
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLignin-derivable, thermoplastic, non-isocyanate polyurethanes with increased hydrogen-bonding content and toughness vs. petroleum-derived analogues†
Jignesh S.
Mahajan
 ab,
Zachary R.
Hinton
c,
Eduardo
Nombera Bueno
ad,
Thomas H.
Epps, III
*abcd and
LaShanda T. J.
Korley
*abcd
ab,
Zachary R.
Hinton
c,
Eduardo
Nombera Bueno
ad,
Thomas H.
Epps, III
*abcd and
LaShanda T. J.
Korley
*abcd
aDepartment of Materials Science & Engineering, University of Delaware, Newark, Delaware 19716, USA. E-mail: thepps@udel.edu; lkorley@udel.edu
bCenter for Research in Soft matter and Polymers, University of Delaware, Newark, Delaware 19716, USA
cCenter for Plastics Innovation, University of Delaware, Newark, Delaware 19716, USA
dDepartment of Chemical & Biomolecular Engineering, University of Delaware, Newark, Delaware 19716, USA
First published on 2nd April 2024
Abstract
The functionality inherent in lignin-derivable bisguaiacols/bissyringols can improve the processability and performance of the resulting polymers. Herein, non-isocyanate polyurethanes (NIPUs) were synthesized from bisguaiacols/bissyringols with varying degrees of methoxy substitution and differing bridging groups. Notably, the presence of increasing numbers of methoxy groups (0, 2, and 4) in bisphenol F (BPF)-, bisguaiacol F (BGF)-, and bissyringol F (BSF)-NIPUs led to higher percentages of hydrogen-bonded –OH/–NH groups (i.e., ∼65%, ∼85%, ∼95%, respectively). Increased hydrogen bonding between chains improved the elongation-at-break (εbreak) and toughness of lignin-derivable NIPUs over their petroleum counterparts without a reduction in Young's moduli and tensile strengths. For example, BSF-NIPU exhibited the highest εbreak ∼210% and toughness ∼62 MJ m−3, followed by BGF-NIPU (εbreak ∼185% and toughness ∼58 MJ m−3), and then BPF-NIPU (εbreak ∼140% and toughness ∼42 MJ m−3). Similar trends were found in the dimethyl-substituted analogues, particularly for the bisphenol A-NIPU and bisguaiacol A-NIPU. Importantly, the melt rheology of the lignin-derivable NIPUs was comparable to that of the petroleum-derived analogues, with a slightly lower viscosity (i.e., improved melt flow) for the bio-derivable NIPUs. These findings suggested that the added functionalities (methoxy groups) derived from lignin precursors improved thermomechanical stability while also offering increased processability. Altogether, the structure–property-processing relationships described in this work can help facilitate the development of sustainable, performance-advantaged polymers.
1. Introduction
Thermoplastic polyurethanes (PUs) are highly desirable materials for numerous applications (e.g., automotive parts, packaging films, medical materials) because they are readily processable and offer highly tunable chemical structures (e.g., compositions of soft and hard segments) that yield a range of sought-after properties (e.g., toughness, modulus, thermal stability, adhesion).1–4 Conventional thermoplastic PUs are synthesized by reacting diisocyanates with diols (and chain extenders in the case of segmented PUs);4,5 however, this route has significant health and environmental challenges. For example, isocyanates are synthesized using highly toxic phosgene,6–8 and the most common aromatic isocyanates, such as methylene diphenyl diisocyanate (MDI) and toluene diisocyanate (TDI), have been classified as carcinogenic, mutagenic, and reprotoxic compounds.6,7 Long-term exposure to isocyanates also can cause respiratory disorders, and the European Union is considering restrictions to isocyanate usage.8Non-isocyanate polyurethanes (NIPUs) are gaining attention in the research community as a means to address the concerns associated with traditional PUs,7–17 yet several synthetic routes for NIPUs reported in the literature use toxic precursors. For instance, NIPU syntheses have been reported that incorporate the rearrangement of acyl azides (i.e., Curtius rearrangement)18 or employ hydroxamic azides (i.e., Lossen rearrangement).19 Additionally, NIPUs have been synthesized through the polycondensation of polycarbamoyl chlorides and polyols.6 These compounds (acyl azides, hydroxamic azides, and polycarbamoyl chlorides) use phosgene or its derivatives as a key building block. Hence, an emerging pathway for NIPU synthesis is via the step-growth polyaddition of cyclic carbonates and diamines, which significantly reduces the use of toxic precursors.6,9–11,20–23 Moreover, CO2 sequestration via the synthesis of cyclic carbonates may provide an environmental benefit.6,8
Another consideration is that most precursors for PU/NIPU synthesis are derived primarily from petroleum-feedstocks, which are limited in quantity and unevenly distributed globally,24 and there is significant opportunity for new building blocks with reduced toxicity and increased sustainability. Most bio-based/bio-derivable NIPU studies have focused on using aliphatic/cycloaliphatic building blocks derived from plant oils and fatty acids,7 but many polymers derived from such aliphatic/cycloaliphatic sources lack the robust structural features necessary to generate materials with properties competitive to those of similar polymers made from conventional aromatics (e.g., TDI, MDI) in terms of glass transition temperatures [Tgs] and moduli.25,26 Petroleum-derived bisphenol A (BPA) has been used to make aromatic NIPUs,21,27 but BPA is a suspected endocrine disruptor, and the potential of BPA to leach out from bisphenol-derived plastics is implicated in numerous health hazards.28–30
Lignin is an attractive feedstock for renewable polymer applications as it is the most abundant potential source of natural aromatics and can act as replacement feedstock for aromatic NIPUs.24–26,31–40 Bulk lignin is mainly comprised of p-hydroxyphenyl (H; no methoxy groups on the aromatic ring), guaiacyl (G; one methoxy group on the aromatic ring), and syringyl (S; two methoxy groups on the aromatic ring) units.31,34,41,42 The relative content of these units varies with the source of biomass; softwoods include a majority of G units, hardwoods contain a mixture of G and S units, and grasses contain a mixture of H, G, and S units.31,41,42 Unfortunately, materials made from bulk lignin can have inconsistent properties due to lignin's structural heterogeneity and limited reactivity.25,41 To overcome this hurdle, lignin can be broken down by catalytic, enzymatic, or pyrolytic pathways into smaller molecules with several inherent functional groups (e.g., aliphatic/aromatic hydroxyl, methoxy, alkyl, allyl), and many of these functionalities can be used to develop renewable materials, such as polycarbonates, polyesters, epoxy resins, and PUs.25,31,34,41 Recently, renewable bisguaiacols/bissyringols have been reported as potential alternatives to commercial bisphenols because of their structural similarity. These renewable alternatives have methoxy groups on their aromatic rings30,43–46 that have the potential to mitigate the toxicity concerns associated with commercial aromatics (e.g., estrogenic activity, genotoxicity, oxidative DNA damage).28,47,48
Thermoplastic NIPUs have gained less attention in the literature in comparison to thermosetting NIPUs. The majority of research has been aimed at NIPU synthesis strategies, examining the role of reaction temperature, solvent, catalyst, etc., on molar mass of the thermoplastic NIPU.6,7 Unique NIPU architectures (i.e., segmented, phase-separated) also have been explored, in which the effect of different chain extenders and soft segments was investigated.49–51 Most studies in the lignin-derivable NIPU domain have focused on synthesizing NIPUs (especially thermosets) using bulk lignin or different building blocks (limonene, vanillin, syringaresinol, etc.),7,12,52,53 and relatively few investigations have focused on understanding the rheological behavior of thermoplastic NIPUs.51,54 Importantly, the inherent pendent hydroxyl and methoxy groups in the lignin-derivable NIPUs offer additional hydrogen-bonding content not accessible in conventional PUs/NIPUs.45 Together, a thorough understanding of these hydrogen-bonding functionalities (i.e., the influence of methoxy moieties on lignin-aromatics with pendent hydroxyl groups) in the NIPU backbone, and the resulting impact of these groups on the thermal, mechanical, and rheological properties of bio-derivable thermoplastic NIPUs, remains under-explored.
Herein, NIPUs were synthesized from bisguaiacols/bissyringols with varying degrees of methoxy substitution and differing bridging groups (Scheme 1) to understand the effect of these structural features on thermal, mechanical, and rheological behavior. Bisphenol-based NIPUs with unsubstituted bridging carbons (bisphenol F [BPF]-/bisguaiacol F [BGF]-/bissyringol F [BSF]) and dimethyl-substituted bridging carbons (BPA-/bisguaiacol A [BGA]-/bissyringol A [BSA]) served as the library of materials for the investigation. First, the effect of key structural features (methoxy-group content and bridging-carbon substitutions) in the polymer repeat unit on the thermal (i.e., Tg, degradation temperature [Td]) and mechanical properties (i.e., Young's modulus, tensile strength, toughness, elongation-at-break [εbreak]) was studied. Second, the impact of these monomer structures on the rheological response was explored to probe the melt behavior of these bio-derivable NIPUs. Importantly, the functionality of lignin-aromatics in NIPU (particularly methoxy groups) improved mechanical properties (particularly εbreak and toughness) in comparison to the petroleum-based counterparts without significant reduction in Td, Young's modulus, and tensile strength. Additionally, the lignin-derivable NIPUs exhibited lower melt viscosities and had faster terminal relaxation times than the petroleum-derived NIPUs.
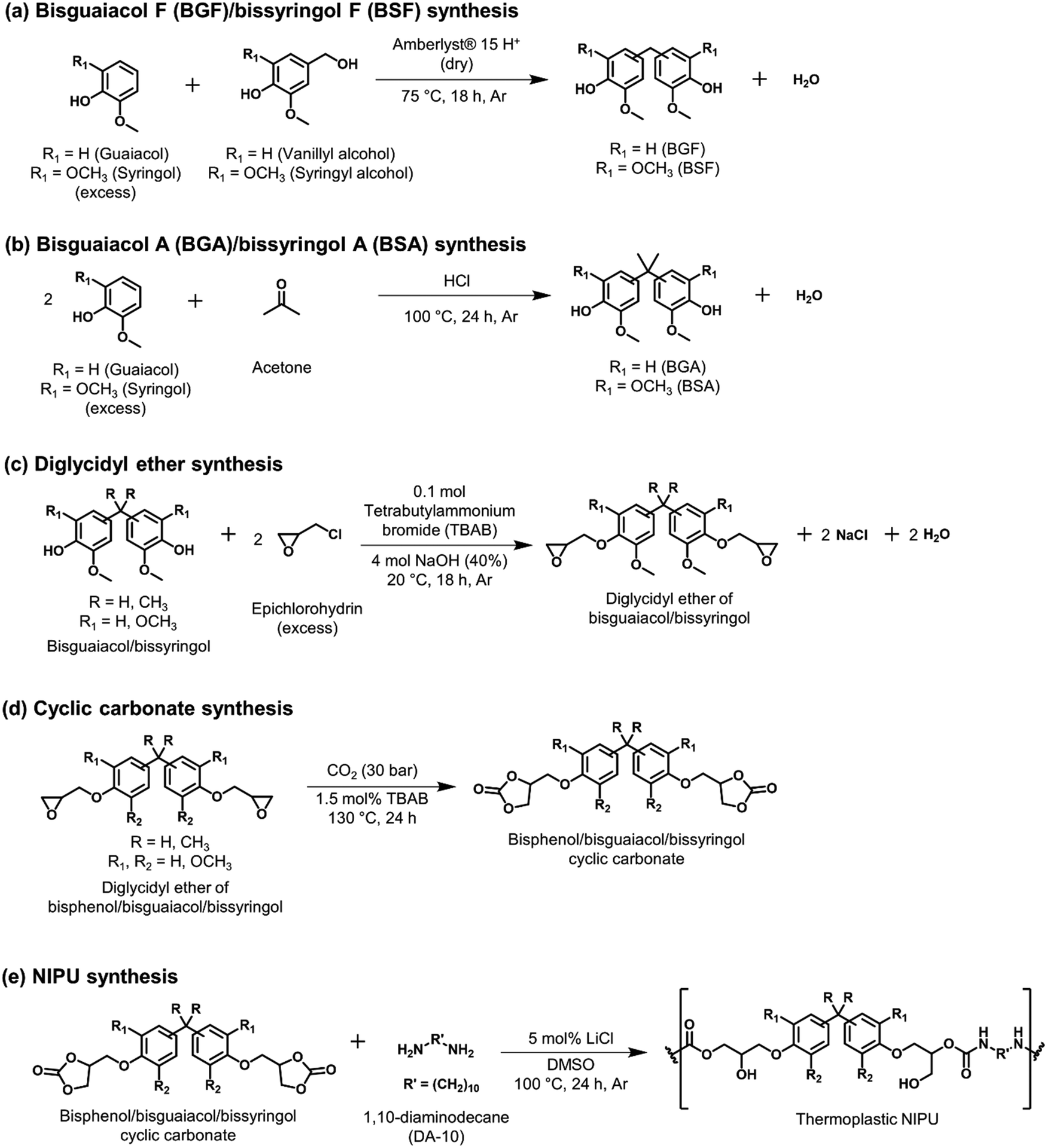 | ||
| Scheme 1 Synthesis of (a) BGF/BSF using guaiacol/syringol and vanillyl/syringyl alcohol, (b) BGA/BSA using guaiacol/syringol and acetone, (c) diglycidyl ether of bisguaiacol/bissyringol from respective bisguaiacol/bissyringol and epichlorohydrin, (d) bisguaiacol/bissyringol cyclic carbonate from respective diglycidyl ether and CO2, and (e) NIPU using respective bisguaiacol/bissyringol cyclic carbonate and 1,10-diaminodecane (DA-10). | ||
2. Experimental
2.1. Materials
BPA diglycidyl ether (extent of oligomerization = ∼0.02, physical form = solid), guaiacol (≥ 98% food grade), syringol (99%), vanillyl alcohol (≥ 98%), thioglycolic acid (98%), LiBr (> 99%), LiCl (> 99%), and Amberlyst® 15 hydrogen form (dry) were purchased from Sigma-Aldrich. DA-10 (> 99%) was purchased from TCI. Syringyl alcohol (97%), deuterated dimethyl sulfoxide (DMSO-d6, ≥99.5%), N,N-dimethylacetamide (DMAc, 99.8+%, HPLC grade), DMSO (anhydrous, 99.7% extra dry), tetrabutylammonium bromide (TBAB, 99+%), epichlorohydrin (99%), and deuterated chloroform (CDCl3, ≥99.75%) were purchased from acros organics. BPF diglycidyl ether (EPONTM 862, extent of oligomerization = ∼0.10, physical form = viscous liquid), a product of Hexion, was purchased from Chemical Marketing Concepts Inc. Acetone (≥99.5%), dichloromethane (DCM, ≥99.5%), N,N-dimethylformamide (DMF, ≥99.8%), HCl (36.5 to 38.0% [w/w]), NaOH (≥97%), magnesium sulfate (anhydrous), sodium bicarbonate (≥99.7%), NaCl (≥99.5%), tetrahydrofuran (THF, ≥99%), hexanes (99%), ethyl acetate (≥99.5%), and acetonitrile (≥99.9%) were purchased from Thermo Fisher Scientific. Ar (grade 5), CO2 (grade 5), and N2 (grade 5) were purchased from Keen Compressed Gas. All chemicals were used as received except acetone, which was passed through a magnesium sulfate column before use.2.2. Monomer synthesis and characterization
BGF synthesis (Scheme 1a). BGF was synthesized as reported in the literature.43 Briefly, guaiacol (40 g, 0.311 mol) and vanillyl alcohol (12 g, 0.078 mol) were loaded into an oven-dried, single-neck, round-bottom flask (250 mL) equipped with a magnetic stir bar. The solution was heated to 70 °C in a silicone oil bath and sparged with Ar gas for 40 min. Then, under Ar flow, Amberlyst® 15 hydrogen form (dry) [3.6 g, 30 wt% relative to the vanillyl alcohol] was added to the reaction mixture, and the reaction was allowed to proceed for 18 h at 70 °C. Next, the reaction mixture was passively cooled to ∼20 °C in air for ∼1.5 h, dissolved in 15 mL of DCM, and the solid catalyst was separated from the crude mixture using a Buchner funnel. The liquid phase was then transferred to a separatory funnel and washed with deionized (DI) water (3 times) and brine solution (2 times). The organic phase was collected, the solvent was removed under reduced pressure, and the product was purified by flash column chromatography (Biotage® Selekt Systems, Biotage® Sfär Silica columns – 60 μm, 100 g) with ethyl acetate (25 v/v%) and hexanes (75 v/v%). The product was dried at 50 °C under vacuum for 24 h, and the dried product was obtained as a white solid (yield ∼50 mol%). Proton (1H) nuclear magnetic resonance (1H NMR) spectra (600 MHz, DMSO-d6) δ 8.68 (s, 2H), 6.80–6.51 (m, 6H), 3.71 (d, J = 4.7 Hz, 8H). Fourier transform mass spectrometry (FTMS) (electrospray ionization [ESI]-FTMS, m/z): calculated for C15H16O4 260.1049; found 260.1041 [Fig. S1, ESI†].
BSF synthesis (Scheme 1a). Syringol (40.1 g, 0.26 mol) and syringyl alcohol (12 g, 0.065 mol) were loaded into an oven-dried, single-neck, round-bottom flask (250 mL) equipped with a magnetic stir bar. The solution was heated to 85 °C in a silicone oil bath and sparged with Ar gas for 40 min. Then, Amberlyst® 15 hydrogen form (dry) [3.6 g, 30 wt% relative to the syringyl alcohol] was added to the reaction mixture, and the reaction was allowed to proceed for 18 h. After the designated time, the reaction mixture was allowed to passively cool to ∼20 °C in air for ∼1.5 h. The reaction mixture was subsequently dissolved in 15 mL of DCM, and the solid catalyst was separated from the crude mixture using a Buchner funnel. Like BGF purification, the liquid phase was washed with DI water (3 times) and brine solution (2 times) in a separatory funnel. The organic phase was collected, and the excess solvent was removed under reduced pressure. Further, the flash column chromatography (Biotage® Selekt Systems, Biotage® Sfär Silica columns – 60 μm, 100 g) with ethyl acetate (25 v/v%) and hexanes (75 v/v%) was used to purify BSF. The product was dried at 50 °C under vacuum for 24 h, and the dried product was obtained as a white solid (yield ∼45 mol%). 1H NMR (600 MHz, DMSO-d6) δ 8.50 (s, 1H), 8.04 (s, 1H), 6.69–6.28 (m, 4H), 3.83–3.54 (m, 14H). FTMS (ESI, m/z): calculated for C17H20O6 320.1260; found 320.1321 [Fig. S2, ESI†].
BGA synthesis (Scheme 1b). Guaiacol (40 g, 0.32 mol) and acetone (3.4 mL, 0.046 mol) were charged in an oven-dried, single-neck, round-bottom flask (250 mL) equipped with a condenser and magnetic stirrer. The reaction was conducted in a large excess of guaiacol to minimize the formation of by-products from the self-condensation of acetone. Next, 5 mL of concentrated HCl (6 g, 15 wt% relative to guaiacol) was slowly added to the above reactants, followed by thioglycolic acid (0.2 vol% with respect to the catalyst) as a promoter. The reaction mixture was sparged with Ar gas for 10 min, and the mixture was heated to 100 °C in a silicone oil bath for 24 h. After that, the reaction mixture was allowed to passively cool to ∼20 °C in air for ∼1.5 h. The crude mixture was dissolved in 15 mL of DCM, and the organic phase was washed with sodium bicarbonate until the aqueous layer reached neutral pH, then washed with DI water (3 times) and brine solution (2 times). The organic phase was collected, the solvent was removed under reduced pressure, and the product was purified by flash column chromatography (Biotage® Selekt Systems, Biotage® Sfär Silica columns – 60 μm, 100 g) with ethyl acetate (25% v/v) and hexanes (75% v/v). The product was dried at 50 °C under vacuum for 24 h, and the dried product was obtained as a white solid (yield ∼40 mol%). 1H NMR (600 MHz, DMSO-d6) δ 8.69 (d, J = 5.7 Hz, 2H), 6.88–6.46 (m, 6H), 3.80–3.51 (m, 6H), 1.54 (d, J = 18.2 Hz, 6H). FTMS (ESI, m/z): calculated for C17H20O4 288.1362; found 288.1354 [Fig. S3, ESI†].
BSA synthesis (Scheme 1b). Syringol (40 g, 0.26 mol) was weighed into an oven-dried, single-neck, round-bottom flask (250 mL) equipped with a condenser and magnetic stirrer. To melt syringol, the round-bottom flask was heated to 85 °C in a silicone oil bath under Ar flow for 30 min. Acetone (2.75 mL, 0.037 mol) and HCl (5 mL, ∼15 wt% relative to syringol) were added dropwise to the reaction flask, and the mixture was heated to 100 °C for 24 h. After 24 h, the reaction mixture was allowed to passively cool to ∼20 °C in air for ∼1.5 h. The reaction mixture was dissolved in 15 mL of DCM and subsequently washed with sodium bicarbonate until the aqueous layer reached neutral pH. The organic phase was then washed with DI water (3 times) and brine solution (2 times). Next, the excess solvent was removed under reduced pressure from the organic phase, and the flash column chromatography (Biotage® Selekt Systems, Biotage® Sfär Silica columns – 60 μm, 100 g) with ethyl acetate (25 v/v%) and hexanes (75 v/v%) was employed to purify BSA. The product was dried at 50 °C under vacuum for 24 h, and the dried product was obtained as a viscous, orangish liquid (yield ∼40 mol%). 1H NMR (600 MHz, DMSO-d6) δ 8.25–7.95 (m, 2H), 6.44 (d, J = 0.9 Hz, 3H), 3.68 (s, 12H), 1.56 (d, J = 27.3 Hz, 6H). FTMS (ESI, m/z): calculated for C19H24O6 348.1573; found 348.1643 [Fig. S4, ESI†].
BPF diglycidyl ether (purchased). Viscous, colorless liquid, 1H NMR (600 MHz, DMSO-d6) δ 7.21–6.71 (m, 9H), 4.39–4.10 (m, 2H), 4.01–3.69 (m, 4H), 2.94–2.57 (m, 4H). FTMS (ESI, m/z): calculated for C19H20O4 312.1362; found 312.1444.
BGF diglycidyl ether. White solid (purity: >99%, yield: ∼85 mol%), 1H NMR (600 MHz, DMSO-d6) δ 6.77 (dd, J = 67.3, 8.0 Hz, 6H), 4.22 (d, J = 8.6 Hz, 2H), 3.91–3.62 (m, 10H), 3.28 (d, J = 4.0 Hz, 2H), 2.88–2.77 (m, 2H), 2.67 (t, J = 4.7 Hz, 2H). FTMS (ESI, m/z): calculated for C21H24O6 372.1573; found 372.1574.
BSF diglycidyl ether. Viscous, light-greenish liquid (purity: >99%, yield: ∼75 mol%),1H NMR (600 MHz, DMSO-d6) δ 6.87 (d, J = 8.5 Hz, 1H), 6.74 (d, J = 7.6 Hz, 1H), 6.49 (s, 2H), 4.20 (d, J = 14.1 Hz, 1H), 4.03 (d, J = 11.4 Hz, 1H), 3.83–3.65 (m, 16H), 3.25 (d, J = 32.8 Hz, 2H), 2.77 (s, 2H), 2.55 (s, 2H). FTMS (ESI, m/z): calculated for C23H28O8 432.1784; found 432.1861.
BPA diglycidyl ether (purchased). White solid, 1H NMR (600 MHz, DMSO-d6) δ 7.22–6.98 (m, 4H), 6.95–6.70 (m, 4H), 4.26 (dd, J = 11.3, 2.8 Hz, 2H), 3.79 (dd, J = 11.3, 6.5 Hz, 2H), 2.82 (dd, J = 5.1, 4.3 Hz, 2H), 2.69 (dd, J = 5.1, 2.7 Hz, 2H), 1.58 (s, 6H). FTMS (ESI, m/z): calculated for C21H24O4 340.1675; found 340.1744.
BGA diglycidyl ether. Viscous, light-yellowish liquid (purity: >99%, yield: ∼75 mol%), 1H NMR (600 MHz, DMSO-d6) δ 6.94–6.64 (m, 6H), 4.21 (ddd, J = 24.1, 11.4, 2.8 Hz, 2H), 3.84–3.61 (m, 8H), 2.80 (ddd, J = 20.0, 5.1, 4.2 Hz, 2H), 2.66 (ddd, J = 13.9, 5.1, 2.6 Hz, 2H), 1.60 (d, J = 7.2 Hz, 6H). FTMS (ESI, m/z): calculated for C23H28O6 400.1886; found 400.1951.
BSA diglycidyl ether. Orangish solid (purity: >99%, yield: ∼80 mol%), 1H NMR (600 MHz, CDCl3) δ 6.36 (s, 4H), 4.11–3.90 (m, 4H), 3.71 (s, 12H), 3.30 (dtd, J = 5.8, 4.1, 2.6 Hz, 2H), 2.74 (dd, J = 5.0, 4.2 Hz, 2H), 2.56 (dd, J = 5.0, 2.7 Hz, 2H), 1.58 (s, 6H). FTMS (ESI, m/z): calculated for C25H32O8 460.2097; found 460.2172.
BPF cyclic carbonate. White solid (purity: >99%, yield: ∼85 mol%), 1H NMR (600 MHz, DMSO-d6) δ 7.21–6.71 (m, 9H), 4.39–4.10 (m, 2H), 4.01–3.69 (m, 4H), 2.94–2.57 (m, 4H). FTMS (ESI, m/z): calculated for C21H20O8 400.1158; found 400.1243.
BGF cyclic carbonate. Off-white solid (purity: >99%, yield: ∼85 mol%), 1H NMR (600 MHz, DMSO-d6) δ 6.95–6.66 (m, 6H), 5.10 (dddd, J = 8.3, 6.2, 4.2, 2.0 Hz, 2H), 4.61 (t, J = 8.5 Hz, 2H), 4.46–4.31 (m, 2H), 4.29–4.02 (m, 4H), 3.73 (d, J = 6.0 Hz, 8H). FTMS (ESI, m/z): calculated for C23H24O10 460.1369; found 460.1443.
BSF cyclic carbonate. Off-white solid (purity: >99%, yield: ∼85 mol%), 1H NMR (600 MHz, DMSO-d6) δ 6.82 (dd, J = 75.1, 8.6 Hz, 2H), 6.50 (s, 2H), 5.11–4.84 (m, 2H), 4.73–4.38 (m, 4H), 4.26–3.87 (m, 4H), 3.82–3.66 (m, 14H). FTMS (ESI, m/z): calculated for C25H28O12 520.1581; found 520.1652.
BPA cyclic carbonate. White solid (purity: >99%, yield: ∼85 mol%), 1H NMR (600 MHz, DMSO-d6) δ 7.21–7.02 (m, 4H), 6.92–6.75 (m, 4H), 5.13 (dtt, J = 8.6, 6.0, 2.6 Hz, 2H), 4.62 (t, J = 8.5 Hz, 2H), 4.37 (dd, J = 8.4, 5.9 Hz, 2H), 4.29–4.11 (m, 4H), 1.58 (s, 6H). FTMS (ESI, m/z): calculated for C23H24O8 428.1471; found 428.1536.
BGA cyclic carbonate. Off-white solid (purity: >99%, yield: ∼85 mol%), 1H NMR (600 MHz, DMSO-d6) δ 6.96–6.67 (m, 6H), 5.20–5.02 (m, 2H), 4.60 (dt, J = 13.8, 8.5 Hz, 2H), 4.50–4.31 (m, 2H), 4.31–4.06 (m, 4H), 3.71 (d, J = 26.7 Hz, 6H), 1.60 (d, J = 6.6 Hz, 6H). FTMS (ESI, m/z): calculated for C25H28O10 488.1682; found 488.1771.
BSA cyclic carbonate. Light-orangish-yellow solid (purity: >99%, yield: ∼85 mol%), 1H NMR (600 MHz, DMSO-d6) δ 6.51 (s, 4H), 5.08–4.91 (m, 2H), 4.69–4.45 (m, 4H), 4.35–4.08 (m, 2H), 4.05–3.86 (m, 2H), 3.71 (s, 12H), 1.64 (s, 6H). FTMS (ESI, m/z): calculated for C27H32O12 548.1894; found 548.1967.
2.3. Polymer synthesis and characterization
BPF-NIPU. 1H NMR (600 MHz, DMSO-d6) δ 7.31–6.58 (m, 11H), 5.20 (s, 1H), 5.03–4.80 (m, 1H), 4.09–3.70 (m, 10H), 3.56 (s, 1H), 2.93 (q, J = 6.5 Hz, 4H), 1.38–1.12 (m, 16H).
BGF-NIPU. 1H NMR (600 MHz, DMSO-d6) δ 7.14 (d, J = 27.5 Hz, 2H), 6.77 (d, J = 59.6 Hz, 6H), 5.31–4.70 (m, 3H), 4.10–3.50 (m, 17H), 2.94 (s, 4H), 1.29 (d, J = 58.5 Hz, 16H).
BSF-NIPU. 1H NMR (600 MHz, DMSO-d6) δ 7.06 (s, 2H), 6.77 (d, J = 53.8 Hz, 2H), 6.47 (d, J = 6.0 Hz, 2H), 5.12–4.66 (m, 3H), 4.17–3.53 (m, 23H), 2.93 (s, 4H), 1.29 (d, J = 58.6 Hz, 16H).
BPA-NIPU. 1H NMR (600 MHz, DMSO-d6) δ 7.09 (d, J = 8.5 Hz, 6H), 6.81 (d, J = 8.6 Hz, 4H), 5.20 (d, J = 5.1 Hz, 1H), 4.95–4.78 (m, 1H), 4.11–3.79 (m, 8H), 3.56 (s, 1H), 2.94 (s, 4H), 1.57 (s, 6H), 1.29 (d, J = 59.7 Hz, 16H).
BGA-NIPU. 1H NMR (600 MHz, DMSO-d6) δ 7.24–6.99 (m, 2H), 6.92–6.64 (m, 6H), 5.18 (s, 1H), 4.87 (d, J = 10.6 Hz, 1H), 4.18–3.48 (m, 15H), 2.94 (s, 4H), 1.59 (s, 6H), 1.29 (d, J = 59.0 Hz, 16H).
BSA-NIPU. 1H NMR (600 MHz, DMSO-d6) δ 7.07 (s, 2H), 6.59 (d, J = 77.9 Hz, 4H), 5.05–4.53 (m, 2H), 4.31–3.45 (m, 21H), 2.93 (s, 4H), 1.61 (d, J = 19.7 Hz, 6H), 1.24 (d, J = 17.3 Hz, 16H).
Linear viscoelastic spectra at a reference temperature of Tref = 150 °C were constructed using the frequency sweep data via time-temperature superposition. Vertical shift factors of Tref/T were applied, and, subsequently, horizontal shift factors were determined using the algorithm in the TRIOS software (TA Instruments).
3. Results and discussion
For this work, NIPUs, synthesized from monomers with varying methoxy-group content on the aromatic rings and the substituents to the bridging carbons, were studied to understand the impact of these structural features on the thermal, mechanical, and rheological behavior of the resulting polymers. The molecular structures of petroleum-based NIPUs and their lignin-derivable analogues are presented in Scheme 2. In the following sections, the synthesis of lignin-derivable precursors and NIPUs is discussed. Additionally, the impact of bridging carbon substituents and methoxy content on the hydrogen-bonding and mechanical properties is presented, and the influence of these structural features on the rheological response is examined.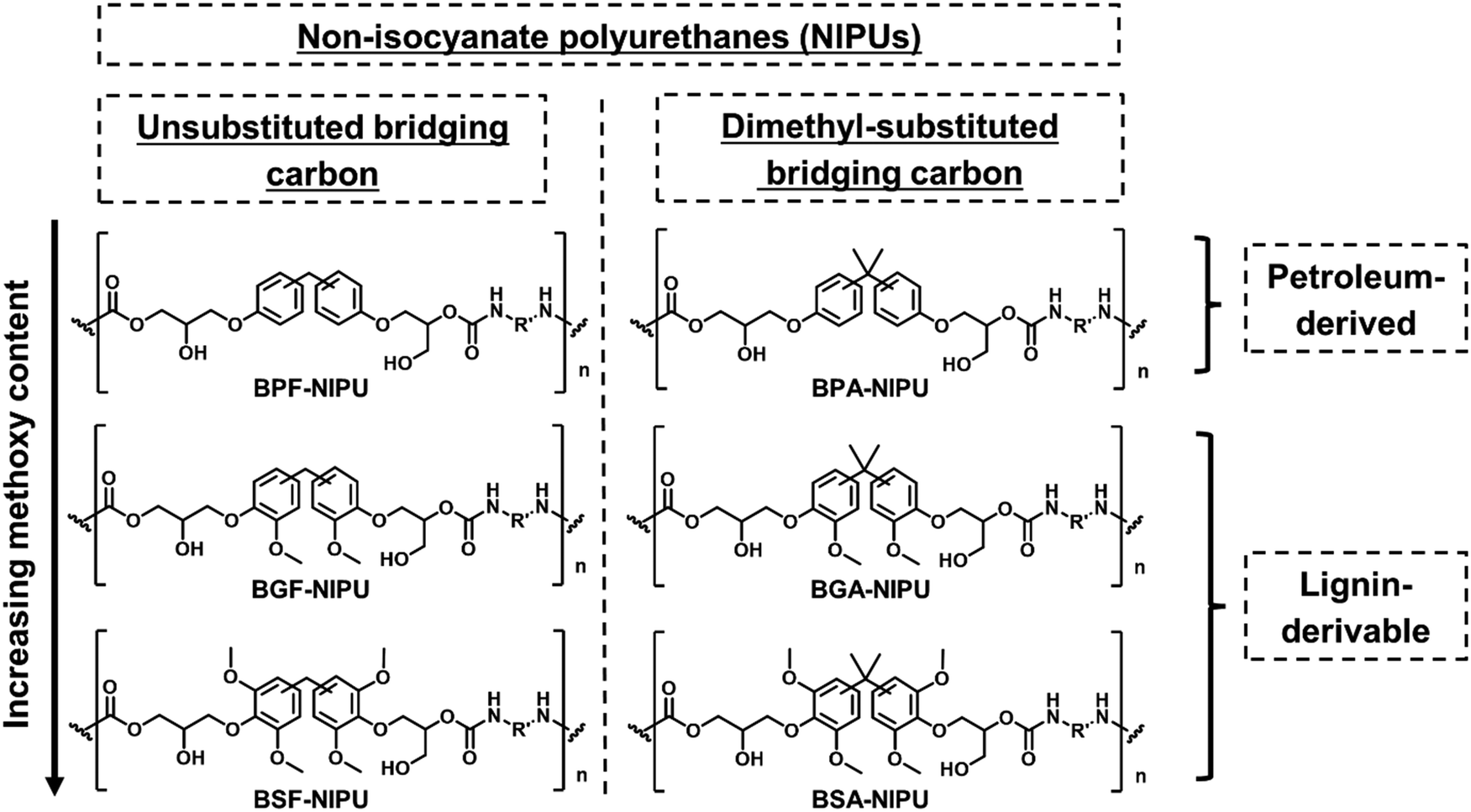 | ||
| Scheme 2 The library of synthesized NIPUs highlighting key features of their chemical structure and source. | ||
3.1. Synthesis of lignin-derivable building blocks and NIPUs
Several lignin-derivable alternatives to commercial bisphenols (BPA/BPF) were synthesized, as shown in Schemes 1a and b. BGF and BSF were synthesized by an acid-catalyzed electrophilic aromatic substitution reaction between guaiacol and vanillyl alcohol (for BGF) or syringol and syringyl alcohol (for BSF) (Scheme 1a).43,46 The direct condensation approach eliminates the use of hazardous formaldehyde inherent in BPF production.41 BGF and BSF do not have a substituted bridging carbon between the two aromatic rings, which makes them more structurally similar to BPF. BGA and BSA were synthesized via an electrophilic aromatic condensation between acetone and guaiacol (for BGA) or acetone and syringol (for BSA) (Scheme 1b).44 The availability of bio-based acetone enables a potentially greener process.44 Both BGA and BSA have dimethyl substituents on the bridging carbon between the two aromatic moieties and more closely resemble the structure of BPA. As mentioned above, the methoxy groups on these lignin-derivable compounds are believed to reduce the toxicity concerns of BSA, BGA, BSF, and BGF vs. BPA and BPF.47The respective bisphenols were converted to diglycidyl ethers and then to cyclic carbonates (Schemes 1c and d). The exact molar masses of the precursors at each step were determined using liquid chromatography–mass spectroscopy, and the chemical structures of the compounds were confirmed using 1H NMR spectroscopy. Both mass spectrometry and 1H NMR spectroscopy results showed excellent agreement with the expected chemical formulae and chemical structures of the compounds, respectively.
These cyclic carbonates were reacted with a bio-derivable diamine (DA-10)53,55,56 to obtain NIPUs (Scheme 1e). All NIPUs exhibited comparable molar masses of ∼15–20 kg mol−1, as shown in Table 1. Although traditional PUs can be obtained with molar masses >20 kg mol−1, most of the NIPUs reported in the literature possess a number-average molecular weight (Mn) <10 kg mol−1 due to the lower reactivity of cyclic carbonates vs. isocyanates,9,53,57 along with numerous possible side reactions (e.g., urea formation, amidification reaction, oxazolidinone formation) that can limit molar mass.6–8 Additionally, the five-membered cyclic carbonates are relatively less reactive than six-, seven-, or eight-membered cyclic carbonates because of the ring strain.6–8 Notably, in this work, higher-than-average Mn values were achieved, and free-standing films were fabricated (Fig. S26, ESI†), which could suggest that the hydrogen bonding in these polymers improves mechanical robustness even in lower-molar-mass NIPUs. As shown in Table 1, most of the NIPUs had dispersities (Đs) ∼2.5–3, except for BPF-NIPU (Đ ∼4). This exception was likely a result of the higher extent of oligomerization (∼0.10) in the commercial BPF diglycidyl ether vs. the other diglycidyl ethers (∼0.01–0.03).
| Polymer | Methoxy groups | M n (kg mol−1) | Đ | ||
|---|---|---|---|---|---|
| a Determined relative to PMMA standards using data from GPC with RI detectors [GPC solvent: DMAc + 0.5 wt% LiBr]. | |||||
| Unsubstituted bridging carbon | BPF-NIPU | 0 | 17 | 4.1 | |
| BGF-NIPU | 2 | 19 | 2.3 | ||
| BSF-NIPU | 4 | 14 | 2.7 | ||
| Dimethyl-substituted bridging carbon | BPA-NIPU | 0 | 19 | 2.6 | |
| BGA-NIPU | 2 | 17 | 2.6 | ||
| BSA-NIPU | 4 | 15 | 3.0 | ||
3.2. Estimation of hydrogen-bonding in NIPUs
Hydrogen bonding plays a crucial role in the properties (mechanical/rheological) of PUs/NIPUs.58,59 Herein, the lignin-derivable NIPUs displayed higher percentages of hydrogen-bonded acceptors (attributable to methoxy moieties) than their petroleum-based analogues. The total number of hydrogen-bond acceptors and donors was calculated from the theoretical structure of the repeat unit. In particular, –OH and –NH were considered the hydrogen-bond donors, and urethane carbonyls (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ether oxygens (–O–), and methoxy groups (–OCH3) were considered the hydrogen-bond acceptors. As shown in Table 2, the hydrogen-bond acceptor count per repeat unit in the NIPUs increased with the number of methoxy groups (0, 2, and 4) in BPF-/BGF-/BSF-NIPUs.
O), ether oxygens (–O–), and methoxy groups (–OCH3) were considered the hydrogen-bond acceptors. As shown in Table 2, the hydrogen-bond acceptor count per repeat unit in the NIPUs increased with the number of methoxy groups (0, 2, and 4) in BPF-/BGF-/BSF-NIPUs.
| Polymer | Methoxy groups | Hydrogen-bond donor count (per repeat unit) | Hydrogen-bond acceptor count (per repeat unit) | Hydrogen-bonded –OH/–NHa (%) | |
|---|---|---|---|---|---|
| a Estimated from ATR-FTIR spectroscopy data. | |||||
| Unsubstituted bridging carbon | BPF-NIPU | 0 | 4 | 6 | ∼65 |
| BGF-NIPU | 2 | 4 | 8 | ∼85 | |
| BSF-NIPU | 4 | 4 | 10 | ∼95 | |
| Dimethyl-substituted bridging carbon | BPA-NIPU | 0 | 4 | 6 | ∼60 |
| BGA-NIPU | 2 | 4 | 8 | ∼90 | |
| BSA-NIPU | 4 | 4 | 10 | ∼95 | |
Semi-quantitative analysis using ATR-FTIR spectroscopy was employed to estimate the amount of hydrogen bonding in these NIPUs. The –OH/–NH absorption bands (3100–3700 cm−1) of the NIPUs samples were deconvoluted using Gaussian distributions.58,60 The –OH/–NH absorption bands were fit to two subpeaks because the stretching vibrations of hydrogen-bonded –OH/–NH were at a slightly lower wavenumber (∼3300–3350 cm−1) than the free –OH/–NH vibrations (∼3450–3550 cm−1), and the relative areas of these two peaks were calculated.58,60 Hydrogen bonding can cause the vibrational frequencies to red-shift (i.e., migrate towards lower energy) for the hydrogen-bonding motifs, with stronger bonds precipitating shifts to lower energies.61,62 The hydrogen-bonded region includes all self- and trans-associations, and the broadness of this region depends upon the number/strength of associations. Herein, the added methoxy groups (hydrogen-bond acceptors) increased the interactions in the hydrogen-bonded region, and therefore, the broadness of this region. FTIR analysis suggested that increasing the number of methoxy groups from 0 to 2 to 4 in BPF-/BGF-/BSF-NIPUs led to higher percentages of hydrogen-bonded –OH/–NH groups (∼65%, ∼85%, ∼95%, respectively). Similar trends were found in the dimethyl-substituted analogues, BPA-/BGA-/BSA-NIPUs (Table 2 and Fig. 1). This finding demonstrated that the added hydrogen-bonding capability of the monomers directly translated to greater secondary interactions between NIPU chains, which was expected to influence mechanical and rheological properties.58,60,63
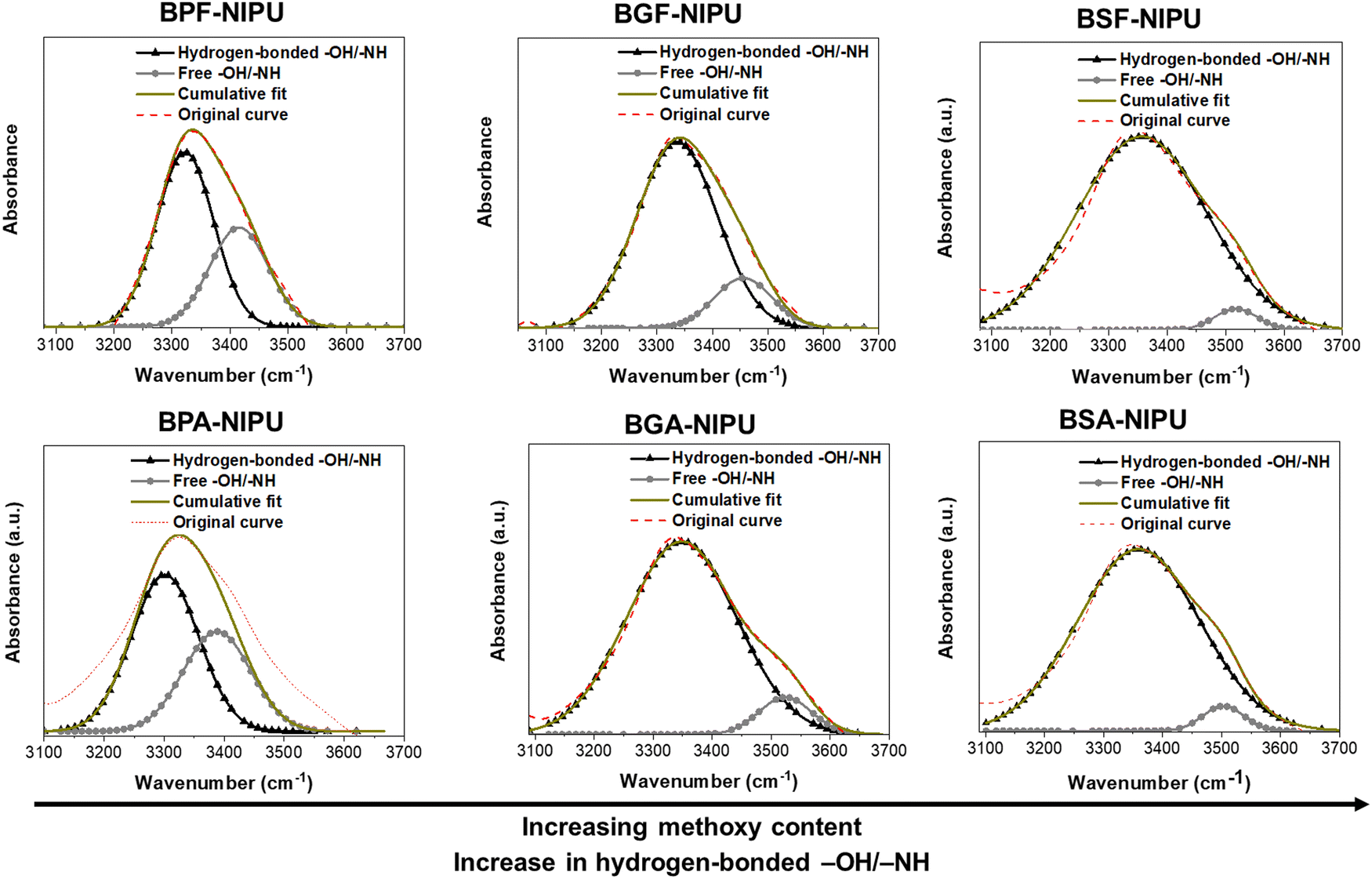 | ||
| Fig. 1 Deconvoluted hydroxyl region in the ATR-FTIR spectra of the NIPUs. The –OH/–NH absorption bands were fit using two Gaussian distributions: one for hydrogen-bonded –OH/–NH groups and another for free –OH/–NH groups. | ||
3.3. Mechanical behavior of NIPUs
The impacts of both hydrogen bonding and substitutive group on the solid-state performance of the NIPUs were quantified using tensile testing. The lignin-derivable NIPUs exhibited improved mechanical properties (εbreak and toughness) over their petroleum-based counterparts without a reduction in Young's modulus or tensile strength (Fig. 2 and Fig. S28 (ESI†) and Table 3). The εbreak and toughness of NIPUs increased with an increase in the methoxy-group content. For example, in the unsubstituted bridging-group case, BSF-NIPU exhibited the highest εbreak (∼210%) and toughness (∼62 MJ m−3), followed by BGF-NIPU (εbreak ∼185%, toughness ∼58 MJ m−3), and then BPF-NIPU (εbreak ∼140%, toughness ∼42 MJ m−3). In the dimethyl-substituted cases, both lignin-derivable versions, BGA-NIPU (εbreak ∼145%, toughness ∼51 MJ m−3) and BSA-NIPU (εbreak ∼120%, toughness ∼44 MJ m−3) exhibited higher εbreak and toughness than petroleum-derived BPA-NIPU (εbreak ∼95%, toughness ∼36 MJ m−3). The increased toughness was attributed to the hydrogen bonds between chains that acted as physical crosslinks. As the petroleum-derived NIPUs had a fixed number of donors for potential hydrogen-bonding interactions, the methoxy moieties in the lignin-derivable systems could provide additional sites for intra- and inter-molecular hydrogen bonding in the NIPU matrix.58,64 The density of these physical crosslinks possibly increased with an increase in methoxy content, which likely led to higher extensibility; however, the increased in the extent of hydrogen bonding in BSA-NIPU did not equate to a higher εbreak and toughness than that found for BGA-NIPU. Thus, more in-depth mechanical investigations of the chain dynamics under stretching might help to understand the phenomenon behind this anomaly.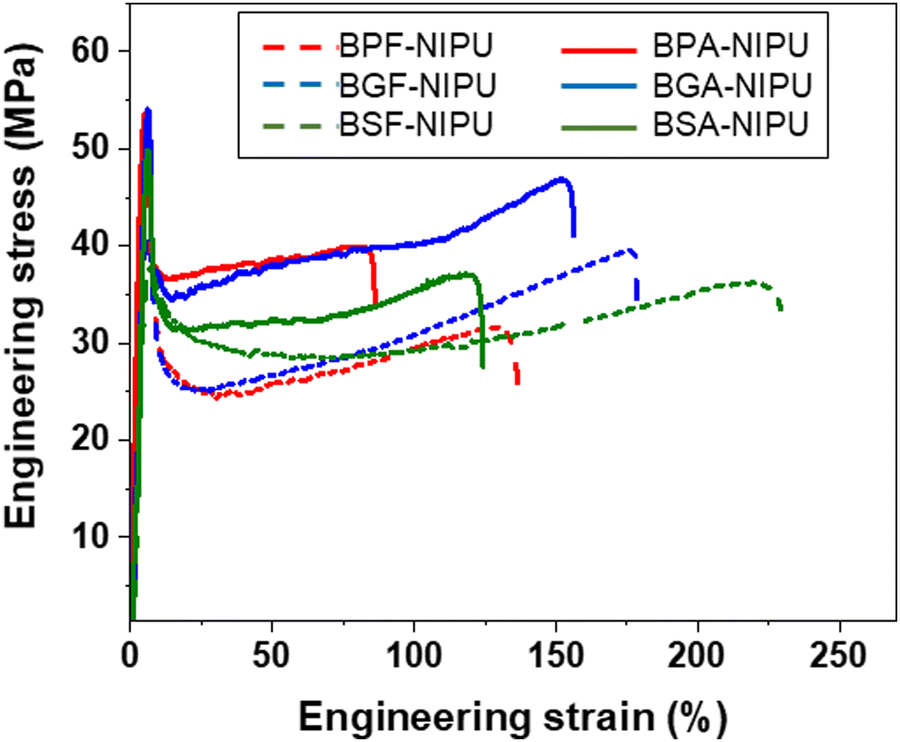 | ||
| Fig. 2 Representative tensile engineering stress vs. engineering strain curves for NIPUs acquired from uniaxial tensile testing using a load cell of 100 N and a strain rate of 10% min−1 at ∼20 °C. | ||
| Polymer | Methoxy groups | Ultimate tensile strength (MPa) | Young's modulus (GPa) | ε break (%) | Toughness (MJ m−3) | |
|---|---|---|---|---|---|---|
| Unsubstituted bridging carbon | BPF-NIPU | 0 | 47 ± 3 | 1.2 ± 0.1 | 140 ± 21 | 42 ± 10 |
| BGF-NIPU | 2 | 44 ± 3 | 1.2 ± 0.1 | 185 ± 17 | 58 ± 5 | |
| BSF-NIPU | 4 | 39 ± 4 | 1.2 ± 0.2 | 210 ± 18 | 62 ± 8 | |
| Dimethyl-substituted bridging carbon | BPA-NIPU | 0 | 55 ± 1 | 1.4 ± 0.2 | 95 ± 13 | 36 ± 6 |
| BGA-NIPU | 2 | 52 ± 2 | 1.4 ± 0.1 | 145 ± 20 | 51 ± 13 | |
| BSA-NIPU | 4 | 55 ± 6 | 1.4 ± 0.1 | 120 ± 16 | 44 ± 8 |
The unsubstituted analogues exhibited a higher εbreak than dimethyl-substituted NIPUs. The εbreak trend was BPF-NIPU > BPA-NIPU; BGF-NIPU > BGA-NIPU; and BSF-NIPU > BSA-NIPU (see Table 3). This trend was probably a result of the absence of substituents on the bridging carbon, allowing free rotation (increasing chain mobility) of the polymer backbone.30,65 Additionally, the Young's moduli for all the unsubstituted bridging carbon-based NIPUs (BPF-/BGF-/BSF-NIPUs) were comparable (∼1.2 GPa) within experimental error. A similar trend was noted for all the dimethyl-substituted bridging carbon-based NIPUs (BPA-/BGA-/BSA-NIPUs) with Young's moduli of ∼1.4 GPa. This phenomenon was likely due to the modulus of the glassy state being dominated by localized motion of the polymer backbone, and therefore, the substituents, had little effect on the modulus. This relationship was expected because of the rigid, aromatic units present in the polymeric backbones could contribute much more to the modulus than inter-chain friction from small side groups.
Next, π–π stacking interactions are expected to be comparable across all NIPUs as it has been reported that lignin-derivable G and S units can form π–π stacking interactions,66,67 and these interactions are almost equivalent in softwood and hardwood lignin samples.38 Thus, the methoxy substituents may not significantly interrupt π–π interactions in lignin-derivable-NIPUs [BGF-/BGA-NIPUs (softwood derivable) and BSF-/BSA-NIPUs (hardwood derivable)] vs. petroleum-derived-NIPUs [BPF-/BPA-NIPUs].
3.4. Thermal properties of NIPUs
The thermal properties of these lignin-derivable NIPUs were benchmarked against their petroleum-derived analogues (Fig. 3). The Tgs of the NIPUs increased with an increase in the methoxy-group content (Fig. 3a). For example, BPF-NIPU with zero methoxy groups had a Tg of ∼36 °C, BGF-NIPU with two methoxy groups had a Tg of ∼39 °C, and BSF-NIPU with four methoxy groups had a Tg of ∼42 °C. Similarly, BPA-NIPU with zero methoxy groups had a Tg of ∼46 °C, BGA-NIPU with two methoxy groups had a Tg of ∼50 °C, and BSA-NIPU with four methoxy groups had a Tg of ∼55 °C. These results were consistent with the literature, as the presence of side groups (i.e., methoxy moieties) on the polymer backbone could increase Tg by limiting the chain mobility.68 Additionally, secondary forces, such as hydrogen-bonding and dipole–dipole interactions,69–71 between the chains also might be responsible for an increase in Tg due to greater constraints on segmental mobility imposed by associations.72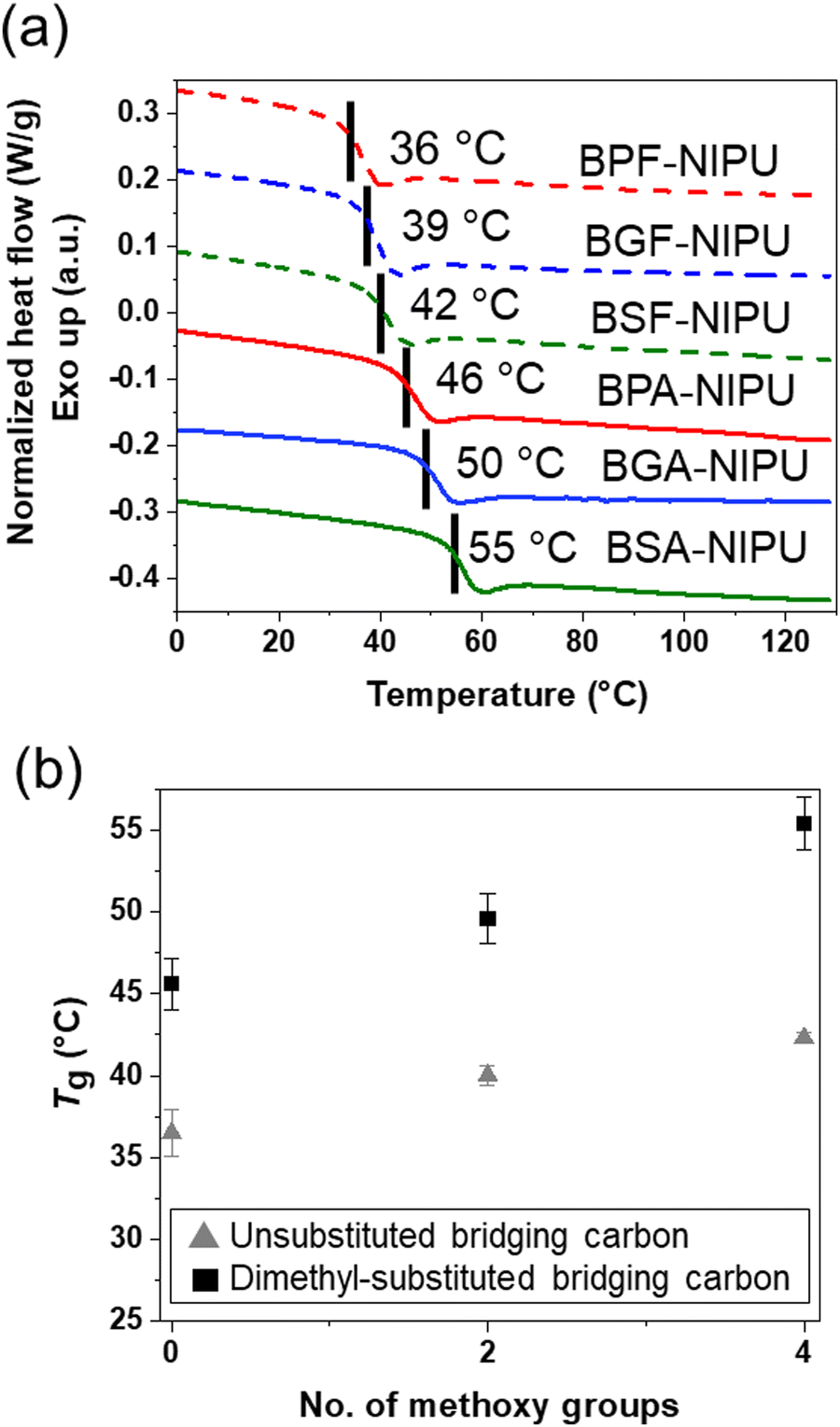 | ||
| Fig. 3 (a) DSC thermograms of NIPUs with a heating rate of 10 °C min−1 in an N2 atmosphere (data from the third heating trace and curves were shifted vertically for clarity with Tg values marked on the respective curves), (b) Tg as a function of the methoxy-group content from 0 to 4: for unsubstituted bridging carbon- and dimethyl-substituted bridging carbon-based NIPUs. [Note: Tg values from the second and third heating traces were identical, and the error bars were determined using Tg values from the third heating traces of two separate samples for each NIPU]. | ||
The NIPUs based on monomers with dimethyl-substituted bridging carbons exhibited higher Tgs than their unsubstituted analogues (Fig. 3b). The Tg values were BPA-NIPU > BPF-NIPU; BGA-NIPU > BGF-NIPU; and BSA-NIPU > BSF-NIPU (see Fig. 3b). The trend was most likely related to the rotational freedom of the aromatic ring around the central carbon, which was lower for the dimethyl group68 as the dimethyl bridge restricted the free rotation of the polymer backbone (BPA-/BGA-/BSA-NIPUs) and led to a comparably higher Tg (BPF-/BGF-/BSF-NIPUs). This finding was important because it had been reported previously that the Tgs of the bisguaiacol/bissyringol-based polymers were lower than BPA-based counterparts.41 In combination with the mechanical responses of the NIPUs, these DSC results demonstrate that hydrogen bonding contributes to the ability to obtain glassy polymers with tunable extensibility at room temperature.
The thermal stabilities of lignin-derivable NIPUs were equivalent to those of the BPA-/BPF-based NIPUs (Td5% ∼280 °C) (Fig. 4a). The Td5% values for BPF-/BGF-/BSF-NIPU were ∼283 °C, 276 °C, and 288 °C, respectively. Similarly, BPA-/BGA-/BSA-NIPUs exhibited Td5% values of ∼275 °C, 285 °C, and 283 °C, respectively. The char contents were nearly identical across all NIPUs. Additionally, the most significant degradation event occurred at similar temperatures (∼350 °C) across all NIPUs (Fig. 4b). The comparable thermal stabilities of all NIPUs (Td5% ∼ 280 °C) were mainly attributed to the initial breakage of the hydroxyurethane linkages, which occurred at ∼250–270 °C.25,45 These results were consistent with our previous findings for lignin-derivable BGF-/BGA-NIPU vs. BPF-/BPA-NIPU thermosets.45 Interestingly, the increased methoxy content in these lignin-derivable NIPUs did not reduce thermal stability. This observation was noteworthy because it had been reported previously that the increased oxygen content in lignin-derivable thermoplastic polymers, such as polycarbonates/polyesters,30,73 reduced thermal stability. Together, the methoxy moieties did not strongly impact the thermal decomposition temperature across the different lignin-derivable NIPUs.
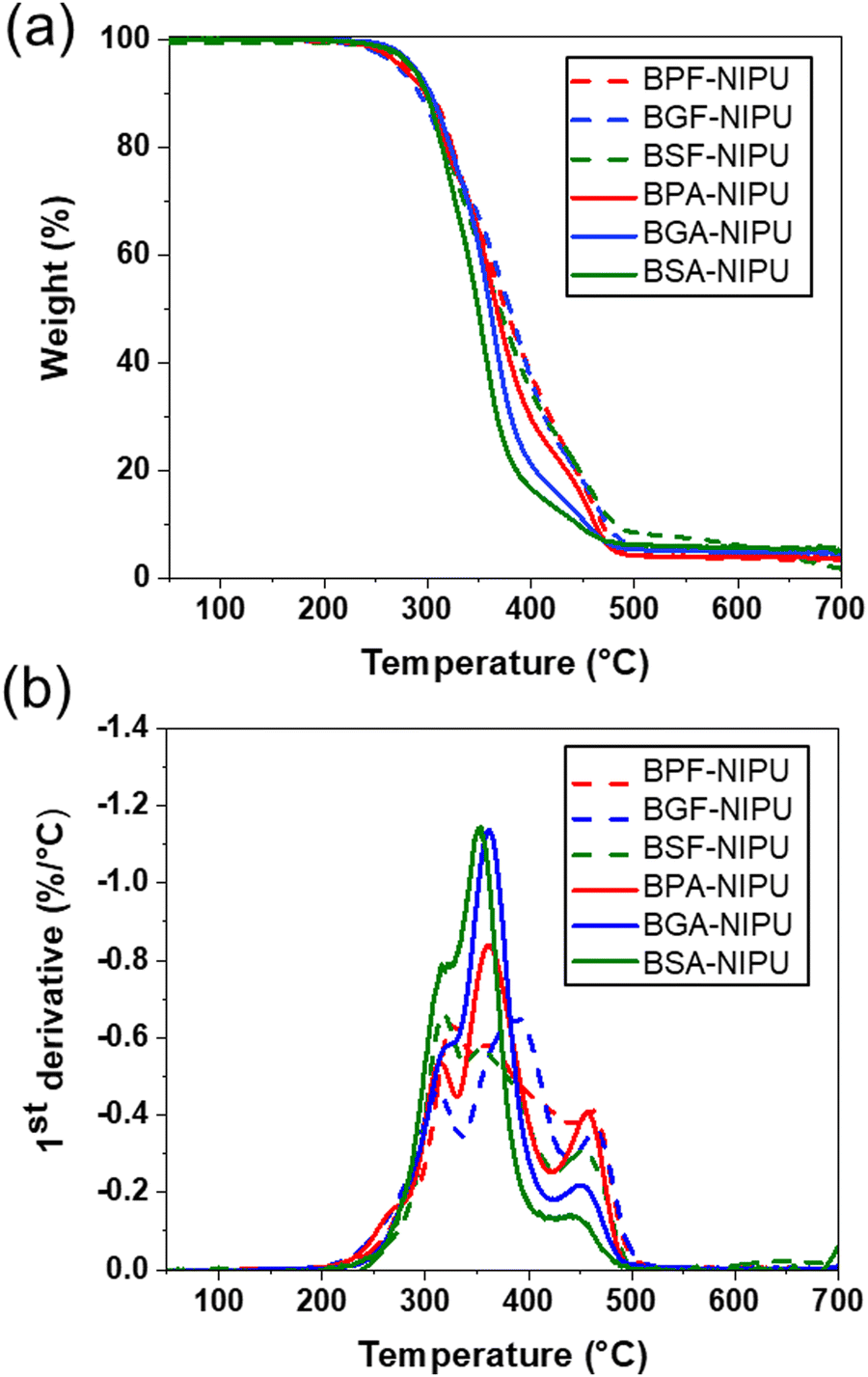 | ||
| Fig. 4 (a) TGA thermograms of NIPUs in N2 atmosphere; sample weight (%) as a function of temperature with a heating rate of 10 °C min−1, and (b) the first derivative of the weight remaining (%/°C) as a function of temperature for all NIPUs. | ||
3.5. Rheological behavior of NIPUs
Thermoplastic PUs are typically melt processed for fabrication of commercial products, and thus, their rheological behavior is key to macromolecular design. It has been well-established that the addition of hydrogen-bonding groups strongly affects melt flow in a wide range of polymers (e.g., polyacrylates, polyolefins, polyethers) and in several complex ways (e.g., increasing terminal relaxation time, altering slope in the terminal regime).69,74–77 Therefore, it is critical to gain an understanding of the contribution of additional hydrogen-bonding capabilities in lignin-derivable NIPUs. For unsubstituted NIPUs, the rheological behavior in the rubbery and glassy states (i.e., frequencies > 105 rad s−1) was similar for all polymers (Fig. 5a). A slight trend in the glassy modulus (i.e., the plateau value of storage modulus, G′, at higher frequencies) was noticed whereby the modulus increased with increasing numbers of methoxy groups. The critical relaxation timescale (τc) (i.e., the inverse of the frequency at which a crossover of G′ and G′′ occurs) when the transitional and rubbery regimes meet is related to localized, fast relaxations.78 Interestingly, τc appeared to be non-monotonic with the number of methoxy groups (Table 4). The value of τc is typically sensitive to inter-monomer friction,79 yet this trend was unexpected given the probable increase in friction with increasing methoxy moieties. This discrepancy might be due to the higher dispersity of BPF-NIPU as result of the extent of oligomerization in comparison to the other NIPUs or relative differences in the frictional contribution of methoxies subject to their position on the aromatic rings.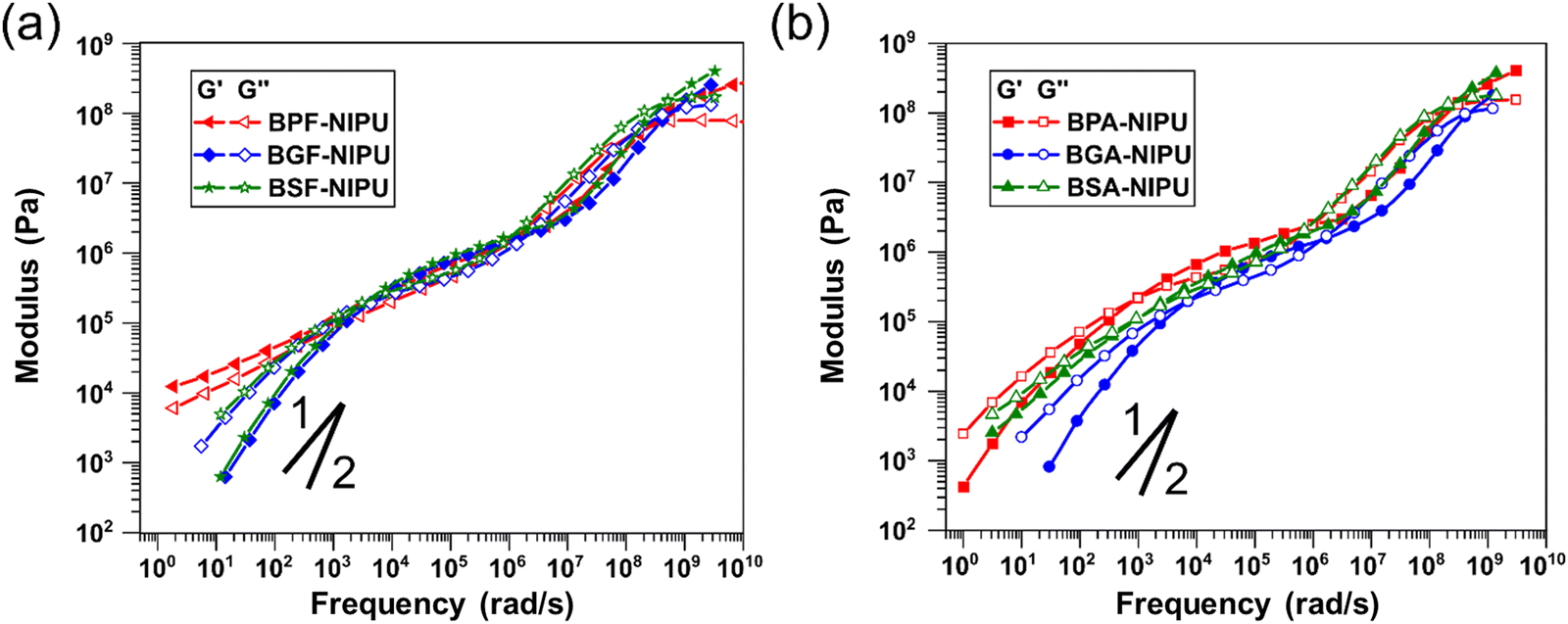 | ||
| Fig. 5 Frequency-dependent storage (G′) and loss (G′′) moduli for (a) unsubstituted bridging carbon- and (b) dimethyl-substituted bridging carbon-based NIPUs. The master curves were constructed using time-temperature superposition and reported at 150 °C. Lines indicate typical power-law scalings of G′ (slope of 2) and G′′ (slope of 1). | ||
| Polymer | Methoxy groups | τ x (s) | τ c (s) | G N,0 (Pa) | C1 | C2 (K) | |
|---|---|---|---|---|---|---|---|
| a Given at the reference temperature of 150 °C. b Calculated as the inverse of the low-frequency crossover. c Calculated as the inverse of the crossover at high frequencies bounded by the rubbery and transitional regimes. d The value of G′ at the minimum of phase angle vs. complex modulus (Fig. S29, ESI). e Approximated as the frequency in the terminal regime at which phase angle vs. complex modulus went through a maximum. | |||||||
| Unsubstituted bridging carbon | BPF-NIPU | 0 | 1.6 × 10−3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e e |
7.6 × 10−7 | 7.2 × 105 | 3.1 | 137 |
| BGF-NIPU | 2 | 2.3 × 10−4 | 4.7 × 10−7 | 8.7 × 105 | 3.1 | 141 | |
| BSF-NIPU | 4 | 2.7 × 10−4 | 9.6 × 10−7 | 8.8 × 105 | 3.2 | 142 | |
| Dimethyl-substituted bridging carbon | BPA-NIPU | 0 | 1.1 × 10−3 | 1.0 × 10−6 | 1.3 × 106 | 3.1 | 128 |
| BGA-NIPU | 2 | 1.3 × 10−4 | 7.1 × 10−7 | 8.2 × 105 | 3.7 | 141 | |
| BSA-NIPU | 4 | 8.0 × 10−4 | 2.5 × 10−6 | 7.8 × 105 | 3.3 | 131 | |
Whereas the high-frequency region of the viscoelastic response provides information regarding the motion of monomer units, the intermediate frequencies reveal long-range interactions between chains. Given the hypothesis that hydrogen bonding was improved for the NIPUs studied in this work, the rubbery response at moderate frequencies could be important for understanding both viscous and elastic behavior. With an increasing number of methoxy groups, a steady increase in the values of the plateau (i.e., rubbery) modulus (GN,0) occurred (Table 4). The GN,0 values for BSF- and BGF-NIPUs were higher (∼8 × 105 Pa) than that of BPF-NIPU (∼7 × 105 Pa). This behavior was directly related to the increase in hydrogen bonding for BSF-NIPU (∼90%) and BGF-NIPU (∼85%) in comparison to BPF-NIPU (∼60%) that was shown by ATR-FTIR spectroscopy. Additionally, as the difference in the hydrogen bonding between BSF- and BGF-NIPUs was not significant, the GN,0 values for both the polymers were equivalent. The most prominent differences in the NIPU rheology were seen in the terminal regime. Between the petroleum-derived and lignin-derivable polymers, there was an order of magnitude decrease in the long relaxation time (τx). Although it would be expected that an increase in hydrogen bonding would lead to an increase in this timescale,69 differences in molar mass and dispersity also would impact this relaxation behavior.78 The BPF-NIPU exhibited a stronger deviation from the classical terminal power-law slopes of G′ (slope of 2) and G′′ (slope of 1) than the BGF- and BSF-NIPUs, indicating a greater strength of association between chains for the former.80 This behavior was unsurprising considering the associations in BPF-NIPU were primarily between hydroxyls, which typically had a stronger hydrogen-bond energy than between methoxy and hydroxyl groups.81,82 Thus, even with a greater number of potential hydrogen-bonding groups, the lignin-derivable NIPUs flowed more easily because these interactions were more easily interrupted.
The transitional and glassy behavior also were similar in the case of the dimethyl-substituted NIPUs (Fig. 5b); however, there was a lesser dependence of the glassy moduli on methoxy content than for unsubstituted NIPUs. This finding might be related to the presence of the bridging group substituents, suggesting these substituents more prominently control solid viscoelasticity. τc followed the same trend as for unsubstituted NIPUs yet was slightly higher in magnitude for BPA-NIPU in comparison to BPF-NIPU, which might be because of the added friction associated with the bridging substituents that was insignificant in the presence of bulkier methoxy substituents on the ring. Although the value of GN,0 for the dimethyl-substituted, lignin-derivable NIPUs was comparable to the unsubstituted counterparts, BPA-NIPU had a higher value than BPF-NIPU. These results suggested that the rigidity of dimethyl-substituted NIPUs increases physical crosslinking; however, this effect had little impact on lignin-derivable NIPUs potentially because the methoxy groups interfere with the hydroxyl–hydroxyl hydrogen bonding, yielding a transient network with more, but weaker, junctions.
Dimethyl-substituted NIPUs exhibited additional features related to hydrogen bonding in the terminal regime.69,80 The expected terminal power-law scaling was followed for all the NIPUs (except BSA-NIPU). This finding demonstrated the strength of hydrogen bonding in BSA-NIPU. Although the impact was small on the plateau modulus, there was a more pronounced influence in the melt state because of the accessibility of hydrogen bonds once backbone features (e.g., chain segments, entanglements) had relaxed. The trend in τx was similar to that seen for unsubstituted NIPUs, except for BSA-NIPU. This relationship could be related to the chain flexibility (i.e., persistence length), as well as the hydrogen-bond strength.69,80 More extensive studies, such as computational investigations into the dynamic conformations of NIPUs under flow and systematic rheological experiments on NIPUs with varying molecular weights, will be required to fully understand the relaxation phenomena in these polymers.
Finally, the thermo-rheological responses of the NIPUs were examined. Time-temperature superposition was performed to construct master curves; however, the quality of the superposition varied. This phenomenon was best visualized using van Gurp-Palmen plots (Fig. S29, ESI†). For all unsubstituted NIPUs, the data did not superimpose to a single curve around the transitional regime, although only BPF-NIPU was poorly superimposed in the terminal regime, which was likely a consequence of a higher molecular-weight dispersity in comparison to the other NIPUs.76 The dimethyl-substituted NIPUs showed good superposition across all temperatures, with only a noticeable spread in the data in the terminal regime caused by the noise related to measuring the low moduli. Vertical shift factors were nearly identical between all of the NIPUs (Fig. S30, ESI†), as demonstrated by the general equivalence of the Williams–Landel–Ferry parameters C1 and C2 (Table 4). The greatest deviation between the NIPUs was noticed at temperatures approaching the glass transition.
Lignin-derivable NIPUs exhibited a similar behavior to their petroleum-derived counterparts in the rubbery and glassy states, yet unexpectedly, the lignin-derivable NIPUs flowed more easily (i.e., exhibited overall lower moduli in the terminal regime) and had faster relaxation times than the petroleum-derived NIPUs. Overall, these findings suggested that the added functionalities derived from lignin precursors were not a hindrance to processability but rather a benefit to modulating the strength of hydrogen bonding from sustainably manufactured NIPUs.
4. Conclusions
This work studied the effect of key structural handles in lignin-derivable bisphenols, such as methoxy-group content and bridging-carbon substitutions, on the thermal, mechanical, and rheological properties on the thermoplastic NIPUs. Notably, the methoxy groups in lignin-derivable thermoplastic NIPUs increased the extent of hydrogen bonding in such polymers, which led to increased εbreak and toughness vs. petroleum-based versions. For example, lignin-derivable, BGF-NIPU (εbreak ∼ 185%, toughness ∼ 58 MJ m−3) and BSF-NIPU (εbreak ∼ 210%, toughness ∼ 62 MJ m−3) exhibited higher εbreak and toughness than BPF-NIPU (εbreak ∼ 140%, toughness ∼ 42 MJ m−3). Furthermore, the Tg of NIPUs increased with an increase in the methoxy-group content as the side groups and secondary interactions arising due to methoxy substituents reduced the chain mobility. These increases in εbreak, toughness, and Tgs were obtained without any adverse effect on other polymer properties, such as modulus (∼1.2–1.4 GPa), tensile strength (∼40–55 MPa), and thermal stability (∼280 °C). Importantly, the melt rheology of the lignin-derivable NIPUs remained favorable, and these methoxy moieties offered improved processability vs. their non-methoxy-containing, petroleum-derived counterparts. Altogether, the thermomechanical properties of lignin-derivable NIPUs can be readily tuned with careful choice of lignin-derivable building blocks, and the structure–property-processing relationships established in this work can aid the development of sustainable polymeric materials.Abbreviations
| ATR-FTIR | Attenuated total reflectance-Fourier transform infrared |
| BGA | Bisguaiacol A |
| BGF | Bisguaiacol F |
| BPA | Bisphenol A |
| BPF | Bisphenol F |
| BSA | Bissyringol A |
| BSF | Bissyringol F |
| C1 | First Williams–Landel–Ferry equation parameter |
| C2 | Second Williams–Landel–Ferry equation parameter |
| Đ | Dispersity |
| ε break | Elongation-at-break |
| DA-10 | 1,10-diaminodecane |
| DCM | Dichloromethane |
| DI | Deionized |
| DMAc | N,N-dimethylacetamide |
| DMF | N,N-dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DMSO-d6 | Deuterated dimethyl sulfoxide |
| DSC | Differential scanning calorimetry |
| DTGS | Deuterated triglycine sulfate |
| ESI | Electrospray ionization |
| FTMS | Fourier transform mass spectrometry |
| G | Guaiacyl |
| G′ | Storage modulus |
| G′′ | Loss modulus |
| G N,0 | Plateau modulus |
| GPC | Gel permeation chromatography |
| H | p-hydroxyphenyl |
| MDI | Methylene diphenyl diisocyanate |
| M n | Number-average molecular weight |
| NIPU | Non-isocyanate polyurethane |
| NMR | Nuclear magnetic resonance |
| PU | Polyurethane |
| RI | Refractive index |
| S | Syringyl |
| TBAB | Tetrabutylammonium bromide |
| τ c | Fast relaxation/critical timescale |
| T d5% | Temperature of 5% weight loss |
| TDI | Toluene diisocyanate |
| T g | Glass transition temperature |
| TGA | Thermogravimetric analysis |
| THF | Tetrahydrofuran |
| T ref | Rheology reference temperature |
| τ x | Longest relaxation time |
Author contributions
Conceptualization: J. S. M., Z. R. H., T. H. E., and L. T. J. K. Investigation and formal analysis: J. S. M, Z. R. H., and E. N. B. Writing (original draft): J. S. M. and Z. R. H. Writing (review and editing): J. S. M., Z. R. H., T. H. E., and L. T. J. K. Funding acquisition, supervision, and project administration: T. H. E. and L. T. J. K.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for financial support from the Army Research Office under Cooperative Agreement Number W911NF-22-2-0257 (thermal characterization and thermomechanical testing). The authors thank the National Science Foundation under award NSF DMR POL 2004682 (synthesis) and the Center for Plastics Innovation, an Energy Frontier Research Center funded by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES) under award DE-SC0021166 (rheology) for partially supporting this work. The authors acknowledge the University of Delaware (UD) Advanced Materials Characterization Laboratory for the use of the ATR-FTIR, DSC, and TGA instruments and the UD Mass Spectrometry facility for the use of the mass spectrometer. The authors also thank the UD NMR laboratory for the use of the NMR spectrometer, which was partially supported by the Delaware COBRE program, with a grant from the National Institute of General Medical Sciences – NIGMS (5 P30 GM110758-02) from the National Institutes of Health (NIH). The views and findings of the authors expressed herein do not necessarily reflect those of the Army Research Office, NSF, DOE, or NIH.References
- BASF Polyurethane, https://polyurethanes.basf.us/, (accessed January 10, 2024).
- Lubrizol, Thermoplastic Polyurethane (TPU), https://www.lubrizol.com/Engineered-Polymers/About/What-is-TPU, (accessed January 10, 2024).
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431–1455 RSC.
- L. T. J. Korley, B. D. Pate, E. L. Thomas and P. T. Hammond, Polymer, 2006, 47, 3073–3082 CrossRef CAS.
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 87, 535–552 CrossRef CAS.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef PubMed.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- N. Fanjul-Mosteirín, L. P. Fonseca, A. P. Dove and H. Sardon, Mater. Adv., 2023, 4, 2437–2448 RSC.
- G. Seychal, C. Ocando, L. Bonnaud, J. De Winter, B. Grignard, C. Detrembleur, H. Sardon, N. Aramburu and J.-M. Raquez, ACS Appl. Polym. Mater., 2023, 5, 5567–5581 CrossRef CAS.
- C. Pronoitis, M. Hakkarainen and K. Odelius, ACS Sustainable Chem. Eng., 2022, 10, 2522–2531 CrossRef CAS.
- G. Hua and K. Odelius, Macromol. Biosci., 2017, 17, 1–9 CrossRef PubMed.
- D. C. Aduba Jr., K. Zhang, A. Kanitkar, J. M. Sirrine, S. S. Verbridge and T. E. Long, J. Appl. Polym. Sci., 2018, 135, 1–11 CrossRef.
- P. B. V. Scholten, J. Cai and R. T. Mathers, Macromol. Rapid Commun., 2021, 42, 1–3 CrossRef PubMed.
- D. V. Palaskar, A. Boyer, E. Cloutet, C. Alfos and H. Cramail, Biomacromolecules, 2010, 11, 1202–1211 CrossRef CAS PubMed.
- O. Kreye, S. Wald and M. A. R. Meier, Adv. Synth. Catal., 2013, 355, 81–86 CrossRef CAS.
- K. Zhang, A. M. Nelson, S. J. Talley, M. Chen, E. Margaretta, A. G. Hudson, R. B. Moore and T. E. Long, Green Chem., 2016, 18, 4667–4681 RSC.
- C. Wulf, M. Reckers, A. Perechodjuk and T. Werner, ACS Sustainable Chem. Eng., 2020, 8, 1651–1658 CrossRef CAS.
- R. H. Lambeth, Polym. Int., 2021, 70, 696–700 CrossRef CAS.
- J. Guan, Y. Song, Y. Lin, X. Yin, M. Zuo, Y. Zhao, X. Tao and Q. Zheng, Ind. Eng. Chem. Res., 2011, 50, 6517–6527 CrossRef CAS.
- G. F. Bass and T. H. Epps, III, Polym. Chem., 2021, 12, 4130–4158 RSC.
- J. S. Mahajan, R. M. O’Dea, J. B. Norris, L. T. J. Korley and T. H. Epps, III, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y. X. Chen, Nat. Rev. Mater., 2022, 7, 83–103 CrossRef CAS.
- R. H. Lambeth and T. J. Henderson, Polymer, 2013, 54, 5568–5573 CrossRef CAS.
- Y. Peng, K. H. Nicastro, T. H. Epps, III and C. Wu, Food Chem., 2021, 338, 1–9 CrossRef.
- Y. Peng, K. H. Nicastro, T. H. Epps, III and C. Wu, J. Agric. Food Chem., 2018, 66, 11775–11783 CrossRef CAS PubMed.
- S.-F. Koelewijn, D. Ruijten, L. Trullemans, T. Renders, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2019, 21, 6622–6633 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- A. J. Shapiro, R. M. O'Dea, S. C. Li, J. C. Ajah, G. F. Bass and T. H. Epps, III, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 109–140 CrossRef PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- S. Zhao and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2021, 9, 1477–1493 CrossRef CAS.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- I. Ribca, B. Sochor, M. Betker, S. V. Roth, M. Lawoko, O. Sevastyanova, M. A. R. Meier and M. Johansson, Eur. Polym. J., 2023, 194, 112141 CrossRef CAS.
- M. Lawoko, L. Berglund and M. Johansson, ACS Sustainable Chem. Eng., 2021, 9, 5481–5485 CrossRef CAS.
- E. M. Maines, M. K. Porwal, C. J. Ellison and T. M. Reineke, Green Chem., 2021, 23, 6863–6897 RSC.
- R. M. O’Dea, J. A. Willie and T. H. Epps, III, ACS Macro Lett., 2020, 9, 476–493 CrossRef PubMed.
- C. Gioia, M. Colonna, A. Tagami, L. Medina, O. Sevastyanova, L. A. Berglund and M. Lawoko, Biomacromolecules, 2020, 21, 1920–1928 CrossRef CAS.
- K. H. Nicastro, C. J. Kloxin and T. H. Epps, III, ACS Sustainable Chem. Eng., 2018, 6, 14812–14819 CrossRef CAS.
- T. H. Epps, III, L. T. J. Korley, M. D. Green, J. S. Mahajan and H. Behbahani, U.S. Patent, App. No. 17/616,245, 2022 Search PubMed.
- S. V. Mhatre, J. S. Mahajan, T. H. Epps, III and L. T. J. Korley, Mater. Adv., 2023, 4, 110–121 RSC.
- K. H. S. Reno, J. F. Stanzione, III, R. P. Wool, J. M. Sadler, J. J. LaScala and E. D. Hernandez; U.S. Pat., No. 10273684, 2020 Search PubMed.
- A. Amitrano, J. S. Mahajan, L. T. J. Korley and T. H. Epps, III, RSC Adv., 2021, 11, 22149–22158 RSC.
- X. Zhang, J. S. Mahajan, L. T. J. Korley, T. H. Epps, III and C. Wu, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2023, 885, 1–9 CrossRef PubMed.
- E. K. Leitsch, G. Beniah, K. Liu, T. Lan, W. H. Heath, K. A. Scheidt and J. M. Torkelson, ACS Macro Lett., 2016, 5, 424–429 CrossRef CAS PubMed.
- S. Hu, X. Chen and J. M. Torkelson, Polymer, 2022, 256, 1–10 CrossRef.
- G. Beniah, D. J. Fortman, W. H. Heath, W. R. Dichtel and J. M. Torkelson, Macromolecules, 2017, 50, 4425–4434 CrossRef CAS.
- V. Schimpf, B. S. Ritter, P. Weis, K. Parison and R. Mülhaupt, Macromolecules, 2017, 50, 944–955 CrossRef CAS.
- M. Janvier, P.-H. Ducrot and F. Allais, ACS Sustainable Chem. Eng., 2017, 5, 8648–8656 CrossRef CAS.
- S. H. Lee and D. S. Lee, Macromol. Res., 2019, 27, 460–469 CrossRef CAS.
- M. A. R. Meier, Macromol. Rapid Commun., 2019, 40, 1–10 CrossRef.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- M. Blain, A. Cornille, B. Boutevin, R. Auvergne, D. Benazet, B. Andrioletti and S. Caillol, J. Appl. Polym. Sci., 2017, 134, 1–13 CrossRef.
- T. Wang, H. Deng, N. Li, F. Xie, H. Shi, M. Wu and C. Zhang, Green Chem., 2022, 24, 8355–8366 RSC.
- R. S. Waletzko, L. T. J. Korley, B. D. Pate, E. L. Thomas and P. T. Hammond, Macromolecules, 2009, 42, 2041–2053 CrossRef CAS.
- Q. Li, H. Zhou, D. A. Wicks, C. E. Hoyle, D. H. Magers and H. R. McAlexander, Macromolecules, 2009, 42, 1824–1833 CrossRef CAS.
- I. S. Ryu, X. Liu, Y. Jin, J. Sun and Y. J. Lee, RSC Adv., 2018, 8, 23481–23488 RSC.
- J. Mattia and P. Painter, Macromolecules, 2007, 40, 1546–1554 CrossRef CAS.
- D. Wołosz and P. G. Parzuchowski, Polymer, 2022, 254, 1–10 CrossRef.
- P. Saha, L. Goswami and B. S. Kim, ACS Sustainable Chem. Eng., 2022, 10, 4623–4633 CrossRef CAS.
- L. Trullemans, S. F. Koelewijn, I. Scodeller, T. Hendrickx, P. Van Puyvelde and B. F. Sels, Polym. Chem., 2021, 12, 5870–5901 RSC.
- I. Ribca, M. E. Jawerth, C. J. Brett, M. Lawoko, M. Schwartzkopf, A. Chumakov, S. V. Roth and M. Johansson, ACS Sustainable Chem. Eng., 2021, 9, 1692–1702 CrossRef CAS.
- X. Chen, X. Ouyang, J. Li and Y.-L. Zhao, ACS Omega, 2021, 6, 22578–22588 CrossRef CAS PubMed.
- R. J. Young and P. A. Lovell, Introduction to polymers, CRC press, 3rd edn, 2011, pp. 1–688 Search PubMed.
- A. Shabbir, H. Goldansaz, O. Hassager, E. van Ruymbeke and N. J. Alvarez, Macromolecules, 2015, 48, 5988–5996 CrossRef CAS.
- L. F. Wang, E. M. Pearce and T. K. Kwei, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 619–626 CrossRef CAS.
- A. L. Agapov, Y. Wang, K. Kunal, C. G. Robertson and A. P. Sokolov, Macromolecules, 2012, 45, 8430–8437 CrossRef CAS.
- Q.-H. Zhou, M. Li, P. Yang and Y. Gu, Macromol. Theory Simul., 2013, 22, 107–114 CrossRef CAS.
- S. F. Koelewijn, C. Cooreman, T. Renders, C. Andecochea Saiz, S. Van den Bosch, W. Schutyser, W. De Leger, M. Smet, P. Van Puyvelde, H. Witters, B. Van der Bruggen and B. F. Sels, Green Chem., 2018, 20, 1050–1058 RSC.
- S. Gupta, X. Yuan, T. C. Mike Chung, M. Cakmak and R. A. Weiss, Polymer, 2016, 107, 223–232 CrossRef CAS.
- C. L. Lewis, K. Stewart and M. Anthamatten, Macromolecules, 2014, 47, 729–740 CrossRef CAS.
- C. Osterwinter, C. Schubert, C. Tonhauser, D. Wilms, H. Frey and C. Friedrich, Macromolecules, 2015, 48, 119–130 CrossRef CAS.
- R. Stadler and L. de Lucca Freitas, Colloid Polym. Sci., 1986, 264, 773–778 CrossRef CAS.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University, New-York, 1st edn, 2003, vol. 100, pp. 1–433 Search PubMed.
- X. Callies, C. Véchambre, C. Fonteneau, S. Pensec, J. M. Chenal, L. Chazeau, L. Bouteiller, G. Ducouret and C. Creton, Macromolecules, 2015, 48, 7320–7326 CrossRef CAS.
- A. Jangizehi, M. Ahmadi and S. Seiffert, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1209–1223 CrossRef CAS.
- M. Palusiak and S. J. Grabowski, J. Mol. Struct., 2002, 642, 97–104 CrossRef CAS.
- K. M. Hambleton and J. F. Stanzione, III, ACS Omega, 2021, 6, 23855–23861 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00039k |
| This journal is © The Royal Society of Chemistry 2024 |