
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photo-responsive organic electrochemical transistor†
Nicholas
Turetta
 a,
Wojciech
Danowski
a,
Luca
Cusin
a,
Pietro Antonio
Livio
a,
Rawad
Hallani
b,
Iain
McCulloch
bc and
Paolo
Samorì
*a
a,
Wojciech
Danowski
a,
Luca
Cusin
a,
Pietro Antonio
Livio
a,
Rawad
Hallani
b,
Iain
McCulloch
bc and
Paolo
Samorì
*a
aUniversity of Strasbourg, CNRS, ISIS UMR 7006, 8 allée Gaspard Monge, F-67000 Strasbourg, France. E-mail: samori@unistra.fr
bUniversity of Oxford, Department of Chemistry, Oxford, OX1 3TA, UK
cKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Thuwal, 23955-6900, Saudi Arabia
First published on 28th February 2023
Abstract
The design of novel organic electrochemical transistor (OECT) channel materials that can be controlled by a whole range of external stimuli is key towards the emergence of unprecedented technologies in bioelectronics. Like the established multiresponsive field-effect transistors, multiresponsive OECTs can in principle be realised via blending, by combining multiple components with each one imparting a specific function to the device. Here we report the first example of an optically switchable OECT which is capable of undergoing a reversible modulation of its ON current by up to 30% upon irradiation with UV and visible light. By investigating the electrical characteristics of the channel material, in conjunction with the electronic characterisation performed by a macroscopic Kelvin probe technique and photoemission yield spectroscopy in air, we gained distinct insight into the electrochemical doping process occurring within the blend upon light irradiation. Such a proof-of-concept work opens perspectives towards the implementation of complex neuromorphic operations and algorithms in OECTs.
10th Anniversary StatementThe Journal of Materials Chemistry C has established itself as a high quality, interdisciplinary platform for reporting innovative concepts in which materials are brought to function through their integration in optical, magnetic and electronic devices. All in all, 10 years of success and a bright future ahead! |
Introduction
Functional diversification in organic materials represents a major research challenge with high potential for disruptive technological applications in tomorrow's optoelectronics and nanoelectronics.1–3 The motion of ionic charge carriers at a solid–liquid (or semisolid) electrolyte interface brings extraordinary functionalities to traditional organic semiconductors (OSCs), thus enabling the future development of these technologies.4,5 Iontronics is a discipline that exploits such concepts to control the ionic–electronic transport in mixed conductors,6 with its most promising application being device interfacing with biological tissues and living systems, i.e., bioelectronics.7,8 In the field of organic bioelectronics, organic electrochemical transistors (OECTs) have concurrently emerged as prototypical devices whose operation, mediated by ionic motion, relies on the electrochemical (EC) doping of an OSC channel material covered by an electrolyte in which the gate electrode is immersed.9Among OSCs, conjugated polymers and polyelectrolytes exhibiting mixed conductivity represent the current state-of-the-art materials for bioelectronics,10,11 and they are thus extremely interesting for the realisation of integrated organic/bio-organic functional systems.12 Recently developed materials have already enabled the sensing of cell parameters,13 the actuation of functions in living cells in vitro,8 and the efficient drug delivery in living tissues as rooted by electrochemical signaling driven by OECTs.14 Such proofs of concept opened the door to new therapeutical approaches, e.g. by locally monitoring disease states.15 The development of biocompatible systems that are fueled by or respond to multiple inputs under physiological conditions such as electronic stimuli, ionic gradients, light pulses and mechanical stresses is a prerequisite for the full integration of man-made electronics in the human body.16,17 During the past decade, the bioelectronics endeavor has mainly been focused on the figures of merit of OECTs, such as transconductance,18 reaching performances that make such devices suitable for functional applications.19 Molecular doping strategies in organic materials have been continuously improved towards the development of reliable electronics.20 Yet, device stability needs further optimisation,21 due to the special environment of living systems, and more testing both in bioelectronic circuits22 and for in vivo applications is required.23
The integration of an optoelectronic control in organic bioelectronics, however, remains relatively unexplored. Preliminary work has been recently reported on photocapacitive and photofaradaic processes in photoelectrodes based on OSC materials,24 the use of photocapacitors to control the activity of living cells by acting as electrodes for extracellular stimulation,25 or the interfacing of traditional light-based devices such as organic LEDs and solar cell photodetectors with living systems.26 These bioelectronics and neuromorphic devices may then be self-powered by transparent photovoltaics, and implemented, for example, as an artificial skin.27 The use of optical tools represents a viable strategy combining minimal invasiveness with the potential to yield fast and low-power photonic devices.28 Light-based smart healthcare for diagnostics and therapeutics is already widespread in clinics, although the realisation of functional tools that can be directly worn by or implanted in patients is still very limited.1 Hence, there are several opportunities to develop new (photo)chemical approaches for the realisation of switchable bioelectronic devices based on OSC materials,29 aiming to mimic and control biological systems with the aid of external stimuli.30 Photoswitchable biomaterials, pioneered by Willner,31,32 can be designed to integrate molecular photoswitches33 into biological systems34 and/or interact with bioelectronic materials as stimuli-responsive blocks to generate dynamic interfaces.30
The use of light to modulate the electrical response of OECTs can provide an additional tool to remotely control the device operation in living systems with minimum invasiveness. Some OSC materials possess bandgap energies of 1–4 eV, thus they are prone to absorbing visible or UV light. This optical absorption determines an increase in conductivity, whereas its lowering occurs much more rarely.35 One possible strategy to achieve a light-triggered bidirectional conductivity modulation consists of the blending of molecular photochromic additives into OSC polymer matrices.36,37 For applications in bioelectronics, the blending can be made by combining molecular photoswitches with an electrochemically active polymer, which is an unprecedented strategy for controlling OECT operation. Among photoswitching molecules that can be used for imparting an additional light responsiveness to the OSC polymer, spiropyran (SP) is a first choice for the design and tuning of dynamic materials.38 SP derivatives exist in two relatively stable isomers under ambient conditions: the closed ring non-polar spiropyran, and the ring-opened conjugated, metastable zwitterionic merocyanine (MC) form.39 It is possible to trigger the isomerisation from SP to MC upon irradiation with UV light (λ ≈ 365 nm), whereas the opposite process can be accomplished by illumination with visible light (λ ≈ 530 nm) or activated thermally.
On the other hand, an OSC polymer matrix is chosen based on its ability to efficiently transport charges and ions as well as its affinity with the side chains of the chosen spiropyran derivative to promote efficient blending. Since a common choice for the side chains of OECT polymers is either oligo(ethylene glycol) (OEG)40 or alkyl chains,41 due to the advantages of the easy polymer synthesis, solution processability and structural arrangement of the polymer, we focused our attention on poly(2-(4,4′-bis(2-methoxyethoxy)-5′-methyl-[2,2′bithiophen]-5-yl)-5-methylthieno[3,2-b]thiophene) (pgBTTT),42 a semiconducting polythiophene derivative whose structure comprises a BTTT backbone decorated by OEG chains in a regiosymmetrical arrangement. To ensure structural similarity for optimal mixing, a SP derivative exposing OEG chains of comparable length (OEG-SP) was also synthesised for improved blending (ESI† – Section S1). Optically switchable OECTs were fabricated by casting solutions containing a mixture of pgBTTT with SP molecular additives to form thin blend films onto Au prepatterned quartz substrates. By coupling a mixed ion-electron conductor with a light-sensitiser, we fabricated OECTs that can toggle between two states of current upon UV or visible light irradiation.
Results and discussion
Multifunctionality in multicomponent molecular films, such as those obtained by blending a polymer with a small molecule additive, is boosted via the subtle control over the interface between the different components.37 The process of blending is aimed at avoiding phase segregation while favoring intermixing towards a maximal degree of intercomponent interfacing, as inspired by van der Waals heterostructures.43 In our photoswitchable blend, the pgBTTT polymer imparts mixed ionic-electronic conductivity whereas the OEG-SP additive confers a photoresponsive nature to the hybrid material. These two components have been mixed in a 20% m/m proportion of OEG-SP in pgBTTT and deposited as drop-cast thin films from CHCl3 solutions at various concentrations. The responsiveness of the OEG-SP and OEG-SP/pgBTTT blends to light stimuli at different wavelengths is investigated by UV-vis absorption spectrophotometry both in solutions and in thin films. Monocomponent films of OEG-SP reveal efficient and reversible photoisomerisation when exposed to UV (365 nm) and visible (530 nm) light (Fig. 1a). The OEG-MC state is characterised by a strong absorption band centred at 569 nm resulting from its conjugated zwitterionic structure. Although partially hidden by the main absorption band of the pgBTTT polymer, this band was also observed in the bicomponent film (red shifted by about 20 nm due to matrix effects)44,45 thus corroborating the efficient photoisomerisation also in the blend (Fig. 1b). Additionally, a weaker band associated with electronic transitions of higher energy46 was ca. 40 nm red shifted in the OEG-MC/pgBTTT blend (from 352 nm of the neat OEG-MC film to ca. 395 nm) further supporting the existence of intermolecular interactions between the MC form and the pgBTTT matrix. The photoisomerisation kinetics of OEG-MC has been studied and the half-life (t1/2) of the metastable OEG-MC form in the dark has been estimated. We found that in spin-coated films it follows a biexponential decay (Fig. S7, ESI†). Among the two thermal relaxations detected, the faster process (t1/2 = 30 min) can be associated with the behaviour of the monomeric form whereas the slower process (t1/2 = 100 min) might be ascribed to aggregated MC species.39 Overall, the average thermal stability of the OEG-MC in the thin film falls between the stability of OEG-MC in low-polarity (THF, t1/2 = 45 s) and high-polarity (MeOH, t1/2 = 80 min) solvents (Fig. S4, ESI†); this is consistent with the stabilisation of the zwitterionic OEG-MC in polar solvents and indicates sufficiently long lifetimes of the MC isomer in the film to enable its integration in light-responsive OECT devices. Details about the modelling equations and the photoswitching experiments performed both in solution and in thin films are provided in ESI† – Section S2.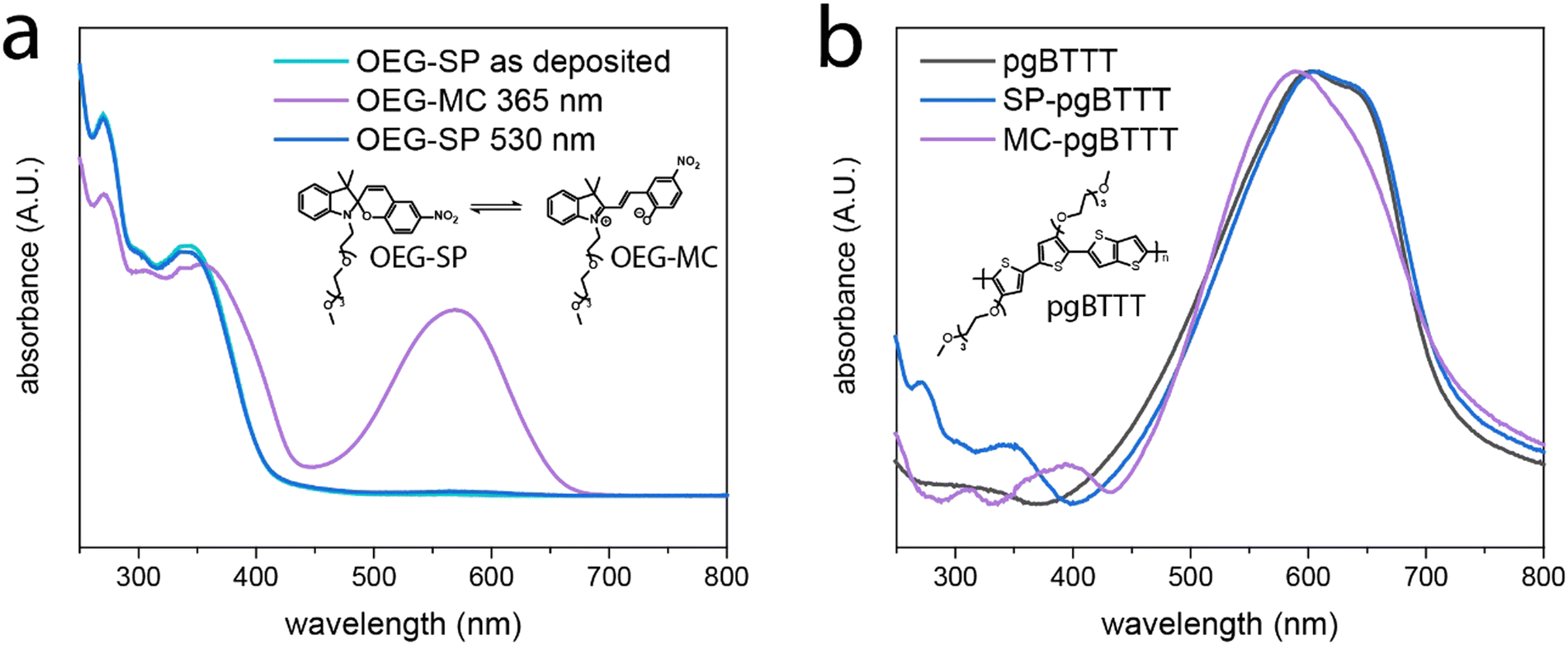 | ||
| Fig. 1 UV-vis photoswitching experiments performed on thin films deposited via drop casting (DC) on quartz substrates. (a) Absorption spectra of OEG-SP in the SP and MC forms. (b) Normalised UV-vis absorption spectra of neat pgBTTTvs. the 20% m/m OEG-SP/pgBTTT blend (SP-pgBTTT). The spectra have been collected before (SP-pgBTTT) and after (MC-pgBTTT) irradiation with a 365 nm UV LED. | ||
Blends of 20% m/m OEG-SP/pgBTTT in CHCl3 were drop-cast onto UV-treated quartz substrates exposing prepatterned Au electrodes with a channel width (W) and length (L) of 300 μm and 30 μm, respectively. The presence of OEG-SP in the solution improves the polymer's processability in the thin films, as evidenced by the smoother surface topography of the blend when compared to the neat pgBTTT polymer films (Fig. 2). The morphology of the blend thin films is characterised by elongated, interconnected, wire-like structures completely covering the substrate. Conversely, neat pgBTTT films exhibit a discontinuous morphology with numerous thick polymer aggregates. Such a morphology limits the further analysis of the surface properties by means of atomic force microscopy (AFM).
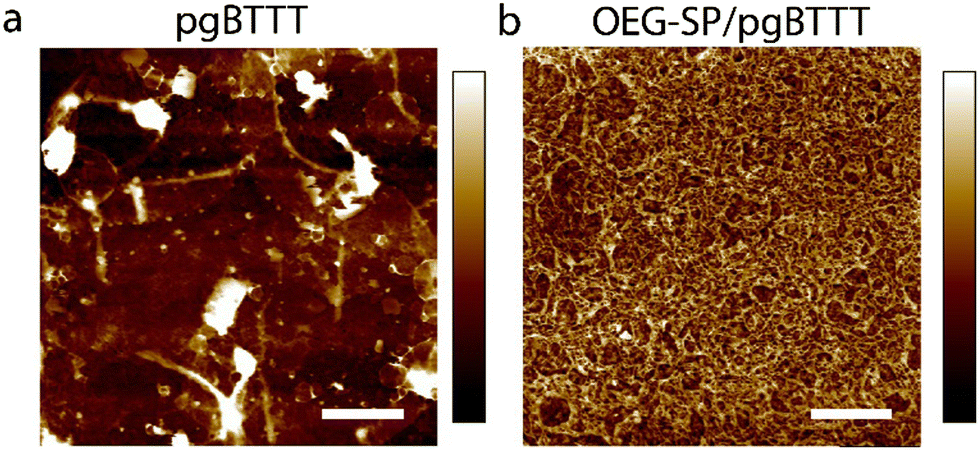 | ||
| Fig. 2 Atomic force microscopy height image of thin films deposited on a UV-SiO2 substate via drop casting from 0.3 mg mL−1 CHCl3 solutions. Image size: 5 × 5 μm2. Lateral scale bar: 5 μm. (a) Neat pgBTTT, Z-scale: 53 nm, nominal thickness: 60 nm, RRMS: 10 nm. (b) OEG-SP/pgBTTT, Z-scale: 21 nm, nominal thickness: 30 nm, RRMS: 3.2 nm. RRMS values are estimated over an area of 2 × 2 μm2. | ||
OECT transfer curves were recorded in a 0.5 M NaCl electrolyte solution (Fig. 3a) by irradiating the devices with either a UV (365 nm, 3 W, 2 min) or a green LED (530 nm, 5 mW, 5 min). OECTs based on the OEG-SP/pgBTTT blend show higher ON currents (ION) when the photoswitch is in the SP form, with a ca. 20% decrease in the ION when the OECT is illuminated with UV light thereby triggering the isomerisation of the photoswitch to the MC form (Fig. 3b). Upon illumination with 530 nm light to induce the back-isomerisation yielding the SP isomer, an ION recovery exceeding 60% of the pristine current variation is observed. The OFF current (IOFF) undergoes analogue changes, while remaining a factor 103 smaller than ION (Fig. 3c). After subsequent cycles of photoswitching, the ION of the OECT experiences an overall negative drift which is probably associated with the partial solubilisation of the photoswitch (Fig. S8, ESI†). ION is stabilised only after the 7th cycle of switching, even though the current recovery associated with the switching to the SP form slowly continues to degrade. In fact, after the 5th cycle of switching between the SP and MC forms, signs of photo-fatigue start to appear as is commonly observed for non-immobilised SP.47 The presence of the aqueous environment is also a factor that may contribute to the MC degradation due to hydrolysis.48 The 66% reduction here determined after the 12th cycle of switching (from 21.5% to 7.2% absolute ION modulation) is comparable to a 57% loss in the initial signal observed in the literature.47 Overall, the ION of the OECT decreases by 40% due to photo-fatigue, with the concomitant gradual loss of the OEG-SP photoswitching capability. Compared to neat pgBTTT OECTs (Fig. 3c) with an identical device configuration and a similar channel depth (60 nm vs. 30 nm for the blend), the product of charge carrier mobility and volumetric capacitance (μC*), extracted from the transfer curves at an overdrive voltage (VOV = VGS − VTH = −0.35 V) is a factor ≈ 15 lower for the blend (7 vs. 113 F cm−1 V−1 s−1). The ION/IOFF ratio is also reduced by a factor of ≈ 10 and VTH is shifted towards more positive values (from −50 mV to +220 mV). To compensate for these performance losses in a commercial device application, VTH and the absolute ION value can both be tuned by controlling the substrate wettability and the thickness of the film, hence providing a tool for further optimisation (see ESI† – Section S3). Remarkably, both pgBTTT and the blend can be processed via spin-coating diluted (0.05 mg mL−1) solutions in CHCl3 to yield continuous ultrathin films whose electrical and interfacial properties can be easily investigated (Fig. S11 and S12, ESI†).
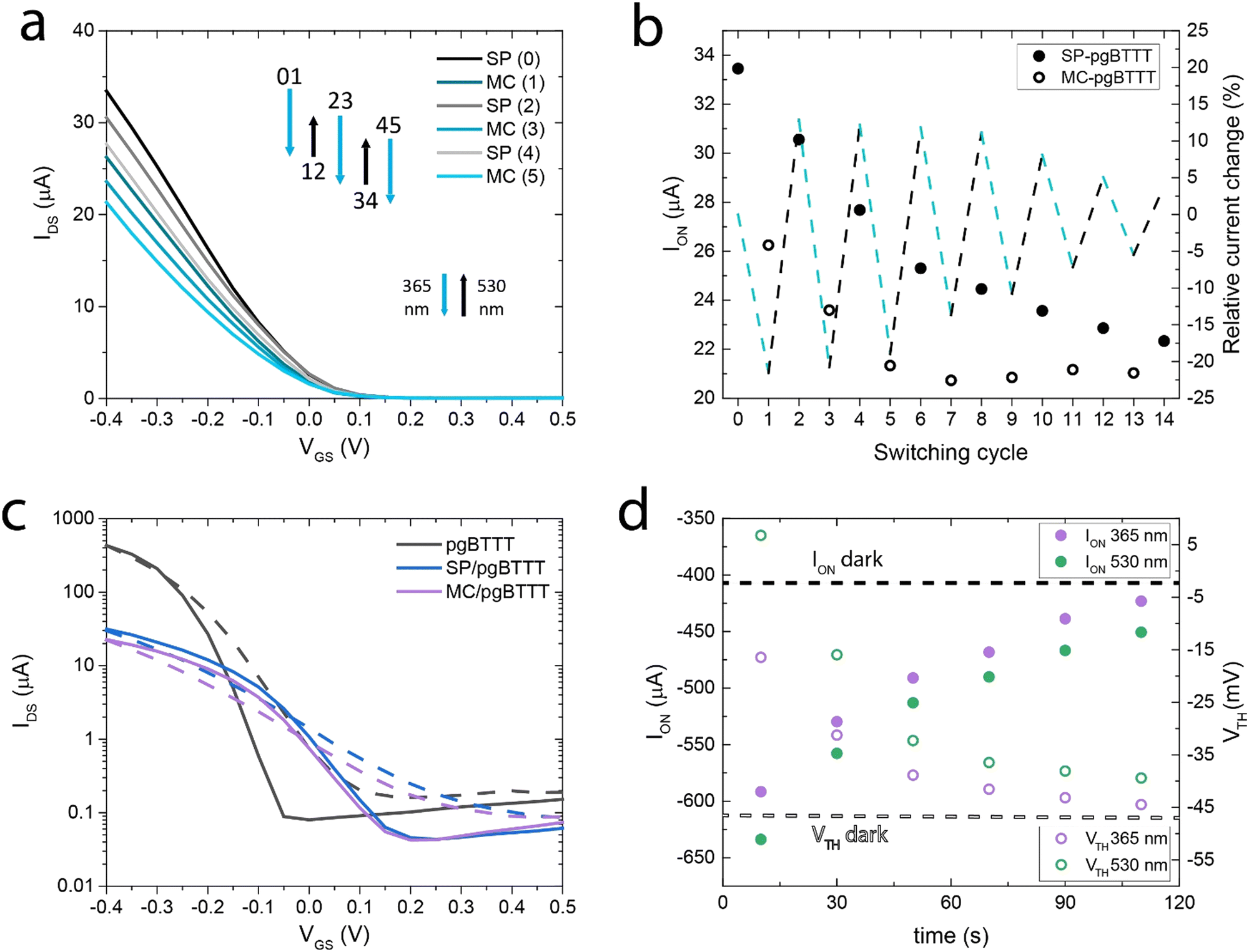 | ||
| Fig. 3 (a) Photoswitching properties of OECT devices based on the OEG-SP/pgBTTT blend. The transfer curves were sequentially collected by performing three complete photoisomerisation cycles. Even and odd numbers refer to the spiropyran (SP) and merocyanine (MC) state, respectively. The blue (downward, 365 nm LED) and black (upward, 530 nm LED) arrows indicate the optical switching between the two states. The length of the arrows is proportional to the change in current caused by the SP switching. (b) ION data for 14 switching operations (full, SP-pgBTTT, and empty, MC-pgBTTT, circles). The relative change in ION after each switching operation is indicated by a dashed line (light blue: conversion to MC, black: conversion to SP). (c) Transfer curves of neat pgBTTT, SP-pgBTTT and MC-pgBTTT bottom contact Au OECTs. The curves (forward, solid, and backward, dashed, sweep) are reported on a logarithmic scale. (d) Recovery of neat pgBTTT device characteristics (ION and VTH) after turning off the irradiation by 365 nm and 530 nm LEDs. The initial ION and VTH values (dashed lines) are prior to light irradiation. All transfer curves were recorded at VDS: −0.5 V, 0.5 M NaCl electrolyte, voltage sweep rate: 100 mV s−1, W/L: 10. | ||
Another important difference between pgBTTT and the OEG-SP/pgBTTT OECTs is that the neat polymer devices exhibit a unidirectional increase in photoconductivity upon both UV and visible light irradiation as a result of the photogeneration of charge carriers (Fig. 3d and Fig. S13, ESI†). In fact, continuous illumination of the thin film causes an increase of ION by about 30% of the photocurrent contribution relative to ION measured in dark conditions, irrespective of the wavelength used for irradiation. A reversible VTH shift of about +50 mV is also observed upon irradiation. While this photoconductive state is transient (Fig. S14, ESI†), the presence of photo-responsive elements in the blend causes the quenching of the photoconductivity and promotes a prolonged subsistence of the electrical properties in one or in the other switching state. Conversely, in the neat pgBTTT devices, both ION and VTH quickly decay as a function of time after irradiation with either UV (365 nm) or visible (530 nm) light (Fig. 3d). The photoexcitation dynamics of the neat pgBTTT system are discussed in more detail in the ESI.†
The optoelectronic properties of neat pgBTTT thin films are sensitive to the EC doping.42 The UV-vis spectrum of these monocomponent films is characterised by a strong absorption due to a π–π* optical transition (maximum at about 602 nm) with an absorption onset at 717 nm, resulting in an optical gap of EOPT = 1.73 eV. The reversible doping of pgBTTT in a NaCl electrolyte solution is characterised by a colour change from an electric blue tint to a pale shade that tends to transparency, as the maximum absorption of the polymer redshifts to about 900 nm.42 To understand the operation of our OECTs, macroscopic Kelvin probe (KP) and photoelectron yield spectroscopy in air (PYSA) measurements have been made aiming to investigate the electrochemically doped (and dedoped) states of the active material.
The ionisation energy (IE) and the Fermi level (EF) of the neat pgBTTT polymer on ITO substrates were measured before and after the processes of doping, and subsequent dedoping (Table 1). No significant variation of IE is observed upon the doping and dedoping processes, suggesting that the presence of Cl− ions within the bulk of pgBTTT is not distorting the energetic landscape of the π-conjugated system in the neat polymer. Conversely, the Fermi level is shifted towards the HOMO by almost 0.3 eV when Cl− ions are intercalated, indicating that the material undergoes p-doping. The incorporation of negative ions via EC intercalation generates hole carriers in the pgBTTT backbone that increase the overall conductivity of the material, enabling the OECT operation.
| Property | ITO | pgBTTT V GS: 0 V | pgBTTT V GS: −0.5 V | pgBTTT V GS: +0.5 V | SP-pgBTTT V GS: 0 V | SP-pgBTTT V GS: −0.5 V | MC-pgBTTT V GS: 0 V | MC-pgBTTT V GS: −0.5 V |
|---|---|---|---|---|---|---|---|---|
| IE (eV) | 5.04 ± 0.03 | 5.29 ± 0.05 | 5.32 ± 0.05 | 5.28 ± 0.03 | 4.96 ± 0.05 | 5.11 ± 0.01 | 4.99 ± 0.04 | 4.95 ± 0.03 |
| E F (eV) | 4.81 ± 0.02 | 4.50 ± 0.01 | 4.79 ± 0.06 | 4.54 ± 0.03 | 4.45 ± 0.03 | 4.59 ± 0.02 | 4.48 ± 0.03 | 4.46 ± 0.01 |
The illumination of pgBTTTvia a UV LED does not significantly affect the energy levels of the polymer, indicating its stability towards 365 nm light irradiation (Table S2, ESI†). The spiropyran itself has IE and EF values higher than the polymer, but, on the contrary, the merocyanine form (OEG-SP after UV, OEG-MC) is more n-doped by about 0.2 eV compared to OEG-SP. Both EC and the UV photoswitching experiments were carried out on the polymer blend (Table 1).
The primary effect of blending is to reduce the IE of the non-EC-doped blend compared to the neat polymer by about 0.3 eV. Since OEG-SP possesses an IE higher than pgBTTT, the lowering of IE in the blend cannot be simply ascribed to electron transfer effects arising from the 20% m/m concentration of the photochrome. The addition of such a compound would not directly affect the minimum IE, but would rather change the doping state of the material. Aside from this major change, the IE − EF difference (related to the doping character of the material) remains around 0.5 eV before and after EC doping of the blend, but also when the blend is phototuned via UV irradiation, indicating that there is already an accumulation of charge carriers in the material at 0 V, in agreement with the VTH values determined previously.
The effects of UV photoswitching are instead more evident in the EC-doped blend. In fact, the 0.15 eV shift in the IE (towards a more oxidised material) observed when SP-pgBTTT undergoes EC doping is compensated by the conversion of the OEG-MC form in the blend (having the EC reduction effects on the channel material). The 0.15 eV change in IE causes a variation in the charge injection (different injection barrier at the Au/OSC interface) that is responsible for the photomodulation observed in the OEG-SP/pgBTTT devices.49 Note here that due to the moderate solubility of the OEG-MC form in the water electrolyte, accurate IE and EF values were collected when the photoswitching experiment was performed after the EC doping step (and not vice versa).
Conclusions and outlook
Optically switchable OECT devices have been fabricated by blending a well-known mixed conductor, pgBTTT, and a versatile photochromic derivative based on a spiropyran core decorated with OEG chains, OEG-SP. Bottom contact devices based on a 20% m/m blend of pgBTTT and OEG-SP exhibited a modulation of the OECT ION by a factor of 30% upon illumination with 365 nm UV light. At the same time, due to the additional capability of the device to respond to light stimuli, we observed a decrease in the OECT mobility and ION/IOFF ratio of about one order of magnitude compared to the neat pgBTTT device. The blending is accompanied by a notable change in the electrical behaviour of the OECT when exposed to light since the neat polymer exhibited solely photocurrent enhancements for both UV and visible light. The UV photocurrent quenching in the blend might suggest that more fundamental processes can be involved in the accumulation/depletion of charge carriers. The response to light stimuli decays after about 6 switching cycles due to photochemical fatigue, causing the irreversible ION negative drift over time which stabilises after about 10 to 12 switching cycles. In fact, the OEG-MC form might easily interact with the water environment and get partly solubilised by the electrolyte, hence being brought into the solution. Further improvements to this system could be implemented by a better chemical design of the OEG-SP core that could involve structural modifications of the switching core to limit the aggregation of molecules in the MC form as well as modifying the side chains to reduce the detrimental interaction with water. An alternative approach to be attempted might involve the incorporation of the photomodulating component directly within the structure of the polymer. The determination of the energy levels (such as IE, WF, EF, optical gap, etc.) offered important insights into the process of doping and dedoping induced by the EC switching as well as the changes in the electronic states due to UV illumination. The decrease in the ON current of the OEG-SP/pgBTTT devices observed upon UV illumination could be explained by a reversible variation of the hole injection barrier resulting from a 0.15 eV shift of the IE of the blend. We believe that this work outlines new opportunities to impart OECT devices with optical switching properties and paves the way towards the integration of these light-sensitive properties in next-generation bioelectronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Actions UHMob (GA-811284), ULTIMATE (GA-813036), BORGES (GA-813863), LAD2DCOFS (GA-101027639), BOOSTER (GA-952911), RoLA-FLEX (GA-862474), CITYSOLAR (GA-101007084) as well as the Agence Nationale de la Recherche through the Interdisciplinary Thematic Institute SysChem via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d’Avenir, the International Center for Frontier Research in Chemistry (icFRC), the Institut Universitaire de France (IUF). The authors also acknowledge financial support from the KAUST Office of Sponsored Research (OSR) CRG 10 award, as well as EPSRC Project EP/T026219/1 and EP/W017091/1.References
- G.-H. Lee, H. Moon, H. Kim, G. H. Lee, W. Kwon, S. Yoo, D. Myung, S. H. Yun, Z. Bao and S. K. Hahn, Nat. Rev. Mater., 2020, 5, 149–165 CrossRef PubMed.
- L. Xiang, L. Liu, F. Zhang, C. Di and D. Zhu, Adv. Funct. Mater., 2021, 31, 2102149 CrossRef CAS.
- Y. Zhao, M. Gobbi, L. E. Hueso and P. Samorì, Chem. Rev., 2022, 122, 50–131 CrossRef CAS PubMed.
- Y. Chang, L. Wang, R. Li, Z. Zhang, Q. Wang, J. Yang, C. F. Guo and T. Pan, Adv. Mater., 2021, 33, 2003464 CrossRef CAS PubMed.
- N. A. Kukhta, A. Marks and C. K. Luscombe, Chem. Rev., 2022, 122, 4325–4355 CrossRef CAS PubMed.
- R. Wu, M. Matta, B. D. Paulsen and J. Rivnay, Chem. Rev., 2022, 122, 4493–4551 CrossRef CAS PubMed.
- Y. Hou and X. Hou, Science, 2021, 373, 628–629 CrossRef CAS PubMed.
- C. Pitsalidis, A.-M. Pappa, A. J. Boys, Y. Fu, C.-M. Moysidou, D. van Niekerk, J. Saez, A. Savva, D. Iandolo and R. M. Owens, Chem. Rev., 2022, 122, 4700–4790 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 1–14 CrossRef.
- I. B. Dimov, M. Moser, G. G. Malliaras and I. McCulloch, Chem. Rev., 2022, 122, 4356–4396 CrossRef CAS PubMed.
- G. Malliaras and I. McCulloch, Chem. Rev., 2022, 122, 4323–4324 CrossRef CAS PubMed.
- M. Fahlman, S. Fabiano, V. Gueskine, D. Simon, M. Berggren and X. Crispin, Nat. Rev. Mater., 2019, 4, 627–650 CrossRef CAS.
- A. Mariano, C. Lubrano, U. Bruno, C. Ausilio, N. B. Dinger and F. Santoro, Chem. Rev., 2022, 122, 4552–4580 CrossRef CAS PubMed.
- R. Avila, C. Li, Y. Xue, J. A. Rogers and Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026405118 CrossRef CAS PubMed.
- G. Yao, C. Yin, Q. Wang, T. Zhang, S. Chen, C. Lu, K. Zhao, W. Xu, T. Pan, M. Gao and Y. Lin, J. Materiomics, 2020, 6, 397–413 CrossRef.
- M. Berggren, E. D. Głowacki, D. T. Simon, E. Stavrinidou and K. Tybrandt, Chem. Rev., 2022, 122, 4826–4846 CrossRef CAS PubMed.
- S.-H. Sunwoo, S. I. Han, H. Joo, G. D. Cha, D. Kim, S. H. Choi, T. Hyeon and D.-H. Kim, Matter, 2020, 3, 1923–1947 CrossRef.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed.
- S. Y. Yeung, A. Veronica, Y. Li and I.-M. Hsing, Adv. Mater. Technol., 2023, 8, 2201116 CrossRef CAS.
- A. D. Scaccabarozzi, A. Basu, F. Aniés, J. Liu, O. Zapata-Arteaga, R. Warren, Y. Firdaus, M. I. Nugraha, Y. Lin, M. Campoy-Quiles, N. Koch, C. Müller, L. Tsetseris, M. Heeney and T. D. Anthopoulos, Chem. Rev., 2022, 122, 4420–4492 CrossRef CAS PubMed.
- J. Chen, W. Huang, D. Zheng, Z. Xie, X. Zhuang, D. Zhao, Y. Chen, N. Su, H. Chen, R. M. Pankow, Z. Gao, J. Yu, X. Guo, Y. Cheng, J. Strzalka, X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2022, 21, 564–571 CrossRef CAS PubMed.
- R. B. Rashid, X. Ji and J. Rivnay, Biosens. Bioelectron., 2021, 190, 113461 CrossRef CAS PubMed.
- A. Nawaz, Q. Liu, W. L. Leong, K. E. Fairfull-Smith and P. Sonar, Adv. Mater., 2021, 33, 2101874 CrossRef CAS PubMed.
- T. Paltrinieri, L. Bondi, V. Đerek, B. Fraboni, E. D. Głowacki and T. Cramer, Adv. Funct. Mater., 2021, 31, 2010116 CrossRef CAS.
- M. Jakešová, M. Silverå Ejneby, V. Đerek, T. Schmidt, M. Gryszel, J. Brask, R. Schindl, D. T. Simon, M. Berggren, F. Elinder and E. D. Głowacki, Sci. Adv., 2019, 5, eaav5265 CrossRef PubMed.
- Z. Chen, S. N. Obaid and L. Lu, Opt. Mater. Express, 2019, 9, 3843 CrossRef CAS.
- P. Bhatnagar, M. Patel, T. T. Nguyen, S. Kim and J. Kim, J. Phys. Chem. Lett., 2021, 12, 12426–12436 CrossRef CAS PubMed.
- L. Hou, X. Zhang, G. F. Cotella, G. Carnicella, M. Herder, B. M. Schmidt, M. Pätzel, S. Hecht, F. Cacialli and P. Samorì, Nat. Nanotechnol., 2019, 14, 347–353 CrossRef CAS PubMed.
- Y. Fang, L. Meng, A. Prominski, E. N. Schaumann, M. Seebald and B. Tian, Chem. Soc. Rev., 2020, 49, 7978–8035 RSC.
- O. Parlak and A. P. F. Turner, Biosens. Bioelectron., 2016, 76, 251–265 CrossRef CAS PubMed.
- I. Willner, Acc. Chem. Res., 1997, 30, 347–356 CrossRef CAS.
- F. Wang, X. Liu and I. Willner, Adv. Mater., 2013, 25, 349–377 CrossRef CAS PubMed.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- I. Tochitsky, M. A. Kienzler, E. Isacoff and R. H. Kramer, Chem. Rev., 2018, 118, 10748–10773 CrossRef CAS PubMed.
- Q. Dai, S. Xu, Y. Peng, W. Lv, L. Sun and Y. Wei, Chem. Phys. Lett., 2020, 742, 137133 CrossRef CAS.
- L. Hou, T. Leydecker, X. Zhang, W. Rekab, M. Herder, C. Cendra, S. Hecht, I. McCulloch, A. Salleo, E. Orgiu and P. Samorì, J. Am. Chem. Soc., 2020, 142, 11050–11059 CrossRef CAS PubMed.
- E. Orgiu and P. Samorì, Adv. Mater., 2014, 26, 1827–1845 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- J. W. Onorato, Z. Wang, Y. Sun, C. Nowak, L. Q. Flagg, R. Li, B. X. Dong, L. J. Richter, F. A. Escobedo, P. F. Nealey, S. N. Patel and C. K. Luscombe, J. Mater. Chem. A, 2021, 9, 21410–21423 RSC.
- M. L. Chabinyc, M. F. Toney, R. J. Kline, I. McCulloch and M. Heeney, J. Am. Chem. Soc., 2007, 129, 3226–3237 CrossRef CAS PubMed.
- R. K. Hallani, B. D. Paulsen, A. J. Petty, R. Sheelamanthula, M. Moser, K. J. Thorley, W. Sohn, R. B. Rashid, A. Savva, S. Moro, J. P. Parker, O. Drury, M. Alsufyani, M. Neophytou, J. Kosco, S. Inal, G. Costantini, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2021, 143, 11007–11018 CrossRef CAS PubMed.
- S. H. Yu, S. Z. Hassan, C. So, M. Kang and D. S. Chung, Adv. Mater., 2023, 35, 2203401 CrossRef CAS PubMed.
- S. Stitzel, R. Byrne and D. Diamond, J. Mater. Sci., 2006, 41, 5841–5844 CrossRef CAS.
- G. Such, R. A. Evans, L. H. Yee and T. P. Davis, J. Macromol. Sci., Part C: Polym. Rev., 2007, 43, 547–579 CrossRef.
- N. W. Tyer and R. S. Becker, J. Am. Chem. Soc., 1970, 92, 1289–1294 CrossRef CAS.
- A. Radu, R. Byrne, N. Alhashimy, M. Fusaro, S. Scarmagnani and D. Diamond, J. Photochem. Photobiol., A, 2009, 206, 109–115 CrossRef CAS.
- J. Kohl-Landgraf, M. Braun, C. Özçoban, D. P. N. Gonçalves, A. Heckel and J. Wachtveitl, J. Am. Chem. Soc., 2012, 134, 14070–14077 CrossRef CAS PubMed.
- T. Mosciatti, M. G. del Rosso, M. Herder, J. Frisch, N. Koch, S. Hecht, E. Orgiu and P. Samorì, Adv. Mater., 2016, 28, 6606–6611 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc05444b |
| This journal is © The Royal Society of Chemistry 2023 |