
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible solid bromine complexation into Ti3C2Tx MXene carriers: a highly active electrode for bromine-based flow batteries with ultralow self-discharge†
Luyin
Tang
ab,
Tianyu
Li
 a,
Wenjing
Lu
*a and
Xianfeng
Li
*a
a,
Wenjing
Lu
*a and
Xianfeng
Li
*a
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Zhongshan Road 457, Dalian 116023, P. R. China. E-mail: luwenjing@dicp.ac.cn; lixianfeng@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 1st April 2024
Abstract
Bromine-based flow batteries (Br-FBs) are appealing for stationary energy storage because of their high energy density and low cost. However, the wider application of Br-FBs is hindered by the sluggish reaction kinetics of the Br2/Br− redox couple and serious bromine crossover. Adding bromine complexing agents (BCAs) into electrolytes can inhibit bromine crossover effectively, but it generally deteriorates the reaction kinetics. Here, we use hexadecyl trimethyl ammonium bromide (CTAB) to intercalate Ti3C2Tx MXene as electrodes for Br-FBs. In this design, CTAB acts as the BCA to form robust solid bromine complexes, which are accompanied by highly active Ti3C2Tx MXene carriers. Soluble bromine species diffusion is perfectly suppressed based on such a strong and reversible solid complexation effect, while the adverse effect of solid complexes on reaction kinetics is well overcome. Thus, the assembled zinc-bromine flow battery delivered a remarkable improvement in suppressing self-discharge, achieving an unprecedently high capacity retention rate of 82.93% after standing for 24 h at 80 mA cm−2. At a high current density of 180 mA cm−2, this battery exhibited the highest voltage efficiency of 66.76% and energy efficiency of 66.06% ever reported, and also showed an outstanding long-term durability for 580 cycles with a high coulombic efficiency of 99.30%. This work provides a new strategy for designing electrodes with ultralow self-discharge, high power density and long cycle life for Br-FBs.
Broader contextBromine-based flow batteries (Br-FBs) are gaining increasing attention due to their high energy density and cost effectiveness. Nevertheless, the sluggish reaction kinetics of the Br2/Br− redox couple and the serious self-discharge caused by the diffusion/crossover of soluble bromine species lead to low power density and severe capacity fading, constraining the widespread application of Br-FBs. Consequently, the robust and reversible solid complexation effect of bromine complexing agents is incorporated into electrodes, endowing Br-FBs with ultralow self-discharge. We use hexadecyl trimethyl ammonium bromide to intercalate highly catalytic Ti3C2Tx MXene carriers, which can form solid complexes with bromine species. Meanwhile, Ti3C2Tx MXene carriers exhibit low adsorption energy to bromine, accelerating the kinetics of Br2/Br− reactions. Remarkably, a record high capacity retention rate of 82.93% after standing for 24 h at 80 mA cm−2 is achieved for a Br-FB. At a high current density of up to 180 mA cm−2, this Br-FB achieves the highest voltage efficiency and energy efficiency ever reported. This unique design breaks the “trade-off” between high bromine retention capacity and fast reaction kinetics. As a result, this work provides valuable insights into the design of high-performance electrodes for Br-FBs with ultralow self-discharge and high power density. |
Introduction
The huge demand for wide application of renewable resources such as solar energy has promoted the rapid development of energy storage technologies, which can ensure a stable electricity output and improve the grid safety and reliability.1–4 Among various energy storage systems, bromine-based flow batteries (Br-FBs) have emerged as compelling candidates for stationary energy storage because of their high energy density and low cost.5–7 Nevertheless, the bromine diffusion/crossover issue leads to severe self-discharge, decreasing the efficiencies, capacity and cycle life of Br-FBs. Additionally, the sluggish reaction kinetics of the Br2/Br− redox couple results in a low working current density and power density of batteries.8–10 To address the bromine diffusion/crossover issue, the common strategy is to introduce bromine complexing agents (BCAs) into electrolytes, which generally contain quaternary ammonium bromide groups, such as N-methylethylpyrrolidinium bromide (MEPBr).11–13 BCAs will complex with soluble bromine species to form an oily polybromide phase, decreasing the bromine concentration in the aqueous phase. Moreover, it is easier for the as-formed bromine complexes with larger sizes to be excluded through a frequently used size-sieving membrane, improving the coulombic efficiency (CE) and capacity retention rate of the battery.14,15However, the formation of the oily polybromide phase lowers the electrolyte conductivity and uniformity, deteriorating the reaction kinetics of the Br2/Br− redox couple.15–17 To address such a “trade-off” between inhibiting bromine diffusion and boosting reaction kinetics, our group has devoted great efforts to constructing highly electrochemically active electrodes with high bromine entrapping/retention capacity.8,18,19 In particular, such electrode materials can play the same roles as BCAs, helping in constructing BCA-free Br-FBs with low self-discharge, high power density and long lifespan. We have used the adsorption and spatial confinement effects of porous carbon nitride nanosheets or vertically aligned MoS2 nanosheet arrays to successfully endow the electrodes with high catalytic activity and high bromine retention capacity at the same time.18,19 Nonetheless, the adsorption effect is not robust enough to retain bromine more sufficiently, while the spatial confinement effect seriously depends on the precise size control.
Herein, we directly introduced BCAs into the electrode, which entrap/retain bromine species in the electrode based on the strong and reversible solid complexation effect (Scheme 1). In this design, Ti3C2Tx MXene served as a carrier to combine hexadecyl trimethyl ammonium bromide (CTAB) with a nonpolar carbon felt (PCF) substrate by intercalation (Scheme 1). CTAB could complex with bromine species to form stable solid complexes instead of conventional oily complexes (Fig. S1 and S2, ESI†).13,20 The interlayer spacing of Ti3C2Tx MXene carriers was enlarged by CTAB intercalation, allowing the free entrance of Br− ions into their interlayers during the charge process. The oxidized and soluble Brn− sequentially complexed with CTA+ to form solid CTA+–Brn− complexes, which were stored in the interlayers of Ti3C2Tx MXenes (Scheme 1). The bromine crossover issue was thereby effectively suppressed. Additionally, the Ti3C2Tx MXene exhibited a strong adsorption effect on bromine species, boosting the rate-determining step of Br2/Br− reactions.21 The “trade-off” between inhibiting bromine diffusion and boosting reaction kinetics was consequently well overcome. Using Ti3C2Tx-CTAB decorated PCF substrate as the cathode, the assembled zinc–bromine flow battery (ZBFB) exhibited an extremely high capacity retention rate of 82.93% after the 24-h standing test at 80 mA cm−2, much higher than those based on the adsorption effect or the spatial confinement effect.18,19 At 180 mA cm−2, this ZBFB exhibited a high voltage efficiency (VE) of 66.76% and a high energy efficiency (EE) of 66.06%, which could also run stably for over 580 cycles. Notably, the VE and EE were the highest values ever reported for ZBFBs (Table S1, ESI†). As a result, the strategy of incorporating the robust solid bromine complexation effect into electrodes can open up a new avenue for the application of Br-FBs with ultralow self-discharge, high power density and long lifespan.
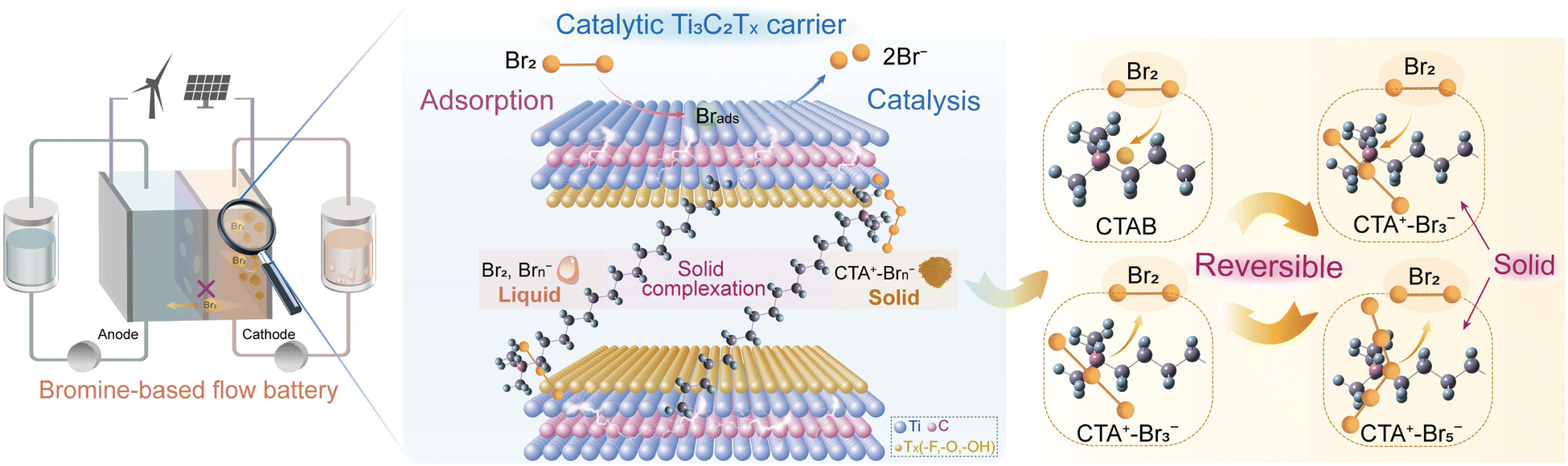 | ||
| Scheme 1 Illustration of the Ti3C2Tx-CTAB decorated electrode with high catalytic activity and excellent bromine retention capacity for a Br-FB with ultralow self-discharge and a long lifespan. | ||
Results and discussion
Rational design of CTAB intercalation into Ti3C2Tx MXene
The intercalation of CTAB into Ti3C2Tx MXene carriers was realized using a one-step method, as shown in Fig. S3 and S4 (ESI†). X-ray diffraction (XRD) patterns revealed that after being intercalated by CTAB, the (002) peak of Ti3C2Tx MXene shifted from 8.55° to 7.2°, indicating the extended interlayer spacing from 1.0 nm to 1.2 nm (Fig. 1a and Fig. S5, ESI†).22,23 The scanning electron microscopy (SEM) images clearly suggested the successful fabrication of Ti3C2Tx MXene nanosheets from Ti3AlC2 powders (Fig. 1b and c). After being intercalated by CTAB, the laminated structure of Ti3C2Tx-CTAB was more obvious with a larger interlayer spacing, supporting XRD analysis (Fig. 1d). Moreover, compared with Ti3C2Tx, the energy dispersive spectra (EDS) images of Ti3C2Tx-CTAB showed the presence of N and Br elements of CTAB (Fig. 1e and Fig. S6 and S7, ESI†). Also, an additional C–N peak appeared in the high-resolution C 1s X-ray photoelectron spectroscopy (XPS) spectrum of Ti3C2Tx-CTAB (Fig. 1f).24,25 In the Ti 2p XPS spectra, the decreased proportion of the Ti–C bond and the increased proportion of Ti2+/Ti3+ and Ti–O bonds in Ti3C2Tx-CTAB also confirmed the intercalation of CTAB (Fig. S8 and Table S2, ESI†).24 All these results confirmed the successful intercalation of CTAB into Ti3C2Tx MXene. | ||
| Fig. 1 Characterization of Ti3C2Tx-CTAB. (a) The XRD patterns of Ti3AlC2, Ti3C2Tx and Ti3C2Tx-CTAB. (b)–(d) SEM images of Ti3AlC2, Ti3C2Tx and Ti3C2Tx-CTAB. (e) EDS mappings of Ti3C2Tx-CTAB. (f) High-resolution C 1s XPS spectra of Ti3C2Tx and Ti3C2Tx-CTAB. | ||
Solid complexation effect between CTAB and bromine
The solid complexation effect between CTAB (namely CTA+-Br−) and bromine was investigated via Raman spectroscopy and XPS test. After the interaction of CTAB with bromine (denoted as CTA+–Brn−, n = 3 or 5), two novel peaks of Br3− (159 cm−1) and Br5− (211 cm−1) appeared in the Raman spectra of CTA+–Brn−, indicating the successful bromine complexation (Fig. 2a and Fig. S9, ESI†).26–28 In the C 1s XPS spectra of CTAB, the peak of C–N+ was found at 285.89 eV, which shifted to a higher energy (286.05 eV) than that of CTA+–Brn− (Fig. 2b). The C–N+ peak in the N 1s XPS spectra also exhibited a positive shift of 0.07 eV (Fig. S10, ESI†).29,30 These positive shifts were because that the electron cloud density of the C–N bond decreased when CTA+ was combined with Brn− (Fig. S9, ESI†). Moreover, in the high-resolution Br 3d XPS spectra of CTA+–Brn−, the emerging peaks at 68.95 and 70.30 eV were assigned to Br3− and Br5−, respectively, further verifying the bromine complexation effect of CTAB (Fig. 2c).21,31 The bromine content in EDS energy spectra obviously increased from 3.52% in CTAB to 7.35% in CTA+–Brn−, confirming Raman and XPS results (Fig. S11 and Table S3, ESI†). As a result, CTAB can play the same role as that of BCAs in inhibiting bromine diffusion/crossover.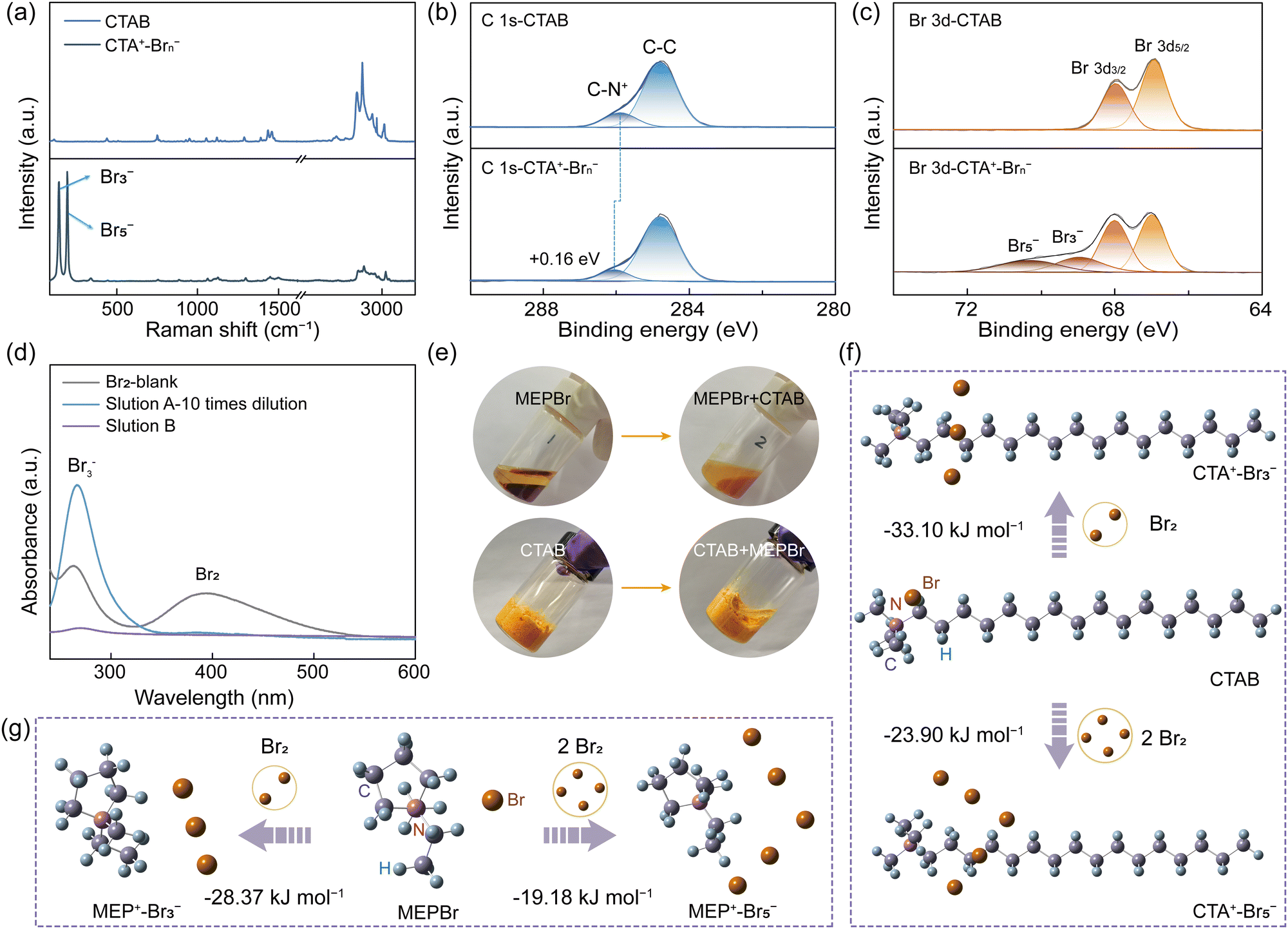 | ||
| Fig. 2 Verification of the solid bromine complexation effect of CTAB. (a) Raman spectra of CTAB and CTA+–Brn−. High-resolution (b) C 1s and (c) Br 3d XPS spectra of CTAB and CTA+–Brn−. (d) UV-vis spectra of different aqueous bromine solutions. (e) Photographs of the bromine complexes before and after adding the aqueous CTAB solution to MEP+–Brn− (up) and the aqueous MEPBr solution to CTA+–Brn− (down). The formation processes of (f) CTA+–Brn− and (g) MEP+–Brn− and the corresponding ΔGs of reactions. | ||
To study the complexing power of CTAB, we used MEPBr for comparison, which is nowadays the most commonly used BCA. When adding MEPBr to a bromine solution, an oily MEP+–Brn− phase was formed and the aqueous solution (labelled as solution A) color turned clear but still yellow after 12 h (Fig. S12, ESI†). However, in the same solution containing CTAB, the yellow solid bromine complexes were formed, while the aqueous solution (labelled as solution B) was almost colorless, indicating the dramatically decreased bromine concentration in the aqueous phase (Fig. S12, ESI†). Then, the UV-vis spectra were utilized to quantitatively analyse the bromine species in solutions A and B, respectively (Fig. 2d and Fig. S12b, ESI†). The intensities of bromine species in solution B were so low as to be almost undetectable, which were much lower than that of solution A even after 10 times dilution (Fig. 2d). Furthermore, CTAB could capture bromine species from the oily MEP+–Brn− phase and then almost completely transferred to the solid CTA+–Brn− phase, however which was an irreversible process (Fig. 2e). All these comparisons indicated that CTAB had a stronger complexing effect on bromine species than that of MEPBr.32,33
To further confirm this, density functional theory (DFT) calculations were carried out to calculate the Gibbs free-energy changes (ΔGs) between MEPBr/CTAB and Br2 (Fig. 2f–g and Table S4, ESI†). The ΔG of MEPBr with Br2 was −28.37 kJ mol−1, higher than that of CTAB (−33.10 kJ mol−1), indicating the stronger bromine complexing capacity of CTAB. Also, the ΔG values of forming MEP+-Br5− and CTA+-Br5− complexes were −19.18 kJ mol−1 and −23.90 kJ mol−1, respectively, further implying the superiority of CTAB (Fig. 2f–g and Table S4, ESI†). Furthermore, at higher temperatures of 50 °C and 70 °C, bromine clearly volatilized from the oily MEP+–Brn− phase (Fig. S13, ESI†). In contrast, very rare bromine vapor could be observed from the solid CTA+–Brn− complexes, proving that this solid complexation effect of CTAB was more robust even at high temperatures.
Effects of the solid complexation on the electrochemical performance
An adverse effect of the solid complexation is the probably decreased reaction kinetics of the Br2/Br− redox couple (Fig. S14, ESI†).13,20 Thus, we used Ti3C2Tx MXene with excellent electronic conductivity, high hydrophilicity and plentiful active sites as a carrier to combine CTAB and a PCF substrate.34–36 Especially, Ti3C2Tx MXene also exhibited very low adsorption energies (ΔEs) to bromine, indicating great bromine adsorption capacity (Fig. 3a and Fig. S15, ESI†). Considering that the rate-determining step of Br2/Br− reactions is to form Brads at the adsorbed state, Ti3C2Tx MXene can thus expedite Br2/Br− reactions, which is a prerequisite for being a carrier of CTAB. Moreover, the expanded interlayer spacing dramatically increased the specific surface area of Ti3C2Tx-CTAB (Fig. S16, ESI†). More active sites were exposed and the mass transfer was accelerated, further enhancing its electrochemical performance (Fig. S17, ESI†).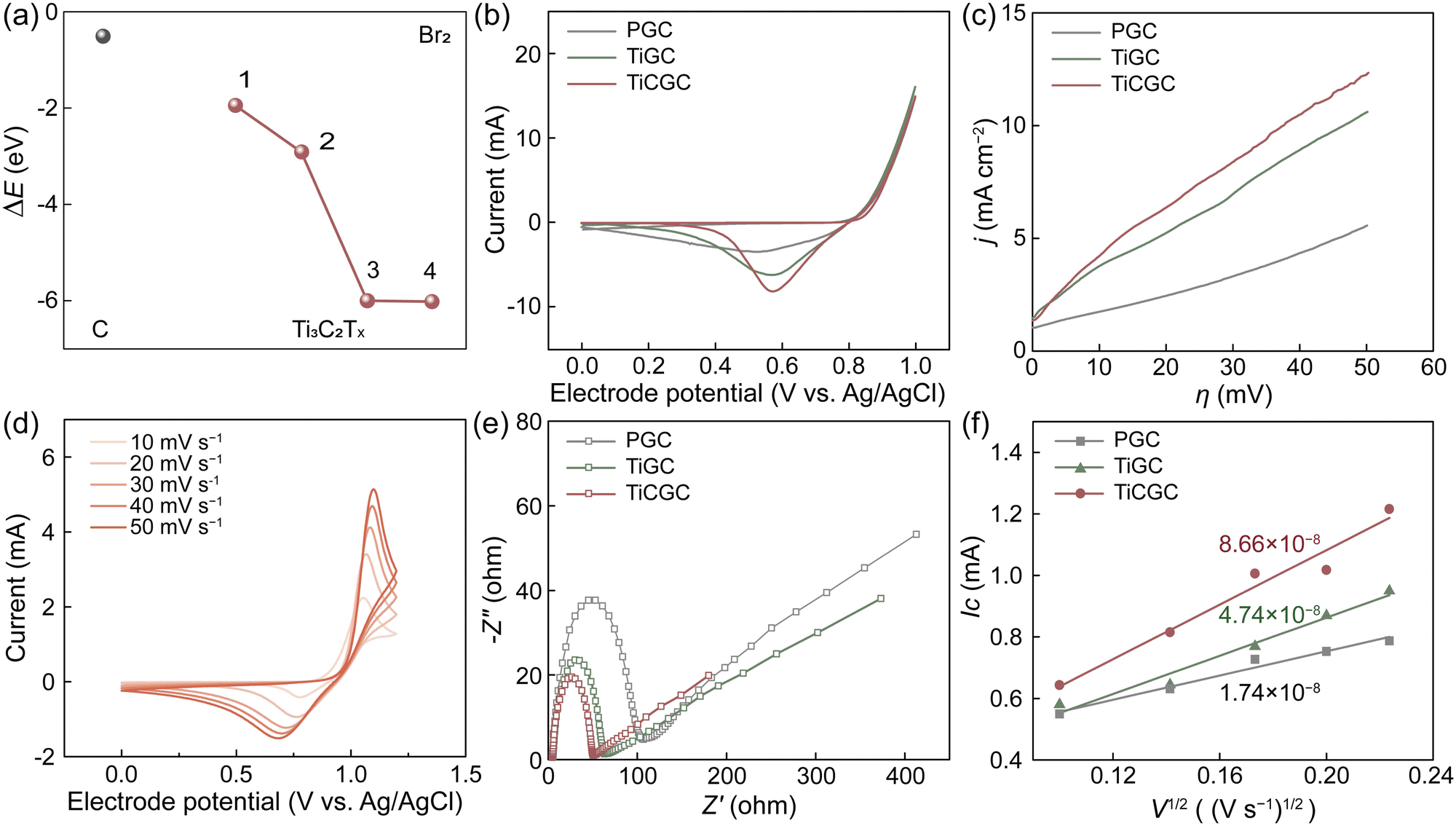 | ||
| Fig. 3 Electrochemical performance and the bromine affinity of Ti3C2Tx-CTAB. (a) The ΔEs of bromine molecules adsorbed on different Ti sites of Ti3C2Tx MXene. (b) CV curves at a scan rate of 10 mV s−1. (c) LSV curves at a scan rate of 1 mV s−1. (d) CV curves of TiCGC at different scan rates. (e) Nyquist plots. (f) Relationships between the Ic and the square root of the scan rate (v1/2). | ||
The electrocatalytic properties of Ti3C2Tx and Ti3C2Tx-CTAB were then evaluated via cyclic voltammetry (CV), linear sweep voltammetry (LSV) and electrochemical impedance spectroscopy (EIS), which were loaded on pristine glassy carbon (PGC) substrates (denoted as TiGC and TiCGC, respectively). As shown in Fig. 3b, the higher cathodic peak current (Ic) of TiGC than that of PGC suggested the high catalytic activity of Ti3C2Tx MXene carriers in Br2/Br− reactions. However, even though the solid CTA+–Brn− complexes were formed, TiCGC still exhibited the highest Ic, namely the best electrochemical activity. Moreover, the slopes of the LSV curves followed the order of TiCGC > TiGC > PGC, also indicating the fastest reaction kinetics of the Br2/Br− redox couple on Ti3C2Tx-CTAB (Fig. 3c). The resultant kinetic parameters, including the exchange current density (i0) and rate constant for the reaction (k0) of TiCGC, were also the highest, quantifying the significant improvement in reaction kinetics (Table S5, ESI†). As a result, in Br-FBs, the reaction kinetics of the Br2/Br− redox couple would not be affected by the solid complexation of CTAB but enhanced based on the high electrochemical activity of Ti3C2Tx MXene carriers. Most importantly, as the scan rate increased, the anodic peak potential shifted positively and the cathodic peak potential shifted negatively, indicating the reversible solid complexation of CTAB with bromine (Fig. 3d). Namely, the solid CTA+–Brn− complexes could release the soluble bromine species during the discharge process (Scheme 1). Moreover, the steady activity for 500 cycles further indicated that Ti3C2Tx-CTAB was electrochemically stable (Fig. S18, ESI†). Meanwhile, the interlayer spacing, structure and electrochemical activity of Ti3C2Tx-CTAB almost did not change before and after reacting with bromine, showing its high oxidization stability and anti-stack ability (Fig. S19 and S20, ESI†).
The Nyquist plots in Fig. 3e all included semicircles and liner parts, attributing to the electron transfer process at high frequencies and the diffusion process at low frequencies, respectively.8 The smallest semicircle radius of TiCGC indicated its lowest charge transfer resistance, supporting the CV and LSV results. Furthermore, the calculated diffusion coefficient of Br2 on TiCGC (8.66 × 10−8 cm s−1) was higher than that of TiGC (4.74 × 10−8 cm s−1), indicating the better mass transfer capacity of Ti3C2Tx-CTAB due to the intercalation (Fig. 3f and Table S6, ESI†). This was another reason why Ti3C2Tx-CTAB displayed better electrochemical performance than Ti3C2Tx.
Bromine capture capacity of Ti3C2Tx-CTAB
Notably, the bromine complexation effect of CTAB and the bromine adsorption effect of Ti3C2Tx MXene carriers could jointly contribute to the excellent bromine entraping/retention capacity of Ti3C2Tx-CTAB, then inhibiting bromine diffusion/crossover. We first applied a stationary visualization experiment to support this. From Fig. 4a, after adding Ti3C2Tx and Ti3C2Tx-CTAB to bromine solution, respectively, the solution color became clearer and clearer as prolonging the time. Nonetheless, the solution containing Ti3C2Tx-CTAB was always the clearest because of the stronger complexation effect of the intercalated CTAB. The higher stability of Ti3C2Tx-CTAB-Br2 than that of Ti3C2Tx-Br2 at a high temperature of 50 °C further proved the effectiveness and robustness of such a solid complexation effect (Fig. S21, ESI†). In another words, the strong and stable bromine complexation capacity of CTAB played the dominant role in entrapping bromine species. Then, the rotating ring-disk electrode technique was used to interpret their bromine entrapping capacities. In Fig. 4b, the |Iring| represented the bromine entrapping capability of different materials, which was determined by the bromine diffused to the ring electrode. Compared with the substrate, the |Iring| of Ti3C2Tx was reduced by adsorbing bromine. The lowest |Iring| of Ti3C2Tx-CTAB validated its best capacity to entrap bromine, which was mainly due to the powerful complexation effect of CTAB.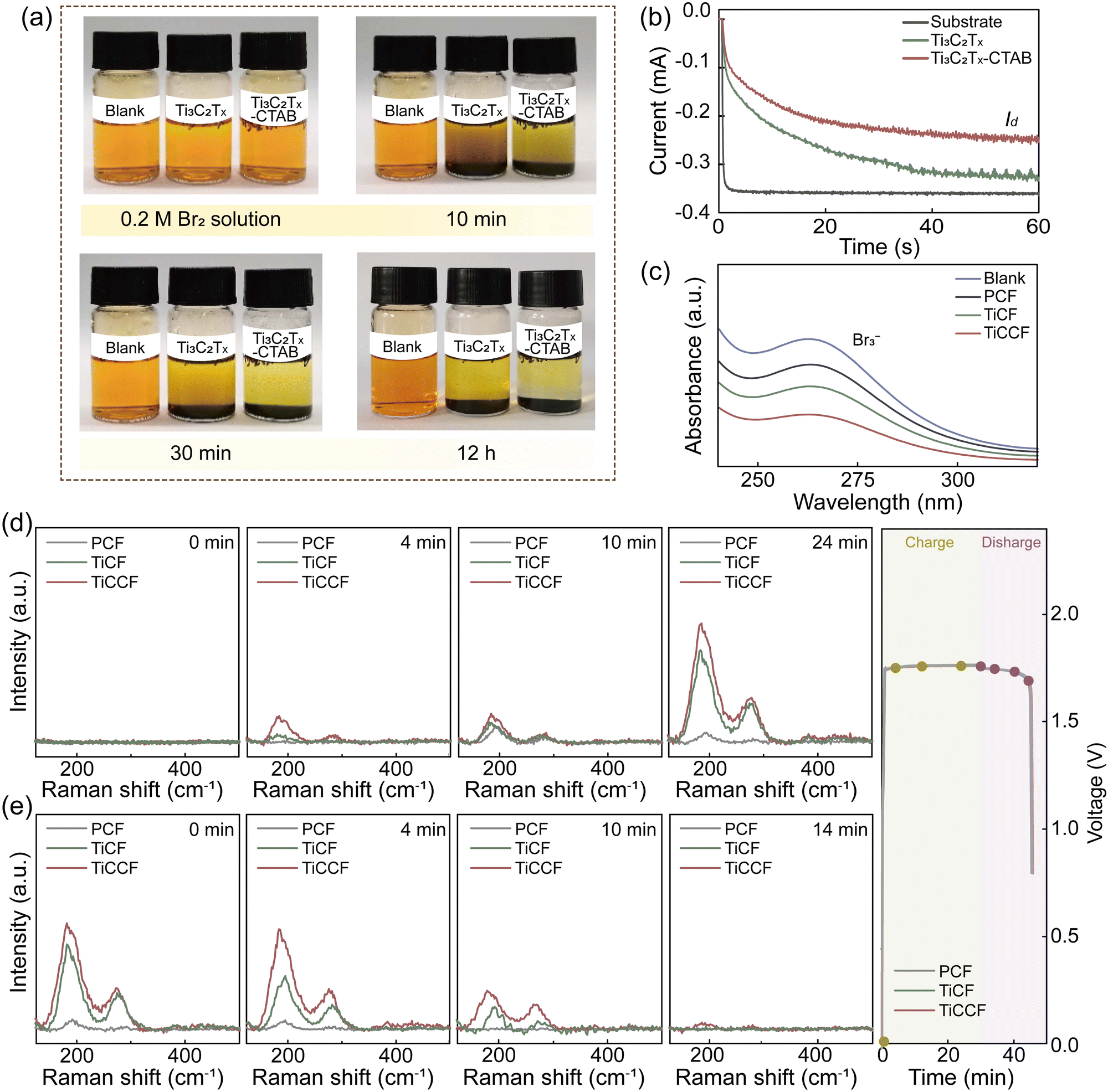 | ||
| Fig. 4 Bromine capturing capacity tests and species changes on electrodes during electrochemical reactions. (a) Photographs of the color changes of bromine solutions over time after adding different materials. (b) Iring–t curves of different materials. (c) UV-vis spectra of bromine solution after immersing PCF, TiCF and TiCCF. In situ Raman spectra on different cathodes of ZBFBs during the (d) charge and (e) discharge processes at different time intervals. | ||
We then loaded Ti3C2Tx-CTAB onto a PCF substrate to construct a composite electrode (denoted as TiCCF) and investigate its application in Br-FBs. The decreased absorbance intensity in UV-vis spectra represented that bromine in the solution was entrapped in different electrodes (Fig. 4c).33 Unsurprisingly, compared with the Ti3C2Tx-modified electrode (denoted as TiCF), TiCCF captured the most bromine (Fig. 4c). Also, from the in situ visualization experiment, bromine diffused rapidly from the PCF cathode to the anode, while the least bromine diffused from the TiCCF cathode no matter with or without BCAs, confirming the outstanding capacity of TiCCF to inhibit bromine diffusion/crossover (Fig. S22–S24, ESI†).
To gain deeper insights into the electrocatalytic behavior and bromine immobilization capacity of TiCCF, in situ Raman spectra were collected during the charge and discharge processes. As shown in Fig. 4d and e, the peaks of symmetrical stretching modes υ1 of Br3− (199 cm−1) and Br5− (291 cm−1) were detected mainly due to the combination of Br2 and Br−.12,37 As charging continued, Br5− appeared and the peak intensities of Br3− and Br5− gradually increased. Compared with PCF and TiCF, TiCCF always exhibited the highest peak intensities of Brn−, confirming its best electrochemical activity and bromine retention capacity (Fig. 4d). Meanwhile, the intensity of Br3− on TiCCF was always much higher than that of Br5−, which was mainly because that more Br2 complexed with CTAB instead of combining with Br3−. Namely, more Br2 could be encapsulated and retained in the TiCCF electrode, effectively preventing the bromine crossover. During the discharge process, the intensities of Br3− and Br5− peaks gradually declined and nearly vanished with the reduction of Br2. Similarly, TiCCF still always displayed the highest peak intensities of Brn− due to the fact that bromine species were entrapped and hardly diffused to the negative side. The nearly disappeared Brn− peaks on TiCCF also indicated that the solid bromine complexation of CTAB was indeed reversible (Fig. 4d and e).
Practical applications of TiCCF in Br-FBs
In general, solid bromine complexes in electrolytes will block the pipeline, which are consequently applied to single-flow or static bromine-based batteries generally (Fig. S14, ESI†).13,20 However, solid CTA+–Brn− complexes in the electrode did not affect the flow of the electrolyte. As a result, TiCCF was used in ZBFBs (double-flow) to study its availability in Br-FBs at 40 mA h cm−2 (Scheme 1). We first constructed a ZBFB using a TiCCF cathode, a PCF anode and electrolytes without BCAs (denoted as BCA-free ZBFB), which showed a high capacity retention rate of 50.53% after a standing test for 24 h at 80 mA cm−2 (Fig. 5a). This much higher capacity retention rate than that of the current BCA-free ZBFB using the PCF cathode (only 9.93%) certified the potential of using the TiCCF cathode to suppress self-discharge. Additionally, in Fig. 5b and Fig. S25 (ESI†), TiCCF enabled the BCA-free ZBFB to maintain higher and more stable efficiencies for 233 cycles (232 h) at 80 mA cm−2, showing its excellent stability. Of note, there was no electrolyte diffusion from the positive side to the negative side or corrosion of the graphite plate at the positive side in this BCA-free ZBFB after long cycling, benefiting from the excellent bromine entrapping capacity of Ti3C2Tx-CTAB on TiCCF (Fig. S26–S27, ESI†). Most importantly, the reversible solid bromine complexation of highly electrochemically active Ti3C2Tx-CTAB would not harm the reaction kinetics of the Br2/Br− couple (Fig. S28 and Table S7, ESI†).38,39 The BCA-free ZBFB based on the TiCCF cathode delivered a CE of 97.38% and a VE of 62.37% at a high current density of 200 mA cm−2. This battery retained stable performance for 400 cycles, in favor of improved power density and capacity maintenance (Fig. S29, ESI†). Most importantly, as shown in Fig. 5c, even though adding MEPBr into electrolytes, the capacity retention rate (39.15%) of the ZBFB based on the PCF cathode was still much lower than that of the BCA-free ZBFB using the TiCCF cathode. These results verified the much stronger bromine complexation capacity of CTAB than that of MEPBr, indicating the potential for CTAB into electrodes to replace MEPBr into electrolytes. Additionally, the electrochemical stability of the TiCCF electrode in the highly corrosive bromine solution without BCAs also confirmed its superiority in practical applications (Fig. S30a, ESI†).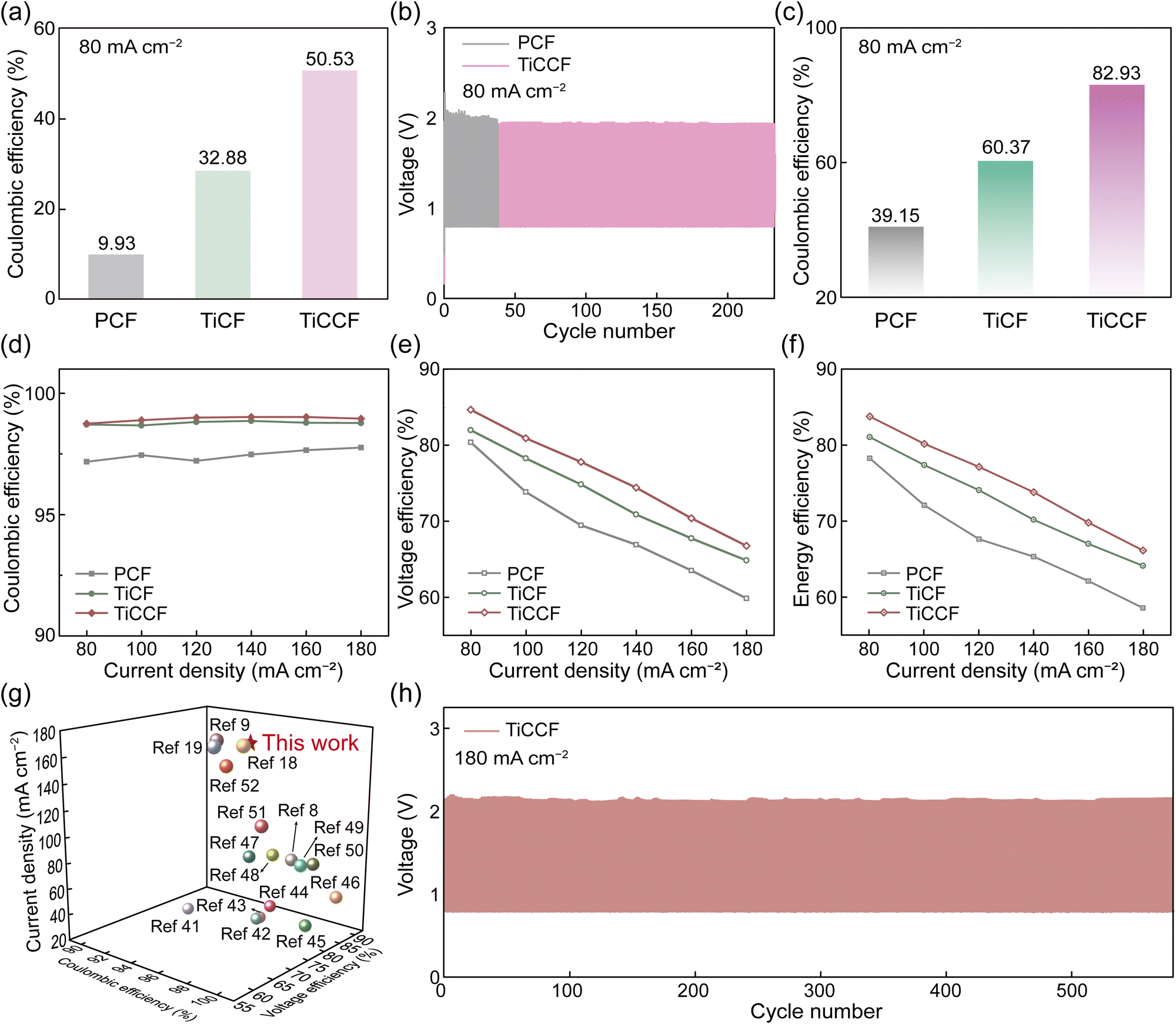 | ||
| Fig. 5 Performance of ZBFBs with different electrodes. (a) The self-discharge performance of BCA-free ZBFBs with different cathodes at 80 mA cm−2. (b) Cycling performance of BCA-free ZBFBs using different cathodes at 80 mA cm−2. (c) The self-discharge performance of ZBFBs assembled with different cathodes at 80 mA cm−2. The performance of ZBFBs assembled with different cathodes at different current densities: (d) CE, (e) VE, and (f) EE. (g) Comparisons of reported current densities and efficiencies of ZBFBs (all values derived from ref. 8, 9, 18, 19 and 41–52). (h) Charge–discharge curves of the ZBFB using the TiCCF cathode at 180 mA cm−2. | ||
It was worth noting that the current ZBFB systems in the demonstration stage utilized MEPBr as the BCA in electrolytes, and the self-discharge of the aforementioned BCA-free ZBFB was still relatively high. Consequently, to improve the feasibility of TiCCF cathodes in practical applications, TiCCF cathodes were utilized in ZBFBs using electrolytes with MEPBr. Notably, the TiCCF electrode also exhibited the stable electrochemical performance in the electrolyte with MEPBr, ensuring its availability (Fig. S30b, ESI†). The assembled ZBFB demonstrated an ultrahigh capacity retention rate of 82.93% at 80 mA cm−2, indicating the ultralow self-discharge of this battery (Fig. 5c). This capacity retention rate was more than twice higher than the current ZBFB system using the PCF cathode and MEPBr in electrolytes (39.15%), further confirming the outstanding bromine complexation capacity of CTAB. For the TiCF cathode, the capacity retention rate of the assembled ZBFB was 60.37%, due to the lack of such a strong CTAB complexation effect. Moreover, over the testing current density range from 80 to 180 mA cm−2, the ZBFB assembled with the TiCCF cathode always delivered the highest efficiencies (Fig. 5d–f). The higher CE was mainly due to the high capacity retention rate and the excellent electrochemical reversibility, which was caused by the superior properties of Ti3C2Tx-CTAB. The higher VE was because of the significantly decreased polarization resulting from the best electrochemical performance of TiCCF (Fig. S31 and S32 and Table S7 and S8, ESI†).40 Especially, its CE, VE and EE reached 98.95%, 66.76% and 66.06% at 180 mA cm−2, respectively. The VE and EE were the highest values for ZBFBs ever reported at such a high current density, contributing to higher power density (Fig. 5g and Table S1, ESI†). Additionally, the remarkable cycling stability for 580 cycles (262 h) with stable efficiencies at 180 mA cm−2 suggested that this ZBFB manifested efficient utilization of bromine species during the reversible electrochemical processes (Fig. 5h and Fig. S33, ESI†). The above results validated the superiority and practicability of TiCCF electrodes with considerable catalytic activity and bromine entrapping capacity in Br-FBs. Notably, compared with BCA-free ZBFBs, the VE and EE decreased at 180 mA cm−2 (Fig. S34, ESI†). These were because of the increased battery polarization caused by introducing MEPBr (Fig. S28, S30, and S31, ESI†). Consequently, more efforts still need to be made on more effective and simple electrode strategies in order to construct BCA-free Br-FBs with lower self-discharge and higher power density.
Conclusions
In summary, we proposed to introduce CTAB as the BCA onto Ti3C2Tx MXene carriers to be electrodes for Br-FBs in order to reduce the self-discharge. The robust and reversible solid complexation effect of CTAB effectively entrapped bromine species into electrodes, inhibiting bromine diffusion/crossover more effectively. Meanwhile, the good bromine adsorption capacity of Ti3C2Tx MXene carriers accelerated the reaction kinetics of the Br2/Br− redox couple. Thus, the assembled ZBFB based on the Ti3C2Tx-CTAB modified cathode achieved an ultrahigh capacity retention rate of 82.93%, much higher than that of the current ZBFB system (39.15%). This battery also exhibited a high VE of 66.76% and a high EE of 66.06% at 180 mA cm−2, in favor of higher power density. Furthermore, this ZBFB exhibited excellent cycling stability for 580 cycles with no obvious efficiency fading at 180 mA cm−2. This work provides a valid access to construct Br-FBs with ultralow self-discharge, high power density and long lifespan based on electrode design, accelerating their further demonstration applications.Author contributions
Luyin Tang performed the experiments, analyzed the data and wrote the initial manuscript draft. Dr Tianyu Li conducted the DFT simulations. Dr Wenjing Lu and Prof. Xianfeng Li designed the experiments, participated in project planning and discussions of the results, and revised and finalized the manuscript for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFB3805300), the National Natural Science Foundation of China (Grant No. 22379141 and 21935003), the International Partnership Program of Chinese Academy of Sciences (121421KYSB20210028), the Dalian Science and Technology Innovation Fund (2022JJ12GX024), and the Youth Innovation Promotion Association CAS (2022184). The authors would like to thank Congzhi Deng at DICP for the morphology test. Prof. Xianfeng Li acknowledged the financial support from XPLORER PRIZE.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
.
- Z. Zhao, X. Liu, M. Zhang, L. Zhang, C. Zhang, X. Li and G. Yu, Chem. Soc. Rev., 2023, 52, 6031–6074 RSC
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
- J. Noack, N. Roznyatovskaya, T. Herr and P. Fischer, Angew. Chem., Int. Ed., 2015, 54, 9776–9809 CrossRef CAS
- L. Zhang, R. Feng, W. Wang and G. Yu, Nat. Rev. Chem., 2022, 6, 524–543 CrossRef PubMed
- Y. Zhang, F. Li, T. Li, M. Zhang, Z. Yuan, G. Hou, J. Fu, C. Zhang and X. Li, Energy Environ. Sci., 2023, 16, 231–240 RSC
- C. Wang, Q. Lai, P. Xu, D. Zheng, X. Li and H. Zhang, Adv. Mater., 2017, 29, 1605815 CrossRef PubMed
- W. Lu, P. Xu, S. Shao, T. Li, H. Zhang and X. Li, Adv. Funct. Mater., 2021, 31, 2102913 CrossRef CAS
- K. Oh, A. Z. Weber and H. Ju, Int. J. Hydrogen Energy, 2017, 42, 3753–3766 CrossRef CAS
- X. Li, C. Xie, T. Li, Y. Zhang and X. Li, Adv. Mater., 2020, 32, 2005036 CrossRef CAS PubMed
- X. Li, T. Li, P. Xu, C. Xie, Y. Zhang and X. Li, Adv. Funct. Mater., 2021, 31, 2100133 CrossRef CAS
- L. Gao, Z. Li, Y. Zou, S. Yin, P. Peng, Y. Shao and X. Liang, iScience, 2020, 23, 101348 CrossRef CAS
- W. Lu, T. Li, C. Yuan, H. Zhang and X. Li, Energy Storage Mater., 2022, 47, 415–423 CrossRef
- L. Hua, W. Lu, T. Li, P. Xu, H. Zhang and X. Li, Mater. Today Energy, 2021, 21, 100763 CrossRef CAS
- M. Küttinger, P. A. Loichet Torres, E. Meyer, P. Fischer and J. Tübke, Molecules, 2021, 26, 2721 CrossRef
- K. Saadi, M. Kuettinger, P. Fischer and D. Zitoun, Energy Technol., 2021, 9, 2000978 CrossRef CAS
- L. Tang, T. Li, W. Lu and X. Li, Sci. Bull., 2022, 67, 1362–1371 CrossRef CAS
- L. Tang, C. Liao, T. Li, C. Yuan, G. Li, W. Lu and X. Li, Adv. Energy Mater., 2024, 14, 2303282 CrossRef CAS
- P. Xu, T. Li, Q. Zheng, H. Zhang, Y. Yin and X. Li, J. Energy Chem., 2022, 65, 89–93 CrossRef CAS
- X. Li, N. Li, Z. Huang, Z. Chen, Y. Zhao, G. Liang, Q. Yang, M. Li, Q. Huang, B. Dong, J. Fan and C. Zhi, ACS Nano, 2021, 15, 1718–1726 CrossRef CAS PubMed
- J. Luo, C. Wang, H. Wang, X. Hu, E. Matios, X. Lu, W. Zhang, X. Tao and W. Li, Adv. Funct. Mater., 2019, 29, 1805946 CrossRef
- J. Luo, J. Zheng, J. Nai, C. Jin, H. Yuan, O. Sheng, Y. Liu, R. Fang, W. Zhang, H. Huang, Y. Gan, Y. Xia, C. Liang, J. Zhang, W. Li and X. Tao, Adv. Funct. Mater., 2019, 29, 1808107 CrossRef
- Z. Li, X. Wang, W. Zhang and S. Yang, Chem. Eng. J., 2020, 398, 125679 CrossRef CAS
- K. Li, J. Zhao, A. Zhussupbekova, C. E. Shuck, L. Hughes, Y. Dong, S. Barwich, S. Vaesen, I. V. Shvets, M. Möbius, W. Schmitt, Y. Gogotsi and V. Nicolosi, Nat. Commun., 2022, 13, 6884 CrossRef CAS PubMed
- G. Bauer, J. Drobits, C. Fabjan, H. Mikosch and P. Schuster, J. Electroanal. Chem., 1997, 427, 123–128 CrossRef CAS
- M. E. Easton, A. J. Ward, B. Chan, L. Radom, A. F. Masters and T. Maschmeyer, Phys. Chem. Chem. Phys., 2016, 18, 7251–7260 RSC
- Y. Xu, C. Xie, T. Li and X. Li, ACS Energy Lett., 2022, 7, 1034–1039 CrossRef CAS
- M. Foss, S. Diplas and E. Gulbrandsen, Electrochim. Acta, 2010, 55, 4851–4857 CrossRef CAS
- C. Wang, G. Ma, J. Zhou, M. Zhang, X. Ma, F. Duo, L. Chu, J. Huang and X. Su, Appl. Organomet. Chem., 2019, 33, e5077 CrossRef
- W. Cai, J. Wang, X. Quan and Z. Wang, J. Appl. Polym. Sci., 2018, 135, 45657 CrossRef
- Q. Liu and D. W. Margerum, Environ. Sci. Technol., 2001, 35, 1127–1133 CrossRef CAS PubMed
- B. N. Grgur, J. Electrochem. Soc., 2019, 166, E50–E61 CrossRef
- Y. An, Y. Tian, H. Shen, Q. Man, S. Xiong and J. Feng, Energy Environ. Sci., 2023, 16, 4191–4250 RSC
- Y. Zhao, J. Zhang, X. Guo, X. Cao, S. Wang, H. Liu and G. Wang, Chem. Soc. Rev., 2023, 52, 3215–3264 RSC
- H. Wang, Z. Du, Z. Cheng, Z. Cao, Y. Ye, Z. Wang, J. Wei, S. Wei, X. Meng, L. Song, Y. Gong, S. Yang and L. Guo, Adv. Mater., 2023, 35, 2301399 CrossRef CAS PubMed
- J.-H. Lee, Y. Byun, G. H. Jeong, C. Choi, J. Kwen, R. Kim, I. H. Kim, S. O. Kim and H.-T. Kim, Adv. Mater., 2019, 31, 1904524 CrossRef CAS PubMed
- M. Schneider, G. P. Rajarathnam, M. E. Easton, A. F. Masters, T. Maschmeyer and A. M. Vassallo, RSC Adv., 2016, 6, 110548–110556 RSC
- S. S. Li, J. R. Sun and J. Q. Guan, Chin. J. Catal., 2021, 42, 511–556 CrossRef CAS
- Y.-Q. Zhang, G.-X. Wang, R.-Y. Liu and T.-H. Wang, Energies, 2023, 16, 3043 CrossRef CAS
- W. I. Jang, J. W. Lee, Y. M. Baek and O. O. Park, Macromol. Res., 2016, 24, 276–281 CrossRef CAS
- S. Suresh, M. Ulaganathan and R. Pitchai, J. Power Sources, 2019, 438, 226998 CrossRef CAS
- S. Suresh, M. Ulaganathan, R. Aswathy and P. Ragupathy, ChemElectroChem, 2018, 5, 3411–3418 CrossRef CAS
- K. Mariyappan, P. Ragupathy and M. Ulaganathan, J. Electrochem. Soc., 2021, 168, 090566 CrossRef CAS
- R. P. Naresh, K. Mariyappan, K. S. Archana, S. Suresh, D. Ditty, M. Ulaganathan and P. Ragupathy, ChemElectroChem, 2019, 6, 5688–5697 CrossRef
- K. Mariyappan, R. Velmurugan, B. Subramanian, P. Ragupathy and M. Ulaganathan, J. Power Sources, 2021, 482, 228912 CrossRef CAS
- K. Mariyappan, T. Mahalakshmi, T. S. Roshni, P. Ragupathy and M. Ulaganathan, Adv. Mater. Interfaces, 2023, 10, 2202007 CrossRef CAS
- C. Wang, X. Li, X. Xi, W. Zhou, Q. Lai and H. Zhang, Nano Energy, 2016, 21, 217–227 CrossRef CAS
- H. Jung, J. Lee, J. Park, K. Shin, H. T. Kim and E. Cho, Small, 2023, 19, 2208280 CrossRef CAS PubMed
- H. X. Xiang, A. D. Tan, J. H. Piao, Z. Y. Fu and Z. X. Liang, Small, 2019, 15, 1901848 CrossRef PubMed
- R. Wang, J. Energy Storage, 2023, 74, 109487 CrossRef
- C. Wang, W. Lu, Q. Lai, P. Xu, H. Zhang and X. Li, Adv. Mater., 2019, 31, 1904690 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00580e |
| This journal is © The Royal Society of Chemistry 2024 |