
Predictive design of multimonomeric polyelectrolytes enables lung-specific gene delivery†
Jeffrey M.
Ting
 *,
John D.
Fisher
,
Tyler
Conyers
,
Suteja
Patil
,
Catherine G.
Robohn
,
Teresa
Tamayo-Mendoza
,
Felipe
Oviedo
and
Shashi K.
Murthy
*,
John D.
Fisher
,
Tyler
Conyers
,
Suteja
Patil
,
Catherine G.
Robohn
,
Teresa
Tamayo-Mendoza
,
Felipe
Oviedo
and
Shashi K.
Murthy
Nanite, Inc., Boston, Massachusetts 02109, USA. E-mail: jeff@nanitebio.com
First published on 22nd May 2024
Abstract
We present a class of programmable polymer nanoparticles capable of intravenous pDNA delivery with ∼350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold enhancement of specificity to the lung by qPCR in mice. Molecular level insight of these polyelectrolytes is connected to bioperformance, exemplifying how robust, data-driven workflows can expedite synthesis/screening campaigns for gene therapy.
000-fold enhancement of specificity to the lung by qPCR in mice. Molecular level insight of these polyelectrolytes is connected to bioperformance, exemplifying how robust, data-driven workflows can expedite synthesis/screening campaigns for gene therapy.
The scarcity of safe, effective delivery technologies is the single largest barrier to the development of a new wave of genetic therapies. Significant shortcomings exist in current modalities including immunogenicity and payload size limitations for viral vectors,1 and manufacturing and intellectual property constraints for lipid nanoparticles.2 Furthermore, the vast majority of nonviral delivery systems are cleared by the liver via hepatic clearance after systemic administration,3 presenting significant challenges for non-hepatic gene delivery. This feature can be somewhat mitigated by controlling the nanocarriers’ physical characteristics (such as size/shape4 or PEGylation5), adopting active targeting strategies,6 or by exploiting circulating plasma protein interactions in vivo.7 However, it remains unclear how a genetic cargo packed within heterogeneous nanocarriers overcomes extracellular barriers to reach the intended site of action. This biodistribution challenge has been well recognized in nanomedicine for decades.8 To expand tropism outcomes to clinically relevant gene therapy targets such as the spleen,9 lungs,10 heart,11 eye,12 or nervous system,13 greater investigation of the biological consequences from the integrated chemical and biophysical features of nanoparticles is needed.
Polymer nanoparticles (PNPs, or polyplexes formed from tailored cationic polymers and therapeutic nucleic acids) offer distinct advantages as multifunctional delivery systems.14,15 Advanced polymer chemistry techniques now offer access to nearly any conceivable chain structure and architecture from a diverse array of building blocks.16,17 Despite these synthetic advances, there remain two major limitations to wider adoption of polymers as gene therapy excipients. First, the chemical design space of organic small molecules relevant to drug delivery18 is estimated to be on the order of 1060— a vast number that, even if reduced to more surmountable subsets via exhaustive data mining, cannot be practically interrogated with the current state of polymer informatics.19 Second, in vitro cellular assays, while amenable to high-throughput screening for highly valuable transfection data across cell types,20,21 often provide poor predictions on the crossing of complex physiological barriers in vivo.22,23 Thus, unclear in vitro–in vivo correlation results represent a difficult obstacle in PNP optimization in translating genetic drugs from cellular level assays to living systems.
In this communication, we report the discovery of a three-component polyelectrolyte system that was identified while developing PNP candidates for localization to the lung. Nanite's proprietary platform SAYER™ couples experimental data generation with artificial intelligence (AI) to identify high-performing PNPs for efficient delivery of diverse genetic cargo to tissues outside of the liver24 (Fig. 1). The details of how PNP complexation, in vitro, and in vivo probabilistic models predict ex-hepatic delivery from the vast polymer design space are described elsewhere.25,26 Large chemical, materials, and biological datasets are stored and used for PNP down-selection and screening in our general workflow. We focus here on developing a mechanistic understanding of this polyelectrolyte candidate, and establish structure–property relationships that support nonintuitive design strategies for potent therapeutic PNPs with lung specificity. This framework further reveals new AI opportunities for universal tissue-specific gene delivery.
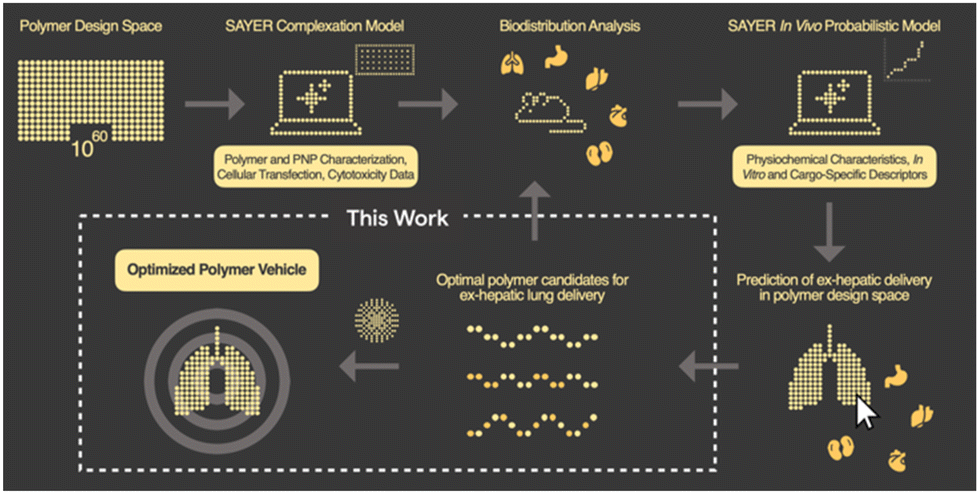 | ||
| Fig. 1 Overview of designing tissue-specific gene delivery polymer nanoparticles using data-driven workflows and prediction from in vivo screening. This work shows how lead polymer candidates are characterized and optimized for lung delivery. | ||
The conventional paradigm to design polymeric systems for gene delivery is to combine cationic interactions offered by alkyl substituted amine,27,28 imidazolium,29,30 guanidinium,31,32 or even non-nitrogenous monomers (e.g., sulfonium,33,33 phosphonium34,35) with hydrophilic, hydrophobic, or stimuli-responsive comonomers.36–39 The general rationale is to balance trends in charge type and density trends against complementary polymer/cargo associations in solution. In the current work, we took a broader approach by generating not only two-component polyelectrolytes to test traditional PNP hypotheses, but also multimonomeric statistical analogs, which are known to offer rich (and sometimes, unintuitive) dynamics in solution.40,41 By scanning a more diverse landscape of multimonomeric polyelectrolytes, we aimed to determine whether unique PNP assemblies can be trafficked to the lung with minimal liver accumulation via systemic administration. Lung-specific delivery is often attributed to the entrapment of large nanoparticles in pulmonary capillaries,4,42 a passive targeting approach that is difficult to recapitulate in vitro. In the present work we aimed to rigorously generate polymers that do not rely solely on passive targeting to deliver pDNA to the lung.
In the current work, aqueous reversible addition–fragmentation chain transfer (RAFT) polymerization43,44 was employed to prepare a prototypical library via automated liquid handling.45,46 After screening over 1300 polymers synthesized in house and digitally labelled for training AI models from PNP characterization, in vitro assays, and in vivo biodistribution data,24 unique monomer candidates emerged for plasmid DNA (pDNA) delivery, based on N-(3-aminopropyl)methacrylamide hydrochloride (APMAm), 2-(trimethylammonio)ethyl methacrylate chloride (TMAEMA), and 3-phenoxy-2-hydroxypropyl methacrylate (PhHPMA). APMAm offers a pH-responsive primary amine that is expected to bind strongly to nucleic acids. TMAEMA exhibits a permanently charged ammonium group that can assist with cellular uptake. PhHPMA provides complementary non-covalent interactions in hydrophobicity and hydrogen bonding through an aromatic ring and hydroxyl group. However, it was unclear how to combine them at the nanoscopic level to assemble with pDNA into PNP candidates for lung delivery.
To further characterize the properties and behavior of the APMAm/TMAEMA/PhHPMA polymer, a series of six representative samples were prepared (Scheme 1). For close consistency with the degree of polymerization (DP) and compositions of a polymer library investigated by Kumar et al.,25 we targeted DP = 80 using thermal initiator VA-044 and RAFT chain transfer agents 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (CTA1) and poly(ethylene glycol) (PEG) 4-cyano-4-(phenylcarbonothioylthio) pentanoate (CTA2) at a 10:1 molar ratio for 18 h. PEG coatings in micellar PNPs are known to inhibit aggregation in the blood and prolong circulation.47 The inclusion of PEG allows for modulation of colloidal nanoparticle sizes from known physical scaling laws of polyelectrolyte complex micelles,48 where the charged block length predominately drives its core size. PEGylated PNPs can condense pDNA payloads into 10–100 nm discrete domains depending on chemistry and solution conditions.49 As shown in Table 1, we varied APMAm/PhHPMA ratios at fixed TMAEMA for non-PEGylated polymers (P1, P2, and P3) and PEGylated block polymers (P4, P5, and P6) so that the total charge (i.e., APMAm + TMAEMA) was 75, 50, and 25 mol% relative to neutral PhHPMA. By aqueous size-exclusion chromatography PEGylated samples show a reasonable molar mass (Mn ∼ 10–20 kg mol−1 relative to PEG standards) with reasonable dispersity (Đ ≤ 1.4). We were unable to quantify the molar mass of P1–P3 due to known solubility challenges43,44 of multicomponent water-soluble polymers and column interactions.
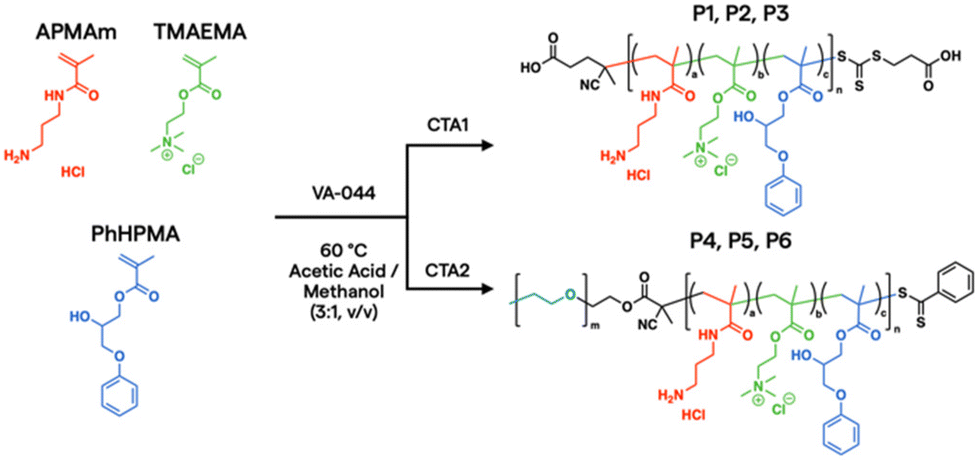 | ||
| Scheme 1 RAFT synthesis of multimonomeric polyelectrolytes. | ||
| ID | PEG (DP) | Polyelectrolyte (DP)a | M n (kg mol−1) | Đ | ||
|---|---|---|---|---|---|---|
| APMAm | TMAEMA | PhHPMA | ||||
| a Based on targeted feed ratio. b Targeted molecular weight; samples not fully soluble in tested aqueous mobile phase. | ||||||
| P1 | 0 | 52 | 8 | 20 | 14.1b | — |
| P2 | 0 | 32 | 8 | 40 | 15.1b | — |
| P3 | 0 | 12 | 8 | 60 | 16.2b | — |
| P4 | 45 | 52 | 8 | 20 | 9.86 | 1.3 |
| P5 | 45 | 32 | 8 | 40 | 10.4 | 1.4 |
| P6 | 45 | 12 | 8 | 60 | 19.9 | 1.3 |
Because reactions were carried out to near complete conversion, we chose to investigate the statistical distribution of monomers by measuring pairwise reactivity ratio (r) values and evaluating composition drift effects.50 This approach has previously demonstrated utility in characterizing multimonomeric RAFT polymers that can impart various non-covalent interactions.51,52 Three standard radical polymerization runs were conducted for each monomer combination, keeping total monomer conversion below ∼15%. The monomers’ conversion from the initial feed composition (f) and polymer composition (F) were assessed by 1H NMR spectroscopy (see ESI†). We fitted the data directly to the Copolymerization equation F1 = (r12f12 + f1f2)/(r12f12 + 2f1f2 + r21f22). We further constructed Mayo–Lewis plots (Fig. S3†) and determined the r values under these specific reaction conditions to be rAPMAm-TMAEMA = 0.38 ± 0.02, rAPMAm-PhHPMA = 1.01 ± 0.32, rTMAEMA-APMAm = 0.14 ± 0.02, rTMAEMA-PhHPMA = 0.52 ± 0.16, rPhHPMA-APMAm = 1.59 ± 0.51, and rPhHPMA-TMAEMA = 0.13 ± 0.08.
Determination of r values allows us to describe the polymers’ microstructure and reveal the consequences of compositional drift from carrying out RAFT polymerizations to high conversion. Smith et al. developed a practical visualization tool using Monte Carlo methods from measured r values.53 This compositional drift program uses the relative reactivities from r values, adding one repeat unit from a large pool of monomers (set to 200000) to growing chains per activation/deactivation cycle. Fig. 2A–C shows the results for P1, P2, and P3. For the charged monomers, we observe that TMAEMA (green) are quickly incorporated into polymer chains as single units, whereas APMAm (red) are distributed throughout the chain. For instance, the instantaneous polymer composition of P2 at 10% and 50% conversion are 0.37/0.26/0.37 and 0.36/0.06/0.58 mole fractions of APMAm/TMAEMA/PhHPMA, respectively. It is also clear that depending on the targeted composition seen in P1 and P3, the APMAm and PhHPMA blockiness run lengths can be modulated. Unreacted TMAEMA monomers are quickly consumed as the target composition decreases, whereas APMAm is added throughout monomer conversion (Fig. 2D). This observation implies that ammonium moieties are physically localized in the resultant chains’ microstructure because of the relative reactivities of monomers under these reaction conditions. For PEGylated polymers, TMAEMA units are incorporated into the chain closer to the PEG block.
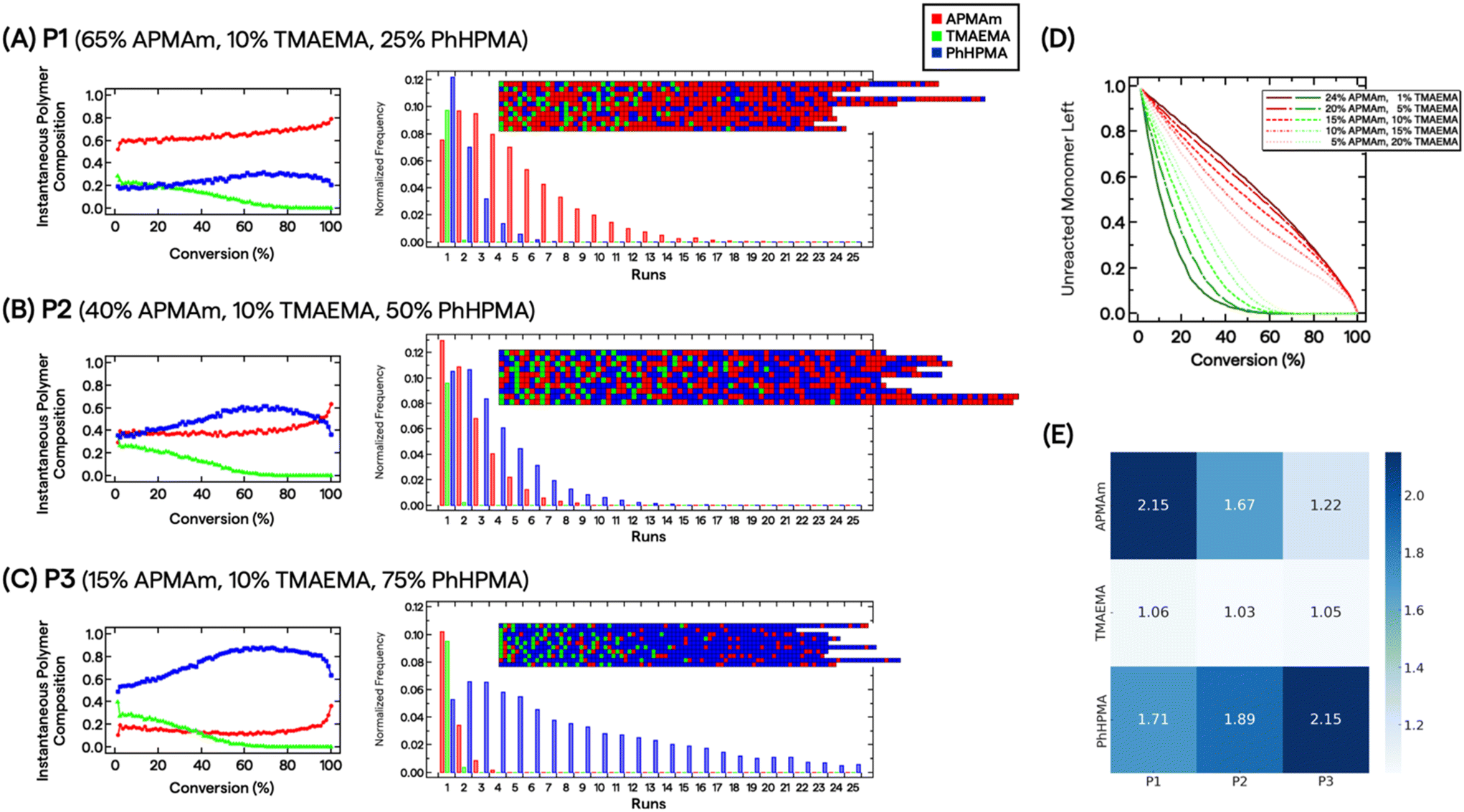 | ||
| Fig. 2 Predicted distribution of APMAm (red), TMAEMA (green), and PhHPMA (blue) using the compositional drift program for (A) P1, (B) P2, and (C) P3 from determined reactivity ratios. In (A)–(C) on the left, plots show the instantaneous polymer composition as a function of monomer conversion; on the right, Monte Carlo simulation results show the normalized frequency of monomers as a function of run lengths; in the inset ten representative simulated chains are shown from a pool of 200000 monomers. (D) For simulated P3 chains, the effect of varying the APMAm/TMAEMA charge composition for the unreacted monomers remains tunable as a function of monomer conversion. (E) Heatmap array plot shows the calculated Shannon entropy of constituent monomers (left) for corresponding polymers P1–P3 (bottom); darker blue indicates higher entropy. | ||
Based on the multiple chain instantiations of the Monte Carlo simulation,53 we computed the probability of a monomer X polymerized in a chain position i as p(Xi). Thus, for monomer j in polymer P, the Shannon entropy of the monomer is given by: 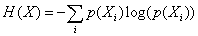 . The Shannon entropy will be higher for constitutional monomers that are randomly distributed across the polymer. Fig. 2E presents the calculated entropies for each monomer in each polymer. TMAEMA shows the lowest entropy across P1, P2 and P3, confirming its physical localization and early depletion in the resultant chain microstructures.
. The Shannon entropy will be higher for constitutional monomers that are randomly distributed across the polymer. Fig. 2E presents the calculated entropies for each monomer in each polymer. TMAEMA shows the lowest entropy across P1, P2 and P3, confirming its physical localization and early depletion in the resultant chain microstructures.
Such insights are challenging to obtain experimentally but may offer a unique feature of multimonomeric polyelectrolytes’ ability to control complexation dynamics. From random phase approximation theory and molecular dynamics simulations, random polyelectrolyte sequences are expected to have a dramatic effect on the cooperativity of Coulomb interactions between oppositely charged macromolecules.54,55 While we do not dwell on polyelectrolyte phase behavior in this current work, compositional drift can potentially offer more fundamental understanding of the statistical distribution of charges in multimonomeric polymers, for not only gene therapy but also wastewater treatment/purification strategies,56 viral vaccine formulation,57 and enhancing viscoelastic response in coacervation.58 Here, we hypothesize that blocky attributes of P1 and P3 (as well as their PEGylated analogs P4 and P6) can influence not only pDNA stabilization, but also blood plasma protein interactions, distinguishing them from random binary polyelectrolytes.
We next formed PNPs with enhanced-green fluorescent protein (EGFP)-encoding pDNA and evaluated complexation. Ionic strength and buffer additives are known to affect polyelectrolyte assembly formation and stability of phase separation over time.49,59 To this end, all PNPs were prepared at N/P (the ratio of the nitrogen on polymer to the phosphate on nucleotide) of 5 as a starting point from previous work25 for structural analysis under various solution conditions. We mixed P1–P6 with pDNA to form nanoparticles PNP1–PNP6. The size distributions of PNP1–PNP6 were characterized using dynamic light scattering (DLS) in an automated plate reader as a function of added salt (50, 100, and 250 mM NaCl) and sugar (0, 5, 10% glucose) after 30 min equilibration. Adding salt and sugar provides annealing60 and isotonicity61 for bioapplications, respectively. The apparent mean hydrodynamic radius (Rh) and polydispersity (PDI) were determined from a cumulant fitting to DLS autocorrelation functions that exhibited a single decay. Fig. 3 shows a representative size comparison of PNPs as a function of NaCl at 25 °C. Non-PEGylated PNP1–3 showed evidence of aggregation from 50 to 100 mM NaCl, followed by size reduction at 250 mM NaCl suggesting complex disassembly. We therefore excluded these from consideration for preliminary in vivo testing. By comparison, the mean Rh of PEGylated PNP4–6 grew by ∼15–20% as NaCl increased from 50 to 250 mM. Of these samples, PNP5 and PNP6 were the smallest (70–90 nm) and most salt resistant (PDI 0.3–0.4). Overall size and stability did not vary strongly as a function of added sugar, temperature, or CaCl2, a divalent salt that disrupts short-range cation–π interactions.59,62 To make a decision on prioritizing PNP testing in vivo, we performed high-content analysis of conventional HEK293T cells across all PNPs as a function of N/P = 1.25 to 40 (Fig. S18†). The mean effective transfection capability by EGFP expression in vitro was demonstrated for PNP5 (14% at N/P = 2.5) and PNP6 (19% at N/P = 20), with PNP6 showing less cytotoxicity at higher N/P (Fig. S19†). Because of the multiplicity of PNP properties that may impact in vitro translation to in vivo delivery to the lung, we limit discussions of transfection here and direct interested readers to the ESI† for full details.
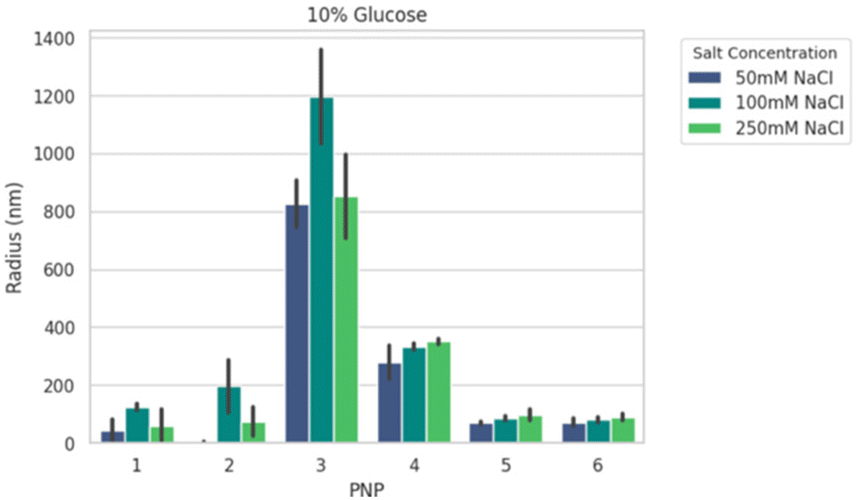 | ||
| Fig. 3 Representative summary of the apparent radius of PNP1–PNP6 with 50, 100, and 250 mM added NaCl at 10% added glucose at 25 °C. All values show the mean + standard deviation of 15 acquisitions from dynamic light scattering. | ||
With an understanding of polymer features, PNP properties, and trained AI models, we down selected PNP6 to be tested as a proof-of-concept for in vivo biodistribution of pDNA (5 μg dose) in 10 mice via intravenous administration. After 6 h, major organs (liver, lung, spleen, heart, and kidney) were harvested and analyzed. Fig. 4A shows the biodistribution from processed organs using real-time quantitative polymerase chain reaction (qPCR). We analyzed gene expression using the conventional relative quantification (RQ) or 2(−ΔΔCT) method.63 Here, 2(−ΔΔCT) is defined as the fold change in pDNA delivered between the treatment group and a control group, in this case, PBS buffer only. See the ESI† for full qPCR analysis details. In vivo-JetPEI, a commercial gold standard polyethylenimine optimized for in vivo gene delivery, served as the positive control. While the in vivo-JetPEI PNP exhibited broad gene delivery in the liver, lung, and spleen, PNP6 delivered pDNA cargo near exclusively to the lung with a ∼350000-fold enhancement of specificity. Fig. 4B presents a linear comparison of this enhancement in the exponential gene delivery process to illustrate the remarkable ex-hepatic delivery of PNP6 to the lung. PNP6 outperforms in vivo-JetPEI by a factor of 4 with statistical significance (p < 0.05). High lung-selectivity is also shown relative to all other major organs (Fig. S17).
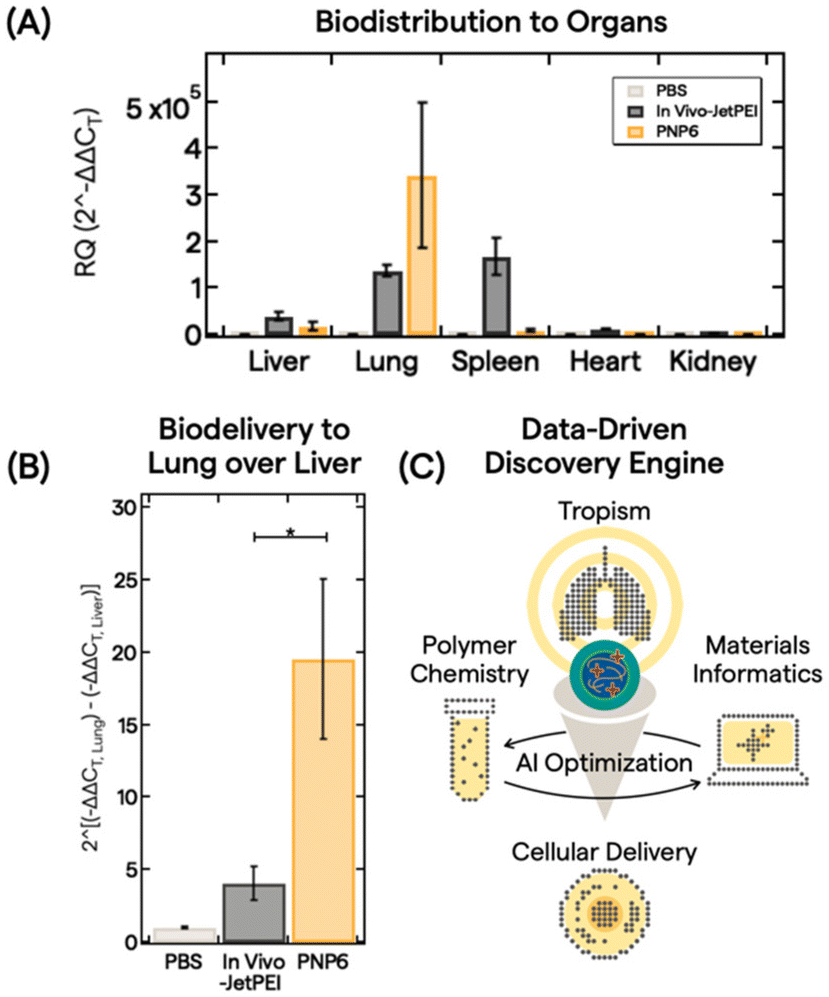 | ||
| Fig. 4 Delivery of PNP6 in vivo. (A) Biodistribution data of PBS (light gray), In Vivo-JetPEI (dark gray), and PNP6 (dark yellow) in major organs (liver, lung, spleen, heart, and kidney) in mice. pDNA tissue accumulation was quantified by qPCR DNA amplification using the 2^(−ΔΔCT) method for analyzing the fold change in pDNA delivery. All values show the mean + standard error of the mean for N = 3 (PBS, JetPEI) or N = 4 (PNP6). (B) Evaluation of ex-hepatic delivery with the 2^(−ΔΔCT) method by taking the mean difference between the −ΔΔCT lung and the −ΔΔCT liver values for PBS, JetPEI, and PNP6. * denotes statistical significance using Student's t-test at p = 0.05. (C) Use of PNP lung specificity in AI optimization with polymer chemistry and MI for downstream cellular delivery. | ||
We trace this exceptional lung specificity to the inherent molecular design of PNP6. From literature ref. 64–66, it appears that polymer- and lipid-based nanocarriers containing ammonium motifs show lung targeting upon intravenous administration. Cao et al. showed exquisite mRNA delivery to the lung using poly(β-amino esters) (PBAEs) as hydrophobic helper polymers with DOPAT (2-(dodecylthiocarbonothioylthio)propanoic acid), a lipid comprising a quaternary ammonium headgroup.64 Analogously, Dilliard et al. demonstrated selective lung targeting of mRNA LNPs by formulating DOPAT into conventional 4-component LNP formulations.65 By altering the surface chemistry of PBAEs or LNPs and analyzing animal plasma proteins, both groups showed how DOTAP promotes vitronectin corona formation, which binds to the αvβ3 integrin present in pulmonary endothelium (but not liver or other vascular cells). We believe that the multimonomeric polyelectrolytes, with carefully tailored APMAm, TMAEMA, and PhHPMA compositions such as in PNP6, can exploit similar design principles as nanocarriers without exhaustive formulation efforts. Further strategies, such as blending polymers for bioenhancement67,68 or bioconjugation to adopt endosomolytic agents,68,69 can then be carried out to selectively increase targeted delivery to cells of interest.
These findings demonstrate an attractive approach to deliver clinically relevant genetic cargo to the lung, thereby expanding potential non-viral treatments for genetic disorders such as cystic fibrosis.70 Using a data-driven discovery engine that combines polymer chemistry, biology, and AI, we have achieved lung selectivity of pDNA payload delivery with a single multimonomeric polyelectrolyte nanocarrier from a vast, generative PNP design space. Further polymer chemistry campaigns combined with explanatory polymer microstructure models can potentially expand structure-tropism relationships developed for PNP6 to optimize PNPs for any nucleic acid cargo for cellular delivery (Fig. 4C). Molecular engineering and bioconjugation initiatives to address cell-specific uptake and mucus penetration in vitro with cystic fibrosis treatment are in progress. mRNA-based therapeutics are also being incorporated into multicomponent PNPs using this workflow. Further reports will exemplify our vision of combining the ever-evolving precision of polymer synthesis with the expanding repertoire of biologic medicines to offer powerful modalities for non-viral gene delivery and to pioneer new eras of gene therapy.
Notes
All animal procedures were performed according to the standards of the Association for Assessment and Accreditation of Laboratory Animal Care and to the Guidelines and Use of Laboratory Animals of Biomedical Research Models, Inc. (Worcester, MA, USA). All mouse experiments were approved by the Institutional Animal Care and Use Committee of Biomere Biomedical Research Models, Inc. (Worcester, MA, USA).Conflicts of interest
The authors declare the following competing financial interest(s): J. M.T., J. D. F., S. P., T. T.-M., F. O., and S. K. M. have an equity interest in Nanite, Inc. Nanite has filed a patent applications covering aspects of the work described.Acknowledgements
We thank Dr Thomas X. Neenan for helpful discussions, Mitch Stern for his support in general automation, Jared Van Reet for his support in general screening, and Dr Shannon R. Petersen for helpful discussions and feedback on graphic design. We thank Joshua McDonald and Cassandra Vongrej for assistance with routine qPCR analysis and describing its procedure in the ESI.† We also gratefully thank Dr Wendy E. Gavin and Seamus O'Brien for assistance running 1H NMR spectroscopy experiments.References
- C. Sheridan, Nat. Biotechnol., 2023, 41, 737–739 CrossRef CAS PubMed.
- R. Burrows and E. Lambrix, Vaccine Insights, 2022, 1, 191–199 CrossRef.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS PubMed.
- S.-D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496–504 CrossRef CAS PubMed.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. Van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CrossRef CAS PubMed.
- J. Li and H. Wang, Nanoscale Horiz., 2023, 8, 1155–1173 RSC.
- M. J. Hajipour, R. Safavi-Sohi, S. Sharifi, N. Mahmoud, A. A. Ashkarran, E. Voke, V. Serpooshan, M. Ramezankhani, A. S. Milani, M. P. Landry and M. Mahmoudi, Small, 2023, 2301838 CrossRef CAS PubMed.
- M. Torrice, ACS Cent. Sci., 2016, 2, 434–437 CrossRef CAS PubMed.
- F. Wang, J. Lou, X. Gao, L. Zhang, F. Sun, Z. Wang, T. Ji and Z. Qin, Nano Today, 2023, 52, 101943 CrossRef CAS.
- U. Griesenbach, D. M. Geddes and E. W. F. W. Alton, Gene Ther., 2004, 11, S43–S50 CrossRef CAS PubMed.
- M. Tranter, Y. Liu, S. He, J. Gulick, X. Ren, J. Robbins, W. K. Jones and T. M. Reineke, Mol. Ther., 2012, 20, 601–608 CrossRef CAS PubMed.
- R. N. Mitra, M. Zheng and Z. Han, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 160–174 CAS.
- C. A. Maguire, S. H. Ramirez, S. F. Merkel, M. Sena-Esteves and X. O. Breakefield, Neurotherapeutics, 2014, 11, 817–839 CrossRef CAS.
- S. Uchida and K. Kataoka, J. Biomed. Mater. Res., 2019, 107, 978–990 CrossRef CAS.
- R. Kumar, C. F. Santa Chalarca, M. R. Bockman, C. V. Bruggen, C. J. Grimme, R. J. Dalal, M. G. Hanson, J. K. Hexum and T. M. Reineke, Chem. Rev., 2021, 121, 11527–11652 CrossRef CAS.
- J. A. Johnson, F. E. Du Prez and E. Elacqua, Polym. Chem., 2022, 13, 2400–2401 RSC.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- J.-L. Reymond, Acc. Chem. Res., 2015, 48, 722–730 CrossRef CAS PubMed.
- T. B. Martin and D. J. Audus, ACS Polym. Au, 2023, 3, 239–258 CrossRef CAS.
- E. C. Day, S. S. Chittari, M. P. Bogen and A. S. Knight, ACS Polym. Au, 2023, 3, 406–427 CrossRef CAS PubMed.
- R. Upadhya, S. Kosuri, M. Tamasi, T. A. Meyer, S. Atta, M. A. Webb and A. J. Gormley, Adv. Drug Delivery Rev., 2021, 171, 1–28 CrossRef CAS PubMed.
- P. Jain, R. S. Pawar, R. S. Pandey, J. Madan, S. Pawar, P. K. Lakshmi and M. S. Sudheesh, Biotechnol. Adv., 2017, 35, 889–904 CrossRef CAS PubMed.
- K. Paunovska, C. D. Sago, C. M. Monaco, W. H. Hudson, M. G. Castro, T. G. Rudoltz, S. Kalathoor, D. A. Vanover, P. J. Santangelo, R. Ahmed, A. V. Bryksin and J. E. Dahlman, Nano Lett., 2018, 18, 2148–2157 CrossRef CAS PubMed.
- J. M. Ting, T. Tamayo-Mendoza, S. R. Petersen, J. Van Reet, U. A. Ahmed, N. J. Snell, J. D. Fisher, M. Stern and F. Oviedo, Chem. Commun., 2023, 59, 14197–14209 RSC.
- R. Kumar, N. Le, F. Oviedo, M. E. Brown and T. M. Reineke, JACS Au, 2022, 2, 428–442 CrossRef CAS PubMed.
- R. J. Dalal, F. Oviedo, M. C. Leyden and T. M. Reineke, Chem. Sci., 2024, 15, 7219–7228 RSC.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS PubMed.
- D. Sprouse and T. M. Reineke, Biomacromolecules, 2014, 15, 2616–2628 CrossRef CAS PubMed.
- S. Asayama, T. Sekine, H. Kawakami and S. Nagaoka, Bioconjugate Chem., 2007, 18, 1662–1667 CrossRef CAS PubMed.
- M. H. Allen, M. D. Green, H. K. Getaneh, K. M. Miller and T. E. Long, Biomacromolecules, 2011, 12, 2243–2250 CrossRef CAS PubMed.
- N. J. Treat, D. Smith, C. Teng, J. D. Flores, B. A. Abel, A. W. York, F. Huang and C. L. McCormick, ACS Macro Lett., 2012, 1, 100–104 CrossRef CAS PubMed.
- A. M. Funhoff, C. F. Van Nostrum, M. C. Lok, M. M. Fretz, D. J. A. Crommelin and W. E. Hennink, Bioconjugate Chem., 2004, 15, 1212–1220 CrossRef CAS.
- S. T. Hemp, M. H. Allen, A. E. Smith and T. E. Long, ACS Macro Lett., 2013, 2, 731–735 CrossRef CAS PubMed.
- S. T. Hemp, M. H. Allen, M. D. Green and T. E. Long, Biomacromolecules, 2012, 13, 231–238 CrossRef CAS PubMed.
- V. Loczenski Rose, F. Mastrotto and G. Mantovani, Polym. Chem., 2017, 8, 353–360 RSC.
- D. Y. Takigawa and D. A. Tirrell, Macromolecules, 1985, 18, 338–342 CrossRef CAS.
- S. Y. Wong, N. Sood and D. Putnam, Mol. Ther., 2009, 17, 480–490 CrossRef CAS PubMed.
- C. Van Bruggen, J. K. Hexum, Z. Tan, R. J. Dalal and T. M. Reineke, Acc. Chem. Res., 2019, 52, 1347–1358 CrossRef CAS PubMed.
- R. Kumar, N. Le, Z. Tan, M. E. Brown, S. Jiang and T. M. Reineke, ACS Nano, 2020, 14, 17626–17639 CrossRef CAS PubMed.
- Z. Ruan, S. Li, A. Grigoropoulos, H. Amiri, S. L. Hilburg, H. Chen, I. Jayapurna, T. Jiang, Z. Gu, A. Alexander-Katz, C. Bustamante, H. Huang and T. Xu, Nature, 2023, 615, 251–258 CrossRef CAS PubMed.
- M. Reis, F. Gusev, N. G. Taylor, S. H. Chung, M. D. Verber, Y. Z. Lee, O. Isayev and F. A. Leibfarth, J. Am. Chem. Soc., 2021, 143, 17677–17689 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312–325 CrossRef CAS PubMed.
- A. W. Fortenberry, P. E. Jankoski, E. K. Stacy, C. L. McCormick, A. E. Smith and T. D. Clemons, Macromol. Rapid Commun., 2022, 43, 2200414 CrossRef CAS PubMed.
- S. Cosson, M. Danial, J. R. Saint-Amans and J. J. Cooper-White, Macromol. Rapid Commun., 2017, 38, 1600780 CrossRef PubMed.
- J. M. Ting, H. Wu, A. Herzog-Arbeitman, S. Srivastava and M. V. Tirrell, ACS Macro Lett., 2018, 7, 726–733 CrossRef CAS PubMed.
- T. A. Tockary, K. Osada, Q. Chen, K. Machitani, A. Dirisala, S. Uchida, T. Nomoto, K. Toh, Y. Matsumoto, K. Itaka, K. Nitta, K. Nagayama and K. Kataoka, Macromolecules, 2013, 46, 6585–6592 CrossRef CAS.
- A. E. Marras, J. M. Ting, K. C. Stevens and M. V. Tirrell, J. Phys. Chem. B, 2021, 125, 7076–7089 CrossRef CAS PubMed.
- A. E. Marras, J. R. Vieregg, J. M. Ting, J. D. Rubien and M. V. Tirrell, Polymers, 2019, 11, 83 CrossRef PubMed.
- I. Skeist, J. Am. Chem. Soc., 1946, 68, 1781–1784 CrossRef CAS PubMed.
- J. M. Ting, T. S. Navale, F. S. Bates and T. M. Reineke, ACS Macro Lett., 2013, 2, 770–774 CrossRef CAS PubMed.
- A. L. Holmberg, M. G. Karavolias and T. H. Epps, Polym. Chem., 2015, 6, 5728–5739 RSC.
- A. A. A. Smith, A. Hall, V. Wu and T. Xu, ACS Macro Lett., 2019, 8, 36–40 CrossRef CAS PubMed.
- A. M. Rumyantsev, N. E. Jackson, B. Yu, J. M. Ting, W. Chen, M. V. Tirrell and J. J. de Pablo, ACS Macro Lett., 2019, 8, 1296–1302 CrossRef CAS PubMed.
- B. Yu, A. M. Rumyantsev, N. E. Jackson, H. Liang, J. M. Ting, S. Meng, M. V. Tirrell and J. J. de Pablo, Mol. Syst. Des. Eng., 2021, 6, 790–804 RSC.
- C. Fick, Z. Khan and S. Srivastava, Mater. Adv., 2023, 4, 4665–4678 RSC.
- P. U. Joshi, C. Decker, X. Zeng, A. Sathyavageeswaran, S. L. Perry and C. L. Heldt, Biomacromolecules, 2024, 25, 741–753 CrossRef CAS.
- M. Yang, Z. A. Digby, Y. Chen and J. B. Schlenoff, Sci. Adv., 2022, 8, eabm4783 CrossRef CAS.
- S. Perry, Y. Li, D. Priftis, L. Leon and M. Tirrell, Polymers, 2014, 6, 1756–1772 CrossRef.
- K. C. Stevens, A. E. Marras, T. R. Campagna, J. M. Ting and M. V. Tirrell, Macromolecules, 2023, 56, 5557–5566 CrossRef CAS PubMed.
- W. Wang, Int. J. Pharm., 2015, 490, 308–315 CrossRef CAS PubMed.
- J. Huang, L. Knight and J. Laaser, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-g13t8.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- Y. Cao, Z. He, Q. Chen, X. He, L. Su, W. Yu, M. Zhang, H. Yang, X. Huang and J. Li, Nano Lett., 2022, 22, 6580–6589 CrossRef CAS PubMed.
- S. A. Dilliard, Q. Cheng and D. J. Siegwart, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109256118 CrossRef CAS PubMed.
- Y. Huang, J. Wu, S. Li, Z. Liu, Z. Li, B. Zhou and B. Li, Theranostics, 2024, 14, 830–842 CrossRef CAS PubMed.
- M. G. Hanson, C. J. Grimme, N. W. Kreofsky, S. Panda and T. M. Reineke, Bioconjugate Chem., 2023, 34, 1418–1428 CrossRef CAS PubMed.
- K. Leer, L. S. Reichel, J. Kimmig, F. Richter, S. Hoeppener, J. C. Brendel, S. Zechel, U. S. Schubert and A. Traeger, Small, 2024, 20, 2306116 CrossRef CAS PubMed; D. Ulkoski, M. J. Munson, M. E. Jacobson, C. R. Palmer, C. S. Carson, A. Sabirsh, J. T. Wilson and V. R. Krishnamurthy, ACS Appl. Bio Mater., 2021, 4, 1640–1654 CrossRef PubMed.
- J. Di, P. Huang and X. Chen, Bioconjugate Chem., 2024, 35, 453–456 CrossRef CAS PubMed.
- T. Montier, P. Delépine, C. Pichon, C. Férec, D. J. Porteous and P. Midoux, Trends Biotechnol., 2004, 22, 586–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00196f |
| This journal is © The Royal Society of Chemistry 2024 |