
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the diastereoselective synthesis of a macrocycle under Curtin–Hammett control†
Angus
Yeung
a,
Martijn A.
Zwijnenburg
 b,
Georgia R. F.
Orton
a,
Jennifer H.
Robertson
a and
Timothy A.
Barendt
*a
b,
Georgia R. F.
Orton
a,
Jennifer H.
Robertson
a and
Timothy A.
Barendt
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: t.a.barendt@bham.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 5th March 2024
Abstract
This work sheds new light on the stereoselective synthesis of chiral macrocycles containing twisted aromatic units, valuable π-conjugated materials for recognition, sensing, and optoelectronics. For the first time, we use the Curtin–Hammett principle to investigate a chiral macrocyclisation reaction, revealing the potential for supramolecular π–π interactions to direct the outcome of a dynamic kinetic resolution, favouring the opposite macrocyclic product to that expected under reversible, thermodynamically controlled conditions. Specifically, a dynamic, racemic perylene diimide dye (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 P:M) is strapped with an enantiopure (S)-1,1′-bi-2-naphthol group (P-BINOL) to form two diastereomeric macrocyclic products, the homochiral macrocycle (PP) and the heterochiral species (PM). We find there is notable selectivity for the PM macrocycle (dr = 4:1), which is rationalised by kinetic templation from intramolecular aromatic non-covalent interactions between the P-BINOL π-donor and the M-PDI π-acceptor during the macrocyclisation reaction.
:1 P:M) is strapped with an enantiopure (S)-1,1′-bi-2-naphthol group (P-BINOL) to form two diastereomeric macrocyclic products, the homochiral macrocycle (PP) and the heterochiral species (PM). We find there is notable selectivity for the PM macrocycle (dr = 4:1), which is rationalised by kinetic templation from intramolecular aromatic non-covalent interactions between the P-BINOL π-donor and the M-PDI π-acceptor during the macrocyclisation reaction.
Introduction
The balance between thermodynamics and kinetics determines the distribution of products in a chemical reaction. An understanding of these fundamental principles in the context of chiral molecules is essential for designing new and efficient stereoselective syntheses, including for pharmaceuticals,1 chemical sensing,2 and chiroptical materials,3 as well as unravelling the origins of single handedness in nature.4 Here, supramolecular chemistry plays a privileged role because the tuning of non-covalent interactions between molecules can be used to direct the stereochemical outcome of a chemical reaction5 or the self-assembly of a chiral material.6 In the latter case, chiral self-assembly mainly operates under reversible conditions (i.e., thermodynamic control),7 which means the selection for homochiral or heterochiral products is dictated by their relative energies.8 However, a deeper understanding of the role of supramolecular chemistry in a dynamic kinetic resolution is essential for directing the stereochemical outcome of a reaction between interacting chiral molecules under irreversible conditions.Non-covalent kinetic templates can favour the formation of a particular product under irreversible reaction conditions,9 including the synthesis of macrocycles which have huge value across supramolecular chemistry.10 While often being challenging to design, the impact of templating interactions in reactions under kinetic control can be interpreted through the Curtin–Hammett principle, which states that the outcome of a dynamic kinetic resolution is dependent on the relative free energy of the interconverting precursors (i.e., ΔG° in Fig. 1) and the relative activation energies (i.e., ΔG‡PP − ΔG‡PM).11 Indeed, the Curtin–Hammett principle explains the distribution of products in irreversible chemical transformations, including stereoselective reactions on dynamic heterocycles,12 as well as understanding the interactions between conformationally flexible macrocycles and proteins13 and, more recently, the operation of Brownian ratches in molecular machines.14 However, to the best of our knowledge, an investigation of Curtin–Hammett control in the context of a chiral macrocyclisation reaction (i.e., the ring-closing step itself) is unprecedented, with such a development having important consequences for the templated stereoselective synthesis of chiral materials under kinetic control.
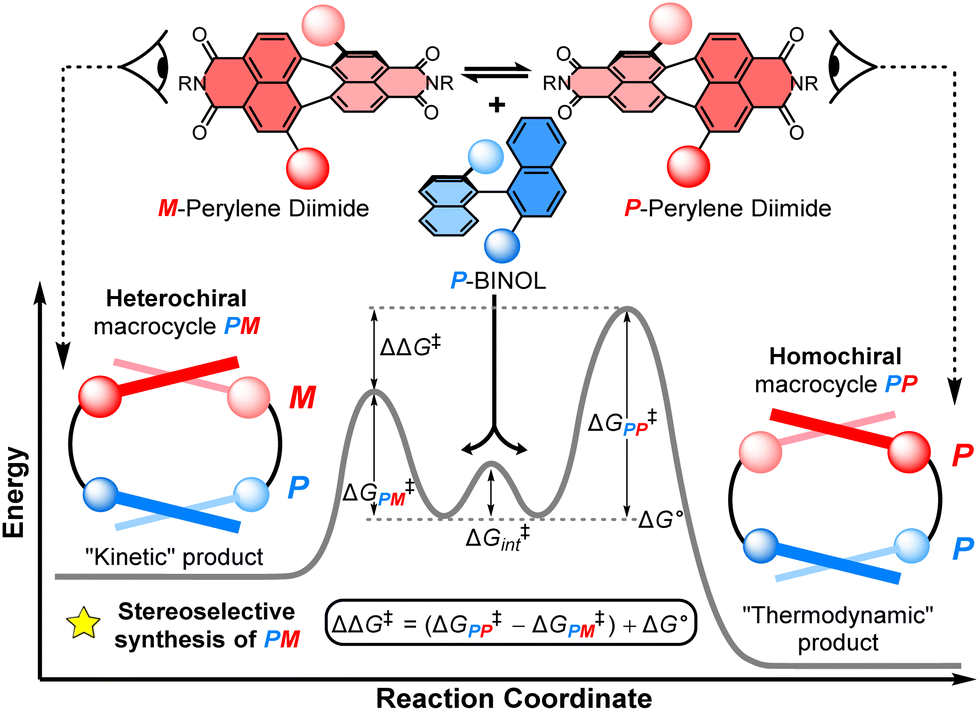 | ||
| Fig. 1 The diastereoselective synthesis of a heterochiral (PM) over a homochiral (PP) macrocycle under Curtin–Hammett control. Here, ΔΔG‡ = (ΔG‡PP − ΔG‡PM) + ΔG°; ΔG‡PP/PM = the activation energy of PP/MM macrocycles; ΔG° = the relative free energy of the interconverting precursors; and ΔG‡int = the activation energy of this interconversion. | ||
Perylene diimides (PDIs) are a class of functional organic dyes and outstanding building blocks for studying chiral molecular systems.15 This is because PDIs may be twisted through substitution at their bay positions (1, 6, 7, 12) to generate atropisomers (P or M)‡ with distinct chiroptical spectra,16 while their potential for π–π aromatic stacking interactions,17 including with complementary π-donors,18 gives rise to an extensive supramolecular chemistry.15 Alongside functional complexes,19 cages,20 and polymers,21 macrocycles containing twisted PDIs have been developed to explore homo- and heterochirality.22 We have reported a bis-PDI macrocycle that is predominantly homochiral (diastereomeric ratio [dr] > 88:12 MM/PP:PM) due to complementary π–π stacking.23 However, in this system, as for most bis-PDI macrocycles,22a–c the PDI units are not configurationally stable (i.e., their atropisomers can freely interconvert), hence the macrocycle's chiral conformation is under thermodynamic control, which makes them unsuitable for investigating kinetic templation under Curtin–Hammett control.
Herein, we report the first stereoselective synthesis of a configurationally stable PDI macrocycle (Fig. 1). We impose configurational stability on a racemic 1,7-disubstituted PDI dye (1:1 P:M) by strapping it with a (S)-1,1′-bi-2-naphthol group (P-BINOL), enabling us to isolate concurrent heterochiral (PM) and homochiral (PP) macrocycles. The contrast between BINOL, which is always chirally locked, and PDI, whose atropisomers rapidly interconvert prior to, but not after, macrocylisation, is critical to a diastereoselective macrocycle synthesis under Curtin–Hammett control (Fig. 1). We discover that the use of P-BINOL impacts the chirality of the PDI during the ring-closing reaction, affording diastereoselectivity for the heterochiral macrocycle (dr = 4:1 PM:PP). This is the opposite outcome to that expected from a reversible, thermodynamically controlled synthesis, since the homochiral macrocycle is predicted, by calculations, to be significantly lower in energy. While BINOL has been integrated into macrocycles,24 including for the dynamic kinetic resolution of 2,2′-bipyridine N,N′-dioxide atropisomers,25 it has never been combined with a twisted PDI dye, which here enables us to explore the possibility of supramolecular templation as an origin of stereoselectivity. These investigations point towards the importance of aromatic non-covalent interactions between the P-BINOL π-donor and M-PDI π-acceptor, demonstrating the potential for π–π templation to be exploited in the stereoselective synthesis of π-conjugated atropisomeric materials under irreversible conditions.26
Results and discussion
Synthesis and characterisation
Several macrocyclic (1, 2) and acyclic (3–5) compounds were prepared using multi step syntheses, the final step of which uses copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) “click” chemistry (Fig. 2a) to connect the PDI π-acceptor to a planar (hydroquinone) or twisted (P-BINOL) π-donor. The macrocycles were synthesised under high dilution conditions to favour macrocyclisation, using stoichiometric amounts of a PDI bis-alkyne and bis-azide n-alkyl linker (Fig. 2a). The desired [1 + 1] macrocycles were formed in good yields (up to 53%), with major side products being larger [2 + 2] and [3 + 3] macrocyclic analogues. All the synthetic procedures and complete characterisation data are provided in the ESI (Section 2†).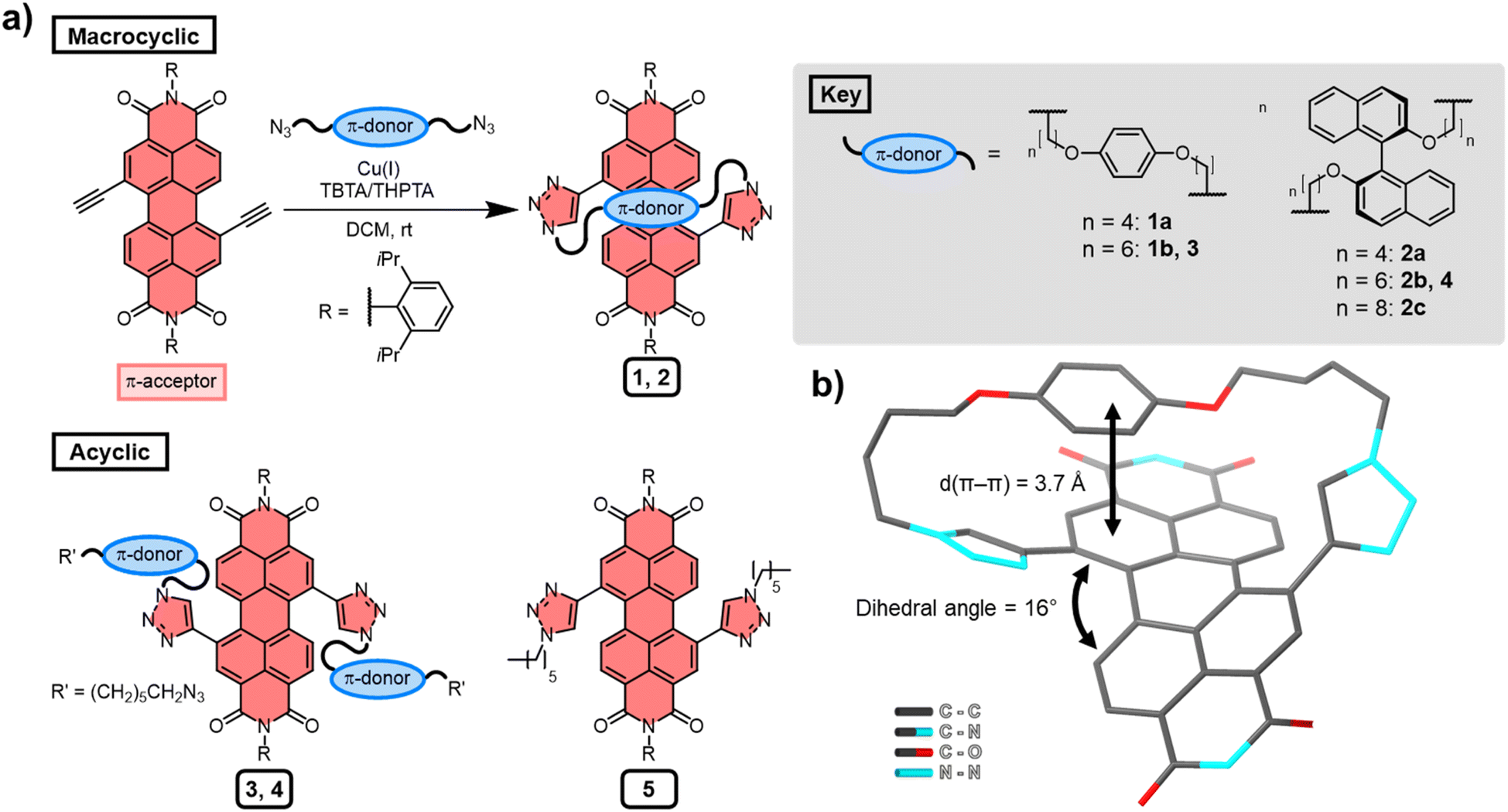 | ||
| Fig. 2 (a) The synthesis of macrocyclic and acyclic compounds 1–5 and (b) X-ray crystal structure of macrocycle 1a. For clarity, hydrogen atoms and the 2,6-diisopropylphenyl imide substituents have been removed from the structure. The Cu(I) coordinating ligands are tris((1-benzyl-4-triazolyl)methyl)amine (TBTA) or tris(3-hydroxypropyltriazolylmethyl)amine (THPTA). | ||
All the derivatives of 1 and 2 were characterised by 1H and 13C NMR spectroscopy, which, alongside high-resolution mass spectrometry, confirmed they are [1 + 1] macrocycles. From this compound library, PDI-hydroquinone macrocycle 1a yielded single crystals that were suitable for X-ray diffraction (ESI, Section 7†). Importantly, the crystal structure of 1a shows the PDI is twisted (16°), with one half of the π-electron deficient unit forming a π–π interaction (d(π–π) = 3.7 Å)27 with the π-electron rich hydroquinone group (Fig. 2b). We note that, in contrast to our previous bis-triazole PDI-based macrocycle,23 the crystal structure of 1a shows that the triazole heterocycles are directed towards the macrocyclic cavity, most likely because this reduces ring strain in the shorter cyclic framework.
The significant (up) downfield shifts of protons (Hd,d′) Ha–c in the 1H NMR spectrum of macrocycles 1a (Fig. 3d) and 1b (Fig. 3c), relative to acyclic analogues 3 and 5 (Fig. 3a and b), indicates the macrocyclic framework preorganises the π-conjugated units in solution, providing the potential for intramolecular aromatic stacking interactions (vide infra). This is also true for macrocycles containing the twisted P-BINOL unit since 2a–c exhibit the same pattern of proton shifts (Fig. 4 and S11†). Moreover, NOESY NMR spectroscopy shows cross-peaks between PDI and P-BINOL protons (Fig. S1†), confirming the proximity of these units in 2 and with P-BINOL adopting a transoid conformer (ESI, Section 8†). This overall conformation is also consistent with that obtained from conformer searches using the CREST code28 and the GFN2-xTB tight-binding DFT method29 for 2a, b (ESI, Section 8†). This approach is supported by the fact that the predicted lowest energy conformer of macrocycle 1a (Fig. S26†), for which we can obtain an X-ray crystal structure, agrees with that in the experimental X-ray crystal structure (Fig. 2b).
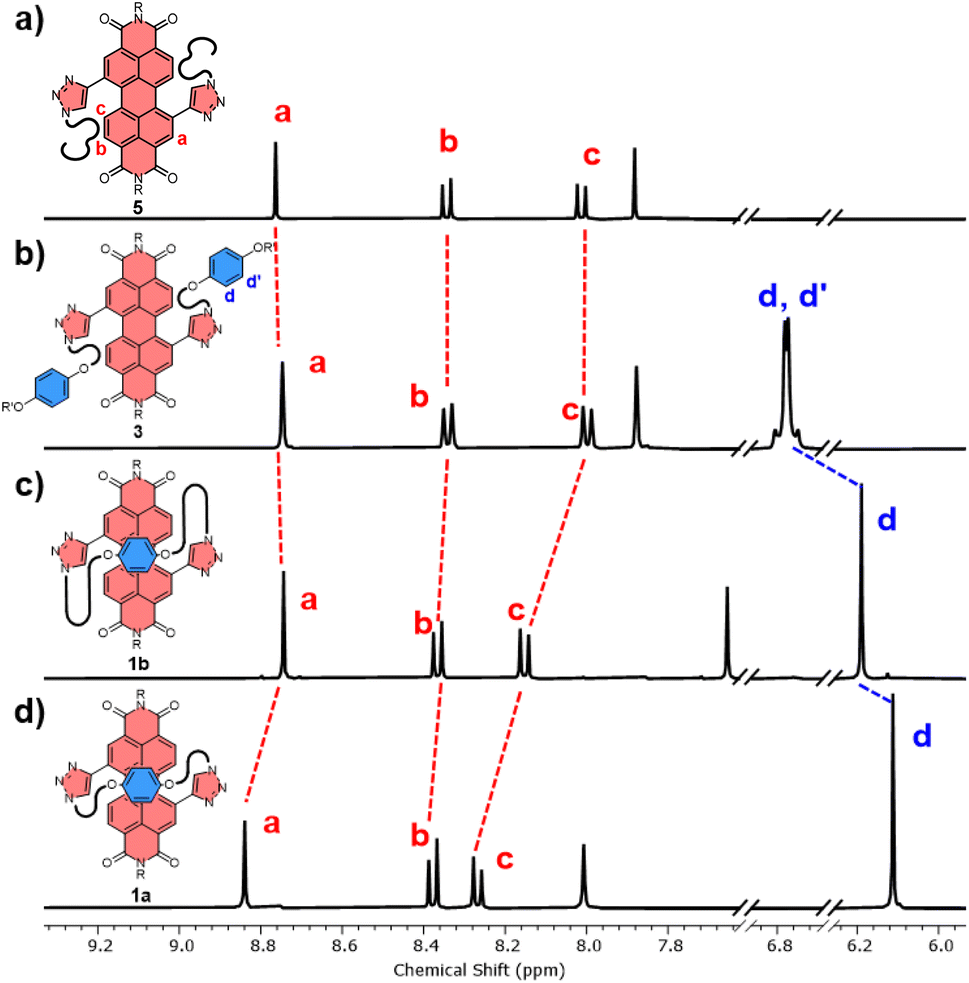 | ||
| Fig. 3 Stacked 1H NMR spectra of (a) acyclic bis-triazole PDI 5, (b) acyclic bis-hydroquinone PDI 3, (c) hydroquinone-PDI macrocycles 1b and, (d) 1a (CDCl3, 298 K, 400 MHz). | ||
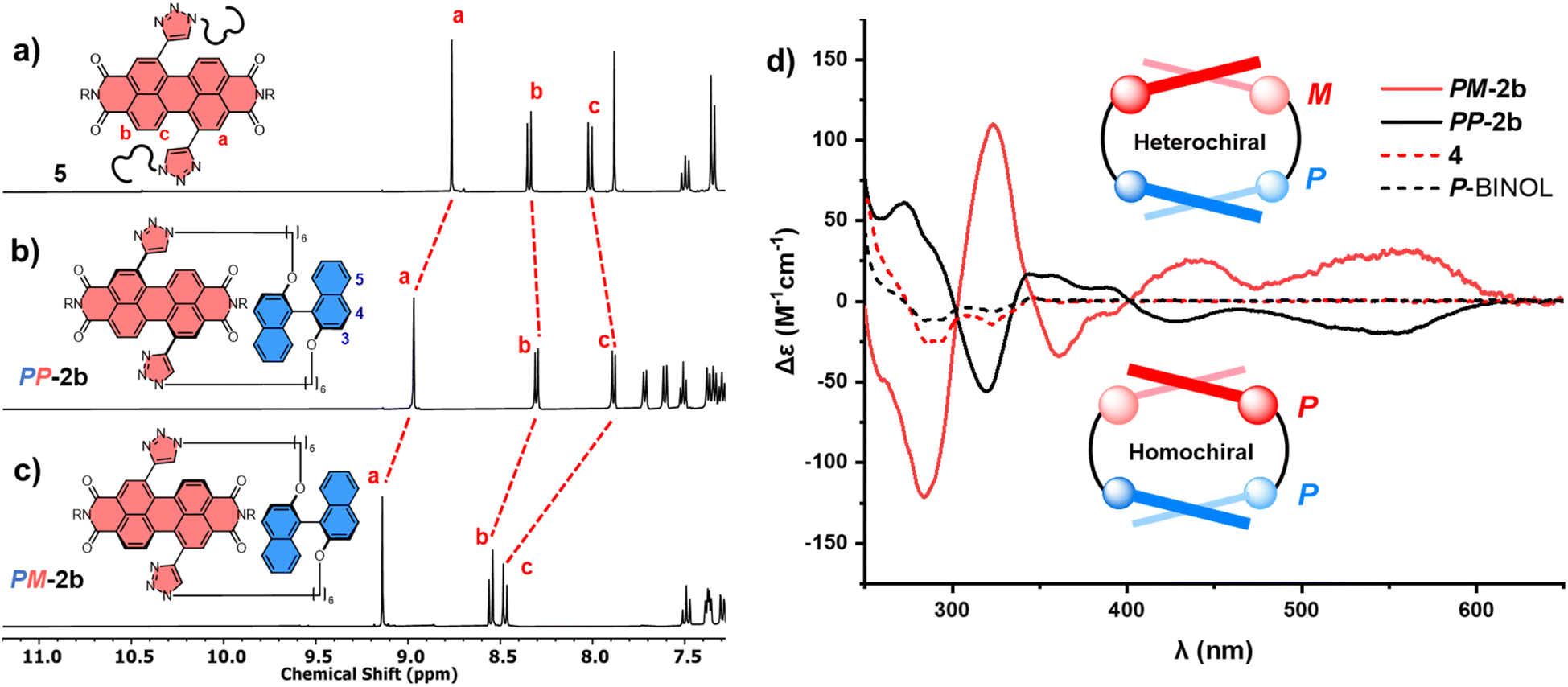 | ||
| Fig. 4 Stacked 1H NMR spectra of (a) acyclic bis-triazole PDI 5, (b) homochiral PDI–P-BINOL macrocycle PP-2b, and (c) heterochiral PDI–P-BINOL macrocycle PM-2b (CDCl3, 298 K, 400 MHz); and (d) the circular dichroism (CD) spectra of PP-2b and PM-2b which are opposite in the PDI-only region (λ = 350–600 nm), where neither P-BINOL nor acyclic control 4 exhibits a CD spectrum in this region (CHCl3, 298 K). | ||
Macrocycle chirality
Our next goal was to assess the impact of the twisted BINOL π-donor on the chirality of the PDI π-acceptor, focussing initially on macrocycle 2b. Importantly, while P-BINOL is configurationally stable at room temperature (ΔG‡ = 158 kJ mol−1),30 the twisted bis-alkyne PDI starting material is conformationally unstable (i.e., chirality is dynamic with ΔG‡ < 39 kJ mol−1)16,31 and is racemic (1:1 P:M).31a,b This provides the potential for two diastereomeric [1 + 1] macrocyclic products, homochiral PP-2b and heterochiral PM-2b. Indeed, upon analysis of the 2b crude mixture, we identified two P-BINOL–PDI macrocycles, formed in different yields. Since the relationship between PP-2b and PM-2b is diastereomeric, these compounds were separable by silica gel chromatography, exhibiting distinct 1H NMR spectra (Fig. 4b and c) and a single peak by chiral high-performance liquid chromatography (HPLC) (Fig. S23†). Therefore, our synthetic strategy enables us to obtain both P and M PDI atropisomers in high enantiopurity (>99%), without the need for chiral HPLC.32 By contrast, there is no stereoselectivity when the planar hydroquinone π-donor is used: macrocycle 1b is isolated as a racemic mixture and requires a chiral stationary phase to resolve the enantiomers P-1b and M-1b (Fig. S18 and S24†). Of course, upon swapping P-BINOL for M-BINOL in the macrocyclisation reaction, we obtain homo- and heterochiral macrocycles in the same diastereomeric ratio, with MM-2b and MP-2b having identical 1H NMR spectra to PP-2b and PM-2b respectively, due to their enantiomeric relationship (Fig. S2†).
To assign the chirality of the two P-BINOL–PDI macrocycle diastereomers, we used a combination of CD spectroscopy and (time-dependent) density functional theory ((TD)-DFT) calculations. In the latter we took the results of the above mentioned conformer search for the macrocycles using the GFN2-xTB tight-binding DFT method, and optimised the lowest-energy conformers with DFT using the B97-3c functional33 before calculating their UV-vis and CD spectra with (TD)-DFT using the ωB97x functional34 (ESI, Section 8†). Examination of the PDI-only region of the CD spectra (λ = 350–600 nm) reveals PP-2b and PM-2b exhibit opposite CD spectra, indicative of the enantiomeric relationship between the PDI units (Fig. 4d). With the aid of TD-DFT (Table S2†) we assign the negative signals in the visible part of the CD spectrum (λ > 400 nm) to the P atropisomer of PDI (i.e., PP-2b), while the positive CD signals are from M-PDI (i.e., PM-2b). Therefore, the higher yielding product is the heterochiral macrocycle (PM) and the lower yielding is homochiral (PP), giving a diastereomeric ratio of 4:1 (Fig. S5†). This is intriguing because previous work on diastereomeric PDI-based macrocycles has always found the major species to be homochiral.22a–c,23 Indeed, calculations suggests that PP-2b is the lowest energy macrocycle. A full GFN2-xTB tight-binding DFT conformer search performed using the heterochiral macrocycle PM-2b as the starting point returned only low-energy homochiral macrocycle conformers, thus PM-2b must lie at least 20–30 kJ mol−1 above that of PP-2b (ESI, Section 8†). As such, PP-2b is akin to the “thermodynamic product” of the macrocyclisation reaction while PM-2b, which is formed in the highest yield, is akin to the “kinetic product” (Fig. 1).
Interestingly, electronic circular dichroism is stronger throughout the PDI-only region for the heterochiral macrocycle relative to the homochiral macrocycle (Fig. 4d). This is indicative of a larger PDI twist angle32d in PM-2b compared to PP-2b, a factor that will contribute to the latter being lower in energy. Indeed, inspection of the DFT optimised structures reveals that the PDI twist angle increases from 23° to 25° on moving from the homochiral (PP-2b) to the heterochiral (PM-2b) macrocycle structure and upon decreasing the linker length from hexyl (2b) to butyl (2a), which in both cases leads to an amplification of electronic circular dichroism (Fig. 4d, S15 and S29†).32d Furthermore, the DFT optimised structure of macrocycle PP-2b shows an additional intramolecular CH–O hydrogen bond (d = 2.5 Å) between the triazole unit and the BINOL's O atom compared to PM-2b, which will also contribute to the former being lower in energy (Fig. S28†).§
The reason that the heterochiral macrocycle does not convert to its lower energy homochiral analogue is because the CuAAC ring-closing reaction is irreversible, and, upon macrocyclisation, the PDI becomes configurationally stable (like P-BINOL). As such, there is no change to the 1H NMR or CD spectra of macrocycle 2 upon heating up to 180 °C, which is just below the isomerisation temperature of BINOL35 (i.e., ΔG‡ > 158 kJ mol−1, Fig. S3 and S19†). The interconversion of macrocycle stereoisomers is inhibited because the macrocyclic cavities of 1 and 2 are too small for the PDI dye to somersault through,22b,d therefore enabling us to isolate a configurationally stable heterochiral mono-PDI macrocycle for the first time. The stereoisomers of acyclic compounds 3–5 could not be resolved at room temperature because, as for all current acyclic disubstituted PDIs,16,36 the interconversion of bis-triazole PDI atropisomers is too fast (ΔG‡int < 39 kJ mol−1, rate kint > 6 × 105 s−1, ESI Section 3.7†), making them “Class 1” atropsiomers under the LaPlante classification system.31c
Curtin–Hammett analysis
We sought to understand the origin of stereoselectivity in the synthesis of macrocycle 2b using the Curtin–Hammett principle. Firstly, it is important to note that, in a macrocyclisation reaction, the rate determining step is typically the one that forms the ring,37 which for 1, 2 is the second ring-closing CuAAC ‘click’ reaction. For macrocycle 2, this reaction fulfils the criteria for Curtin–Hammett control because, while the CuAAC chemistry is irreversible and the macrocycle products are configurationally stable, the diastereomers of the mono-clicked macrocycle intermediate (PP and PM, Fig. 5) are in thermodynamic equilibrium and able to rapidly interconvert (ΔG‡int < 39 kJ mol−1), being acyclic disubstituted PDIs (vide supra). Indeed, the diastereomers of acyclic BINOL–PDI–BINOL control compound 4 (PMP and PPP) could not be isolated or observed by 1H NMR spectroscopy due to their rapid exchange (see ESI Section 3.7†).38 The Curtin–Hammett principle states that the reaction outcome is solely dependent on the difference in transition state energies of the products (ΔΔG‡, Fig. 1). By synthesising macrocycle 2b at different temperatures, we showed that diastereoselectivity decreases upon increasing the macrocylisation reaction temperature (Fig. S9†) and hence calculated the difference in free energy between the transition states of the PM and PP macrocyclic products, |ΔΔG‡| = 6.3 ± 0.4 kJ mol−1 (Fig. 6). This ΔΔG‡ is dependent on the relative free energy of the interconverting intermediate (ΔG°) and the relative activation energies (ΔG‡PP − ΔG‡PM). For 2b, ΔG° ≈ 0, since the acyclic BINOL–PDI–BINOL compound 4 does not exhibit a CD spectrum in the PDI-only region (Fig. 4d), which indicates that the mono-clicked PP and PM diastereomeric intermediates of 2b are very close in energy prior to macrocyclisation. Therefore, the difference in transition state energies ΔΔG‡ (and hence the kinetic resolution of 2b) does not occur from any inherent stability of the heterochiral mono-clicked intermediate over the homochiral intermediate (ΔG°), and instead arises purely on the difference in activation energies (ΔG‡PP − ΔG‡PM, Fig. 5).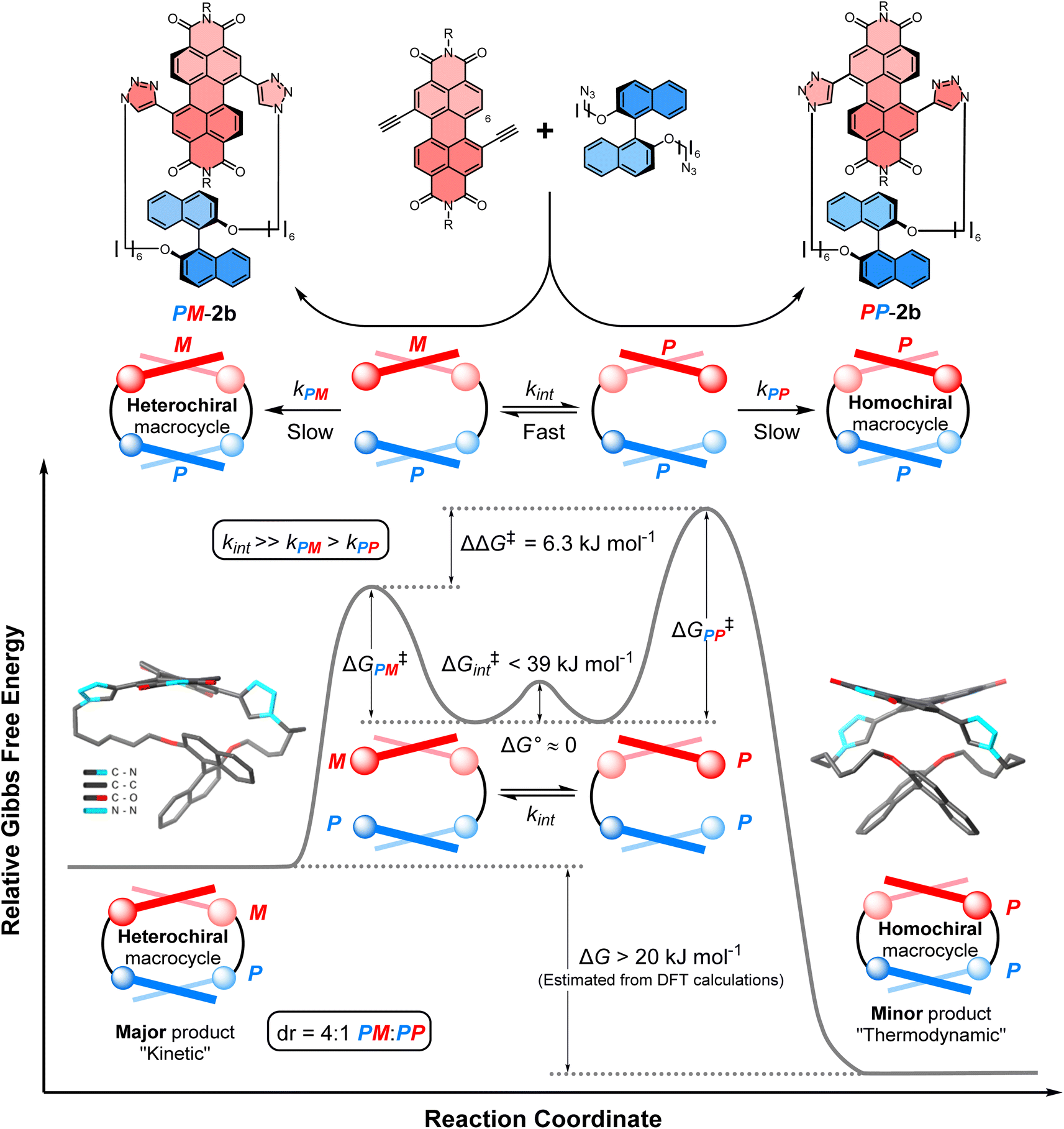 | ||
| Fig. 5 The energy profile diagram for the diastereoselective synthesis of macrocycle PM-2b under Curtin–Hammett control. Values for the free energies ΔΔG‡, ΔG° and ΔG‡int have been determined experimentally, while the free energy difference between macrocycles PM-2b and PP-2b (ΔG) has been estimated computationally using the DFT (B973c) optimized geometries of the likely lowest energy conformer of macrocycles PM-2b and PP-2b, as found in the tight-binding DFT (GFN2-xTB) conformer searches (ESI, Section 8†). These computational structures are displayed on the diagram, with hydrogen atoms and the 2,6 diisopropylphenyl imide substituents removed for clarity. | ||
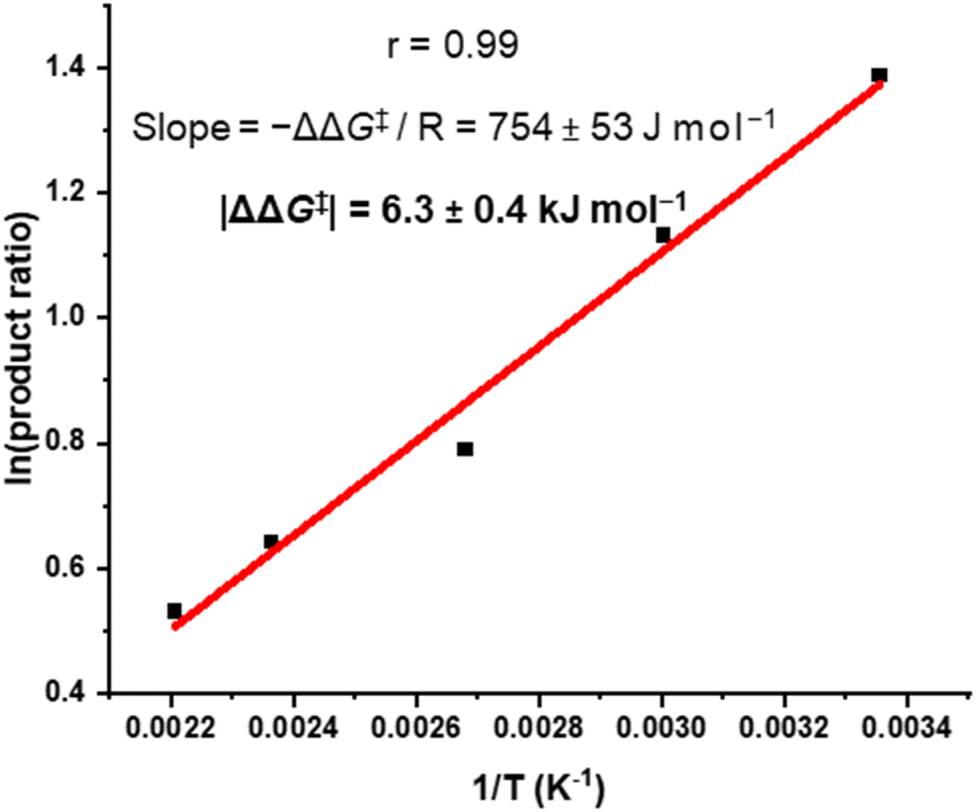 | ||
| Fig. 6 Curtin–Hammett plot of macrocycle 2b, where 2b was synthesised at a range of temperatures (T) and the product ratio ([PM-2b]/[PP-2b]) determined by 1H NMR spectroscopy. | ||
To investigate the potential steric and electronic factors behind this difference in activation energies, we prepared two further PDI–BINOL macrocycles with shorter (n-butyl, 2a) and longer (n-octyl, 2c) strap lengths in comparison to 2b (n-hexyl).¶ Interestingly, neither of these macrocycles exhibited any diastereoselectivity (i.e., PP-2a,c:PM-2a,c = 1:1). Therefore, stabilisation of the heterochiral transition state is unique to macrocycle 2b, suggesting that the trend in diastereoselectivity does not correlate with the change in ring strain that would be anticipated upon changing the linker length. As such, in light of the evidence for intramolecular π–π stacking in our macrocycles (vide supra), we also sought to identify any connection between diastereoselectivity and the potential differences in electronic properties.
To investigate this, we measured the UV-vis absorption spectra of the diastereomers of macrocycles 2a–c and the acyclic control compounds 4 and 5. Here it is important to clarify that π–π interactions primarily consist of electrostatic and van der Waals contributions,39 with the former readily probed by UV-vis spectroscopy since red-shifted charge transfer absorbances can be a consequence of donor–acceptor interactions.18b,40 Relative to acyclic bis-triazole PDIs, the main PDI absorption band (S0–S1) of PM-2b is significantly red-shifted (Δλ = 10 nm), indicative of new donor–acceptor π–π interactions in this heterochiral macrocycle (Fig. 7a).|| However, the absorption spectra of PP-2b is relatively unchanged (Fig. 7b), indicating any donor–acceptor π–π interactions in the homochiral macrocycle are significantly weaker.** The importance of donor–acceptor π–π interactions is also demonstrated by UV-vis analysis of the shorter- (2a) and longer-linked (2c) macrocycles. With both macrocycles exhibiting no diastereoselectivity (Fig. S4 and S8†), it is notable that the diastereomers PP-2c and PM-2c have similarly red-shifted PDI absorption bands (ΔΔλ ≈ 0 nm, Fig. 7c), as do diastereomers PP-2a and PM-2a (ΔΔλ = 1 nm, Fig. S22†).†† Moreover, this trend is in line with our evidence for intramolecular aromatic interactions by 1H NMR spectroscopy, for which CH–π as well as π–π interactions may contribute. Indicative of π–π interactions, the heterochiral macrocycle PM-2b exhibits a larger downfield shift of PDI protons relative to homochiral PP-2b (for Hc, Δδ = 8.47 vs. 7.88 ppm, ΔΔδ = 0.59 ppm, Fig. 4a–c), while the difference between diastereomers is much smaller for macrocycles 2a and 2c (ΔΔδ = 0.1 and 0.07 ppm respectively, ESI Section 1†). Indicative of BINOL–PDI CH–π interactions,41 relative to acyclic P-BINOL, PM-2b also exhibits a larger average upfield shift of BINOL protons H3–5, compared to PP-2b (Δδ = 0.7 vs. 0.4 ppm, Fig. S12†). These findings are supported by the computational structures, which show that BINOL–PDI π–π and CH–π distances are overall shorter, and in the latter case also more numerous, in the heterochiral vs. homochiral structures of macrocycle 2b (Fig. S28†).
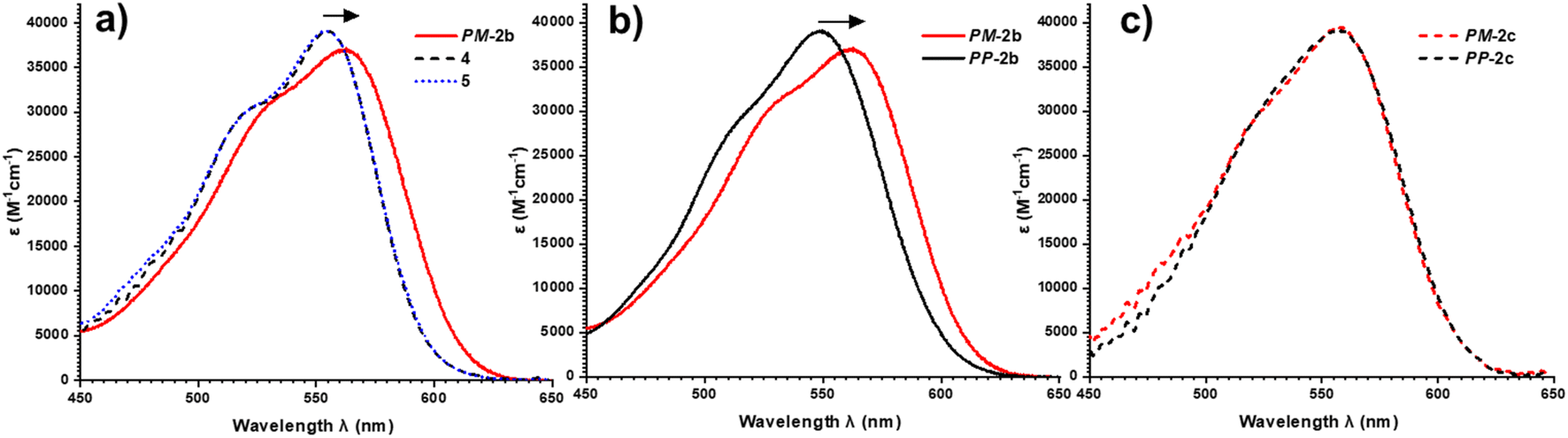 | ||
| Fig. 7 UV-vis spectra (CHCl3, 298 K, 26 μM) of: (a) macrocycle PM-2b, which is bathochromically shifted relative to acyclic bis-triazole PDIs 4, 5; (b) heterochiral macrocycle PM-2b, which is bathochromically shifted relative to homochiral macrocycle PP-2b; (c) heterochiral and homochiral macrocycles PM-2c and PP-2c, with no relative bathochromic shift. | ||
Overall, diastereomers PM-2b and PP-2b are the only pair of macrocyclic products to show significantly different PDI absorption bands (ΔΔλ = 14 nm) and have the largest discrepancy between their 1H NMR PDI aromatic protons (ΔΔδ = 0.59 ppm) and are the only pair to show diastereoselectivity. Taken together, these results show that the geometry of macrocycle 2b is unique in facilitating stronger non-covalent interactions, including π–π and CH–π interactions, in PM-2b relative to PP-2b. These stronger aromatic interactions provide a potential kinetic template effect during the formation of PM-2b and help to rationalise why the heterochiral transition state of 2b is stabilised relative to the homochiral transition state in the Curtin–Hammett-controlled macrocycle synthesis (Fig. 5). Indeed, a ΔΔG‡ = −6.3 kJ mol−1 is of the order of the binding free energy of donor–acceptor pairs such as dialkoxy naphthalene–naphthalene diimide in a chlorinated solvent (ΔG = −9.3 kJ mol−1),42 which supports our theory of the importance of aromatic-based non-covalent interactions. To provide further evidence for supramolecular templation, we repeated the synthesis of macrocycle 2b in competitive solvents for π–π donor–acceptor interactions and, as expected, found that the 4:1 PP:PP ratio decreases both upon increasing solvent polarity (dr = 3:1 in DMF, Fig. S7†), which tempers electrostatic contributions, and upon the addition of an aromatic solvent (dr = 3:1 in 1:1 toluene:DCM, Fig. S6†).‡‡
Conclusions
We have used the Curtin–Hammett principle to investigate a chiral macrocyclisation reaction, revealing the potential for intramolecular aromatic non-covalent interactions to direct the outcome of a dynamic kinetic resolution, in this case the diastereoselective synthesis of a macrocycle containing twisted aromatic units (2b). A racemic and dynamically chiral PDI dye (1:1 P:M) is made configurationally stable by its strapping with an enantiopure P-BINOL derivative under irreversible reaction conditions. Therefore, in contrast to previous diastereomeric PDI-based macrocycles,22a–c,23 both the macrocyclisation reaction and products are under kinetic control, enabling the facile isolation of heterochiral and homochiral macrocycles in high enantiopurities. Notably, instead of a 1:1 mixture of macrocycle diastereomers,19a,43 there is diastereoselectivity for the heterochiral (PM-2b) over homochiral (PP-2b) product (dr = 4:1). From Curtin–Hammett analysis, the origin of this diastereoselectivity is the lower transition state energy of the hetero- vs. homo-chiral macrocycle (ΔΔG‡ = 6.3 kJ mol−1), with experimental and computational studies revealing that stabilising π–π and CH–π interactions are predominant in PM-2b, indicative of a kinetic template effect.9a
While homochirality appears to dominate in dimeric assemblies of PDIs,22a,23,32a,d a preference for heterochiral π–π stacking has recently been reported in PDI-based complexes of polycyclic aromatic hydrocarbons19e and a BINOL-perylene cyclophane22c where, as in our work here, it is notable that the π-conjugated units in the π–π stack are different. Overall, this suggests that the selection of equivalent or distinct aromatic units is an important consideration when designing new templated syntheses of homo- or hetero-chiral π-conjugated materials. The use of aromatic non-covalent interactions to direct the synthesis of such atropisomeric π-conjugated materials22a,44 under irreversible conditions is desirable for preparing preorganised chiral receptors45 or persistent chiroptical materials3 or, as found here, for tuning selectivity for a chiral product that may not be favoured under thermodynamically controlled conditions. This is leading us to develop the stereoselective synthesis of new supramolecular architectures for chiral sensing and the emission/detection of circularly polarised light.
Data availability
All synthetic method descriptions, product characterisation data, spectroscopic data and other supplementary figures and tables (NMR spectra, UV-vis spectra, circular dichroism spectra, chiral HPLC chromatograms, etc.), crystallographic information, and computational data can be found in the ESI.† The relevant computational structures are available in the folder named “Hetero_homochiral_macrocycles_structures”.Author contributions
A. Y. and T. A. B. conceptualisation, writing and editing; A. Y. investigation and analysis of all synthetic and spectroscopic data (NMR, UV-vis, CD, chiral HPLC), visualisation (figures), and writing of ESI;† M. A. Z. performed all computational studies and wrote the computational sections; G. R. F. O. processed and solved all crystallographic data, and wrote the crystallographic sections; J. H. R. crystallographic data collection; and T. A. B. writing of original manuscript draft, supervision and funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. A. B. acknowledges support from the University of Birmingham, an RSC Research Enablement Grant (E20-4166) and the UK Engineering and Physical Sciences Research Council (EP/W037661/1). G. R. F. O. acknowledges the support of the UK Engineering and Physical Sciences Research Council (EP/S002995/2).Notes and references
- (a) E. Francotte and W. Lindner, Chirality in Drug Research, John Wiley & Sons, Weinheim, Germany, 2007 Search PubMed; (b) V. Andrushko and N. V. Andrushko, Stereoselective synthesis of drugs and natural products, John Wiley & Sons, Inc., Hoboken, NJ, 2013 Search PubMed; (c) T. T. Talele, J. Med. Chem., 2020, 63, 13291–13315 CrossRef CAS PubMed.
- (a) T. Hirao, S. Kishino and T. Haino, Chem. Commun., 2023, 59, 2421–2424 RSC; (b) M. Quan, X. Y. Pang and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201258 CrossRef CAS PubMed; (c) L. Pu, Acc. Chem. Res., 2012, 45, 150–163 CrossRef CAS PubMed; (d) Z. Chen, Q. Wang, X. Wu, Z. Li and Y. B. Jiang, Chem. Soc. Rev., 2015, 44, 4249–4263 RSC.
- (a) M. C. Carreno, A. Enriquez, S. Garcia-Cerrada, M. J. Sanz-Cuesta, A. Urbano, F. Maseras and A. Nonell-Canals, Chem.–Eur. J., 2008, 14, 603–620 CrossRef CAS PubMed; (b) J. Wade, F. Salerno, R. C. Kilbride, D. K. Kim, J. A. Schmidt, J. A. Smith, L. M. LeBlanc, E. H. Wolpert, A. A. Adeleke, E. R. Johnson, J. Nelson, T. Mori, K. E. Jelfs, S. Heutz and M. J. Fuchter, Nat. Chem., 2022, 14, 1383–1389 CrossRef CAS PubMed; (c) C. Liu, J. C. Yang, J. W. Y. Lam, H. T. Feng and B. Z. Tang, Chem. Sci., 2022, 13, 611–632 RSC; (d) J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS; (e) T. Ikai, K. Oki, S. Yamakawa and E. Yashima, Angew. Chem., Int. Ed., 2023, 62, e202301836 CrossRef CAS PubMed; (f) J. Crassous, M. J. Fuchter, D. E. Freedman, N. A. Kotov, J. Moon, M. C. Beard and S. Feldmann, Nat. Rev. Mater., 2023, 8, 365–371 CrossRef.
- S. F. Ozturk and D. D. Sasselov, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204765119 CrossRef CAS PubMed.
- (a) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed; (b) J. M. Ovian, P. Vojackova and E. N. Jacobsen, Nature, 2023, 616, 84–89 CrossRef CAS PubMed; (c) S. Das, C. Zhu, D. Demirbas, E. Bill, C. K. De and B. List, Science, 2023, 379, 494–499 CrossRef CAS PubMed.
- (a) F. S. Menke, B. Wicher, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2023, 62, e202217325 CrossRef CAS PubMed; (b) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- R. L. Furlan, S. Otto and J. K. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801–4804 CrossRef CAS PubMed.
- (a) F. S. Menke, B. Wicher, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2023, 62, e202217325 CrossRef CAS PubMed; (b) Y. Zuo, X. Liu, E. Fu and S. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202217225 CrossRef CAS PubMed.
- (a) S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469–475 CrossRef CAS; (b) J. H. van Maarseveen, M. D. Cornelissen and S. Pilon, Synthesis, 2021, 53, 4527–4548 CrossRef; (c) J.-C. Chambron and J.-P. Sauvage, New J. Chem., 2013, 37, 49–57 RSC.
- Macrocyclic and Supramolecular Chemistry: How Izatt–Christensen Award Winners Shaped the Field, ed. R. M. Izatt, John Wiley & Sons, 2016 Search PubMed.
- (a) J. I. Seeman, Chem. Rev., 2002, 83, 83–134 CrossRef; (b) J. I. Seeman, J. Chem. Educ., 1986, 63, 42 CrossRef CAS.
- (a) P. Beak, A. Basu, D. J. Gallagher, Y. S. Park and S. Thayumanavan, Acc. Chem. Res., 1996, 29, 552–560 CrossRef CAS; (b) G. Fodor, R. V. Chastain, D. Frehel, M. J. Copper, N. Mandava and E. L. Gooden, J. Am. Chem. Soc., 2002, 93, 403–413 CrossRef.
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- (a) S. Amano, M. Esposito, E. Kreidt, D. A. Leigh, E. Penocchio and B. M. W. Roberts, J. Am. Chem. Soc., 2022, 144, 20153–20164 CrossRef CAS PubMed; (b) L. Binks, S. Borsley, T. R. Gingrich, D. A. Leigh, E. Penocchio and B. M. W. Roberts, Chem, 2023, 9, 2902–2917 CrossRef CAS; (c) M. Alvarez-Perez, S. M. Goldup, D. A. Leigh and A. M. Slawin, J. Am. Chem. Soc., 2008, 130, 1836–1838 CrossRef CAS PubMed.
- J. Li, P. Li, M. Fan, X. Zheng, J. Guan and M. Yin, Angew. Chem., Int. Ed., 2022, 61, e202202532 CrossRef CAS PubMed.
- P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129, 14319–14326 CrossRef CAS PubMed.
- (a) Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845–5855 RSC; (b) F. Würthner, J. Org. Chem., 2022, 87, 1602–1615 CrossRef PubMed.
- (a) N. Pearce, K. E. A. Reynolds, S. Kayal, X. Z. Sun, E. S. Davies, F. Malagreca, C. J. Schurmann, S. Ito, A. Yamano, S. P. Argent, M. W. George and N. R. Champness, Nat. Commun., 2022, 13, 415 CrossRef CAS PubMed; (b) P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- (a) M. Weh, J. Ruhe, B. Herbert, A. M. Krause and F. Würthner, Angew. Chem., Int. Ed., 2021, 60, 15323–15327 CrossRef CAS PubMed; (b) M. Sapotta, P. Spenst, C. R. Saha-Möller and F. Würthner, Org. Chem. Front., 2019, 6, 892–899 RSC; (c) T. A. Barendt, M. L. Ball, Q. Xu, B. Zhang, B. Fowler, A. Schattman, V. C. Ritter, M. L. Steigerwald and C. Nuckolls, Chem.–Eur. J., 2020, 26, 3744–3748 CrossRef CAS PubMed; (d) T. A. Barendt, W. K. Myers, S. P. Cornes, M. A. Lebedeva, K. Porfyrakis, I. Marques, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2020, 142, 349–364 CrossRef CAS PubMed; (e) M. Weh, K. Shoyama and F. Würthner, Nat. Commun., 2023, 14, 243 CrossRef CAS PubMed; (f) T. Turel, S. Bhargava and S. Valiyaveettil, J. Org. Chem., 2020, 85, 3092–3100 CrossRef CAS PubMed.
- (a) Y. Hou, Z. Zhang, S. Lu, J. Yuan, Q. Zhu, W. P. Chen, S. Ling, X. Li, Y. Z. Zheng, K. Zhu and M. Zhang, J. Am. Chem. Soc., 2020, 142, 18763–18768 CrossRef CAS PubMed; (b) Y. Hou, Z. Zhang, L. Ma, R. Shi, S. Ling, X. Li, G. He and M. Zhang, CCS Chem., 2022, 4, 2604–2611 CrossRef CAS; (c) I. Heckelmann, Z. Lu, J. C. A. Prentice, F. Auras, T. K. Ronson, R. H. Friend, J. R. Nitschke and S. Feldmann, Angew. Chem., Int. Ed., 2023, 62, e202216729 CrossRef CAS PubMed.
- (a) M. Wehner, M. I. S. Rohr, V. Stepanenko and F. Würthner, Nat. Commun., 2020, 11, 5460 CrossRef CAS PubMed; (b) T. Pal and D. Chaudhuri, J. Am. Chem. Soc., 2023, 145, 2532–2543 CrossRef CAS PubMed.
- (a) I. Solymosi, S. Krishna, E. Nuin, H. Maid, B. Scholz, D. M. Guldi, M. E. Perez-Ojeda and A. Hirsch, Chem. Sci., 2021, 12, 15491–15502 RSC; (b) M. Ball, B. Fowler, P. Li, L. A. Joyce, F. Li, T. Liu, D. Paley, Y. Zhong, H. Li, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2015, 137, 9982–9987 CrossRef CAS PubMed; (c) G. Ouyang, J. Ruhe, Y. Zhang, M. J. Lin, M. Liu and F. Würthner, Angew. Chem., Int. Ed., 2022, 61, e202206706 CrossRef CAS PubMed; (d) M. Weh, A. A. Kroeger, K. Shoyama, M. Grüne, A. Karton and F. Würthner, Angew. Chem., Int. Ed., 2023, 62, e202301301 CrossRef CAS PubMed.
- S. E. Penty, M. A. Zwijnenburg, G. R. F. Orton, P. Stachelek, R. Pal, Y. Xie, S. L. Griffin and T. A. Barendt, J. Am. Chem. Soc., 2022, 144, 12290–12298 CrossRef CAS PubMed.
- (a) S. Anderson, U. Neidlein, V. Gramlich and F. Diederich, Angew. Chem., Int. Ed., 2003, 34, 1596–1600 CrossRef; (b) J.-T. Hou, Q.-F. Zhang, B.-Y. Xu, Q.-S. Lu, Q. Liu, J. Zhang and X.-Q. Yu, Tetrahedron Lett., 2011, 52, 4927–4930 CrossRef CAS.
- T. Shimada, A. Kina and T. Hayashi, J. Org. Chem., 2003, 68, 6329–6337 CrossRef CAS PubMed.
- (a) C. B. Roos, C. H. Chiang, L. A. M. Murray, D. Yang, L. Schulert and A. R. H. Narayan, Chem. Rev., 2023, 123, 10641–10727 CrossRef CAS PubMed; (b) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- L. Meca, D. Reha and Z. Havlas, J. Org. Chem., 2003, 68, 5677–5680 CrossRef CAS PubMed.
- (a) S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed; (b) M. Mancinelli, G. Bencivenni, D. Pecorari and A. Mazzanti, Eur. J. Org Chem., 2020, 2020, 4070–4086 CrossRef CAS; (c) J. P. Heeb, J. Clayden, M. D. Smith and R. J. Armstrong, Nat. Protoc., 2023, 18, 2745–2771 CrossRef CAS PubMed.
- (a) M. M. Safont-Sempere, P. Osswald, K. Radacki and F. Würthner, Chem.–Eur. J., 2010, 16, 7380–7384 CrossRef CAS PubMed; (b) R. Renner, B. Mahlmeister, O. Anhalt, M. Stolte and F. Würthner, Chem.–Eur. J., 2021, 27, 11997–12006 CrossRef CAS PubMed; (c) Z. Xie and F. Würthner, Org. Lett., 2010, 12, 3204–3207 CrossRef CAS PubMed; (d) M. M. Safont-Sempere, P. Osswald, M. Stolte, M. Grüne, M. Renz, M. Kaupp, K. Radacki, H. Braunschweig and F. Würthner, J. Am. Chem. Soc., 2011, 133, 9580–9591 CrossRef CAS PubMed; (e) P. Osswald, M. Reichert, G. Bringmann and F. Würthner, J. Org. Chem., 2007, 72, 3403–3411 CrossRef CAS PubMed.
- J. G. Brandenburg, C. Bannwarth, A. Hansen and S. Grimme, J. Chem. Phys., 2018, 148, 064104 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- D. C. Patel, R. M. Woods, Z. S. Breitbach, A. Berthod and D. W. Armstrong, Tetrahedron: Asymmetry, 2017, 28, 1557–1561 CrossRef CAS.
- (a) D. Ley, C. X. Guzman, K. H. Adolfsson, A. M. Scott and A. B. Braunschweig, J. Am. Chem. Soc., 2014, 136, 7809–7812 CrossRef CAS PubMed; (b) S. Rieth, Z. Li, C. E. Hinkle, C. X. Guzman, J. J. Lee, S. I. Nehme and A. B. Braunschweig, J. Phys. Chem. C, 2013, 117, 11347–11356 CrossRef CAS; (c) A. G. Phillips, L. M. Perdigao, P. H. Beton and N. R. Champness, Chem. Commun., 2010, 46, 2775–2777 RSC.
- (a) V. Marti-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef CAS PubMed; (b) M. A. Casadei, C. Galli and L. Mandolini, J. Am. Chem. Soc., 2002, 106, 1051–1056 CrossRef.
- S. J. Howell, P. R. Ashton, N. Spencer and D. Philp, Org. Lett., 2001, 3, 353–356 CrossRef CAS PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- (a) T. A. Barendt, L. Ferreira, I. Marques, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 9026–9037 CrossRef CAS PubMed; (b) P. Spenst, A. Sieblist and F. Würthner, Chem.–Eur. J., 2017, 23, 1667–1675 CrossRef CAS PubMed; (c) Z. Wang, X. Gou, Q. Shi, K. Liu, X. Chang, G. Wang, W. Xu, S. Lin, T. Liu and Y. Fang, Angew. Chem., Int. Ed., 2022, 61, e202207619 CrossRef CAS PubMed.
- M. Žabka, L. Naviri and R. M. Gschwind, Angew. Chem., Int. Ed., 2021, 60, 25832–25838 CrossRef PubMed.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- A. Mateo Alonso, R. Horcajada, H. J. Groombridge, R. Chudasama Nee Mandalia, M. Motevalli, J. H. Utley and P. B. Wyatt, Org. Biomol. Chem., 2005, 3, 2832–2841 RSC.
- (a) K. Uehara, H. Kano, K. Matsuo, H. Hayashi, M. Fujiki, H. Yamada and N. Aratani, ChemPhotoChem, 2021, 5, 974–978 CrossRef CAS; (b) L. A. de Ceuninck van Capelle, J. M. Macdonald and C. J. T. Hyland, Org. Biomol. Chem., 2021, 19, 7098–7115 RSC.
- (a) S. M. Wang, Y. F. Wang, L. Huang, L. S. Zheng, H. Nian, Y. T. Zheng, H. Yao, W. Jiang, X. Wang and L. P. Yang, Nat. Commun., 2023, 14, 5645 CrossRef CAS PubMed; (b) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed.
- A. Li, X. Zhang, S. Wang, K. Wei and P. Du, Org. Lett., 2023, 25, 1183–1187 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2249179. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05715a |
| ‡ Throughout, we use the chiral nomenclature of P/M for PDIs, which is based on the Cahn–Ingold–Prelog priority rules for molecules with axial chirality.23,46 This ensures clear comparisons against the axial chirality of BINOL (P/M). |
| § Since this hydrogen bond involves a triazole heterocycle which is formed during the ring-closing of the macrocycle it is less likely to contribute towards differences in stability of the macrocyclisation transition state (Fig. S28†). |
| ¶ Unfortunately, the use of shorter or longer linkers outside of this window did not lead to the formation of stable [1 + 1] macrocycles in an isolable yield. |
| || We note that the absorption spectra of 2b follow the Beer–Lambert law (Fig. S21†), indicating that the π–π donor–acceptor interaction in PM-2b is intramolecular in origin. |
| ** We note that improved π-conjugation arising from increased chromophore planarity would also lead to red-shifted UV-vis spectra, yet this is not consistent with the computational structures which indicate that the bis-triazole PDI is more twisted in PM-2b relative to PP-2b (Fig. S29†). |
| †† Relative to 5, the acyclic control compound 4 does not exhibit a red-shifted PDI absorption band (Fig. 7a), which explains the absence of a CD spectrum in the PDI-only region of 4 (Fig. 4d). |
| ‡‡ The solubility of starting materials dictated the need for a 1:1 toluene:DCM solvent mixture. |
| This journal is © The Royal Society of Chemistry 2024 |