spin three-coordinate iron(II) alkyl complexes 4a–4c.37,40–42 Previous work has shown that an analogous β-diketiminatoiron(II) alkyl complex can be reduced to an iron(I) complex, which upon cooling can bind N2 at the iron center.34,37 Accordingly, we prepared the iron(I) complexes 5a–5c by reduction of the corresponding iron(II) alkyl complexes with KC8 in the presence of 18-crown-6, and isolated them in ca. 80% yield (Scheme 2). Complex 5c was isolated and fully characterized previously.37 Crystals of the new complexes 5a and 5b, grown from THF/hexanes, yielded X-ray crystallographic structures (Fig. 2). The average Fe–N bond lengths of 5a (1.928(5) Å) and 5b (1.918(3) Å) are equivalent to the distance in 5c (1.922(4) Å). The Fe–C bond length of 5b (2.063(4) Å) is longer than that in the starting iron(II) complex 4b (2.041(2) Å),42 consistent with the lower oxidation state, while the Fe–C bond lengths of 5a and 4a are indistinguishable (5a, avg. 2.015(2) Å; 4a, 2.022(2) Å).42 The Mössbauer parameters of 5a (δ = 0.44 mm s−1, |ΔEQ| = 1.90 mm s−1) and 5b (δ = 0.27 mm s−1, |ΔEQ| = 1.75 mm s−1) are similar to those reported for high-spin 5c (δ = 0.38 mm s−1, |ΔEQ| = 2.06 mm s−1).43 Consistent with this assignment, solution magnetic susceptibilities indicate high-spin (S = 3/2) ground states for 5a (μeff = 4.3(1) μB) and 5b (μeff = 4.3(1) μB).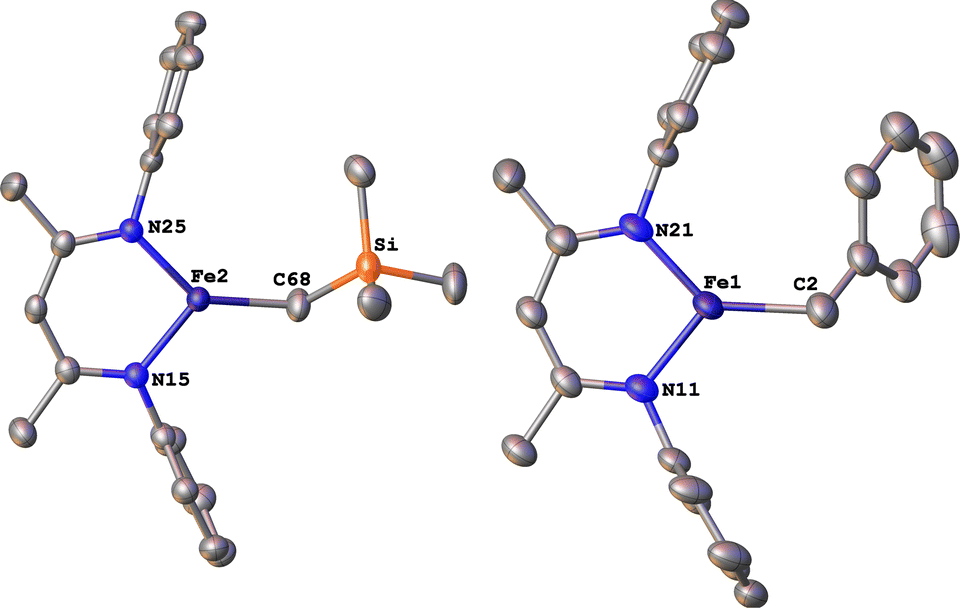 | ||
| Fig. 2 ORTEP diagrams of the iron(I) complexes 5a (left) and 5b (right) with thermal ellipsoids shown at 50% probability. H atoms, iPr groups, and K(18-crown-6)(THF)2+ cations are omitted for clarity, as well as a molecule of THF in the asymmetric unit of 5a. | ||
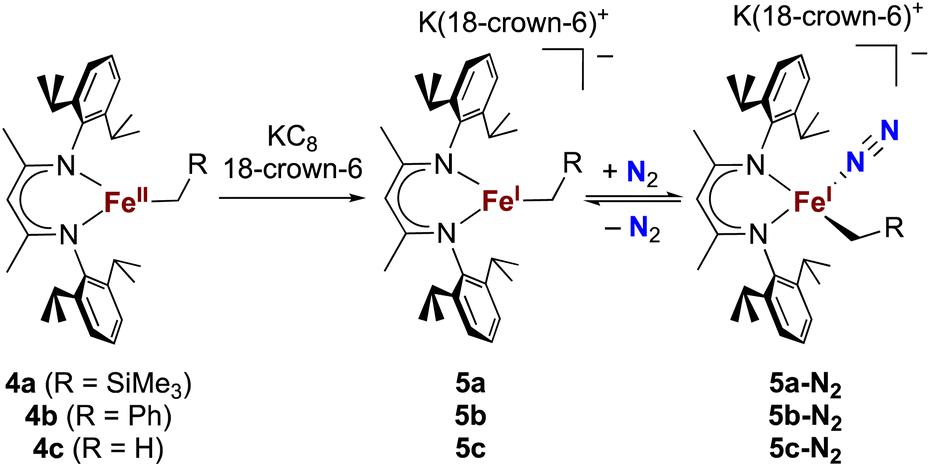 | ||
| Scheme 2 Synthesis of iron(I) alkyl complexes 5a–5c and reversible N2 binding. | ||
We then tested N2 binding at the iron(I) alkyl complexes at low temperature. Freezing solutions of 5a–5c in THF under an atmosphere of N2 led to a color change of the solutions from green to magenta, and thawing the solutions gave back the original green color. These color changes were not observed when freezing solutions under an atmosphere of Ar. van't Hoff analysis of the variable-temperature 1H NMR (5a and 5b) and UV-vis (5c) spectra gave the thermodynamic parameters shown in Table 1. The negative enthalpy and entropy values for each complex are consistent with N2 binding at lower temperatures, likely in an end-on fashion as proposed in the analogous iron β-diketiminate systems mentioned above.34–37
| Alkyl group | ΔH (kcal mol−1) | ΔS (e.u.) | |
|---|---|---|---|
| 5a | CH2SiMe3 | −8.8(6) | −52(3) |
| 5b | CH2Ph | −9.5(6) | −57(2) |
| 5c | CH3 | −4.3(7) | −33(3) |
Silylation of N2 in the trimethylsilylmethyliron complex leads to an isolable iron(IV) alkyl complex
Next we explored the silylation reactions of the N2-bound iron(I) complexes to form formally iron(IV) complexes (Scheme 3). Addition of the bis(silyl) reagent 6 to a mixture of 5a-N2 and K(18-crown-6)(C10H8) (used as an external reductant) in Et2O at −116 °C led to an immediate color change from magenta to brown. The 1H NMR spectrum of the crude reaction mixture showed the formation of a new Cs symmetric complex in 73% spectroscopic yield. Cooling a concentrated hexamethyldisiloxane (HMDSO) solution at −35 °C overnight led to the isolation of brown crystals in 32% yield, which were identified by X-ray diffraction as the formally iron(IV) complex 7a (Fig. 3, top). The N–N bond length is 1.326(3) Å, which lies between the values for a N–N single bond (1.45 Å) and double bond (1.25 Å) in the corresponding organic N2Hx compounds, and is comparable to those in other four-coordinate iron hydrazido(2−) complexes as well as the phenyl complex 2 (1.340(4) Å).35,44,45 However, the bond lengths to iron in 7a are significantly different than those in 2. The Fe–Nhyd bond length (1.749(2) Å) and average Fe–Nnacnac bond length (2.051(1) Å) in 7a are ∼0.08 Å longer than those in 2 (1.673(3) and 1.970(2) Å, respectively). Additionally, the iron center in 7a adopts a distorted tetrahedral geometry (τ4 = 0.88) rather than the distorted trigonal pyramidal geometry in 2 (τ4 = 0.75).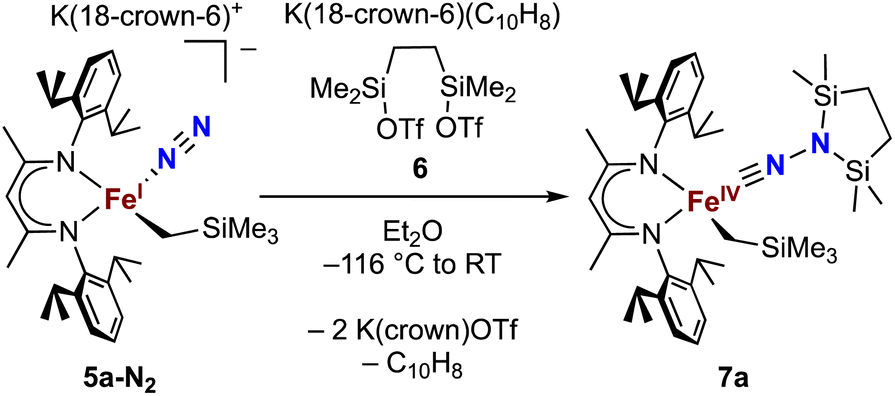 | ||
| Scheme 3 Synthesis of the formally iron(IV) complex 7a. | ||
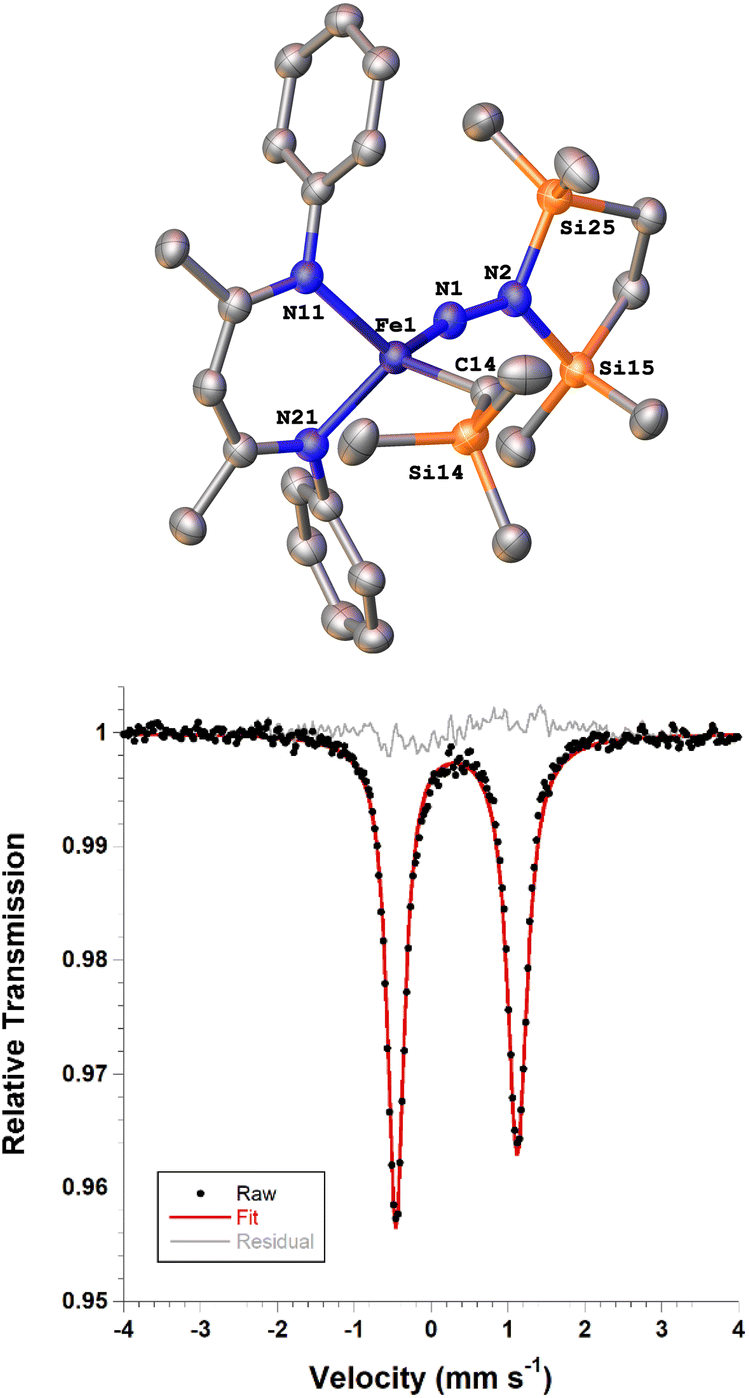 | ||
| Fig. 3 (Top) ORTEP diagram of 7a with thermal ellipsoids shown at 50% probability, with H atoms and iPr groups omitted for clarity. (Bottom) Solid state zero-field Mössbauer spectrum of 7a at 80 K. The black circles are the data, the red line is the fit, and the grey line is the residual (data – fit). | ||
The zero-field Mössbauer spectrum of 7a consists of a doublet with an isomer shift of δ = 0.33 mm s−1, which is much higher than the value of δ = 0.17 mm s−1 in 2 (Fig. 3, bottom). The higher isomer shift in 7a may indicate that the complex has a different ground spin state, and the longer bonds in 7a noted above suggest the higher spin state of S = 2. Furthermore, the bond distances to the iron center in 7a closely resemble those observed in a DFT model of 2 in an S = 2 state (see ESI†).35 Finally, a solution magnetic susceptibility measurement gave μeff = 5.0(2) μB, which confirms the high-spin ground state.
Electronic structure of the alkyl hydrazido(2−) complex
DFT calculations were performed for greater insight into the electronic structure of 7a. The geometry of 7a in an S = 2 ground state was optimized at the B3LYP/def2-TZVP level, giving bond metrics that agree with those in the experimental X-ray structure of 7a (see ESI for details†). This computational model was further validated by computing46 the expected Mössbauer parameters of δ = 0.37 mm s−1 and |ΔEQ| = 1.63 mm s−1, which are close to the experimental values of 0.33 and 1.58 mm s−1, respectively. The quasi-restricted orbitals (QROs) from the model of 7a are shown in Fig. 4, with the z axis along the Fe–hydrazido(2−) bond. The formal bond order of the Fe–Nhyd bond is 2.5, as the Fe dyz π* orbital is singly occupied. This is consistent with the longer Fe–Nhyd bond distance in 7a compared with that of 2, which has an S = 1 ground state and a formal Fe–Nhyd bond order of 3.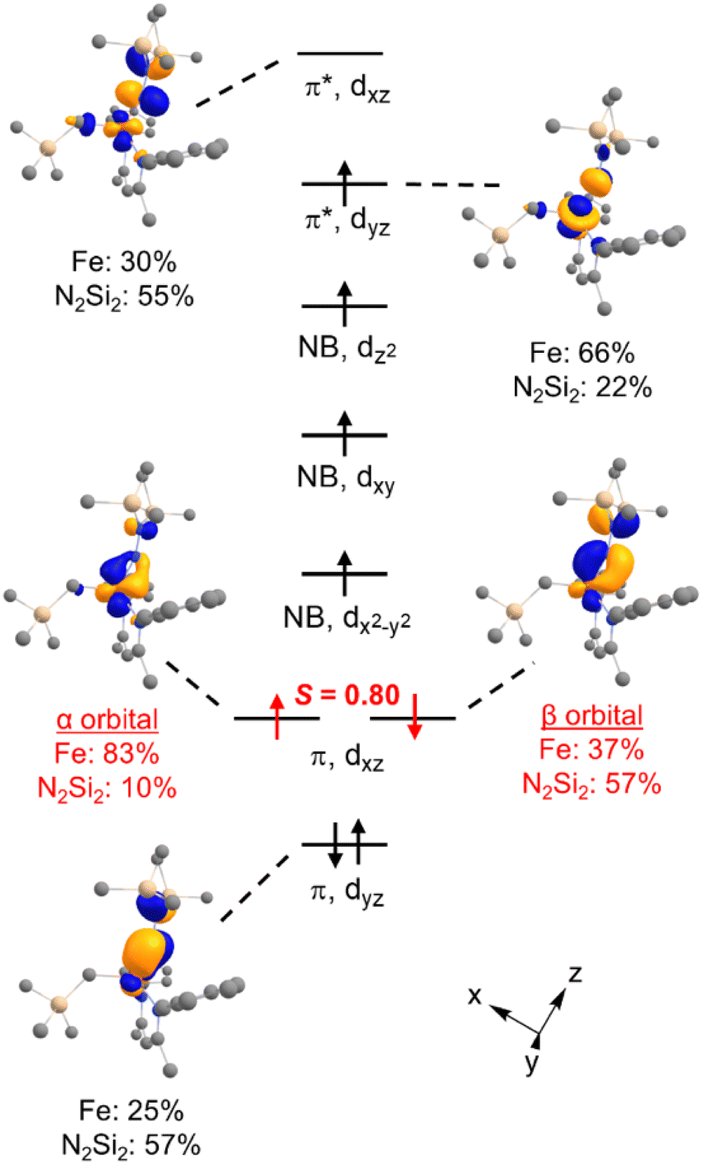 | ||
| Fig. 4 Qualitative molecular orbital diagram showing the QROs of 7a (S = 2) with selected QRO plots shown at an isovalue of 0.05 au. In red are the one-electron orbitals for the “corresponding pair” of electrons in the spin-polarized Fe–N π-bond. | ||
The Fe–Nhyd π bonding interaction involving the Fe dyz orbital has more hydrazido than iron character, typical of a normal π bond. The π bonding interaction involving the Fe dxz orbital, however, is more complex. The doubly-occupied Fe–Nhyd π bonding orbital has a relatively low orbital overlap of 〈α|β〉 = 0.80, and thus it is spin-polarized with the α electron lying 83% on the Fe and the β electron lying only 37% on Fe. Additionally, the unoccupied Fe–Nhyd π* orbital has significantly less Fe character than N2Si2 ligand character, indicative of an “inverted ligand field.”47,48 Taken together, the large spin polarization of one of the Fe–Nhyd π bonding orbitals and “inverted” ligand character suggests that the formally hydrazido(2−) ligand may be alternatively described as a neutral NNR2 (isodiazene) implying an iron(II) oxidation state. This situation is analogous to that described for the metastable formally iron(IV) phenyl complex 2,35 with one spin-polarized orbital having Fe–N π character.
From the available data, it is difficult to discern why 7a has a different ground spin state than 2, as the energetic differences between the two are small. It is possible that the somewhat lower relative energy of the S = 2 state in 7a arises because the geometry is distorted from trigonal pyramidal toward tetrahedral, leading to a weaker ligand field.
To further probe the redox noninnocence of the NNR2 ligand in 7a, multireference CASSCF(8,7) calculations were performed on the DFT-optimized S = 2 structure; details are shown in the SI. The dominant configuration (72%) has the expected Hund filling, corresponding to a hydrazido(2−) ligand. The next two most important configurations have single (16%) and double (10%) occupation of the Fe–N π antibonding orbital involving the Fe dxz orbital, which correspond to iron(III) hydrazido and iron(II) isodiazene descriptions of 7a, respectively. Thus, both single reference and multireference calculations point to ligand noninnocence of the NNR2 ligand, to an extent that is comparable to the phenyl and alkynyl analogues.35,36
The trimethylsilylmethyl group does not migrate
We then turned to the solution behavior of 7a. Complex 7a was much more stable in solution than 2, showing only about 25% decomposition after 4 days in C6D6 solution at room temperature (whereas 2 is completely consumed within a few hours at room temperature). Heating a C6D6 solution of 7a at 80 °C for 2 hours led to its complete consumption. However, the product was not alkyl migration (in analogy to 2) but rather formation of the iron(II) alkyl complex 4a (24%), the hydrazido(2−) product 8 (29%), and the amido complex LFeN(Me2Si(CH2)2SiMe2) (36%) (see ESI for details†). By analogy to a related N–Si bond cleavage in an iron silyldiazenido complex by Ashley,49 we speculate that the mechanism for the formation of 4a might involve homolytic cleavage of N–Si bonds, though there is also cleavage of Fe–C, Fe–N, and N–N bonds to yield the other observed products.Why does the trimethylsilylmethyl group in 7a not migrate as previously observed for the phenyl group in 2? Though it is tempting to attribute this to the difference in the spin state, we were not able to optimize transition states for migration of the CH2SiMe3 group (triplet or quintet states) to assess the impact of spin state and TS geometry with DFT. However, since the electronically similar methyl group does migrate (see below), there is some evidence that steric effects play a role.
Attempts to generate the iron(IV)-benzyl lead to homolysis
We also explored the N2 silylation reactivity of the other iron(I) alkyl complexes. In contrast with the silylation of 5a-N2, the use of K(18-crown-6)(C10H8) as an external reductant in the reaction of the iron(I) benzyl complex 5b-N2 and the silyl triflate 6 led to an intractable mixture of unidentified species. However, it has previously been shown in the synthesis of a related aryl diazenido species that the starting iron(I) aryl complex can provide the necessary electron equivalent in the reaction (unfortunately limiting the yield of silylated product to a maximum of 50% based on iron, since part of it is a sacrificial reductant).34 Thus, addition of 6 to a solution of 5b-N2 in Et2O at −116 °C without an external reductant led to the formation of the oxidized iron(II) product 4b in 64% spectroscopic yield (yield based on the stoichiometry in Scheme 4, quantified by 1H NMR spectroscopy), as well as a new C2v symmetric species 8 in 22% spectroscopic yield (Scheme 4, middle). This same species 8 was also identified in the decomposition mixture of 7a in 27% spectroscopic yield, indicating that it had lost the alkyl group. Indeed, X-ray diffraction revealed 8 to be an iron(III) hydrazido(2−) complex (Fig. 5, top).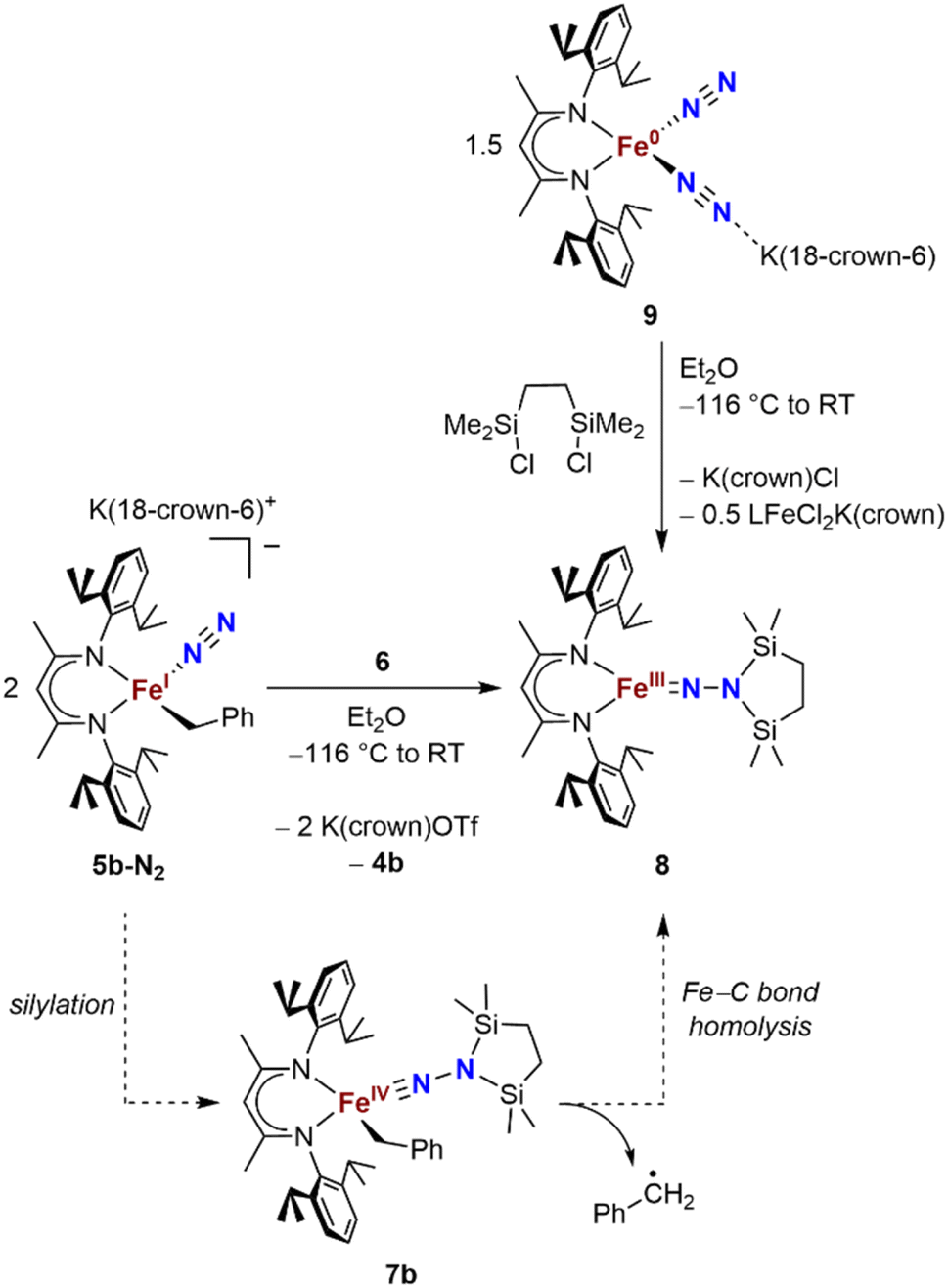 | ||
| Scheme 4 Syntheses of the iron(III) hydrazido(2−) complex 8, and proposed mechanism of formation from 5b-N2. | ||
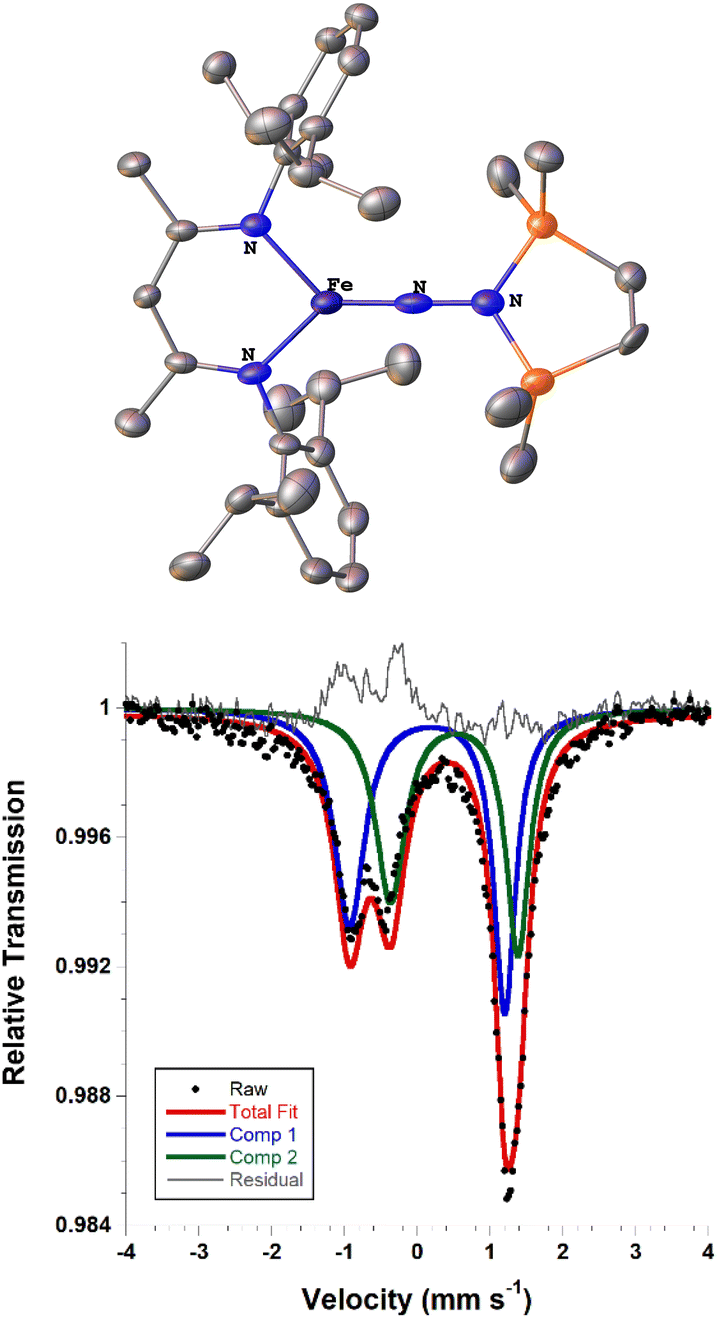 | ||
| Fig. 5 (Top) ORTEP diagram of 8 with thermal ellipsoids shown at 50% probability and H atoms omitted for clarity; due to apparent disorder in the core, the structure is for connectivity only. (Bottom) Solid state zero-field Mössbauer spectrum of 8 at 80 K. The black circles are the data, the blue and green lines are the components of the fit, the red line is the sum of the components, and the grey line is the residual (data – fit). | ||
Compound 8 is closely related to an iron(III) hydrazido(2−) complex we previously reported, the trimethylsilyl analogue LFeNN(SiMe3)2 (L = 2,4-bis(2,6-diisopropylphenylimido)pentyl).50 Accordingly, complex 8 could be prepared independently from the reaction of the iron(0)-bis(dinitrogen) complex 9 and 1,2-bis(chlorodimethylsilyl)ethane in 22% isolated yield (Scheme 4, upper right). The products from the two synthetic methods had identical 1H NMR spectra, further supporting the proposed composition of 8.
We propose that the route from 5b-N2 to 8 begins with silylation to give the formally iron(IV) complex 7b followed by Fe–C bond homolysis,17,26,51,52 producing a benzyl radical (Scheme 4, bottom). In order to test this idea, we performed trapping experiments using the radical scavenger TEMPO (TEMPO = 2,2,6,6-tetramethylpiperidine 1-oxyl). When TEMPO was added to the reaction mixture immediately after silane addition at −116 °C, 1H NMR spectroscopy showed >80% formation of TEMPO-Bn.53 This implies the formation of a transient intermediate that can release benzyl radicals.
Unfortunately, the X-ray crystal structure solution of 8 had a second component in the core, and the disorder prevents us from deriving reliable metrical parameters, and we were unable to obtain satisfactory results from CHN analysis. Despite these problems that prevent deep study of 8, we note in passing several intriguing aspects of its spectroscopic properties. Similar to the previously characterized trimethylsilyl analogue LFeNN(SiMe3)2,50 the solid state Mössbauer spectrum of crystalline 8 collected at 80 K shows two doublets in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with isomer shifts of 0.13 and 0.51 mm s−1 (Fig. 5, bottom). In the previous work, the 1:1 ratio was explained by the presence of two molecules with significantly different bond distances, which coexist in alternating positions in the crystals. Comparison to calculations and fitting of magnetic susceptibility data supported our conclusion that the two molecules had different spin states (S = 1/2 and S = 3/2).50 In order to determine whether this might be the case in 8 as well, DFT calculations were carried out to determine the relative energies of the S = 1/2 and S = 3/2 states using geometry-optimized structures of 8 (Table 2). Calculations using both the BP86 and B3LYP functionals show a small energy difference between the doublet and quartet states: using BP86 the low-spin conformer is lower in energy by 9 kcal mol−1, while B3LYP predicts the high-spin state to be lower in energy by 2 kcal mol−1. This difference in lowest energy calculated spin conformer is not surprising, as hybrid functionals such as B3LYP have been shown to favor higher spin states.54 These small calculated differences in energy suggest that the actual spin isomers could indeed be isoenergetic. Importantly, the calculated Mössbauer parameters of the geometry-optimized doublet and quartet DFT models are in excellent agreement with the two signals in the spectrum of 8 (Table 2).46
:1 ratio with isomer shifts of 0.13 and 0.51 mm s−1 (Fig. 5, bottom). In the previous work, the 1:1 ratio was explained by the presence of two molecules with significantly different bond distances, which coexist in alternating positions in the crystals. Comparison to calculations and fitting of magnetic susceptibility data supported our conclusion that the two molecules had different spin states (S = 1/2 and S = 3/2).50 In order to determine whether this might be the case in 8 as well, DFT calculations were carried out to determine the relative energies of the S = 1/2 and S = 3/2 states using geometry-optimized structures of 8 (Table 2). Calculations using both the BP86 and B3LYP functionals show a small energy difference between the doublet and quartet states: using BP86 the low-spin conformer is lower in energy by 9 kcal mol−1, while B3LYP predicts the high-spin state to be lower in energy by 2 kcal mol−1. This difference in lowest energy calculated spin conformer is not surprising, as hybrid functionals such as B3LYP have been shown to favor higher spin states.54 These small calculated differences in energy suggest that the actual spin isomers could indeed be isoenergetic. Importantly, the calculated Mössbauer parameters of the geometry-optimized doublet and quartet DFT models are in excellent agreement with the two signals in the spectrum of 8 (Table 2).46
| Compound | Functional | Spin state | Rel. energy (kcal mol−1) | δ (mm s−1) | |ΔEQ| (mm s−1) |
|---|---|---|---|---|---|
| 8 | Exp. | 1/2 | 0.13 | 2.13 | |
| 3/2 | 0.51 | 1.75 | |||
| BP86 | 1/2 | 0 | 0.12 | 2.04 | |
| 3/2 | 9 | 0.48 | 1.83 | ||
| B3LYP | 1/2 | 2 | 0.24 | 1.76 | |
| 3/2 | 0 | 0.67 | 1.36 | ||
| LFeNN(SiMe3)2 (ref. 55) | Exp. | 1/2 | 0.22 | 1.99 | |
| 3/2 | 0.46 | 1.16 | |||
| BP86 | 1/2 | 0 | 0.14 | 1.93 | |
| 3/2 | 9 | 0.51 | 1.72 | ||
| 5/2 | 31 | 0.61 | 3.69 | ||
| B3LYP | 1/2 | 1 | |||
| 3/2 | 0 | ||||
| 5/2 | 15 |
Additionally, the electron paramagnetic resonance (EPR) spectrum of a frozen toluene solution of 8 collected at 5 K shows two overlapping signals consistent with coexisting S = 1/2 and S = 3/2 species (Fig. 6). Simulating the spectrum gives an S = 1/2 species with g = [1.80, 1.84, 3.54]. The large anisotropy of g values was similarly observed in the previously reported trimethylsilyl analogue which had g = [1.79, 1.84, 3.61].50 The small signals with geff values of 6.21 and 2.32 are reminiscent of a three-coordinate, intermediate-spin (S = 3/2) iron(III) imido complex with the same diketiminate supporting ligand,55,56 supporting this assignment. Overall, it appears that spin isomerism is present in this three-coordinate iron(III) hydrazido(2−) complex. Future studies will aim to unravel the reason why the 1:1 ratio is observed even without apparent crystal constraints.
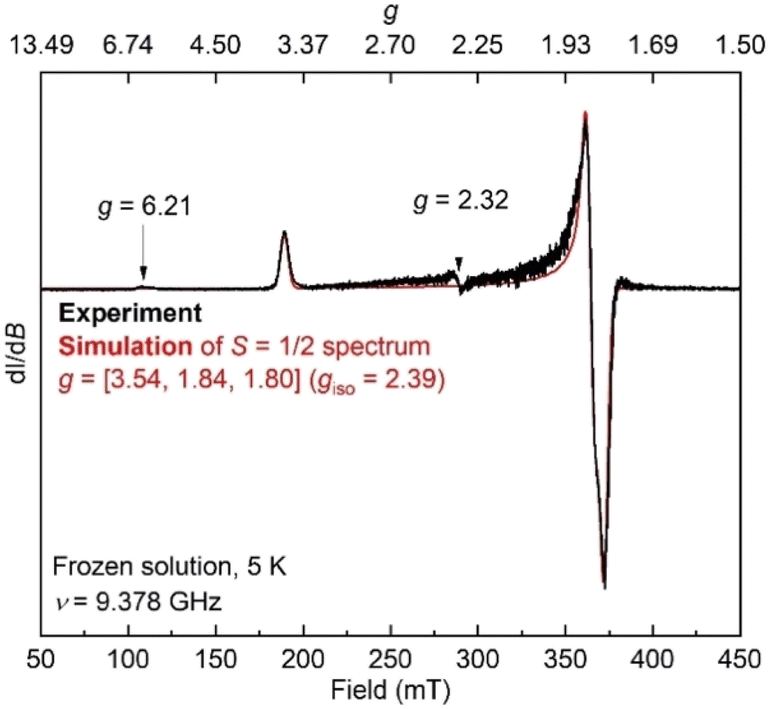 | ||
| Fig. 6 EPR spectrum of 8 in toluene at 5 K (black) and simulation of the S = 1/2 component (red). | ||
Methyl migration followed by silyl migration leads to a different kind of complex, with spin and coordination isomers
Finally, we explored the silylation of the iron(I) methyl complex 5c (Scheme 5). Addition of the disilyl electrophile 6 to a solution of 5c-N2 in Et2O at −116 °C led to an immediate color change from magenta to brown, and then the mixture turned yellow upon warming to ambient temperature. The 1H NMR spectrum of the crude reaction mixture showed the presence of two species: the oxidized iron(II) complex 4c (57%) and a new Cs symmetric species 10 (37%). In this case, no formation of 8 (which would result from loss of a methyl radical) was observed in the crude mixture. X-ray crystallography identified 10 as a 1,2-bis(silyl)methylhydrazido complex (Fig. 7), with disorder that indicates co-crystallization of η1 and η2 isomers (described in more detail below). A reasonable mechanism for the formation of 10 is through the initial formation of the formally iron(IV) complex 7c, followed by methyl migration to form the expected methylhydrazido complex 11 (Scheme 5, bottom). This complex could then isomerize through a silyl shift to give the observed product 10. Shifting of the bis(silyl)methylhydrazido ligand in this way has precedent in the isomerization of free bis(trimethylsilyl)methylhydrazine, which can shift its silyl groups in the presence of catalytic amounts of base.57 Most relevantly, Peters has reported an example of a disilylhydrazido(2−) complex in which the silyl group is proposed to go through an intermediate that resembles 10.58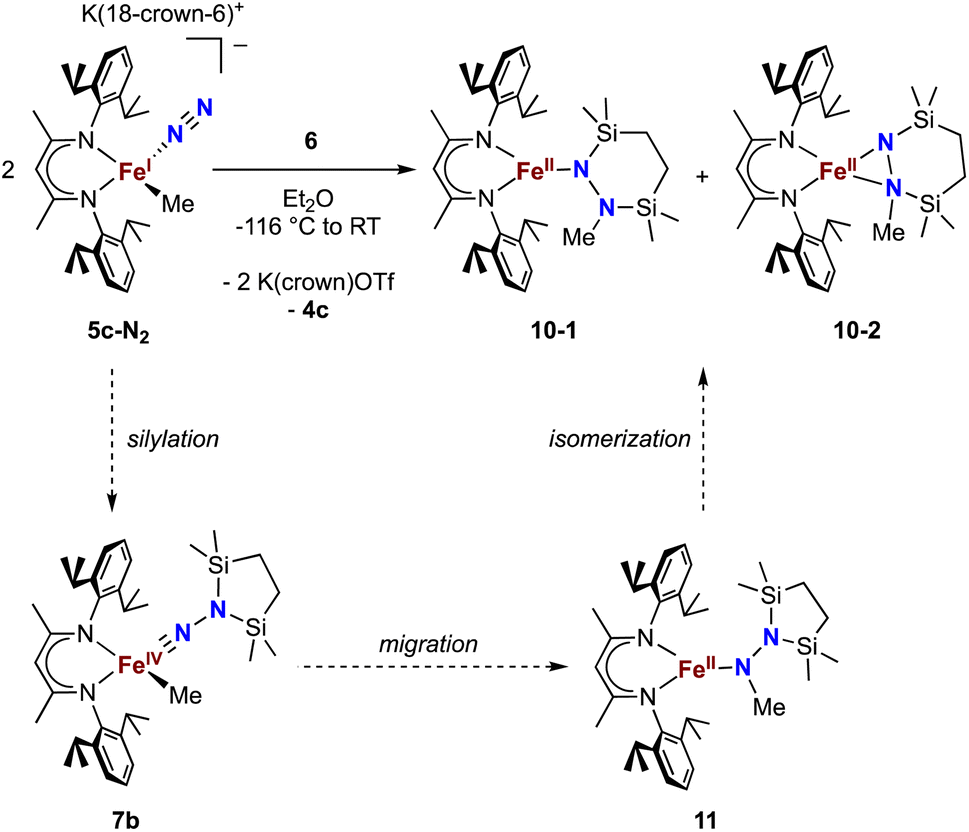 | ||
| Scheme 5 Synthesis of the methylhydrazido complex 10, which crystallizes as two isomers 10-1 and 10-2. Along the bottom is shown the proposed mechanism of the reaction. | ||
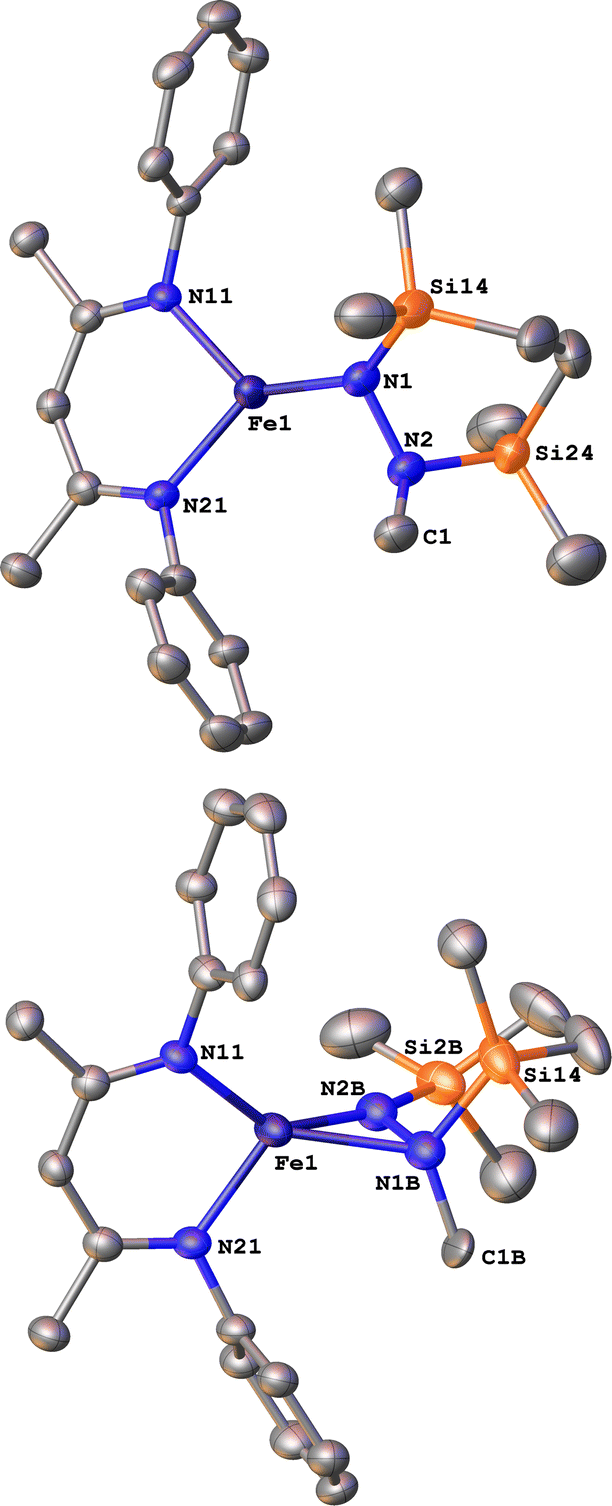 | ||
| Fig. 7 ORTEP diagrams of 10-1 and 10-2 with thermal ellipsoids shown at 50% probability and H atoms and iPr groups omitted for clarity. The η1 isomer (major component 10-1, 75%) is shown on the top, and the η2 isomer (minor component 10-2, 25%) is shown on the bottom. | ||
To test whether the ability of the silyl to shift arises somehow from the change from trimethylsilyl to the bis(silyl) reagent 6, we prepared the bis(silyl) analogue of 2. Specifically, the iron(I) phenyl complex 1-N2 was treated with 6 in the presence of K(18-crown-6)(C10H8) (Scheme 6),34 which led to the expected 1,1-bis(silyl)phenylhydrazido complex 12 (Fig. 8) without the silyl shift observed in the methyl system. It is unclear whether the lack of isomerization by 12 to give the 1,2-bis(silyl)phenylhydrazido complex is the result of a high kinetic barrier or a thermodynamically unfavorable reaction. Regardless, this result suggests that the use of the bis(silyl) reagent is not the sole reason for hydrazido isomerization, and that the identity of the hydrocarbyl group on the hydrazido ligand influences its ability to take part in the silyl shift.
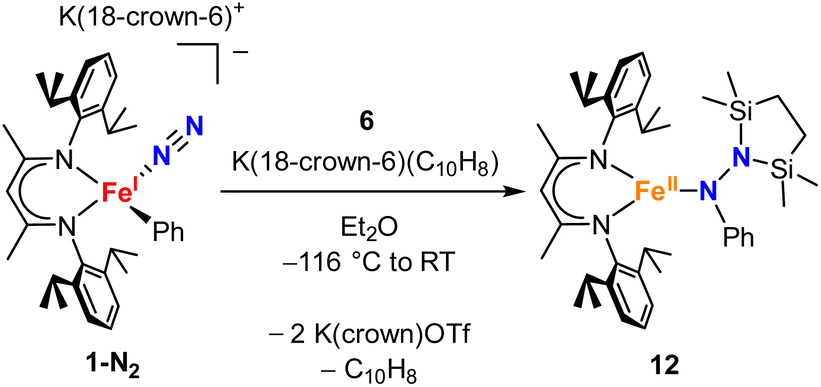 | ||
| Scheme 6 Synthesis of the phenyl-migrated complex 12. | ||
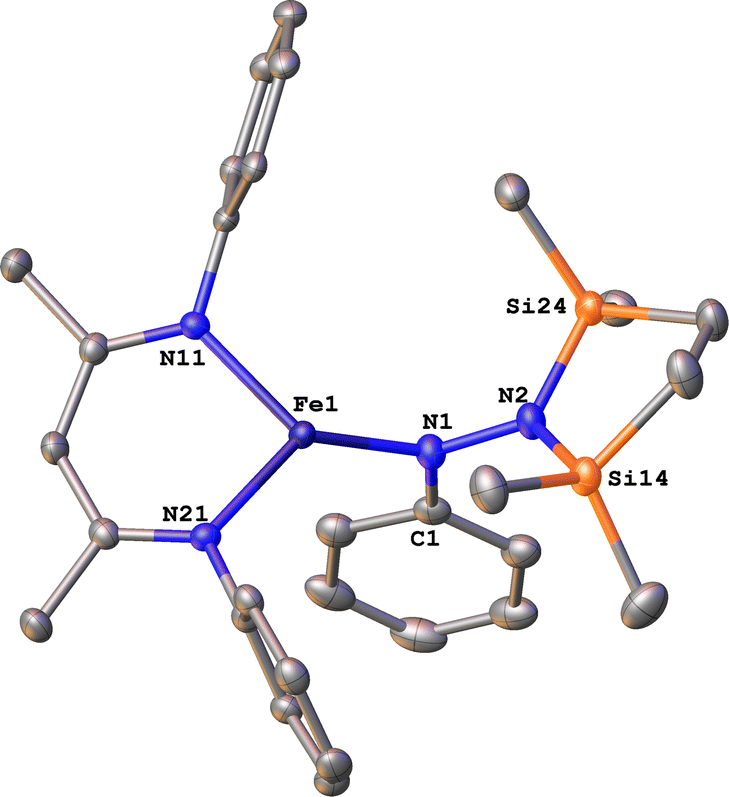 | ||
| Fig. 8 ORTEP diagram of 12 with thermal ellipsoids shown at 50% probability. H atoms and iPr groups omitted for clarity. | ||
Isomers of compound 10
The X-ray crystal structure of 10 was disordered, and the best-fit model has two components, as mentioned above. Though both components contain the same cyclic ligand, its coordination to iron in one of the components (10-1) is η1 (roughly 75% occupancy), whereas the other (10-2) is η2 through both nitrogen atoms (roughly 25% occupancy). Accordingly, the Mössbauer spectrum of 10 has a shoulder that is indicative of multiple components, and the spectrum could be fit with two or three Mössbauer doublets (Fig. S21 and S22†). Because both X-ray and Mössbauer methods suggested multiple isomers in samples that were pure (as judged by CHN analysis), we explored the energies and geometries of both isomers in triplet and quintet states using DFT geometry optimizations (Table S10†). These indicated that there are three forms that have low energies (within 4 kcal mol−1 of one another): an η1 isomer in a quintet state, and η2 isomers in triplet or quintet states. Further, a superposition of signals with the calculated isomer shift and quadrupole splitting values from these three models fits well to the experimental Mössbauer spectrum (Table S11†). Finally, the predominance of quintet states agrees with the experimental solution magnetic moment of 10 of μeff = 5.1(2) μB. Though the limited amount of experimental information hinders our ability to delve further, this combination of spectroscopy, crystallography, and computations is consistent with the idea that multiple isomers could coexist and be distinct in the solid-state structure (though equilibrating on a subsecond timescale such that one set of resonances is observed by 1H NMR spectroscopy).DFT computations give insight into product selectivity
The difference in product speciation between the silylation reactions of the benzyl complex 5b and the methyl complex 5c suggests that there may be two competing reaction pathways upon formation of the resulting formally iron(IV) alkyl complexes: Fe–C homolysis and hydrocarbyl migration. These two competing reaction pathways were investigated using DFT calculations (Fig. 9, next page). Geometries were optimized using DFT (B3LYP/def2-TZVP) in both the triplet and quintet states to probe possible spin crossover as observed in the aryl migration mechanism.35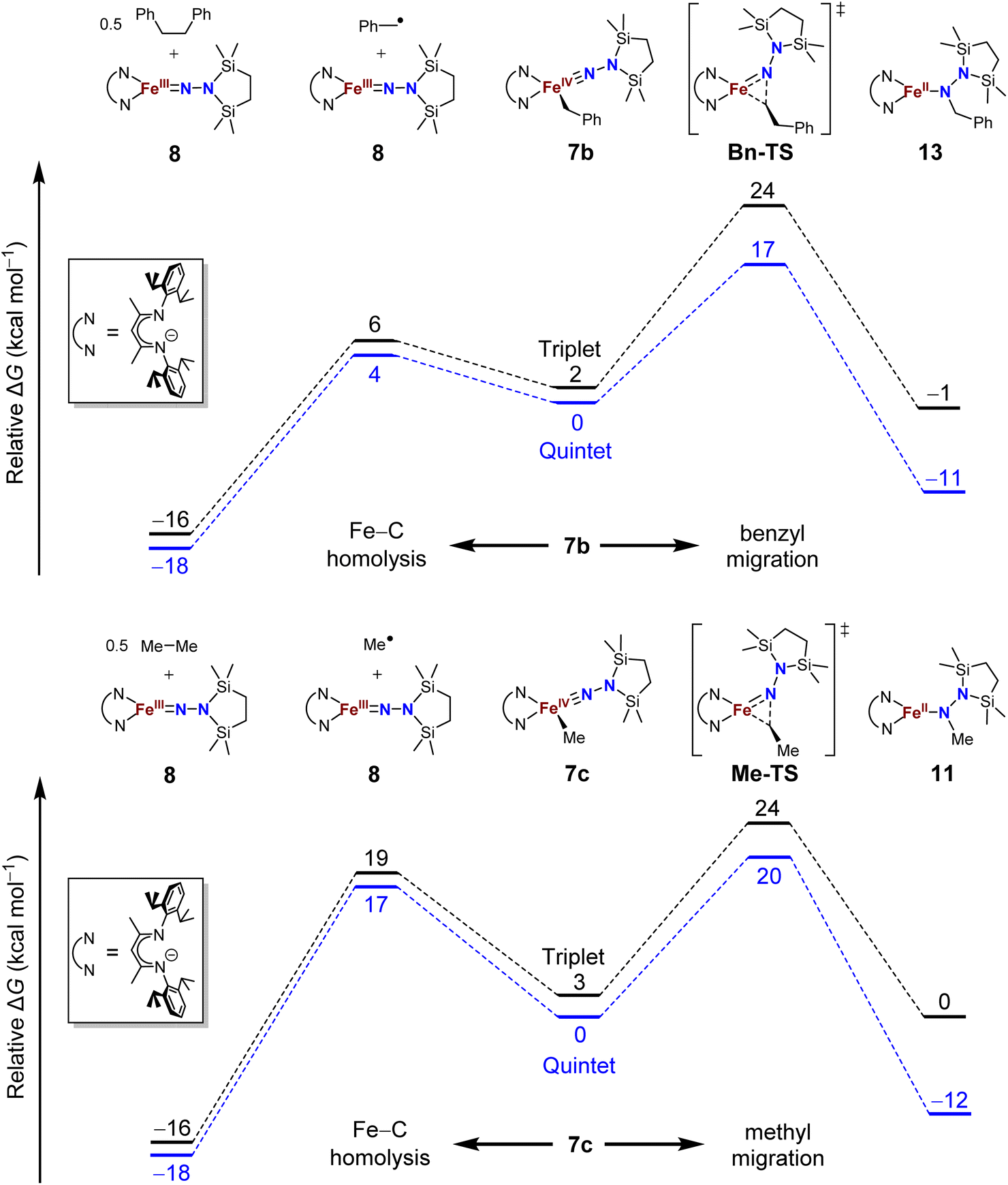 | ||
| Fig. 9 Potential energy surfaces of Fe–C bond homolysis (to the left) versus alkyl migration (to the right) starting from the proposed iron(IV) benzyl (top) and methyl (bottom) complexes from DFT calculations (B3LYP/def2-TZVP). | ||
No transition states were found for the Fe–Calkyl bond homolysis steps in either the benzyl (Fig. 9, top) or methyl (Fig. 9, bottom) system. This is not surprising, because bond homolysis reactions often have barriers very close to the BDFE if uncomplicated by steric constraints or spin state changes.59 The quintet surface was calculated to be the lowest energy pathway for Fe–C homolysis in both systems. The products of Fe–benzyl bond homolysis are only 4 kcal mol−1 uphill from 7b, while products of Fe–methyl homolysis are higher at 17 kcal mol−1 relative to 7c. This difference likely stems from the greater stability of a benzyl radical relative to a methyl radical.60 Therefore, alkyl radical formation is expected to be much more rapid for the benzyl complex.
To assess the alkyl migrations, the nudged elastic band method was used to find transition states, which were then optimized. The lowest energy pathways for both benzyl and methyl migration were found on the quintet surfaces. The calculated barriers for benzyl (17 kcal mol−1) and methyl (20 kcal mol−1) migration are similar to the experimentally measured activation barriers for aryl migration reactions (21–23 kcal mol−1).35
The significantly lower energy for Fe–CBn bond homolysis compared to the barrier for benzyl migration is consistent with the observation that silylation of 5b-N2 gave homolysis to the iron(III) complex 8 rather than migration. Meanwhile, the energy for Fe–CMe bond homolysis starting from 7c is similar to the barrier for methyl migration, suggesting that after the silylation of 5c-N2, homolysis could potentially compete with the migration pathway. We cannot rule out the possibility that radical recombination after Fe–CMe bond homolysis forms 11 in a two-step migration mechanism.21
Conclusions
In summary, the above studies have demonstrated that silylation of iron(I) alkyl N2 complexes can give formally iron(IV) alkyl hydrazido(2−) species, with interesting differences in the subsequent reactivity. A methyl group does migrate, though it is followed by a silyl migration. With the bulkier trimethylsilylmethyl, the alkyl hydrazido(2−) product can be isolated, indicating that steric effects slow the migration. The resistence to migration in the iron(IV) compound in this case allowed us to characterize it in detail, and show that it has a high-spin ground state. The benzyl compound contrasts with the others, with a weak Fe–C bond leading to homolysis. DFT calculations support the feasibility of each proposed pathway and show that they are indeed expected to be kinetically competitive.An important conclusion is that the use of formally iron(IV) centers enables migrations that can form N–Calkyl as well as N–Caryl bonds to a hydrazido(2−) ligand derived from N2, showing the generalizability of this novel approach for forming N–C bonds that come from organometallic fragments and N2. However, attempted migration of alkyl ligands is problematic when there is the potential to form a stabilized alkyl radical, because homolysis may result in rapid Fe–C bond cleavage. Thus, while alkyl migration was feasible for one case of N–C bond formation from N2, the selectivity of the reaction was poorer for alkyl than the previously observed aryl migration.
Data availability
The data are in the ESI.†Author contributions
S. F. M. and P. L. H. conceptualized the project, S. M. B. and R. X. H. and B. Q. M. collected and analyzed the data, S. M. B. wrote the original draft, and P. L. H. supervised the research and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was solely supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science program, under Award DE-SC0020315. We thank the Yale Center for Research Computing for guidance and use of the research computing infrastructure.Notes and references
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- J. J. Scepaniak, C. G. Margarit, J. N. Harvey and J. M. Smith, Inorg. Chem., 2011, 50, 9508–9517 CrossRef CAS PubMed.
- J. M. Smith and D. Subedi, Dalton Trans., 2012, 41, 1423–1429 RSC.
- S. A. Cramer, R. Hernández Sánchez, D. F. Brakhage and D. M. Jenkins, Chem. Commun., 2014, 50, 13967–13970 RSC.
- W.-T. Lee, R. A. Juarez, J. J. Scepaniak, S. B. Muñoz, D. A. Dickie, H. Wang and J. M. Smith, Inorg. Chem., 2014, 53, 8425–8430 CrossRef CAS PubMed.
- S. B. Muñoz III, W.-T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Angew. Chem., Int. Ed., 2015, 54, 10600–10603 CrossRef PubMed.
- A. K. Maity, J. Murillo, A. J. Metta-Magaña, B. Pinter and S. Fortier, J. Am. Chem. Soc., 2017, 139, 15691–15700 CrossRef CAS PubMed.
- I. S. Golovanov, A. V. Leonov, V. K. Lesnikov, E. V. Pospelov, K. V. Frolov, A. A. Korlyukov, Y. V. Nelyubina, V. V. Novikov and A. Y. Sukhorukov, Dalton Trans., 2022, 51, 4284–4296 RSC.
- R. L. Lucas, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2005, 127, 11596–11597 CrossRef CAS PubMed.
- S. Kumar, A. S. Faponle, P. Barman, A. K. Vardhaman, C. V. Sastri, D. Kumar and S. P. de Visser, J. Am. Chem. Soc., 2014, 136, 17102–17115 CrossRef CAS PubMed.
- L. Wang, L. Hu, H. Zhang, H. Chen and L. Deng, J. Am. Chem. Soc., 2015, 137, 14196–14207 CrossRef CAS PubMed.
- B. Mondal, L. Roy, F. Neese and S. Ye, Isr. J. Chem., 2016, 56, 763–772 CrossRef CAS.
- J. L. Lee, D. L. Ross, S. K. Barman, J. W. Ziller and A. S. Borovik, Inorg. Chem., 2021, 60, 13759–13783 CrossRef CAS PubMed.
- D. Mansuy, J. P. Battioni, D. Dupre, E. Sartori and G. Chottard, J. Am. Chem. Soc., 1982, 104, 6159–6161 CrossRef CAS.
- A. Casitas, J. A. Rees, R. Goddard, E. Bill, S. DeBeer and A. Fürstner, Angew. Chem., Int. Ed., 2017, 56, 10108–10113 CrossRef CAS PubMed.
- B. P. Jacobs, P. T. Wolczanski, Q. Jiang, T. R. Cundari and S. N. MacMillan, J. Am. Chem. Soc., 2017, 139, 12145–12148 CrossRef CAS PubMed.
- K. P. Caulfield and Z. J. Tonzetich, Organometallics, 2022, 41, 155–160 CrossRef CAS.
- B. K. Bower and H. G. Tennent, J. Am. Chem. Soc., 1972, 94, 2512–2514 CrossRef CAS.
- R. A. Lewis, D. E. Smiles, J. M. Darmon, S. C. E. Stieber, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 8218–8227 CrossRef CAS PubMed.
- Z. J. Tonzetich, F. Héroguel, L. H. Do and S. J. Lippard, Inorg. Chem., 2011, 50, 1570–1579 CrossRef CAS PubMed.
- I. M. Arafa, K. Shin and H. M. Goff, J. Am. Chem. Soc., 1988, 110, 5228–5229 CrossRef CAS.
- Z. Li and H. M. Goff, Inorg. Chem., 1992, 31, 1547–1548 CrossRef CAS.
- A. L. Balch, M. M. Olmstead, N. Safari and T. N. St. Claire, Inorg. Chem., 1994, 33, 2815–2822 CrossRef CAS.
- S. Byungho and H. M. Goff, Inorg. Chim. Acta, 1994, 226, 231–235 CrossRef.
- B. Song and H. M. Goff, Inorg. Chem., 1994, 33, 5979–5980 CrossRef CAS.
- C. G. Riordan and J. Halpern, Inorg. Chim. Acta, 1996, 243, 19–24 CrossRef CAS.
- P. T. Matsunaga, C. R. Hess and G. L. Hillhouse, J. Am. Chem. Soc., 1994, 116, 3665–3666 CrossRef CAS.
- K. Koo and G. L. Hillhouse, Organometallics, 1996, 15, 2669–2671 CrossRef CAS.
- X. Hu and K. Meyer, J. Am. Chem. Soc., 2004, 126, 16322–16323 CrossRef CAS PubMed.
- B. Chevrier, R. Weiss, M. Lange, J. C. Chottard and D. Mansuy, J. Am. Chem. Soc., 1981, 103, 2899–2901 CrossRef CAS.
- M. M. Olmstead, R. J. Cheng and A. L. Balch, Inorg. Chem., 1982, 21, 4143–4148 CrossRef CAS.
- I. Artaud, N. Gregoire, J. P. Battioni, D. Dupre and D. Mansuy, J. Am. Chem. Soc., 1988, 110, 8714–8716 CrossRef CAS.
- B. M. Hakey, D. C. Leary, J. C. Martinez, J. M. Darmon, N. G. Akhmedov, J. L. Petersen and C. Milsmann, Organometallics, 2022, 41, 2268–2280 CrossRef CAS.
- S. F. McWilliams, D. L. J. Broere, C. J. V. Halliday, S. M. Bhutto, B. Q. Mercado and P. L. Holland, Nature, 2020, 584, 221–226 CrossRef CAS PubMed.
- S. M. Bhutto, R. X. Hooper, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2023, 145, 4626–4637 CrossRef CAS PubMed.
- S. M. Bhutto, B. Q. Mercado and P. L. Holland, Inorg. Chem., 2023, 62, 9335–9342 CrossRef CAS PubMed.
- A. L. Nagelski, M. S. Fataftah, M. M. Bollmeyer, S. F. McWilliams, S. N. MacMillan, B. Q. Mercado, K. M. Lancaster and P. L. Holland, Chem. Sci., 2020, 11, 12710–12720 RSC.
- M. M. Deegan and J. C. Peters, Chem. Sci., 2018, 9, 6264–6270 RSC.
- S. Bhutto, S. McWilliams, R. Hooper, B. Mercado and P. Holland, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-55shl.
- J. M. Smith, R. J. Lachicotte and P. L. Holland, Organometallics, 2002, 21, 4808–4814 CrossRef CAS.
- J. Vela, S. Vaddadi, T. R. Cundari, J. M. Smith, E. A. Gregory, R. J. Lachicotte, C. J. Flaschenriem and P. L. Holland, Organometallics, 2004, 23, 5226–5239 CrossRef CAS.
- T. J. J. Sciarone, A. Meetsma and B. Hessen, Inorg. Chim. Acta, 2006, 359, 1815–1825 CrossRef CAS.
- K. C. MacLeod, I. M. DiMucci, E. P. Zovinka, S. F. McWilliams, B. Q. Mercado, K. M. Lancaster and P. L. Holland, Organometallics, 2019, 38, 4224–4232 CrossRef CAS PubMed.
- M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118–18121 CrossRef CAS PubMed.
- P. A. Rudd, N. Planas, E. Bill, L. Gagliardi and C. C. Lu, Eur. J. Inorg. Chem., 2013, 2013, 3898–3906 CrossRef CAS.
- S. F. McWilliams, E. Brennan-Wydra, K. C. MacLeod and P. L. Holland, ACS Omega, 2017, 2, 2594–2606 CrossRef CAS PubMed.
- R. Hoffmann, S. Alvarez, C. Mealli, A. Falceto, T. J. Cahill, T. Zeng and G. Manca, Chem. Rev., 2016, 116, 8173–8192 CrossRef CAS PubMed.
- I. M. DiMucci, J. T. Lukens, S. Chatterjee, K. M. Carsch, C. J. Titus, S. J. Lee, D. Nordlund, T. A. Betley, S. N. MacMillan and K. M. Lancaster, J. Am. Chem. Soc., 2019, 141, 18508–18520 CrossRef CAS PubMed.
- A. D. Piascik, R. Li, H. J. Wilkinson, J. C. Green and A. E. Ashley, J. Am. Chem. Soc., 2018, 140, 10691–10694 CrossRef CAS.
- S. F. McWilliams, E. Bill, G. Lukat-Rodgers, K. R. Rodgers, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2018, 140, 8586–8598 CrossRef CAS PubMed.
- K. P. Caulfield, J. Conradie, H. D. Arman, A. Ghosh and Z. J. Tonzetich, Inorg. Chem., 2019, 58, 15225–15235 CrossRef CAS PubMed.
- D. Kim, S. M. W. Rahaman, B. Q. Mercado, R. Poli and P. L. Holland, J. Am. Chem. Soc., 2019, 141, 7473–7485 CrossRef CAS PubMed.
- S. Barroso, A. M. Coelho, P. Adão, M. J. Calhorda and A. M. Martins, Dalton Trans., 2017, 46, 9692–9704 RSC.
- H. Paulsen and A. X. Trautwein, in Spin Crossover in Transition Metal Compounds III, ed. P. Gütlich and H. A. Goodwin, Springer, Berlin, 2004, pp. 197–219 Search PubMed.
- R. E. Cowley, N. A. Eckert, J. Elhaïk and P. L. Holland, Chem. Commun., 2009, 1760–1762 RSC.
- N. A. Eckert, S. Vaddadi, S. Stoian, R. J. Lachicotte, T. R. Cundari and P. L. Holland, Angew. Chem., Int. Ed., 2006, 45, 6868–6871 CrossRef CAS PubMed.
- R. West, M. Ishikawa and R. E. Bailey, J. Am. Chem. Soc., 1967, 89, 4068–4072 CrossRef CAS.
- D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 4938–4941 CrossRef CAS PubMed.
- R. Poli, C. R. Chim., 2021, 24, 147–175 CrossRef CAS.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental methods, spectroscopy, crystallography, and computations. CCDC 2226338–2226342, 2226344, and 2303073. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05939a |
| This journal is © The Royal Society of Chemistry 2024 |