
Recent advances in iridium-based catalysts with different dimensions for the acidic oxygen evolution reaction
Chunyan
Wang
,
Fulin
Yang
and
Ligang
Feng
 *
*
School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, P. R. China. E-mail: ligang.feng@yzu.edu.cn
First published on 20th June 2023
Abstract
Proton exchange membrane (PEM) water electrolysis is considered a promising technology for green hydrogen production, and iridium (Ir)-based catalysts are the best materials for anodic oxygen evolution reactions (OER) owing to their high stability and anti-corrosion ability in a strong acid electrolyte. The properties of Ir-based nanocatalysts can be tuned by rational dimension engineering, which has received intensive attention recently for catalysis ability boosting. To achieve a comprehensive understanding of the structural and catalysis performance, herein, an overview of the recent progress was provided for Ir-based catalysts with different dimensions for the acidic OER. The promotional effect was first presented in terms of the nano-size effect, synergistic effect, and electronic effect based on the dimensional effect, then the latest progress of Ir-based catalysts classified into zero-dimensional (0D), one-dimensional (1D), two-dimensional (2D) and three-dimensional (3D) catalysts was introduced in detail; and the practical application of some typical examples in the real PEM water electrolyzers (PEMWE) was also presented. Finally, the problems and challenges faced by current dimensionally engineered Ir-based catalysts in acidic electrolytes were discussed. It is concluded that the increased surface area and catalytic active sites can be realized by dimensional engineering strategies, while the controllable synthesis of different dimensional structured catalysts is still a great challenge, and the correlation between structure and performance, especially for the structural evolution during the electrochemical operation process, should be probed in depth. Hopefully, this effort could help understand the progress of dimensional engineering of Ir-based catalysts in OER catalysis and contribute to the design and preparation of novel efficient Ir-based catalysts.
 Chunyan Wang | Chunyan Wang received her bachelor's degree in chemical engineering and technology from Xi’an University of Science and Technology in 2021. She is currently pursuing a master's degree at the School of Chemistry and Chemical Engineering, Yangzhou University. Her research interests focus on the design and synthesis of nanostructured catalysts for electrochemical energy conversion, including water electrolysis and small molecule oxidation. |
 Fulin Yang | Fulin Yang received his PhD degree from the College of Chemistry and Molecular Sciences, Wuhan University. He started as a lecturer in 2022 at the School of Chemistry and Chemical Engineering, Yangzhou University. His research interests focus on energy conversion and storage, including water splitting, anion exchange, membranes, alkaline fuel cells, zinc–air batteries, and lithium batteries. |
 Ligang Feng | Prof. Ligang Feng received his PhD degree from Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences in 2012 and worked shortly at CIAC and Ecole Polytechnique Fédérale de Lausanne (EPFL) and Chalmers University of Technology from 2012 to 2016. He joined Yangzhou University as a professor in April 2016. His research interests focus on energy conversion and storage, particularly novel and cost-effective catalyst materials from earth-abundant materials and their applications in fuel cells and water splitting. |
1. Introduction
Renewable energy generation technologies such as wind and solar are effective candidates for promoting the development of clean and sustainable energy infrastructure.1,2 However, it is difficult to provide continuous and stable power output due to their nature of intermittency and discontinuity.3,4 Green hydrogen produced through electrochemical water splitting driven by these renewable energy sources is considered the ideal energy carrier to replace fossil fuels; it has high energy density and no pollution that can effectively enable new integrated energy systems.5–7 The water-splitting reactions can be conducted in alkaline water electrolyzers (AWE) and proton exchange membrane water electrolyzers (PEMWE).2,8 Compared with AWE, PEMWE has more industrial development prospects, albeit still in the experimental stage, due to the more compact system design, lower ohmic loss, higher current density, and faster dynamic response.5,9,10 Compared to the half-reaction of water reduction, water oxidation (also known as the oxygen evolution reaction, OER) involves four-electron transfer (2H2O → O2 + 4H+ + 4e−), which is one of the most critical electron-donating reactions with multiple electrochemical steps. The slow reaction kinetics including the breaking of H–O bonds and the formation of O–O bonds dominate the energy consumption for the electrochemical water splitting technique.11,12 Therefore, much effort has been directed to the research and development of acidic OER catalysts for the proton exchange membrane (PEM) water electrolysis technique.13,14Due to the harsh corrosion in an acidic environment, the noble metals of iridium (Ir), ruthenium (Ru), and their oxides are currently the most attractive electrocatalysts for the acidic OER.15–17 Although Ru-based catalysts show slightly higher catalytic activity, Ir-based catalysts generally exhibit much stronger catalytic stability in the acidic electrolyte, which can flexibly balance the limiting relationship between the catalytic activity and stability.18,19 Because of the high cost and scarcity of metallic Ir for practical applications, the development of cost-effective Ir-based catalysts has become a consensus for their commercialization,18,20 and great effort has been made to improve the catalytic activity, reduce the amount of metal Ir and narrow the gap between the laboratory-scale catalytic performance and industrial requirements.21 Rational dimensional engineering is an effective approach to endow Ir-based nanocatalysts with unique properties by maximizing the number of active sites, facilitating electron transport, and enhancing the intrinsic activity.22 Moreover, dimensional engineering can cause lattice expansion or contraction, resulting in a lattice strain effect that will change the electronic structure and greatly improve the electrocatalytic performance.23 For example, one-dimensional (1D) IrxCu1−xOy nanotubes with varied composition ratios (x = 0.64, 0.48, and 0.33) were synthesized by electrospinning and thermal annealing methods,24 and the nanotube morphology enlarged the catalyst surface area and promoted the catalytic performance for the OER by facile electronic structure modification and surface adsorption energy optimization for the reaction intermediates. Two-dimensional (2D) ultrathin Ir hydroxide nanosheets (HxIrO3 NSs) with a distinct honeycomb structure were prepared by an electrochemically assisted exfoliation process which had an average thickness of about 1.6 nm with excellent acidic OER performance.25 Its high performance was attributed to the abundant Ir sites and the enriched surface defects caused, which effectively suppressed the formation of high-valent Ir species and reduced the Ir dissolution during the OER process. In addition, the ultrathin shell for three-dimensional (3D) IrCo hollow nanospheres with 11 atomic layers (2.26 nm) exhibited very high activity for the acidic OER with an overpotential of 284 mV to provide 10 mA cm−2.26 The lattice contraction induced by coupling Co to the Ir crystal increased the d-orbital overlap of Ir atoms and reduced the d-band center energy, thereby pushing more antibonding states below the Fermi level and weakening the oxygen adsorption energy. Furthermore, the hollow structure could provide a large specific surface area and favorable electron transfer, which improved the catalytic performance of the acidic OER. It can be concluded that the Ir-based nanocatalysts based on dimensional engineering are significant in the performance improvement in the field of acidic OER.
Considering the exciting and inspiring achievements of dimensional-engineered Ir-based catalysts for the OER, an overview of the current effort would be necessary to help develop new and efficient catalysts and understand the catalytic mechanism for the OER in acidic electrolytes. To the best of our knowledge, there are several reports on the development of OER catalysts under acidic conditions, while efforts based on the dimensional effect of the relevant catalyst are still very rare.27,28 For example, the recent progress of noble metal-based perovskite oxides as electrocatalysts for acidic water oxidation was reported; Ir- and Ru-based metal nanostructures, noble metal oxides, non-noble metal oxides, and carbon-based nanomaterials have been summarized in the progress of electrocatalyst design for the acidic OER;28 the deactivation mode and the efforts on solving the stability problem were concluded in the research progress of Ir-based electrocatalysts for the acidic OER focusing on the durability evaluation.29 However, they were insufficient to update the new contributions of the intensive effort made on Ir-based catalysts. Moreover, dimensional effect induced high-performance Ir-based catalysts are still not well overviewed. Therefore, this review was proposed that focused on the recent progress of high-performance Ir-based catalysts from the perspective of different dimensions. We first concluded the catalytic performance enhancement effect in terms of the nano-size effect, synergistic effect, and electronic effect. Then the latest progress of Ir-based catalysts was summarized and discussed by combining with some typical examples, where the Ir-based catalysts were classified based on their dimensional scale into 0D, 1D, 2D, and 3D Ir-based nanocatalysts. In addition, the application of Ir-based catalysts in PEMWE was also briefly summarized and their practical application in water electrolysis technology was discussed. Finally, a summary and outlook on the dimensional engineering of Ir-based catalysts that can be used in the acidic OER were given, which, hopefully, inspires the future development of the catalysts.
2. Dimensional effect
As mentioned in the introduction, Ir-based catalysts are currently one of the most efficient acidic OER catalysts.30,31 The dimension of Ir-based catalysts is closely related to their activity and stability.32 To successfully design high-performance nanocatalysts, the dimensional effect on catalytic activity boosting has been extensively studied.33,34 Dimensional engineering can increase the number of unsaturated coordination atoms by changing the size of the catalyst and improve the specific surface area and atom utilization of the electrocatalyst;35 it can also change the surface and electronic structure of the catalyst through morphology engineering which affects the electron transfer rate and the adsorption energy of reactants.36 In addition, catalysts of different dimensions can be combined with interface engineering to generate a strong metal–support interaction (SMSI) between the active metal and the support, thereby enhancing the activity.37,38 To our knowledge, the dimensional effect usually affects catalytic performance from the aspect of the size effect, electronic effect, and synergistic effect. In this section, the dimensional effect will be briefly discussed by combining it with some typical catalyst systems. Table 1 summarizes the performance of different dimensions of Ir-based catalysts discussed in this review.| Types | Catalysts | Electrolyte | Overpotential (mV)@10 mA cm−2 | Tafel slope (mV dec−1) | Specific activity (mA cm−2)@potentiala | Ref. |
|---|---|---|---|---|---|---|
| a The potentials in the table are all relative to the reversible hydrogen electrode (RHE). | ||||||
| Dimension effect | IrNi0.57Fe0.82 NPs | 0.5 M H2SO4 | 284 | 48.6 | — | 35 |
| Ir–Pd NWs | 0.5 M H2SO4 | 297 | 60 | 0.0485@1.48 V | 46 | |
| Ir–NiCo2O4 NSs | 0.5 M H2SO4 | 240 | 60 | — | 47 | |
| Ir-ITO film | 0.1 M HClO4 | 340 | 52 | — | 48 | |
| IrCo0.14 nanorings | 0.1 M HClO4 | 278 | 41.7 | 0.434@1.53 V | 49 | |
| Ir0.48Cu0.52Oy | 0.5 M H2SO4 | 258 | 40.6 | — | 24 | |
| IrO2 DLN | 0.5 M H2SO4 | 270 | 43 | — | 50 | |
| 0D | Ir NPs | 0.5 M HClO4 | 290 | 51.4 | — | 51 |
| Rh22Ir78 NPs | 0.5 M H2SO4 | 292 | 101 | — | 6 | |
| P–Ir | 0.1 M HClO4 | 266 | — | — | 52 | |
| Ir@N-G-600 | 0.5 M H2SO4 | 314 | 108 | — | 53 | |
| AuIr@CNT | 0.5 M H2SO4 | 257 | 77.6 | 0.156@1.53 V | 54 | |
| IrOx–TiO2–Ti | 0.5 M H2SO4 | 200 | 48.66 | — | 55 | |
| IrRu@Te | 0.5 M H2SO4 | 220 | 35 | 0.015@1.53 V | 56 | |
| Ir-SA@Fe@NCNT | 0.5 M H2SO4 | 250 | 58.2 | — | 57 | |
| Ir(20)/Fe@NCNT-900 | 0.5 M H2SO4 | 300 | 64.2 | — | 58 | |
| Ir0.08Co2.92O4 NWs | 0.5 M H2SO4 | 189.5 | 22.32 | — | 59 | |
| 1D | Ir–Te NWs | 0.5 M H2SO4 | 284 | 66.3 | — | 60 |
| IrOx/CeO2 NWs | 0.5 M H2SO4 | 220 | 63 | — | 61 | |
| SrIrO3 NFs | 0.5 M H2SO4 | 280 | 44.2 | — | 62 | |
| Ir-NR/C | 0.5 M H2SO4 | 290 | 72.4 | 0.0272@1.5 V | 63 | |
| (Mn0.8Ir0.2)O2:10F | 1 M H2SO4 | 200 | 38 | 0.0056@1.45 V | 64 | |
| Ir NTs | 0.1 M HClO4 | 245 | 49.02 | — | 65 | |
| Ir AC/NN | 1 M H2SO4 | 296 | 97.3 | 0.051@1.55 V | 66 | |
| PdPtIr PNTs/C | 0.1 M HClO4 | 337 | 90.2 | — | 67 | |
| RuIrTe NTs | 0.5 M H2SO4 | 205 | 41.2 | — | 68 | |
| 2D | IrOx nanosheets | 0.5 M H2SO4 | 250 | 47 | 69 | |
| Ir–IrOx/C nanosheets | 0.5 M H2SO4 | 198 | 106.3 | 0.0567@1.428 V | 70 | |
| HIONs | 0.1 M HClO4 | 300 | 46.9 | 0.0425@1.53 V | 71 | |
| S-doped M-SrIrO3 | 0.5 M H2SO4 | 228 | 58.4 | — | 72 | |
| IrCr | 0.5 M H2SO4 | 400 | 59 | — | 73 | |
| IrRe–IrOx | 0.1 M HClO4 | 248 | 52 | — | 74 | |
| CoIr nanoframes | 0.1 M HClO4 | 290 | 40 | 0.80@1.51 V | 75 | |
| 3D | IrOx network | 0.1 M HClO4 | 305 | — | 1.1@1.53 V | 76 |
| IrxPb network | 0.1 M HClO4 | 307 | 60.7 | 2.8@1.53 V | 77 | |
| np-Ir70Ni15Co15 | 0.1 M HClO4 | 220 | 44.1 | 0.25@1.5 V | 78 | |
| IrRuMn spheres | 0.1 M HClO4 | 260 | 45.6 | — | 79 | |
| Ir p-HNs | 0.5 M H2SO4 | 243 | — | — | 80 | |
| IrGa IMC | 0.1 M HClO4 | 272 | 57.2 | — | 81 | |
| Ir–W@Ir–WO3−x | 0.5 M H2SO4 | 261 | 65 | — | 82 | |
| Ir0.16Co0.84Ox | 0.1 M HClO4 | 262 | 45.2 | 0.077@1.53 V | 83 | |
| Ir0.5Ru0.5 nanocages | 0.5 M H2SO4 | 219 | 56 | 22.1@1.48 V | 84 | |
| Co–IrCu ONCs | 0.1 M HClO4 | 293 | 50 | — | 36 | |
| IrNi-RF | 0.1 M HClO4 | 300 | 48.6 | — | 85 | |
| IrNiCu DNFs | 0.1 M HClO4 | 300 | — | — | 86 | |
2.1. Nano-size effect
The size effect is the phenomenon wherein the macroscopic physical and chemical properties substantially change due to the reduction in the size of the catalyst, which has a significant impact on the catalytic performance.22 Particularly, as particles are scaled down to the nanoscale, the related geometric and electrical properties undergo a significant change with particle size, which is quite different from their bulk equivalents, significantly improving the catalytic efficacy of nanoscale catalysts.39–41 Geometrically, metal nanoparticles consist of different sorts of surface metal sites, for example, low-coordinating metal atoms at the edges and corners and high-coordinating metal atoms on the planes.40 Different geometric positions of metal atoms can significantly change the chemical bond-breaking and catalysis mechanism.42 As a result, changing the particle size alters the coordination of surface metal atoms as well as the metal dispersion, resulting in size-dependent catalytic activity.43 In electronics, as the particle size decreases to the nanoscale, the valence bands of the continuous bulk metal begin to split into discrete states, similar to the valence bands of semiconductors, and this so-called “quantum size effect” is determined by the energy of the entire particle.44,45 Controlled by the drastic variation in order with size, the orbital hybridization and overall charge transfer between the metal and the reactants are strongly disturbed, thereby significantly modulating the intrinsic activity of all surface metal sites.87By decreasing the particle size, the ratio of surface atoms to the total number of nanoparticles increases sharply, which can expose more surface-active sites to a certain extent, thereby enhancing the catalytic activity.88 For example, the ultra-small IrNiFe nanoparticles (IrNi0.57Fe0.82 NPs) with an average diameter of 2.2 nm and a narrow size distribution showed good catalytic activity with an overpotential of 284 mV to reach 10 mA cm−2 in 0.5 M HClO4 electrolyte.35 The small size and monodispersity of nanoparticles played an important role in the catalysis reaction. The colloidal Ir nanoparticles (Ir NPs) with an average diameter of 1.5 nm uniformly distributed on the porous indium tin oxide (ITO) films were obtained by the solvothermal deposition approach.48 Among them, the crystalline Ir NPs had about 150 atoms, of which 90 Ir atoms were located on the surface, which greatly improved the atom utilization rate and catalytic performance. They required an overpotential of 340 mV to drive a current of 10 mA cm−2 for the OER, and a core–shell structure was formed for the Ir NPs after the OER to improve the stability of the catalyst, in which the particle core was metal and the outer layer was amorphous Ir-oxo-hydroxide. Even in the nanoscale, the catalytic performance can also be largely influenced by the different morphology that determines the exposure of the active site, the facets, mass and electron transfer, etc.89,90 For example, a series of shape-tunable Pt–Ir alloy nanocrystals can be obtained by the introduction of halide ions as crystal face selectors through a simple solvothermal method, which changed the crystal facet state (such as (111), (100), and (110) and step crystal planes) of the alloy nanocrystals.91 Among them, Pt–Ir nano-short-chains (Pt–Ir NSCs) exposing a large number of (110) crystal facets of irregular step exhibited excellent catalytic performance for the OER. The surface effect study found that the catalytic activity of the OER was positively correlated with the proportion of surface IrOx species, and this unique crystal facet could prevent the adsorption of bisulfate anions on the catalyst surface during mass transfer, making the surface easy to form oxides. In addition, the alloying effect on the surface of Pt–Ir nanocrystals changed the rate-determining step of the OER during the reaction process to the dissociation of water instead of the adsorption states of surface hydroxyl species.
The electrocatalytic reaction occurs on the surface of the catalyst, and a larger surface area can expose more active sites to the electrolyte.92 The surface area can be obtained by physical measurements and electrochemical technology.93 The surface area measured using the Brunauer–Emmett–Teller method showed the whole physical surface area but not the entire surface is effective for electrochemical reactions.94 Basically, the surface area expressed by electrochemical surface area (ECSA) obtained by electrochemical technology is more reliable in evaluating the effective active area.95 The method of calculating ECSA by measuring the Coulomb charge of a specific surface faradaic reaction through hydrogen underpotential deposition,96 CO dissolution,97 and metal underpotential deposition98 is generally employed. Currently, the common method for the ECSA test of Ir-based catalysts for the acidic OER is the electric double-layer capacitance (Cdl) calculated from CV curves.99 ECSA can be calculated using the following equation: ECSA = Cdl/Cs, where Cs is the specific capacitance of a smooth electrode (e.g. 0.04 mF cm−2). Then, the specific activity of different catalysts can be calculated and fairly compared by normalizing the current to the ECSA to evaluate the catalytic efficiency. Ir–Pd alloy nano-hollow spheres (NHSs), worm-like nanowires (NWs), and nano-tetrahedrons (NTs) were compared to probe the morphology effect for acidic OER catalysts.46 The surface-microstructure-sensitive enhancement effect was revealed by the electrochemical study for the OER and the specific activity of Ir–Pd nanocatalysts was positively correlated with the surface roughness of NWs > NHSs > NTs. It was found that the specific activity was ordered as Ir–Pd NWs (48.5 μA cm−2) > NHSs (24.6 μA cm−2) > NTs (11.1 μA cm−2) and the specific activity of Ir–Pd NWs was even much higher than that of commercial Ir/C catalysts (8.1 μA cm−2) at an overpotential of 0.25 V. The surface Ir(VI) oxide generated at the surface defect sites of Ir–Pd nanocatalysts was the key intermediate of the OER, and the excellent catalytic performance of Ir–Pd NWs was attributed to its roughest surface and many surface defect sites for Ir(VI) oxide formation due to the oriented attachment polycrystalline structure.
Theoretically, the final size limitation of metal particles was a single atom, where metal atoms were scattered on a specific support and isolated from each other; this could impart high atom utilization and unique coordination and electronic structures, which could significantly promote catalytic reactions.100 For example, Ir–NiCo2O4 nanosheets (Ir–NiCo2O4 NSs) were successfully prepared by electrodepositing Ir single atoms on corrugated ultrathin porous NiCo2O4 nanosheets (Fig. 1a and b).47 The coupling of surface-exposed Ir single atoms to oxygen vacancies (VO) in acidic media enhanced the OER activity and stability, and a low overpotential of 240 mV was required to reach 10 mA cm−2, with good stability in 0.5 M H2SO4 for 70 h. The anchored Ir–Ox sites acted as a partition to reduce the inactivation due to overbound O and H species covering the active site on the Co site. The atomic rearrangement in the system would also improve the catalytic performance because the numerous Ir single atoms in Ir–NiCo2O4 NSs coupled with VO enabled efficient electron transport by lowering the energy barrier for the transition of *O to *OOH. In particular, the formation of OOH* species is generally regarded as a potentially limiting step in the oxygen adsorption mechanism of the acidic OER. The properties of Ir-based nanocatalysts are significantly affected by the active metal size and surface atoms; achieving good control over the size of monodisperse nanoparticles and maximization of each active site would be an effective strategy to improve the catalytic performance of Ir-based catalysts for the acidic OER.
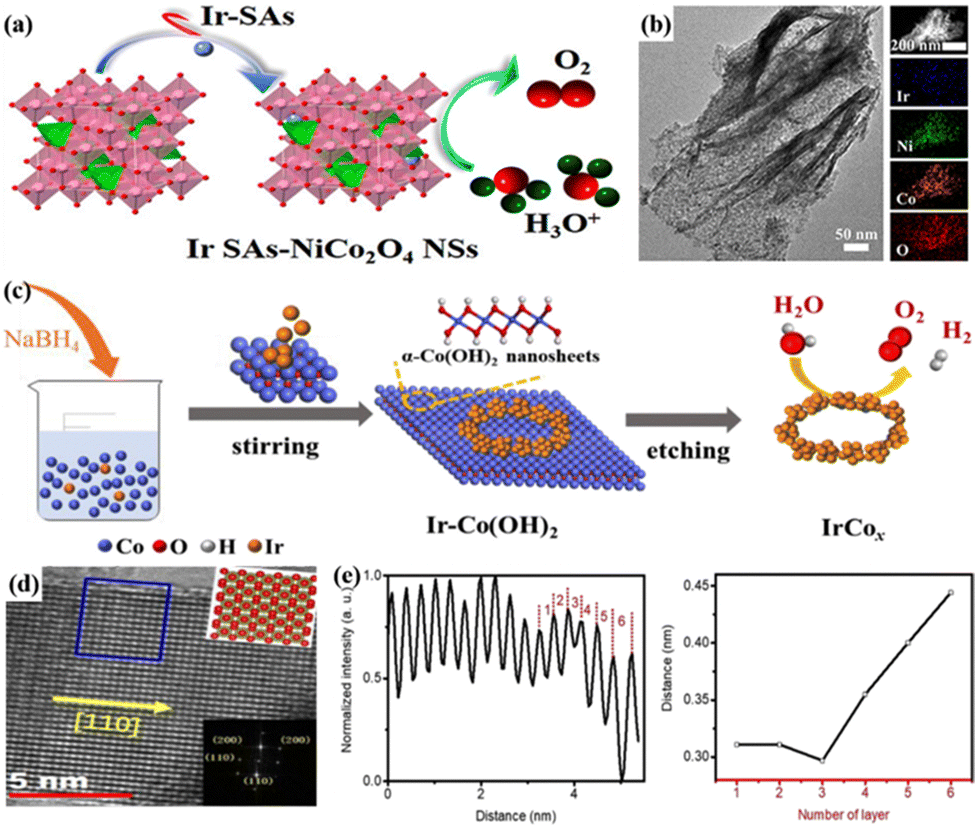 | ||
| Fig. 1 (a) Structural characterization and OER mechanism of Ir–NiCo2O4 NSs. (b) High-resolution transmission electron microscopy (HRTEM) images of Ir–NiCo2O4 NSs. Reprinted with permission from ref. 47 Copyright 2020, American Chemical Society. (c) Schematic diagram of the synthesis of IrCox symmetry-breaking bracelet-like nanorings with special structures. Reprinted with permission from ref. 49 Copyright 2020, Elsevier Ltd. (d) HRTEM image and lattice distribution at the surface of IrO2 DLNs. (e) Interlayer distance distribution and distance variation of IrO2 DLNs. Reprinted with permission from ref. 50 Copyright 2020, Royal Society of Chemistry. | ||
2.2. Synergistic effect
The synergistic effect means an interaction or cooperation of different parts in a catalyst system giving rise to a whole that is greater than the simple sum of its parts.101 Moreover, in electrocatalysis, it generally refers to two or more active sites playing different roles in the catalysis process, thus leading to a higher activity than simply the sum of them.102 The synergistic effect is a common principle to enhance the catalytic performance of Ir-based catalysts in the OER.35,103 The low efficiency of a single active site in the catalyst can thus be overcome through the synergy of two or more components, and the catalytic ability can be also boosted by the synergism of the different active sites.104 It can occur in different catalyst systems, such as different dopants, metal–support interactions, hybrid structures, composite materials, etc.105,106Currently, multiple components have been intensively reported to increase the intrinsic activity of catalysts based on the synergistic effect.107–110 For example, low-Ir-doped Co3O4 (Ir–Co3O4) catalysts only required an overpotential of 225 mV to drive 10 mA cm−2, much lower than those of Co3O4 (382 mV) and IrO2 (314 mV).111 Its high activity was attributed to the fact that both the doped Ir species and defective Co3O4 could serve as active sites for the OER that synergistically accelerated reaction kinetics. Moreover, the abundant defect sites on the surface of Ir–Co3O4 improved the adsorption of intermediates such as OH* and OOH* and lowered the catalytic energy barrier. Ca2YxIr1−xO4 (x = 0.1, 0.2, 0.3) nanocrystals fabricated by replacing some iridium sites with yttrium (Y) in Ca2IrO4 were optimized for the acidic OER.112 The proportion of lattice oxygen in Ca2Y0.2Ir0.8O4 nanocrystals was about 21.5%, much higher than that in Ca2IrO4 nanocrystals (14.9%); the abundant O(II−δ)− (electrophilic lattice oxygen) species favored the formation of *O–O during the acidic OER process and significantly lowered the kinetic barriers. The formation of *O–O bonds was generally important to the lattice oxygen mechanism rate-limiting step. Furthermore, its structural changes revealed the synergistic effects between high-valence Ir sites and increased lattice oxygen concentration induced by Y3+ substitution, which greatly enhance the intrinsic OER activity where the optimized system of Ca2Y0.2Ir0.8O4 only required a low overpotential of 213 mV to reach 10 mA cm−2, and the mass activity and turnover frequency value were approximately 203.7-fold and 204.4-fold improvements in comparison to those of IrO2 at 1.5 V vs. RHE.
The heterostructure of amorphous and well-formed crystals with disordered arrangement and an unsaturated coordination structure can also induce a synergistic effect to enhance the catalytic activity and stability.113,114 For example, the amorphization–crystallization of IrOx was regulated in the hybrid catalyst of IrO2@LnIr1−nOx(OH)y (Ln = La−Lu) by introducing lanthanides to suppress the crystallization of Ir atoms during the calcination process.115 They all exhibited an overpotential of 290–300 mV at 10 mA cmgeo−2; among them, the mass activity of Ir in the IrO2@LuIr1−nOx(OH)y structure reached 128.3 A gIr−1, which was 14.6 times that of the benchmark IrO2. The lattice oxygen atoms in amorphous IrOx are more available for combination with water, and the nucleophilic *O species caused by the deprotonation of IrIIIOOH can directly promote O–O bond formation. Moreover, the crystalline–amorphous interface induced efficient charge transfer capability was beneficial to the stability of reactive oxygen species, and the surface reactive oxygen species and the tensile strain of the [IrO6] octahedron mediated by lanthanides could synergistically enhance the intrinsic OER activity. Ir0.16Co0.84Ox nanospheres with an abundant crystalline/amorphous heterostructure required a low overpotential of only 262 mV in acidic media to provide a good OER performance of 10 mA cm−2, much lower than that of crystallized IrO2 (338 mV).83 In addition, the rich homogeneous interface and the synergistic alloying effect between Ir and Co were considered to be the origin of the high catalytic activity in the IrCox nanorings (NRs) with a unique 2D bracelet-like structure.49 The catalyst was derived from the self-assembled Ir-doped Co(OH)2 nanosheet precursors by the redox reaction between the Co and Ir3+ precursor to form metallic Ir nanoparticles in a bracelet-like structure, and Co(OH)2 nanosheets were removed by acid etching to obtain well-dispersed IrCox NRs (Fig. 2c). The synergistic effect of the metal–support interaction can also significantly improve the catalytic performance.116 For example, by depositing Ir nanoparticles and titanium oxynitride (TiONx) nanoflakes on the surface of reduced graphene oxide nanoribbons (rGONRs), the synergy of SMSI between Ir and TiONx, as well as the dual contact between Ir and the supports on both sides greatly enhanced the catalytic performance for the OER.117 For the appropriate amount of TiONx deposited on rGONRs, the mass activity could reach 4822 A gIr−1, which was much higher than that of Ti-free samples (2450 A gIr−1). Therefore, the catalytic performance can be improved by optimizing the synergistic effect between different characteristic components, which changed the surface structure of the catalyst and enhanced the interaction between components.
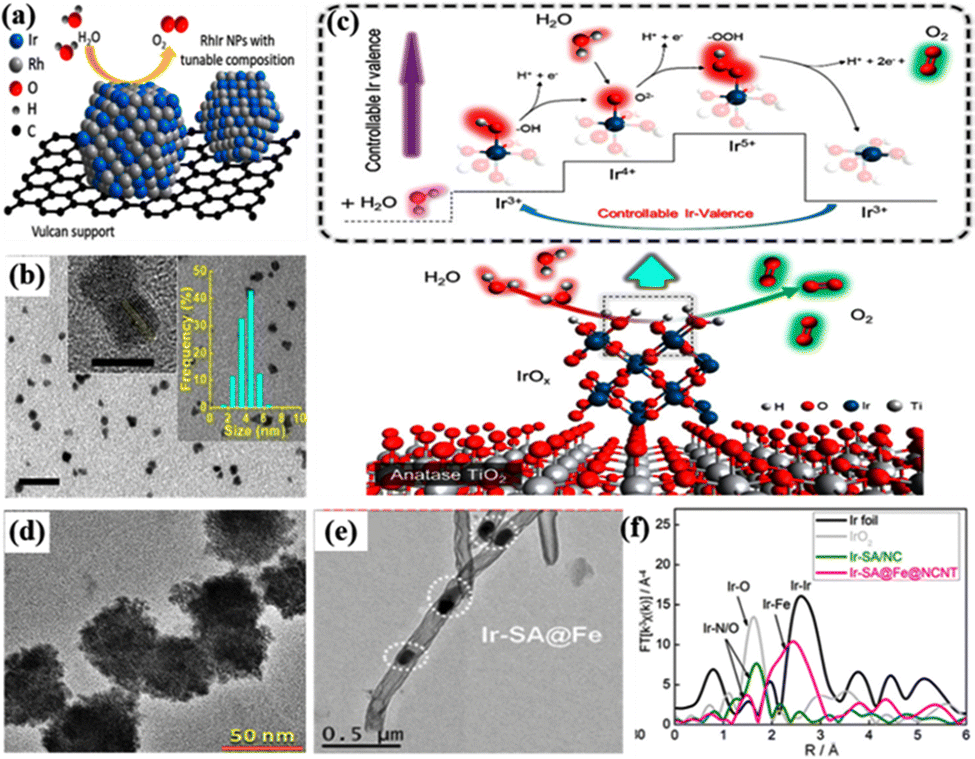 | ||
| Fig. 2 (a) Schematic of the Vulcan XC-72R carbon-supported RhxIr(100−x) NP structure. (b) Transmission electron microscopy (TEM) image, HRTEM image of (111) planar lattice fringes and size distribution histogram of Rh49Ir51 NPs. Reprinted with permission from ref. 6 Copyright 2019, American Chemical Society. (c) OER mechanism and performance map of ITOT catalysts. Reprinted with permission from ref. 55 Copyright 2019, American Chemical Society. (d) Morphological and structural characterization of IrRu@Te catalysts. Reprinted with permission from ref. 56 Copyright 2020, American Chemical Society. (e) TEM images of the Ir-SA@Fe@NCNT electrocatalyst. (f) Fourier-transformed k3-weighted extended X-ray absorption fine structure (EXAFS) spectra of the Ir-L3 edge for Ir foil, IrO2, Ir-SA/NC, and Ir-SA@Fe@NCNT. Reprinted with permission from ref. 57 Copyright 2020, American Chemical Society. | ||
2.3. Electronic effect
The electronic effect is also one of the principles to improve the catalytic performance, usually due to changes in the electronic structure of the catalyst system.102,118 The dimensional engineering and morphological tuning induced electronic structure change and electron re-distribution can lower the energy barrier and speed up the reaction.41,119 In general, diverse components with variable electronegativities, such as multimetallic components, supports, and ligands, can generate electronic effects.35,120,121 Among them, the preparation of multimetallic Ir-based nanocatalysts is an effective strategy to tune the local electronic structure of active centers (including orbitals, charge density, and degrees of freedom of active sites) through charge redistribution.28,122 For example, ultrasmall IrW nanoparticles dispersed on carbon nanotubes (IrW/CNT) were prepared by introducing tungsten (W) atoms with flexible valence states into metallic Ir, and Ir atoms donated electrons on d orbitals to W atoms due to the different electronegativity.123 The density of states (DOS) calculation results showed that the strong electronic interaction made the d-band center of Ir atoms upshift after doping with W atoms. In addition, after the introduction of W atoms, the weakening of the s–d hybridization of the Ir–Ir bond made the adsorbed *O species prone to formation of *OOH structures via nucleophilic attack in the acidic OER with a decrease in free energy from 2.21 eV to 2 eV. This result well demonstrated the origin of the excellent catalytic performance of the IrW/CNT catalyst. FeCoNiIrRu high-entropy alloy nanocrystals required a low overpotential of 241 mV to drive 10 mA cm−2 for the acidic OER.124 Its DOS and charge density results showed that electron density redistribution occurs from low electronegativity elements (Fe, Co, and Ni) to high electronegativity elements (Ir and Ru), making Ir sites more active. Furthermore, according to the calculation results of the Gibbs free energy of each metal, the Ir site significantly reduced the adsorption of *OH and promoted the conversion of *OOH and the generation of O2 at the same time. In addition, the electronic effect between the support and the active metal would also impact the catalytic performance.125 For example, IrOx nanoparticles supported on F-doped titanium dioxide (F-TiO2) were an efficient catalyst for the acidic OER, and the electron transfer between F-TiO2 and IrOx effectively inhibited the oxidative dissolution of Ir active species, thereby significantly improving the stability of the catalyst.126Besides, the introduction of defects in the catalyst could disturb the regular arrangement of the crystal lattice, leading to the redistribution of atoms or electrons, which produces electronic effects to improve the catalytic activity.13,127 Defects are generated due to the difference between ideal and actual structures, which includes the types of vacancies, dislocations, voids, disorders, etc.128 A vacancy is the simplest type of defect due to the absence of atoms at a lattice site, and the catalyst vacancies are usually located on metallic sites (cations) or non-metallic sites (anions).24 The introduction of VO into Ir-based catalysts can enhance the acidic OER performance.129,130 For example, 3D IrO2 with a dendritic-like nanostructure (IrO2 DLN) containing a large amount of VO was prepared by the molten salt method, and the crystals were growing along the (110) crystal direction to ensure a high exposure rate of the (110) plane.50 The lattice O and sublayer Ir cations on this facet participated in the OER in acidic electrolytes, which exhibited much better OER activity than commercial IrO2 (Fig. 1d). The appearance of VO was caused by the corrosion of Ir and the adsorption of O/OH− during the OER process, which greatly stretched the outer layer of IrO2 DLN, and it was discovered that the lattice spacing of the outermost layer was 0.44 nm, which was much larger than that of the inner layer (0.31 nm) (Fig. 1e). The self-reconstruction process of the chemically active surface from the compressed lattice to the stretched lattice changed the atomic arrangement and electron configurations of the active Ir, which effectively improved the acidic OER performance. In conclusion, the combination of dimensional engineering with heteroatom doping and the introduction of vacancies can adjust the electronic structure of the catalyst by electronic redistribution and electron transfer, thus improving the catalytic performance.
3. Recent progress of Ir-based catalysts with different dimensions
It is well known that the morphology and structure of catalysts have an important impact on their activity. Morphology, as an important component of the surface structure, has attracted extensive attention in recent years.131 Using suitable preparation methods such as the solvothermal approach, thermal annealing method, chemical etching method, template-assisted method, and chemical deposition method, the size and shape of the catalyst can be adjusted to form zero-dimensional (0D) nanoparticles, nanoclusters, one-dimensional (1D) nanoneedles, nanotube structures, two-dimensional (2D) nanosheet structures and three-dimensional (3D) nanostructures.68,132,133 Furthermore, small changes in the preparation details can also affect the final properties of the catalysts. We here first summarized and compared some commonly used synthetic methods for the preparation of Ir-based catalysts in Table 2, where the features of the methods are highlighted. In addition, catalysts with different dimensions have different characteristics, as well as different effects on catalytic performance.134 Therefore, it is important to control the morphology and structure of catalysts to improve the acidic OER performance of Ir-based catalysts.79 Based on the dimensional engineering for the catalysts as 0D, 1D, 2D, and 3D, in this section, the high-performance Ir-based catalysts for the acidic OER are presented and discussed.| Synthetic methods | Introduction | Advantages/disadvantages | Catalysts | Ref. |
|---|---|---|---|---|
| Solvothermal approach | A process takes place in a closed system that requires high temperatures and pressures to induce a chemical reaction of the precursor materials to form the desired compound directly from a solution. | Good uniformity; easy operation/invisible reaction process; risk of high pressure | IrRu@Te | 56 |
| Ir-ITO film | 48 | |||
| Pt–Ir | 91 | |||
| (Mn0.8Ir0.2)O2:10F | 64 | |||
| IrRuMn spheres | 79 | |||
| Thermal annealing | The process of heating a material at a predetermined temperature in a certain gas atmosphere to realize the specific functionality by oxidation, reduction, carbonization, or decomposition. | Easy operation; temperature control; large-scale fabrication/risk of high temperature; poisoning gas | IrOx/CeO2 NWs | 61 |
| SrIrO3 NFs | 62 | |||
| Ir-NR/C | 63 | |||
| Ir-SA@Fe@NCNT | 57 | |||
| Ir/Fe@NCNT | 58 | |||
| Ir0.08Co2.92O4 NWs | 59 | |||
| Chemical etching | The method of changing the surface environment by chemical technology, removing inert components to form a porous or hollow structure, mainly includes acid etching, oxide etching, and plasma etching. | Low cost; easy access to hollow structures/high-temperature sensitivity; chemical corrosion and chemical poisoning | CoIr nanoframes | 75 |
| IrOx network | 76 | |||
| np-Ir70Ni15Co15 | 78 | |||
| Ir p-HNs | 80 | |||
| Ir0.5Ru0.5 nanocages | 84 | |||
| Co–IrCu ONCs | 36 | |||
| IrNi-RF | 85 | |||
| IrNiCu DNFs | 86 | |||
| Template-assisted | The method of selecting specific materials with well-defined structures as templates to guide the synthesis and assembly of nanomaterials. | Easy to control morphology; good reproducibility/limited choice of templates; complicated process | Ir–Te NWs | 60 |
| Ir NTs | 65 | |||
| RuIrTe NTs | 68 | |||
| PdPtIr PNTs/C | 67 | |||
| Chemical deposition | The process of using a suitable reducing agent to reduce the metal ions in the solution and deposit them on the surface of the support, mainly includes chemical vapor deposition, liquid phase deposition, and electrochemical deposition. | Easy to form supported catalysts/uneven distribution of active metals | Ir@N-G-600 | 53 |
| AuIr@CNT | 54 | |||
| Ir–NiCo2O4 NSs | 47 | |||
| IrCr | 73 | |||
| Ir/TiO2–MoOx | 136 | |||
3.1. 0D catalysts
0D catalysts refer to functional materials with a size between 1 and 100 nm in each dimension (x, y, and z axes) and the surrounding interfacial layer for nanoparticles, nanoclusters, single atoms, etc.88 They have attracted much attention in the field of electrocatalysis reactions due to the large specific surface area, short effective charge transfer length, and abundantly exposed atoms.32,135 Ir-based nanoparticles are widely used as the simplest and most common acidic OER catalysts. For example, Ir nanoparticles with narrow size distribution and high dispersibility were synthesized by the colloidal method, which exhibited excellent acidic OER performance with an overpotential of 290 mV to reach 10 mA cm−2, lower than those of state-of-the-art commercial catalysts.51 To further improve the catalytic performance, multi-metal alloy NPs have been developed by alloying Ir with other metals. For instance, RhxIr(100−x) alloy NPs with different contents (x = 22–73) were prepared by the microwave-assisted reduction method (Fig. 2a and b).6 Among them, Rh22Ir78 NPs had a mass activity of about 1.17 A mgIr−1, which was 3 times that of pure Ir NPs. The faster kinetics and enhanced OER activity were due to the alloying of a small amount of Rh with Ir and the synergism of electronic effects to reduce the binding energy of O and OOH intermediates. Ir can also form IrX (X = P, Te) alloy nanocrystals with some non-metallic elements, and the catalytic performance is greatly influenced by the size, bonding properties, and coordination number of the anions X.137 Furthermore, by changing the valence of X in the alloy, the phase and structure can be varied and tuned to produce a desirable performance for the OER.102 For example, phosphorus–iridium (P–Ir) nanocrystalline catalysts were synthesized by the solvothermal method and the catalyst with an atomic ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (P:Ir) showed a narrow size distribution of 1.8–3.6 nm for the nanoparticles.52 An onset potential of 190 mV was required to drive the OER in 0.1 M HClO4 solution, which was about 30 mV less compared with Ir NPs. In addition, the catalyst also exhibited ultra-high stability under acidic conditions. The catalytic activity was enhanced because the introduction of P changed the metal surface and its affinity for water molecules.
:1 (P:Ir) showed a narrow size distribution of 1.8–3.6 nm for the nanoparticles.52 An onset potential of 190 mV was required to drive the OER in 0.1 M HClO4 solution, which was about 30 mV less compared with Ir NPs. In addition, the catalyst also exhibited ultra-high stability under acidic conditions. The catalytic activity was enhanced because the introduction of P changed the metal surface and its affinity for water molecules.
In addition, Ir NPs can also be combined with suitable support materials to reduce the amount of noble metal Ir and improve their intrinsic activity for the OER.138 The unique coordination electronic structure between metal Ir and the support is conducive to the rapid electron transfer of metals that improves the performance of the OER.139 Moreover, the strong interaction between the Ir NPs and the support material helps the dispersion of the metal particles and prevents the aggregation of the nanoparticles, which is also very important for stability improvement.140–142 Carbon-based materials are often used as supports due to their excellent electrical conductivity, inherently large specific surface area, and excellent chemical stability.135 For example, ultra-small Ir NPs were loaded on graphene sheets doped with a large number of N atoms (Ir@N-G-600) by a radiofrequency N2 plasma process.53 At an extremely low Ir loading of 6.98 μg cm−2, the OER activity was remarkably better than that of commercial RuO2 and Ir/C catalysts. The uniform distribution of Ir NPs and the strong coordination between Ir and N changed the electron arrangement around the Ir atoms that produced the ideal electron configuration which was responsible for the increased electrocatalytic activity. Correspondingly, the Ir alloy NPs were also deposited onto the carbon-based supports to improve the catalytic performance. The ultrasmall AuIr alloy NPs with an average particle size of 1.7 nm supported on carbon nanotubes (AuIr@CNT) showed only a 257 mV overpotential to drive 10 mA cm−2 in 0.5 M H2SO4, significantly lower than that for commercial IrO2 and Ir@CNT.54 A cell voltage of only 1.51 V vs. RHE was needed to drive 10 mA cm−2 in the acidic water-splitting device, and the performance degradation was negligible after 50 h of reaction. As discussed in the report, the positive charge of the Ir atoms adjacent to the doped Au atoms caused the electron delocalization of Ir, which helped reduce the energy barrier of the rate-limiting step (O* → OOH*) in the acidic OER; moreover, the strong interaction between Au and Ir suppressed the coalescence phenomenon during cycling. As a result, largely enhanced catalytic activity and stability were observed. However, it should be noted that the carbon-based support would be easily oxidized and corroded in the acidic medium under the high potentials, which caused a seriously degradation in OER performance.143 Although the carbon-supported catalyst showed some high performance, these catalysts should not be recommended as OER catalysts in practical applications.
Metal oxides like titanium dioxide (TiO2) and tin oxide (SnO2) were also employed to support nanoparticles due to their high surface area and good conductivity.144,145 The SMSI between metals and metal oxide supports played an important role in the catalytic stability improvement for the OER.37 For example, the IrOx–TiO2–Ti (ITOT) composite catalyst was prepared by uniform deposition of IrOx nanoparticles on the surface of TiO2 nanorods constructed on the 3D Ti mesh, which exhibited good OER activity and long-term stability under acidic conditions.55 X-ray photoelectron spectroscopy (XPS) and in situ X-ray absorption spectroscopy (XAS) revealed that the TiO2 support favored the formation of IrIII and the mixed-valence IrIII/IV hydrated amorphous oxyhydroxides, which generated a high concentration of OH*ad species on the surface and a higher valence state (higher than IrIV) from the unstable IrIII during the OER process (Fig. 2c). The increased number of surface-active OH species and mixed-valence IrOx as well as the interaction between the metal/metal oxide and the support made the active sites much easier to be attacked by protons or H2O, thereby adjusting the adsorption energy of O* on the surface and improving the catalytic activity. The hybrid metal oxides of TiO2–MoOx were used to support Ir nanoparticles for the OER;136 high-valent Mo(V) and Mo(VI) were found to be incorporated into TiO2, which significantly improved the electrical conductivity and altered the electronic structure of the active Ir species. The positive binding energy changes of Ti(IV) and lattice O and the oxidation of Mo(V) to Mo(VI) could enable significant interaction between TiO2–MoOx and Ir, and, as a result, they facilitated the production of OER-active Ir(III) species and prevented further oxidation of Ir(III) to Ir(IV) during the OER process. By creating and holding onto Ir(III) during the OER, the Mo–Ir interaction could provide materials with enhanced activity and durability.
Nanoclusters, a transitional state between the conventional nanoparticle and a single atom, have countable atomic numbers and well-defined arrangements; the limited size range is generally less than 2 nm.146 The number of atoms ranges from a few to several hundreds, which determines the physicochemical properties, and most of the atoms are directly exposed on the surface or subsurface, which possesses much higher utilization of atoms in catalytic reactions than conventional nanoparticles.88,147,148 In addition, nanoclusters are usually combined with supports to form supported catalysts, showing great potential in various electrocatalytic reactions.129,149 For example, porous IrOx nanoclusters supported on defective graphene (P–IrOx@DG) were prepared by Ar plasma treatment and acid leaching to remove Cu atoms in IrCuOx nanoclusters dispersed on defective graphene, which was an efficient OER electrocatalyst for pH general-purpose.149 When constructed in the electrolyzer using the commercial 20 wt% Pt/C as the cathode, a cell voltage of only 1.48 V was required to drive 10 mA cm−2 in 0.5 M H2SO4 electrolyte. The high activity was discussed based on the special geometric structure and electronic effects; the surface defects of DG provided abundant anchor sites for direct bonding with metal atoms, and the large number of unsaturated coordinated Ir sites induced by the porous structure could optimize the electronic structure and adsorption energy of oxygen-containing intermediates. In addition, IrRu@Te obtained by loading ultrafine IrRu alloy nanoclusters on amorphous Te nanoparticles showed good OER catalytic performance in 0.5 M H2SO4, and only 220 and 303 mV overpotentials were required to drive 10 and 100 mA cm−2, respectively (Fig. 2d).56 The ultrafine IrO2 nanoclusters with an average diameter of 1.4 nm uniformly dispersed on titanium nitride (TiN) with a mesoporous structure exhibited a much higher active surface area, which could expose more unsaturated Ir atoms to participate in the OER catalytic process.150 The strong interaction between TiN and IrO2 was beneficial for the TiN support to obtain some electrons from the active components and improve the OER catalytic activity.
The limiting size of the metal particle is a single atom, which can be dispersed on a specific support to prepare single atom catalysts (SACs), and the use of Ir-based SACs can decrease the usage of noble metals to reduce the catalyst cost. SACs are claimed to have high activity and 100% atom utilization for potential chemical reactions, which is an excellent choice for the preparation of acidic OER catalysts.151,152 For example, Ir-SA@Fe@NCNT was prepared by dispersing Ir atoms in/on Fe nanoparticles through a pyrolysis process and then further encapsulating Ir@Fe nanoparticles in nitrogen-doped carbon nanotubes (NCNTs) (Fig. 2e).57 The catalyst exhibited highly durable and effective acidic OER catalytic performance, which required an overpotential of only 250 mV to drive a current density of 10 mA cm−2 at an ultra-low Ir loading of 1.14 μg cm−2. Moreover, the overpotential was increased by only 10 mV after 1000 cycles with potentials between 1.3 and 1.7 V, and the performance did not decay significantly after 12 h of the chronoamperometry test at 10 mA cm−2. The high catalytic performance resulted from the strong interaction between Fe and Ir atoms to form Fe–Ir bonds and the Fe nanoparticles embedded in NCNTs stabilized Ir-SAs that inhibited the migration and dissolution of Ir-SAs (Fig. 2f). Based on the above research, Ir/Fe@NCNT was prepared by a pyrolysis process of a mixture of melamine, ferric chloride, and Ir trichloride (IrCl3).58 Two states of Ir species were formed in this catalyst, where Ir nanoclusters (1–2 nm) were distributed on the walls of NCNTs and Ir atoms were distributed on Fe nanoparticles wrapped by NCNTs. The dual confinement of Fe nanoparticles and NCNTs ensured the activity of two Ir species, facilitated the chemisorption of O intermediates and H2O molecules, and thus enhanced the OER performance. The catalysts prepared using different Ir contents and temperatures were compared and Ir (20)/Fe@NCNT-900 (IrCl3 content of 20 mg and pyrolysis temperature at 900 °C) had the best catalyst performance. In 0.5 M H2SO4, they exhibited an overpotential of only 300 mV for the OER at 10 mA cm−2. In addition, Ir-SAs can also be introduced into transition metal oxides to promote acidic OER activity by adjusting the chemical environment of IrO6.153 For example, Ir0.08Co2.92O4 nanowires (Ir0.08Co2.92O4 NWs) were prepared by incorporation of Ir-SAs into the surface lattice structure of Co3O4 by the sol-flame annealing method, and they exhibited outstanding acidic OER performance with a low overpotential of only 189.5 mV to reach 10 mA cm−2 and maintained excellent stability for more than 100 h.59 The reason was the modulated chemical environment of Ir and its adjacent lattice oxygen provided lone pair electrons and charge balance to stabilize Ir-SAs. Compared with IrO2, Ir0.08Co2.92O4 nanowires showed optimized d/p band centers near the Fermi level and extended Ir–O bonds, and reduced energy barriers for the absorption/desorption of reaction intermediates.
3.2. 1D catalysts
1D nanomaterials refer to materials with a size in one of the three dimensions, not between 1 and 100 nm, including nanowires, nanorods, nanotubes, etc. 1D nanomaterials can effectively improve the utilization of Ir atoms and increase the specific surface area of catalysts by adjusting the adsorption energy of the reaction intermediate species at the active site.64,67,154 For the 1D nanowires, they represent a distinct class of material structures with strong electrical conductivity, high aspect ratios, and unsaturated surface coordination.16,155 Inspired by this, 1D metal nanowires have been widely studied and used as advanced acidic OER catalysts. For example, 1D Ir–Te porous nanowires (Ir–Te NWs) with an average diameter of 22.8 nm and high porosity were synthesized by a template-assisted synthesis approach using Te NWs as the template (Fig. 3a and b).60 In 0.5 M H2SO4, the OER overpotential was 284 mV to reach 10 mA cm−2, much lower than those of commercial IrO2 and Ir/C, due to the increased electrochemical surface area and lower electrical resistance of Ir–Te NWs. In addition, amorphous IrOx/CeO2 nanowires (IrOx/CeO2 NWs) composed of intimately mixed sub-nanometer-sized IrOx and CeO2 particles were prepared by electrospinning and continuous high-temperature heat treatment, and they had an average diameter of about 50 nm and a length of several micrometers.61 The close nanoscale features of IrOx/CeO2 created a rich binary interface, where CeO2 acted as an electron buffer to mediate the adsorption of oxygen intermediates; the lowered activation energy barrier for the OER could suppress the over-oxidation and Ir dissolution, thereby significantly improving OER activity and stability, thus exhibiting a low overpotential of 220 mV at 10 mA cm−2 with good stability for 300 h. In addition, nanofibers (NFs) have similar properties to nanowires that are also used as a unique nanomaterial for electrocatalytic reactions.156 Single-phase perovskite SrIrO3 NFs were prepared through electrospinning and calcination processes by combining perovskite materials that can effectively reduce the Ir content.62 Due to the high active surface area induced by the morphology of NFs and the highly stable Ir cations occupying the B sites of the perovskite structure, they exhibited excellent activity and stability during the continuous 20000 s operation for the OER in acidic electrolytes.
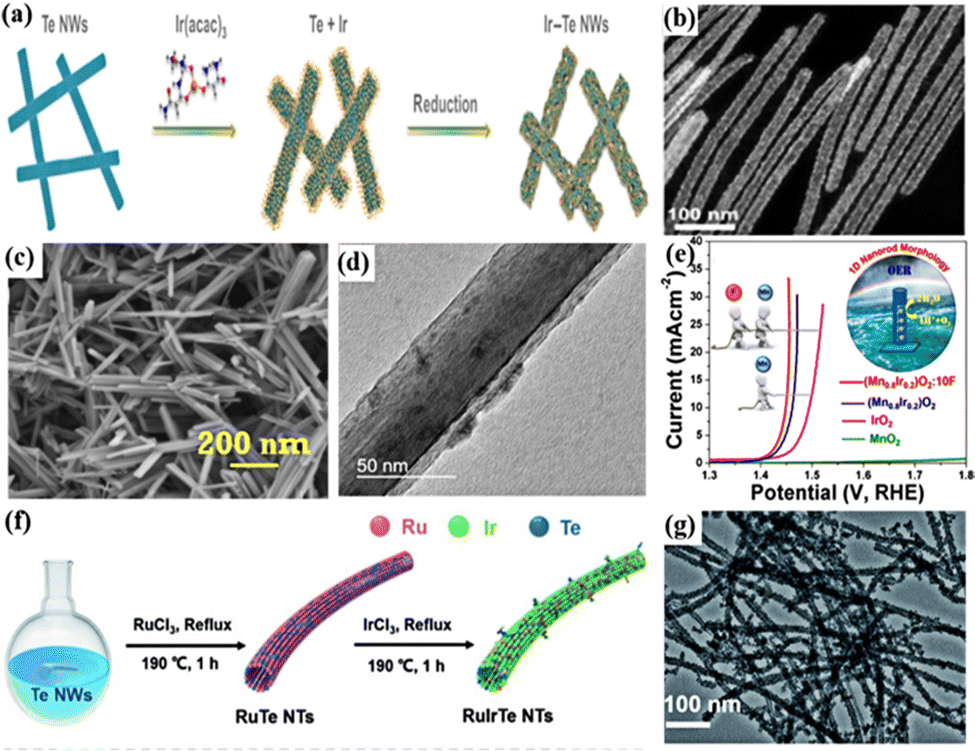 | ||
| Fig. 3 (a) Schematic of the synthesis of 1D porous Ir–Te NWs. (b) TEM image of porous Ir–Te NWs at a high magnification. Reprinted with permission from ref. 60 Copyright 2021, Tsinghua University Press and Springer-Verlag GmbH Germany, part of Springer Nature. (c) Scanning electron microscopy (SEM) image and (b) TEM image of the as-synthesized (Mn0.8Ir0.2) O2:10F NRs. (e) OER polarization curves of (Mn0.8Ir0.2)O2, (Mn0.8Ir0.2)O2:10F NRs and MnO2 with standard IrO2. Reprinted with permission from ref. 64 Copyright 2019, American Chemical Society. (f) Schematic diagram of the synthetic route of the RuIrTe NTs. (g) TEM images of the RuIrTe NTs. Reprinted with permission from ref. 68 Copyright 2022, Royal Society of Chemistry. | ||
Nanorods as a kind of 1D nanomaterials have a smaller aspect ratio of 3–5 compared with nanowires; they also have fast charge transport paths, low charge transfer resistance, and high conductivity.157,158 The acidic OER performance can also be increased for the metal Ir constructed with this unique structure. For example, Ir nanorods (Ir-NR/C) prepared by annealing Ir nanoparticles at 300 °C were a stable and efficient electrocatalyst for wide-pH water splitting reactions.63 In 0.5 M H2SO4, an overpotential of 290 mV was required to drive a current density of 10 mA cm−2. The high performance was attributed to the unique nanorod structure that slowed down the coalescence of Ir and to the formation of an amorphous IrOx layer during annealing which inhibited the electrochemical dissolution of metallic Ir and the formation of soluble IrO42− species. In addition, Ir combined with metal oxides and non-metal element F to form solid solutions was reported to enhance the activity and stability of the catalyst for the OER. 1D (Mn0.8Ir0.2)O2:10F nanorods synthesized by hydrothermal and wet chemical methods had a mesoporous structure that was conducive to offering an efficient transport path for the electrolyte to permeate the inner surface of the electrocatalyst; as a result, they could be used as an efficient electrocatalyst for the acidic OER (Fig. 3c and d).64 During the catalytic process, they exhibited low charge-transfer resistance and a high electrochemically active surface area for catalysis, and the roughness factor can be as high as ∼2114. For the acidic OER performance, they required an overpotential of 200 mV to drive 10 mA cm−2 and exhibited good long-term durability (Fig. 3e). The analysis from the experiments and density functional theory (DFT) calculations proved that the catalytic activity originated from the 1D channel existing in the nanorod structure and the unique electronic structure obtained when forming a solid solution containing F.
Nanotubes are promising 1D nanomaterials with interesting morphological features, confinement effects, and high chemical stability.147,159 The electrochemical surface area and the number of active sites can be further increased by forming a hollow structure, which promoted the rapid transport of electroactive substances.67 For example, hollow Ir nanotubes (Ir NTs) with rough porous surfaces were fabricated by a hydrothermal self-templating method, which consisted of small heterogeneous nanocrystals with inner and outer diameters of 50–80 and 100–150 nm, respectively, in the length range of ca. 3–5 μm.65 They required an overpotential of 245 mV to drive 10 mA cm−2 for the acidic OER, which was 52 mV lower than that of commercial Ir nanocrystals, and the reason for enhanced performance was that the rough, porous surface and 1D hollow structure generated more available active sites, higher surface area and faster kinetics. In addition, other metal or non-metal elements can also be introduced into Ir-based catalysts to synthesize Ir-based alloy nanotubes. For example, PdPtIr porous nanotubes (PdPtIr PNTs/C) with abundant double defects and high refractive index crystal facets were prepared by annealing at 400 °C using Pd nanowires as a template; in an acidic electrolyte, an overpotential of 337 mV was required to drive 10 mA cm−2 for the OER.67 The surface strain and coordination effects of its double grain boundary defects significantly reduced the activation energy of the acidic OER, and the ultra-high surface area/volume ratio increased the number of active sites, thereby enhancing the catalytic performance. The introduction of semimetal Te into Ir-based catalysts can also effectively improve the catalytic activity. For example, the 1D hollow dendritic structure for RuIrTe nanotubes (RuIrTe NTs) was synthesized by a displacement reaction of Ir, Ru elements and Te nanowires, and they exhibited outstanding catalytic activity for the acidic OER, requiring a low overpotential of 205 mV to generate 10 mA cm−2 (Fig. 3f and g).68 The enhanced catalytic activity was attributed to the rough surface structure of 1D nanotubes which can provide a large active surface area and fast mass and charge transfer and to the strong electronic effect between Ru, Ir, and Te which reduced the d-band center of Ir and optimized the adsorption energy of the intermediates on the catalyst. In addition, the heterostructure catalyst synthesized from Ir atomic clusters (AC) anchored on ultrathin 1D rutile IrO2 nanoneedles (NNs) exhibited high activity for the acidic OER, which required only a low overpotential of 296 mV to drive 10 mA cm−2.66 The reason was that the robustness of IrO2 NNs stabilized the surface Ir clusters during the OER, and the crystalline IrO2 NN framework helped facilitate electron transfer to the surface Ir ACs. Therefore, it can be concluded that the 1D Ir-based nanocatalyst can provide a large specific surface area for the reaction and facilitate the rapid transfer of active species and charges due to its special structure. In addition, the 1D nanocatalyst prepared by combining Ir with other elements can reduce the Ir content and improve the catalytic performance.
3.3. 2D catalysts
2D nanomaterials are materials in which two of the three dimensions are not between 1 and 100 nm in size.33 These 2D structures have excellent conductivity and high stability; they have high atom utilization efficiency by exploiting the interfacial electronic effect of exposed surface atoms and the surface coordination of unsaturated metal sites; and the unique edge structure can also help enhance the intrinsic activity.160,161 Nanosheets are the most common 2D nanomaterials with unique electronic structures and high specific surface area for active species exposure.25 For example, 2D mesoporous Ir nanosheets were prepared using formic acid as a reducing agent and shape-directing agent, and they could enhance the acidic OER performance by maximizing the exposure of surface atoms, which exhibited an overpotential of 240 mV at 10 mA cm−2 for the OER.162 Furthermore, inspired by the study of 2D mesoporous metal Ir nanosheets, amorphous IrOx mesoporous nanosheets were formed by heating the mesoporous nanosheets to obtain a diameter of 13 nm and semi-regularly arranged uniform pores on a single nanosheet (Fig. 4a).69 A large number of OH groups were formed on the surface of amorphous IrOx nanosheets under acidic conditions, which were transformed into electrophilic oxygen species that were easily attacked by H2O or OH− species through surface remodeling, and the accelerated formation of O–O bonds greatly increased the reaction rate due to the synergistic effect of the 2D mesoporous structure (Fig. 4b). Furthermore, 2D ordered mesoporous layered Ir–IrOx/C nanosheet catalysts with abundant electrophilic oxygen (O(II−δ)−) species were prepared via a nanoconfined self-assembly strategy.70 They exhibited a low OER overpotential of 198 mV at 10 mA cm−2 in 0.5 M H2SO4. The mixed valence state of Ir reduced the adsorption free energy of *OOH which effectively balanced the interaction of oxygen-containing intermediates; the increased specific surface area and open interlayer channels provided more active sites and enhanced the mass transfer performance of water oxidation; additionally, the special coordination environment for oxygen (O(II−δ)−) on the surface promoted the nucleophilic attack of H2O, which was beneficial to the quick creation of O–O bonds.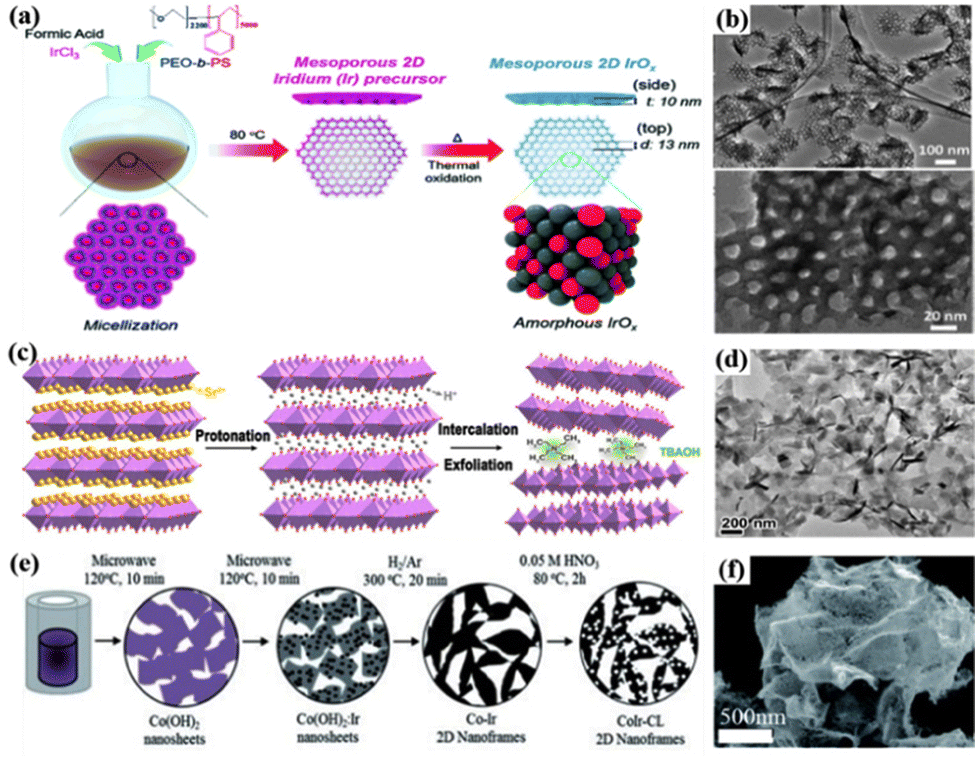 | ||
| Fig. 4 (a) A diagram depicting the process of producing mesoporous IrOx nanosheets. (b) TEM images showing the nanosheets with mesopores. Reprinted with permission from ref. 69 Copyright 2019, Royal Society of Chemistry. (c) Schematic of liquid-phase exfoliation of Sr2IrO4. (d) TEM image of HION. Reprinted with permission from ref. 71 Copyright 2022, American Chemical Society. (e) Schematic representation of the experimental stages for the fabrication of 2D CoIr nanoframes. (f) SEM image of CoIr nanoframes. Reprinted with permission from ref. 75 Copyright 2021, Royal Society of Chemistry. | ||
Ir-based perovskite oxides can also be used to prepare 2D nanosheet catalysts for the acidic OER.163 For example, high-yield liquid-phase exfoliation of Ruddlesden–Popper Sr2IrO4 layered perovskite monolayer protonated iridate nanosheets (HIONs) was about 10 times more active than that of an IrO2 catalyst film (Fig. 4c).71 The reason was that the fully protonated surface, layered perovskite framework, and high colloidal dispersibility of HION provided effective Ir active sites with suitable electronic structures to enhance the intrinsic activity (Fig. 4d). In addition, anion doping can also tune the surface and electronic structures of Ir-based perovskite nanosheets. For example, S-doped M-SrIrO3 perovskite nanosheets were successfully synthesized by an anion engineering strategy combining the sol–gel method and sulfidation process.72 The OER catalytic effect of this catalyst in acidic electrolytes was remarkable, with an overpotential of 228 mV at 10 mA cm−2, which was significantly better than the pristine M-SrIrO3 nanosheets. Through DFT calculations, the incorporation of S could lower the free energy barrier of the rate-limiting step, thereby lowering the activation energy of OER kinetics.
In addition, nanofilms, similar to nanosheets but with slightly lower thickness, are also important 2D materials in catalysis reactions.164 For example, dense thin films of Ir, IrSn2, IrCr, IrTi, and IrNi were prepared by electron beam evaporation co-deposition, and the binary IrCr film had the best catalytic activity and stability for the acidic OER because of the easy generation of IrCrOx during the reaction.73 The theoretical OER activities of Cr-doped IrO2 and pure IrO2 were calculated by establishing a DFT model, and it was found that the existence of high-valence Cr endowed the catalyst with weaker and better oxygen binding energy, which was considered to be the reason for the enhanced activity of the catalyst. IrRe–IrOx nanofilms prepared by electro-oxidation of magnetron sputtered Ir–Re films showed high activity for the acidic OER because the dissolved Re atoms promoted the oxidation of metal Ir to a highly active amorphous IrOx phase.74 As a result, the catalyst exhibited a low overpotential of 248 mV and a mass activity of 4400 mA mg−1 at a current density of 10 mA cm−2 for the OER. Furthermore, a novel 2D hydrated Co–Ir oxide nanoframework was fabricated by thermal reduction of Ir-decorated cobalt hydroxide nanosheets and nitric acid etching to remove the unstable metallic Co (Fig. 4e and f).75 The catalyst had a mass activity of 243 A gIr−1 for the OER at 1.51 V vs. RHE, about 17 times higher than that of commercial IrO2. Moreover, after electrochemical oxidation in the OER potential range, the surface of the bimetallic alloy was transformed into an oxide/hydroxide surface with a large amount of Ir hydroxide active species, which changed the atomic and electronic structures of the surface, thereby further enhancing the OER activity. However, research on the influence of geometric structure changes on catalytic activity is still not clear, and clarifying the relationship between structure and performance to promote the development of electrochemical water splitting would be necessary.
3.4. 3D catalysts
3D nanomaterials, assembled or combined in different ways by nanomaterials of basic structural units in 0D, 1D, and 2D, have some similar properties to other types of nanomaterials, such as a large specific surface area and porous structure.165,166 3D nanomaterials with interconnect structures can effectively improve the electron transport rate and structure–mechanical stability.167,168 Among them, 3D nanonetworks are intensively studied in various electrocatalytic reactions due to the increased number of electroactive sites and high surface area.169,170 In addition, they can provide an inner surface through a continuous porous structure, which increases the contact between the catalyst and the electrolyte solution and accelerates the mass transfer in the reaction.171 For example, the 3D continuous nanoporous Ir (np-Ir) electrocatalyst obtained by the dealloying approach could provide an overpotential of 259 mV to reach a current density of 10 mA cm−2 for the acidic OER, much lower than that of IrOx nanoparticles.172 The high-aspect-ratio interconnected porous metal framework provided an uninterrupted transport path for electron mobility, which offered high electrical conductivity as well as low charge-transfer resistance. The self-supporting 3D nanoporous IrOx network consisted of small nanobones of 2–5 nm with small micro/mesopores of 1–10 nm and macropores of 30–150 nm size, which were prepared by selective leaching and alternating sputtering of Ir and Co.76 3D porous interconnected nanostructure achieved much higher porosity and better Ir dispersion, which increased the high intrinsic activity. As a result, the mass activity of the 3D nanoporous IrOx network was 8505 A gIr−1 at 1.60 V vs. RHE, 8 times that of commercial Ir-black nanoparticles (1030 A gIr−1) for the acidic OER.In addition, the metal nanowire network has attracted attention due to its 3D highly porous structure and a large number of exposed active sites, which can accelerate electron transfer and improve the utilization of noble metals, thereby improving the catalytic efficiency.173,174 For example, the 3D IrxPb bimetallic nanowire network with high surface area and tunable composition were fabricated via a sol–gel process which exhibited enhanced OER performance with an overpotential of 307 mV at 10 mA cm−2, compared with commercial IrO2 (Fig. 5a).77 The high activity was attributed to the introduction of Pb, which optimized the composition of the catalyst, adjusted the electronic structure, and facilitated the interconnection of ultrafine nanowires to form a strong network structure. A series of composition-tunable 3D nanoporous Ir70Ni30−xCox (x = 0, 15, 30) ternary alloy microwires (np-Ir70Ni30−xCox) were prepared by selectively dealloying Ni and Co from the precursor Ir3Ni97−xCox (x = 0, 50, 97) in HCl solution.78 The obtained np-Ir70Ni15Co15 alloy with well-connected grains and an aligned 3D microwire structure showed the lowest overpotential of 220 mV in 0.1 M HClO4 to drive 10 mA cm−2 for the OER. The nanoporous structure and the alloying effect of Ir with 3d transition metals were believed to be responsible for the enhanced catalyst activity, which promoted the permeation of electrolyte and accelerated electron transport during the OER process.
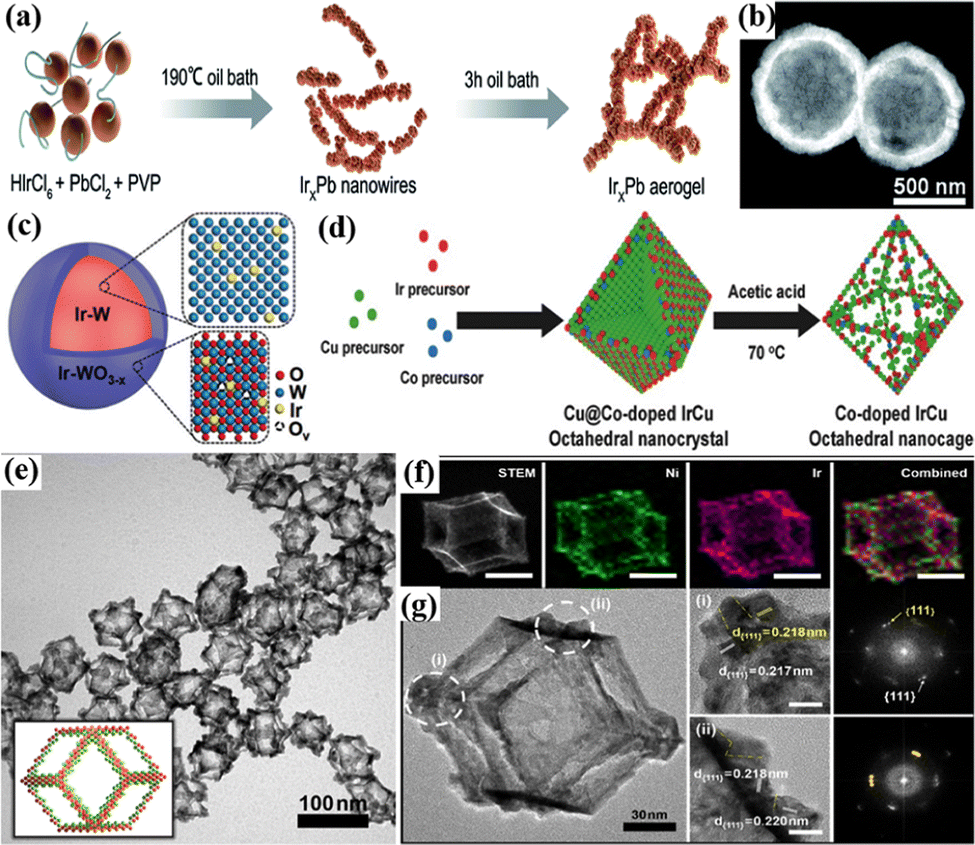 | ||
| Fig. 5 (a) Schematic illustration of the formation of the IrxPb nanowire network. Reprinted with permission from ref. 77 Copyright 2022, Royal Society of Chemistry. (b) Structural characterization of the special Ir p-HNs. Reprinted with permission from ref. 80 Copyright 2022, Royal Society of Chemistry. (c) The schematic illustration of the core–shell structure of Ir–W@Ir–WO3−x. Reprinted with permission from ref. 82 Copyright 2022, Royal Society of Chemistry. (d) Schematic illustration of the production of Co-doped IrCu octahedral nanocages. Reprinted with permission from ref. 36 Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (e) TEM images of IrNi-RF with graphical models in the insets. (f) STEM and elemental mapping images of IrNi-RF for each element, as well as a combined image of Ni (green) and Ir (magenta). (g) HRTEM image of IrNi-RF. Reprinted with permission from ref. 85 Copyright 2017, Elsevier Ltd. | ||
Furthermore, well-structured nanospheres with 3D accessible sites and a large surface area can enhance electrochemical performance.175,176 For example, 3D IrRuMn nanospheres prepared by the wet chemical method exhibited outstanding OER performance in an acidic environment, and they required an overpotential of 260 mV to drive 10 mA cm−2 due to the high surface area of the 3D nanospheres, which exposed more active sites.79 In addition, the hollow structure exposes a unique inner cavity and a shorter transport path, which can further expose the active surface area and accelerate mass transfer and electron transfer.177,178 For example, Ir porous hollow spheres (Ir p-HNs) were fabricated using the Kirkendall effect where in situ etching exhibited a low acidic OER overpotential of 243 mV at a current density of 10 mA cm−2 (Fig. 5b).80 In addition, the core–shell structure can also be considered a special kind of nanosphere fabricated by interfacial engineering, which has been widely used for the OER in electrocatalysis due to the strong interaction between core–shell layers and tunable composition.82,179 For example, the IrRux@Ir catalyst with an IrRu core and an Ir shell was synthesized by a CO-induced phase separation strategy, which required an overpotential of 288 mV to drive 10 mA cm−2 for the acidic OER.180 The enhanced activity was attributed to the strong electronic interaction between different Ir layers leading to charge transfer from the Ir shell to the IrRux core, and the protection of the Ir shell prevents the dissolution of the inner Ru atoms during electrolysis. An IrGa–IrOx core–shell catalyst composed of a structurally ordered IrGa intermetallic compound (IrGa IMC) core and a partially oxidized Ir(IrOx) shell exhibited excellent OER activity, which required an overpotential of only 272 mV to drive 10 mA cm−2.81 The enhanced activity originated from the electronic structure optimization of the surface IrOx sites induced by the IrGa IMC layer, which enhanced the degeneracy of Ir 5d electrons on the IrOx surface, leading to the increased overlap of Ir 5d and O 2p orbitals relative to pure Ir. Therefore, the adsorption capacity of the IrOx layer for O and OH species was improved which lowered the energy barrier for the rate-determining step of the OER. Furthermore, Ir–W@Ir–WO3−x core–shell nanospheres with an Ir–W metal core and an Ir-doped WO3−x (Ir–WO3−x) shell were constructed (Fig. 5c), which provided a low overpotential of 261 mV at 10 mA cm−2 for the acidic OER.82 XAS analysis revealed that Ir–W@Ir–WO3−x greatly reduced peroxide species formation and efficiently avoided peroxide-induced corrosion during the OER, and theoretical studies demonstrated that the modest O binding ability not only enhanced catalytic kinetics but also limited the generation of hydroperoxides.
Nanocages and nanoframes with hollow structures can expose high-density outer surfaces to reactants, thereby enhancing the catalytic activity.12,181 For example, IrxRu1−x cubic nanocages with ultrathin walls of six atomic layers (∼1.21 nm) were prepared by the seed-mediated growth and etching method.84 By adjusting the ratio of Ir and Ru, it was found that the Ir0.5Ru0.5 nanocages exhibited significantly low overpotentials of 219 mV for the OER at 10 mA cm−2. Their high activity was attributed to the highly open structure of the nanocages and the possible electronic coupling between Ir and Ru atoms. Hollow Co-doped IrCu octahedral nanocages (Co–IrCu ONCs) were fabricated by selectively removing the Cu core in octahedral Cu@IrCu core–shell nanocrystals via Co substitution under acidic conditions (Fig. 5d).36 The activities of Co–IrCu ONC with different acid leaching times (1 h, 10 h, and 24 h) on the catalyst were studied, where the activity of Co–IrCu ONC/C-10 h was best due to efficient electron transfer of the 3D interconnected framework structure generated using an appropriate acid leaching time. Furthermore, IrNi nanoframes (IrNi-RF) were generated by selective passivation with lanthanide metal chlorides to form rhombohedral dodecahedral IrNi nanoparticles, followed by selective etching away of the Ni phase planes (Fig. 5e–g).85 They exhibited good electrocatalytic activity for the acidic OER, which required an overpotential of only 313.6 mV at 10 mA cm−2, and after 5000 cycles the performance loss was small with an overpotential increase of only 16 mV, indicating excellent catalytic stability. The exceptional electrocatalytic activity and stability were attributed to the existence of highly active grain boundaries, an agglomeration-free framework structure, and the contact between IrNi and IrOx. Based on the study of single nanoframes, double-layer nanoframes (DNFs) were further developed.182 Taking advantage of the different kinetics of the dual Ir precursors and the dual transition metal Ni and Cu precursors, core–shell alloy@alloy structures were generated using a simple one-step synthesis method, followed by partial removal of Cu and Ni elements by a selective etching strategy to transform the rhombic dodecahedral alloy structure into multimetallic IrNiCu DNFs.86 They exhibited high electrocatalytic activity for the OER in 0.1 M HClO4, requiring an overpotential of 300 mV to drive 10 mA cm−2, significantly better than those of Ir/C catalysts. In addition, the framework structure could minimize the growth and agglomeration of particles and slow down the in situ formation of the rutile IrO2 phase during the long-term operation of the OER. In addition, a special modified SrIrO was prepared by combining layered quasi-two-dimensional Sr2IrO4 with interlacing perovskite slabs and spacer cations and a 3D 6H-SrIrO3 perovskite with a distorted monoclinic hexagonal structure.183 In this catalyst system, Sr could be extracted from the Sr2IrO4 crystal interlayer to maintain the structural stability of the in-plane IrO6 framework; 6H-SrIrO3 with 3D corner-sharing and face-sharing IrO6 networks had proven to be more active for the OER; these two special structures synergistically improved the catalytic performance. Benefiting from the above advantages, OER overpotentials of 245 and 263 mV were required to drive 10 mA cm−2 in 0.1 M HClO4 and 0.5 M H2SO4, respectively; a cell potential of only 1.5 V vs. RHE was required to deliver 10 mA cm−2 in a two-electrode electrolyzer. In summary, 3D Ir-based nanocatalysts including nanonetworks, nanospheres, nanocages, and nanoframes, usually with mesoporous or hollow structures, can enlarge the surface area of the material and improve the utilization of active species. In addition, alloying will reduce the amount of Ir and change the electronic structure of the catalyst, thereby improving the catalytic activity.
4. Application in PEMWE
As mentioned in the introduction, PEM water electrolysis hydrogen production technology has the advantages of high operating current density, low energy consumption, high hydrogen production pressure, adapting to the fluctuation of renewable energy power generation, and compact design. It is considered to be the green hydrogen production technology with the most application potential at present.184 The main components of PEMWE from inside to outside are the PEM, catalyst layer, gas diffusion layer, end plate, and so on.8 Among them, the catalyst layer as the important component of the water electrolyzer occupies a large share of its total cost.29 Therefore, it is necessary to apply the currently developed high-performance OER catalysts to the actual PEMWE to further evaluate the catalytic and practical application capabilities. Generally, the operating temperature, energy efficiency, and current density of PEMWE are expected to be between 60 and 80 °C, 75% and 80%, and 1.5 and 3 A cm−2 in practical applications, respectively.185 Although various catalysts have shown excellent catalytic performance in the acidic OER, they may not be available in the real PEMWE application.29As a result, researchers should put in a lot of effort to address the application of Ir-based catalysts in PEMWE, while, to our knowledge, reports of the use of these catalysts in real devices are still very rare, and more effort is still required.186,187 The Ir AC/NN catalyst mentioned above could also be used as an anode catalyst to evaluate its catalytic performance in actual PEMWE.66 The anode and cathode catalyst layers were prepared by using the catalyst-coated membrane method for spray-coating Ir AC/NN catalyst ink and 40 wt% commercial Pt/C on Ti felt and carbon paper, respectively; the Nafion 212 membrane was used as the PEM and the Ti block and graphite block were used as the gas diffusion layer, which together constituted the core membrane electrode assembly (MEA) (Fig. 6a). The device showed a current density of 3 A cm−2 at only 1.82 V and stable operation for more than 90 h with significantly better activity and stability than individual IrO2 NNs and IrO2 particles, and the synergy between AC and 1D NN was further demonstrated. De-alloyed nanoporous IrNi (DNP-IrNi) with an independent porous interconnected structure was fabricated by adsorption hydrogen-induced co-electrodeposition and dealloying of the IrNi–Ir core–shell structure, which could serve as a bifunctional electrode in PEMWE.14 The DNP-IrNi (loaded at 0.67 mg cm−2) coated on highly conductive carbon paper was assembled in the real device for PEMWE, and the current could reach 6.5 A cm−2 at a cell voltage of 2.0 V (Fig. 6b); the degradation rate was only 1.58 mV h−1 at 2 A cm−2 for a 100 h stability test, indicating the excellent stability (Fig. 6c).
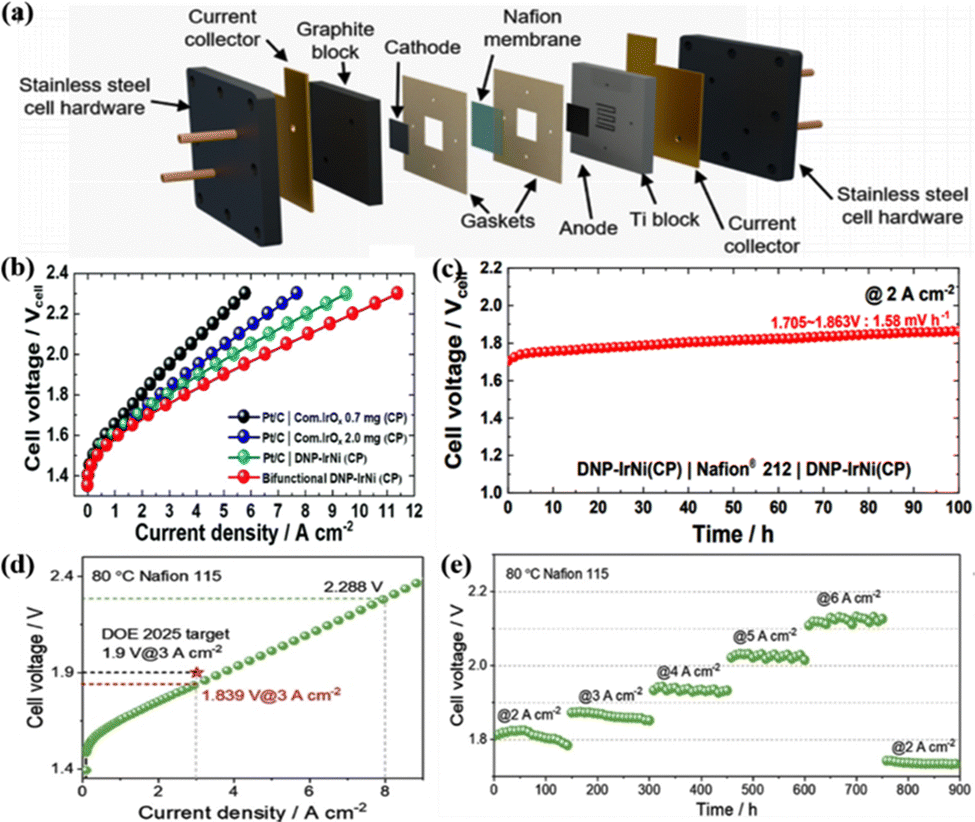 | ||
| Fig. 6 (a) Photographic representation of a single cell for PEMWE. Reprinted with permission from ref. 66 Copyright 2022, Elsevier B.V. (b) Polarization curves of the single cell using carbon paper. (c) Stability test using carbon paper felt at 2 A cm−2 for 100 h. Reprinted with permission from ref. 14 Copyright 2022, Royal Society of Chemistry. (d) The polarization curve of the PEM electrolyzer was obtained with the Nafion 115 membrane at 80 °C. (e) Chronopotentiometric curves of the PEM electrolyzer using Ir/Nb2O5−x at a different current density. Reprinted with permission from ref. 188 Copyright 2022, Wiley-VCH GmbH. | ||
In addition, Ir/Nb2O5−x was prepared by loading metal Ir nanoparticles on Nb2O5−x, in which Ir atoms were closely arranged along the [001] crystal plane of Nb2O5−x, forming a thick quasi-Ir shell around 1 nm.188 The MEA composed of Ir/Nb2O5−x as the anode catalyst, commercial Pt/C as the cathode catalyst and a Nafion 115 membrane was tested at 80 °C, and the cell voltage was found to be 1.839 V at 3 A cm−2 (Fig. 6d); the stability at 2 A cm−2 was maintained for 2000 h with no significant activity decay. In addition, Ir/Nb2O5−x also had good stability under the gradient increasing current density (from 2 to 6 A cm−2); the cell voltage could be maintained at 1.937, 2.026 and 2.123 V for 150 h at a current density of 4, 5, and 6 A cm−2, respectively (Fig. 6e). Its activity and stability were attributed to the tight fusion between Ir and Nb2O5−x and the unique chemical environment of Ir at the interface, and the Ir lattice beyond the grain boundaries and extending to Nb2O5−x facilitated the dynamic migration of oxygen species and reduced the electron transfer resistance at the interface. Furthermore, self-assembled Ir/IrOx catalysts with a layered structure and controllable Ir oxidation state ratio were successfully prepared by a solution reduction method to enhance the efficiency of PEMWE.189 The oxidation state of the catalyst was reduced gradually from the shell to the core, which helped overcome the trade-off between OER activity and stability. A HT-Ir/IrOx MEA and reference MEA were assembled using HT-Ir/IrOx (heat-treated at 398 K for 20 min in an N2 atmosphere) and unheated Ir/IrOx as the anode catalysts, respectively, and the performance of HT-Ir/IrOx MEA was excellent, requiring a cell voltage of 1.73 V to drive 1.0 A cm−2 at 353 K. Furthermore, after 48 h of stability test at a constant current of 1 A cm−2, a significant increase in cell voltage of 370 mV was found for the reference MEA, while only a slight increase of 70 mV was observed for the HT-Ir/IrOx-MEA, further demonstrating the promising application of this catalyst. Ir-ND/ATO was constructed with short rod-like Ir nano-dendrites (Ir-ND) with a porous nanostructure supported on antimony-doped tin oxide (ATO), which could be used as an efficient and stable water-splitting catalyst in PEMWE.190 In single-cell tests, Ir-ND/ATO outperformed Ir-ND/C and Ir-ND, and exhibited a current density of 1.50 A cm−2 at 1.8 V. Its enhanced performance was attributed to the small average particle size, large surface area and high porosity (34.2%) of Ir-ND, and the electronic effects caused by the strong charge redistribution between Ir metal and metal oxide supports.
In conclusion, a great deal of effort has been devoted to solving the problems of high cost and instability of catalysts for practical application in PEMWE. However, regarding the practical application, in addition to the need for high-performance electrocatalysts, other electrolyzer components must also be optimized. There are also some issues that should be considered: (a) improving the ionic conductivity of the membrane materials to reduce membrane resistance and electrolysis energy consumption, (b) optimizing the integration of catalysts and current collectors to ensure effective mass transport and electron transfer, and (c) improving the bipolar plate surface treatment process and developing cheap anti-corrosion coating technology. In addition, the gas bubbles generated during the catalytic process significantly affect the performance of PEMWE, but studies on the influence of gas bubble evolution and transport processes on the performance of electrolyzers are extremely rare. In short, future research needs to comprehensively improve electrolysis efficiency and reduce costs in multiple aspects to accelerate the industrialization of PEMWE technology.
5. Conclusions and outlook
PEM water electrolysis combined with renewable energy is efficient for clean hydrogen production and a crucial component of setting up a future clean energy cycle system. Herein, we provide a systematic and comprehensive review of Ir-based nanocatalysts for the acidic OER from the perspective of different dimensions, aiming to elucidate the relationship between dimensionality, structure, and activity, and to inspire the development of superior catalysts. Based on the impact of the dimensional effect, the promotion mechanisms of the nano-size effect, synergistic effect, and electronic effect were first discussed, and the latest progress of Ir-based catalysts based on the aspects of 0D, 1D, 2D, and 3D was overviewed to understand the dimensional effect and catalytic performance.It can be concluded that the varied size and structure of catalysts significantly affect the catalytic performance by changing their surface properties and electronic structure. Dimensional engineering can increase the specific surface area and the number of active sites, and the unsaturated coordination atoms by changing the size of the catalyst could further increase the atomic utilization of the electrocatalyst. For example, when the size of the active metal Ir is continuously reduced to a single atom, a 100% utilization rate of the catalyst atom can be reached; the surface and electronic structure of the catalyst can be modified through morphology engineering which affects the electron transfer rate and the adsorption energy of reactants, and the dimensional engineering combined with interface engineering could generate a strong metal–support interaction to significantly reduce the agglomeration of active species and improve the catalytic stability. While some challenges are still faced in the study of dimensional engineering of Ir-based catalysts, some research might be considered in the following fields.
5.1. Controllable synthesis of catalyst structure
The controllable design and synthesis of the catalyst with a proper dimension and structure is an effective and necessary way to realize its application in the field of electrocatalysts. However, it is still a great challenge to rationally design Ir-based catalysts of different dimensions in a highly controlled manner, such as 1D, 2D, and 3D catalyst synthesis, which usually involves multiple components, such as reducing agents, surfactants, and solvents (even mixed solvents). In addition, factors such as synthesis conditions (temperature and time) will also have a significant impact on the growth of metal nanocatalysts, and the complex interactions between these components and synthesis conditions make the synthesis of most catalysts in a continuous trial process. Therefore, it is very necessary to establish a database, which can combine the properties of different components. To realize clean surface fabrication on a large scale is still very difficult, which should also be carefully considered in further study. Although a small amount of catalyst with special morphology can be obtained, the surface cleaning process and the morphology control cannot be well conducted on a large scale.5.2. Structure–activity in-depth understanding
As we all know, the design of SACs in 0D materials has been a hot spot in the design of noble metal-based catalysts in recent years. It is necessary to observe and track the reaction process using in situ electron microscopy or in situ synchrotron radiation, as well as other in situ characterization methods; to combine theoretical calculations to study the structure–activity relationship between its structure and catalytic performance might further reveal the possible catalytic reaction mechanism. For 1D nanocatalysts, the effect of an appropriate diameter–length ratio on their electrochemical performance has not been pointed out and verified; for 2D nanomaterials, how to adjust the layer spacing to effectively solve the interlayer agglomeration and stacking effects, and maintain the stability of the 2D structure is still an urgent problem. Therefore, an in-depth study of the structure–activity correlation between the size, structure, porosity, and electrochemical performance of catalysts is likely to find a way to solve these problems. In addition, the structural changes of Ir-based catalysts during the OER can also affect the catalytic activity and stability, and an in-depth understanding of the dynamic dissolution, morphology, and structural evolution of electrocatalysts is the basis for slowing degradation under scientific guidance. Therefore, follow-up studies urgently need more direct and accurate characterization methods to explore the structure–activity relationships of catalysts.5.3. Design and development of 3D catalysts
3D Ir-based nanocatalysts play a very important role in the acidic OER; they have a highly open 3D structure, a high specific surface area, high porosity, and a high density of corner and edge sites, which can improve mass transfer and gas diffusion, which in turn makes the catalyst more fully contacted with the reactant molecules in a narrow space, thereby effectively promoting the reaction kinetics, and the 3D structure is more stable and can resist the harsh reaction environment. Therefore, 3D catalysts containing interconnected network structures can overcome the problems of the aggregation effect of active sites and poor structural stability in other low-dimensional materials to a certain extent, effectively promote the improvement of catalytic activity and stability, and reduce the amount of the noble metal Ir. However, their porous structure and exposed crystal planes with special structures are very difficult to control and usually require complex preparation processes. Therefore, in order to construct a reasonable porous structure and optimize the reaction and transfer of ions and electrons in the catalyst, follow-up studies should pay more attention to 3D catalyst development.5.4. Catalytic stability probing and improvement
The development of Ir-based catalysts that operate smoothly at high current densities is crucial for practical applications. However, many studies on stability were done by only simple continuous cyclic voltammetry tests or long-term stability tests from tens of hours to hundreds of hours, and the research and evaluation of catalyst corrosion and deactivation are extremely lacking. Therefore, it is necessary to establish more reasonable standards to quantitatively evaluate the stability of different catalysts, paying attention to the life, decay rate, and recyclability of catalysts. In addition, it is also necessary to prevent the dissolution of the active centers and the detachment of the catalyst from the electrode during the catalytic process. Therefore, follow-up studies should introduce sacrificial agents and construct a sealing layer to protect the internal active materials, or achieve a dynamic balance of the active site in dissolution–redeposition to maintain stability. Furthermore, the stability of catalysts should be carefully considered for the catalyst design by introducing VO, regulating crystallinity, heteroatom doping and other methods to develop support materials with anti-oxidation and anti-corrosion potential to strengthen the interaction between catalysts and supports.5.5. Performance evaluation in the PEMWE device
Although many advanced Ir-based catalysts of various dimensions suitable for the acidic OER have been produced, their potential application is only achieved by reporting their electrochemical test in a three- or two-electrode system and has not gone from fundamental research to practical application. The present three-electrode devices, which are mostly utilized for performance evaluation, are incapable of accurately reflecting the complexity of genuine catalytic systems. As a result, it is critical to establish generally accepted testing standards to objectively compare the practicability of different Ir-based catalysts. For example, a more rigorous evaluation of the catalyst performance is carried out from the electrode structure, reactants, product transport and operating conditions (temperature, time, current density, operating pressure difference, etc.) of the actual system. In addition, to realize the large-scale application of the catalyst, it is necessary to consider the imbalance between low yield and high cost and the cumbersome preparation process in the development of the catalyst. Therefore, follow-up research should simplify the process steps by developing simple and scalable synthetic routes, thereby reducing catalyst costs. With the gradual unravelling of the mystery of the OER under acidic conditions and the further reduction of catalyst costs, the large-scale application of PEMWE in hydrogen production and implementation has broad prospects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21972124 and 22272148), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institution was also appreciated by the authors.Notes and references
- S. Hu, S. Ge, H. Liu, X. Kang, Q. Yu and B. Liu, Adv. Funct. Mater., 2022, 32, 2201726 CrossRef CAS.
- Y. Liu, X. Liang, H. Chen, R. Gao, L. Shi, L. Yang and X. Zou, Chin. J. Catal., 2021, 42, 1054–1077 CrossRef CAS.
- H. Su, W. Zhou, W. Zhou, Y. Li, L. Zheng, H. Zhang, M. Liu, X. Zhang, X. Sun, Y. Xu, F. Hu, J. Zhang, T. Hu, Q. Liu and S. Wei, Nat. Commun., 2021, 12, 6118 CrossRef CAS PubMed.
- R. He, X. Huang and L. Feng, Energy Fuels, 2022, 36, 6675–6694 CrossRef CAS.
- X. Gu, J. C. A. Camayang, S. Samira and E. Nikolla, J. Catal., 2020, 388, 130–140 CrossRef CAS.
- H. Guo, Z. Fang, H. Li, D. Fernandez, G. Henkelman, S. M. Humphrey and G. Yu, ACS Nano, 2019, 13, 13225–13234 CrossRef CAS PubMed.
- M. Li and L. Feng, J. Electrochem., 2022, 28, 2106211 Search PubMed.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Nano Res. Energy, 2022, 1, e9120032 CrossRef.
- J. A. Arminio Ravelo, J. Quinson, M. A. Pedersen, J. J. K. Kirkensgaard, M. Arenz and M. EscuderoEscribano, ChemCatChem, 2020, 12, 1282–1287 CrossRef CAS.
- H. Wu, Y. Wang, Z. Shi, X. Wang, J. Yang, M. Xiao, J. Ge, W. Xing and C. Liu, J. Mater. Chem. A, 2022, 10, 13170–13189 RSC.
- L. Zhang, Q. Fan, K. Li, S. Zhang and X. Ma, Sustainable Energy Fuels, 2020, 4, 5417–5432 RSC.
- Y. Pi, Q. Shao, X. Zhu and X. Huang, ACS Nano, 2018, 12, 7371–7379 CrossRef CAS PubMed.
- Z. L. Zhao, Q. Wang, X. Huang, Q. Feng, S. Gu, Z. Zhang, H. Xu, L. Zeng, M. Gu and H. Li, Energy Environ. Sci., 2020, 13, 5143–5151 RSC.
- K. Yeo, K. Lee, H. Kim, J. Lee and S. Kim, Energy Environ. Sci., 2022, 15, 3449–3461 RSC.
- X. Yang, Z. Zhao, X. Yu and L. Feng, Chem. Commun., 2019, 55, 1490–1493 RSC.
- W. Gou, M. Zhang, Y. Zou, X. Zhou and Y. Qu, ChemCatChem, 2019, 11, 6008–6014 CrossRef CAS.
- D. Li, M. Zha, L. Feng, G. Hu, C. Hu, X. Wu and X. Wang, Nanoscale, 2022, 14, 790–796 RSC.
- J. He, X. Zhou, P. Xu and J. Sun, Adv. Energy Mater., 2021, 11, 2102883 CrossRef CAS.
- M. A. Hubert, A. M. Patel, A. Gallo, Y. Liu, E. Valle, M. BenNaim, J. Sanchez, D. Sokaras, R. Sinclair, J. K. Nørskov, L. A. King, M. Bajdich and T. F. Jaramillo, ACS Catal., 2020, 10, 12182–12196 CrossRef CAS.
- M. EscuderoEscribano, A. F. Pedersen, E. A. Paoli, R. Frydendal, D. Friebel, P. Malacrida, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Phys. Chem. B, 2018, 122, 947–955 CrossRef CAS PubMed.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- X. Yang, Y. Ouyang, R. Guo and Z. Yao, Chem. Rec., 2023, 23, e202200212 CAS.
- J. Chen, P. Cui, G. Zhao, K. Rui, M. Lao, Y. Chen, X. Zheng, Y. Jiang, H. Pan, S. X. Dou and W. Sun, Angew. Chem., Int. Ed., 2019, 58, 12540–12544 CrossRef CAS PubMed.
- Y. Kim, D. Jin, C. Lee and Y. Lee, J. Alloys Compd., 2022, 909, 164813 CrossRef CAS.
- Z. Liu, J. Qi, H. Zeng, Y. Zeng, J. Wang, L. Gu, E. Hong, M. Yang, Q. Fu, J. Chen and C. Yang, ACS Appl. Energy Mater., 2022, 5, 6869–6877 CrossRef CAS.
- J. Zhu, M. Wei, Q. Meng, Z. Chen, Y. Fan, S. W. Hasan, X. Zhang, D. Lyu, Z. Q. Tian and P. K. Shen, Nanoscale, 2020, 12, 24070–24078 RSC.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- L. She, G. Zhao, T. Ma, J. Chen, W. Sun and H. Pan, Adv. Funct. Mater., 2022, 32, 2108465 CrossRef CAS.
- W. Sun, Y. Song, X. Gong, L. Cao and J. Yang, Chem. Sci., 2015, 6, 4993–4999 RSC.
- J. Zhu, Z. Lyu, Z. Chen, M. Xie, M. Chi, W. Jin and Y. Xia, Chem. Mater., 2019, 31, 5867–5875 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- X. Kong and Z. Peng, Mater. Today Chem., 2019, 11, 119–132 CrossRef CAS.
- F. Gao, Y. Zhang, Z. Wu, H. You and Y. Du, Coord. Chem. Rev., 2021, 436, 213825 CrossRef CAS.
- L. Fu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 24836–24841 RSC.
- T. Kwon, H. Hwang, Y. J. Sa, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2017, 27, 1604688 CrossRef.
- A. Hartig Weiss, M. Miller, H. Beyer, A. Schmitt, A. Siebel, A. T. S. Freiberg, H. A. Gasteiger and H. A. ElSayed, ACS Appl. Nano Mater., 2020, 3, 2185–2196 CrossRef CAS.
- Y. Bao and L. Feng, Acta Phys.-Chim. Sin., 2021, 37, 2008031 Search PubMed.
- J. K. Nørskov, T. Bligaard, B. Hvolbæk, F. Abild Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC.
- H. Wang and J. Lu, Chin. J. Chem., 2020, 38, 1422–1444 CrossRef CAS.
- Y. Bao, M. Zha, P. Sun, G. Hu and L. Feng, J. Energy Chem., 2021, 59, 748–754 CrossRef CAS.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- G. C. Bond, Surf. Sci., 1985, 156, 966–981 CrossRef CAS.
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas and P. P. Edwards, Chem. – Eur. J., 2002, 8, 28–35 CrossRef CAS PubMed.
- W. P. Halperin, Rev. Mod. Phys., 1986, 58, 533–606 CrossRef CAS.
- T. Zhang, S. Liao, L. Dai, J. Yu, W. Zhu and Y. Zhang, Sci. China Mater., 2018, 61, 926–938 CrossRef CAS.
- J. Yin, J. Jin, M. Lu, B. Huang, H. Zhang, Y. Peng, P. Xi and C. Yan, J. Am. Chem. Soc., 2020, 142, 18378–18386 CrossRef CAS PubMed.
- D. Lebedev and C. Copéret, ACS Appl. Energy Mater., 2019, 2, 196–200 CrossRef CAS.
- W. Gao, Q. Xu, Z. Wang, M. Wang, X. Ren, G. Yuan and Q. Wang, Electrochim. Acta, 2020, 337, 135738 CrossRef CAS.
- C. Cai, S. Han and Y. Tang, Sustainable Energy Fuels, 2020, 4, 2462–2468 RSC.
- L. Fu, X. Zeng, C. Huang, P. Cai, G. Cheng and W. Luo, Inorg. Chem. Front., 2018, 5, 1121–1125 RSC.
- L. Yin, T. Yang, X. Ding, M. He, W. Wei, T. Yu and H. Zhao, Electrochem. Commun., 2018, 94, 59–63 CrossRef CAS.
- L. Yi, B. Feng, N. Chen, W. Li, J. Li, C. Fang, Y. Yao and W. Hu, Chem. Eng. J., 2021, 415, 129034 CrossRef CAS.
- H. Hu, F. M. D. Kazim, Z. Ye, Y. Xie, Q. Zhang, K. Qu, J. Xu, W. Cai, S. Xiao and Z. Yang, J. Mater. Chem. A, 2020, 8, 20168–20174 RSC.
- J. Cheng, J. Yang, S. Kitano, G. Juhasz, M. Higashi, M. Sadakiyo, K. Kato, S. Yoshioka, T. Sugiyama, M. Yamauchi and N. Nakashima, ACS Catal., 2019, 9, 6974–6986 CrossRef CAS.
- J. Xu, Z. Lian, B. Wei, Y. Li, O. Bondarchuk, N. Zhang, Z. Yu, A. Araujo, I. Amorim, Z. Wang, B. Li and L. Liu, ACS Catal., 2020, 10, 3571–3579 CrossRef CAS.
- F. Luo, H. Hu, X. Zhao, Z. Yang, Q. Zhang, J. Xu, T. Kaneko, Y. Yoshida, C. Zhu and W. Cai, Nano Lett., 2020, 20, 2120–2128 CrossRef CAS PubMed.
- Z. Zhang, Y. Xia, M. Ye, D. Wen, W. Zhang, W. Peng, L. Tian and W. Hu, Int. J. Hydrogen Energy, 2022, 47, 13371–13385 CrossRef CAS.
- W. Zhao, F. Xu, Z. Wang, Z. Pan, Y. Ye, S. Hu, B. Weng and R. Zhu, Small, 2022, 18, 2205495 CrossRef CAS PubMed.
- L. Li, P. Wang, Z. Cheng, Q. Shao and X. Huang, Nano Res., 2022, 15, 1087–1093 CrossRef CAS.
- W. Gou, Z. Xia, X. Tan, Q. Xue, F. Ye, S. Dai, M. Zhang, R. Si, Y. Zou, Y. Ma, J. C. Ho and Y. Qu, Nano Energy, 2022, 104, 107960 CrossRef CAS.
- S. Shin, T. Kwon, K. Kim, M. Kim, M. H. Kim and Y. Lee, ACS Appl. Energy Mater., 2022, 5, 6146–6154 CrossRef CAS.
- F. Luo, L. Guo, Y. Xie, J. Xu, K. Qu and Z. Yang, Appl. Catal., B, 2020, 279, 119394 CrossRef CAS.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, P. M. Shanthi, S. Tan, K. Damodaran and P. N. Kumta, ACS Catal., 2019, 9, 2134–2157 CrossRef CAS.
- J. Zhu, Q. Xue, Y. Xue, Y. Ding, F. Li, P. Jin, P. Chen and Y. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 14064–14070 CrossRef CAS PubMed.
- J. Lim, G. Kang, J. W. Lee, S. S. Jeon, H. Jeon, P. W. Kang and H. Lee, J. Power Sources, 2022, 524, 231069 CrossRef CAS.
- D. Yu, Q. Liu, B. Chen, Y. Zhao, P. Jia, K. Sun and F. Gao, J. Mater. Chem. A, 2022, 10, 11354–11362 RSC.
- M. Liu, S. Liu, Q. Mao, S. Yin, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 2021–2026 RSC.
- B. Jiang, J. Kim, Y. Guo, K. C. W. Wu, S. M. Alshehri, T. Ahamad, N. Alhokbany, J. Henzie and Y. Yamachi, Catal. Sci. Technol., 2019, 9, 3697–3702 RSC.
- L. Zu, X. Qian, S. Zhao, Q. Liang, Y. E. Chen, M. Liu, B. Su, K. Wu, L. Qu, L. Duan, H. Zhan, J. Zhang, C. Li, W. Li, J. Y. Juang, J. Zhu, D. Li, A. Yu and D. Zhao, J. Am. Chem. Soc., 2022, 144, 2208–2217 CrossRef CAS PubMed.
- H. Chen, L. Shi, K. Sun, K. Zhang, Q. Liu, J. Ge, X. Liang, B. Tian, Y. Huang, Z. Shi, Z. Wang, W. Zhang, M. Liu and X. Zou, ACS Catal., 2022, 12, 8658–8666 CrossRef CAS.
- M. You, L. Gui, X. Ma, Z. Wang, Y. Xu, J. Zhang, J. Sun, B. He and L. Zhao, Appl. Catal., B, 2021, 298, 120562 CrossRef CAS.
- A. L. Strickler, R. A. Flores, L. A. King, J. K. Nørskov, M. Bajdich and T. F. Jaramillo, ACS Appl. Mater. Interfaces, 2019, 11, 34059–34066 CrossRef CAS PubMed.
- V. Latyshev, S. Vorobiov, R. Bodnarova, O. Shylenko, M. Lisnichuk, A. Kovalcikova, M. Gregor and V. Komanicky, Electrochim. Acta, 2021, 398, 139248 CrossRef CAS.
- Y. Ying, J. F. Godínez Salomón, L. LartundoRojas, A. Moreno, R. Meyer, C. A. Damin and C. P. Rhodes, Nanoscale Adv., 2021, 3, 1976–1996 RSC.
- A. W. Jensen, G. W. Sievers, K. D. Jensen, J. Quinson, J. A. ArminioRavelo, V. Brüser, M. Arenz and M. EscuderoEscribano, J. Mater. Chem. A, 2020, 8, 1066–1071 RSC.
- H. Tian, W. Zhu, Q. Shi, S. Ding, Z. Lyu, M. Xu, X. Pan, M. H. Engelhard, D. Dan and Y. Lin, J. Mater. Chem. A, 2022, 10, 11196–11204 RSC.
- Y. Zhao, M. Luo, S. Chu, M. Peng, B. Liu, Q. Wu, P. Liu, F. M. F. de Groot and Y. Tan, Nano Energy, 2019, 59, 146–153 CrossRef CAS.
- M. Aizaz Ud Din, S. Irfan, S. U. Dar and S. Rizwan, Chem. – Eur. J., 2020, 26, 5662–5666 CrossRef CAS PubMed.
- X. Bao, S. Li, C. Hao, Y. Qin, Y. Gong, Y. Yang, A. Xu and M. Luo, J. Mater. Chem. A, 2022, 10, 20005–20010 RSC.
- L. Chen, F. He, R. Shao, Q. Yan, P. Yin, W. Zeng, M. Zuo, L. He and H. Liang, Nano Res., 2022, 15, 1853–1860 CrossRef CAS.
- Z. Lu, C. Wei, X. Liu, Y. Fang, X. Hao, Y. Zang, Z. Pei, J. Cai, Y. Wu, D. Niu, A. Mosallanezhad, D. Sun, J. Ye, S. Niu and G. Wang, Mater. Chem. Front., 2021, 5, 6092–6100 RSC.
- J. Sun, R. Zhao, X. Niu, M. Xu, Z. Xu, Y. Qin, W. Zhao, X. Yang, Y. Han and Q. Wang, Electrochim. Acta, 2022, 432, 141199 CrossRef CAS.
- S. Wang, S. Yang, Z. Wei, Y. Liang, J. Zhu, Y. Tang and X. Qiu, J. Mater. Chem. A, 2022, 10, 25556–25563 RSC.
- H. Jin, Y. Hong, J. Yoon, A. Oh, N. K. Chaudhari, H. Baik, S. H. Joo and K. Lee, Nano Energy, 2017, 42, 17–25 CrossRef CAS.
- J. Park, Y. J. Sa, H. Baik, T. Kwon, S. H. Joo and K. Lee, ACS Nano, 2017, 11, 5500–5509 CrossRef CAS PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- L. Wang, H. Liu, J. Zhuang and D. Wang, Small Sci., 2022, 2, 2200036 CrossRef CAS.
- L. Chen and H. Liang, Catal. Sci. Technol., 2021, 11, 4673–4689 RSC.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- T. Zhang, S. Li, W. Zhu, Z. Zhang, J. Gu and Y. Zhang, Nanoscale, 2017, 9, 1154–1165 RSC.
- D. Li, H. Liu and L. Feng, Energy Fuels, 2020, 34, 13491–13522 CrossRef CAS.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- J. Li, S. Wang, J. Chang and L. Feng, Adv. Powder Mater., 2022, 1, 100030 CrossRef.
- Z. Liu, D. Liu, L. Zhao, J. Tian, J. Yang and L. Feng, J. Mater. Chem. A, 2021, 9, 7750–7758 RSC.
- M. EscuderoEscribano, A. VerdaguerCasadevall, P. Malacrida, U. Grønbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Gronbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 RSC.
- C. L. Green and A. Kucernak, J. Phys. Chem. B, 2002, 106, 1036–1047 CrossRef CAS.
- X. Gu, Z. Liu, M. Li, J. Tian and L. Feng, Appl. Catal., B, 2021, 297, 120462 CrossRef CAS.
- K. Jiang, M. Luo, M. Peng, Y. Yu, Y. Lu, T. Chan, P. Liu, F. M. F. de Groot and Y. Tan, Nat. Commun., 2020, 11, 2701 CrossRef CAS PubMed.
- S. Ma, J. Deng, Y. Xu, W. Tao, X. Wang, Z. Lin, Q. Zhang, L. Gu and W. Zhong, J. Energy Chem., 2022, 66, 560–565 CrossRef CAS.
- J. Li, S. Wang, S. Sun, X. Wu, B. Zhang and L. Feng, J. Mater. Chem. A, 2022, 10, 9308–9326 RSC.
- R. K. Pittkowski, D. F. Abbott, R. Nebel, S. Divanis, E. Fabbri, I. E. Castelli, T. J. Schmidt, J. Rossmeisl and P. Krtil, Electrochim. Acta, 2021, 366, 137327 CrossRef CAS.
- F. Wang, C. Zhang and H. Yang, J. Energy Chem., 2022, 70, 623–629 CrossRef CAS.
- Y. Zhou, Q. Wang, X. Tian, J. Chang and L. Feng, J. Energy Chem., 2022, 75, 46–54 CrossRef CAS.
- Z. Li, X. Wu, X. Jiang, B. Shen, Z. Teng, D. Sun, G. Fu and Y. Tang, Adv. Powder Mater., 2022, 1, 100020 CrossRef.
- Y. Zhou, Y. Kuang, G. Hu, X. Wang and L. Feng, Mater. Today Phys., 2022, 27, 100831 CrossRef CAS.
- X. Gu, Y. Ji, J. Tian, X. Wu and L. Feng, Chem. Eng. J., 2022, 427, 131576 CrossRef CAS.
- H. Liu, Z. Liu, F. Wang and L. Feng, Chem. Eng. J., 2020, 397, 125507 CrossRef CAS.
- Y. Zhou, H. Liu, X. Gu, X. Wu and L. Feng, Carbon Energy, 2022, 4, 924–938 CrossRef CAS.
- Y. Xie, Y. Su, H. Qin, Z. Cao, H. Wei, F. Wu and G. Ou, Int. J. Hydrogen Energy, 2023, 48, 14642–14649 CrossRef CAS.
- Y. Liu, L. Cai, Q. Ji, C. Wang, Z. Liu, L. Lv, B. Tang, H. Duan, F. Hu, H. Wang, N. Li, Z. Sun and W. Yan, ACS Energy Lett., 2022, 7, 3798–3806 CrossRef CAS.
- R. Zhao, Z. Wang, Q. Xu, X. Niu, Y. Han, Y. Qin and Q. Wang, Electrochim. Acta, 2021, 391, 138955 CrossRef CAS.
- R. He, C. Wang and L. Feng, Chin. Chem. Lett., 2023, 34, 107241 CrossRef CAS.
- C. Ma, W. Sun, W. Qamar Zaman, Z. Zhou, H. Zhang, Q. Shen, L. Cao and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 34980–34989 CrossRef CAS PubMed.
- L. Hou, H. Jang, H. Liu, Z. Li, M. G. Kim, Q. Qin and X. Liu, ACS Sustainable Chem. Eng., 2022, 10, 15950–15957 CrossRef CAS.
- L. Moriau, G. Koderman Podboršek, A. K. Surca, S. Semsari Parpari, M. Šala, U. Petek, M. Bele, P. Jovanovič, B. Genorio and N. Hodnik, Adv. Mater. Interfaces, 2021, 8, 2100900 CrossRef CAS.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- J. Sun, N. Guo, T. Song, Y.-R. Hao, J. Sun, H. Xue and Q. Wang, Adv. Powder Mater., 2022, 1, 100023 CrossRef.
- Y. Zhou, L. Yu, J. Chang, L. Feng and J. Zhang, Green Energy Environ., 2023 DOI:10.1016/j.gee.2022.08.007.
- W. Xu, H. Huang, X. Wu, Y. Yuan, Y. Liu, Z. Wang, D. Zhang, Y. Qin, J. Lai and L. Wang, Composites, Part B, 2022, 242, 110013 CrossRef CAS.
- D. He, L. Cao, L. Feng, S. Li, Y. Feng, G. Li, Y. Zhang, J. Li and J. Huang, Chin. Chem. Lett., 2022, 33, 4781–4785 CrossRef CAS.
- C. Chang, Y. Xie, F. Luo, S. Zhou and Z. Yang, Appl. Catal., A, 2023, 649, 118941 CrossRef CAS.
- H. Zhu, Z. Zhu, J. Hao, S. Sun, S. Lu, C. Wang, P. Ma, W. Dong and M. Du, Chem. Eng. J., 2022, 431, 133251 CrossRef CAS.
- X. Liu, S. Xi, H. Kim, A. Kumar, J. Lee, J. Wang, N. Q. Tran, T. Yang, X. Shao, M. Liang, M. G. Kim and H. Lee, Nat. Commun., 2021, 12, 5676 CrossRef CAS PubMed.
- G. Li, X. Xu, H. Liu, X. Yang and M. Lin, ChemCatChem, 2022, 14, e202201039 CAS.
- S. Wang, H. Lv, S. Bi, T. Li, Y. Sun, W. Ji, C. Feng and C. Zhang, Mater. Chem. Front., 2021, 5, 8047–8055 RSC.
- C. Wang, A. Schechter and L. Feng, Nano Res. Energy, 2023, 2, e9120056 CrossRef.
- Z. Yu, J. Xu, Y. Li, B. Wei, N. Zhang, Y. Li, O. Bondarchuk, H. Miao, A. Araujo, Z. Wang, J. L. Faria, Y. Liu and L. Liu, J. Mater. Chem. A, 2020, 8, 24743–24751 RSC.
- Y. Wei, A. Liao, Y. Zhou and Z. Zou, Mater. Today Phys., 2021, 16, 100317 CrossRef CAS.
- W. Sun, C. Ma, X. Tian, J. Liao, J. Yang, C. Ge and W. Huang, J. Mater. Chem. A, 2020, 8, 12518–12525 RSC.
- C. Pei, H. Chen, B. Dong, X. Yu and L. Feng, J. Power Sources, 2019, 424, 131–137 CrossRef CAS.
- Z. Sun, E. Luo, Q. Meng, X. Wang, J. Ge, C. Liu and W. Xing, Acta Phys.-Chim. Sin., 2022, 38, 2003035 Search PubMed.
- Z. Lu, Y. Shi, P. Gupta, X. Min, H. Tan, Z. Wang, C. Guo, Z. Zou, H. Yang, S. Mukerjee and C. Yan, Electrochim. Acta, 2020, 348, 136302 CrossRef CAS.
- W. Qiao, X. Huang and L. Feng, Chin. J. Struct. Chem., 2022, 41, 2207016–2207034 CAS.
- E. J. Kim, J. Shin, J. Bak, S. J. Lee, K. H. Kim, D. Song, J. Roh, Y. Lee, H. Kim, K. S. Lee and E. Cho, Appl. Catal., B, 2021, 280, 119433 CrossRef CAS.
- X. Yang, J. Xue and L. Feng, Chem. Commun., 2019, 55, 11247–11250 RSC.
- C. Massué, V. Pfeifer, X. Huang, J. Noack, A. Tarasov, S. Cap and R. Schlögl, ChemSusChem, 2017, 10, 1943–1957 CrossRef PubMed.
- J. Wang, J. Kim, S. Choi, H. Wang and J. Lim, Small Methods, 2020, 4, 2000621 CrossRef CAS.
- J. Huang, S. B. Scott, I. Chorkendorff and Z. Wen, ACS Catal., 2021, 11, 12745–12753 CrossRef CAS.
- J. Sun, S. E. Lowe, L. Zhang, Y. Wang, K. Pang, Y. Wang, Y. Zhong, P. Liu, K. Zhao, Z. Tang and H. Zhao, Angew. Chem., Int. Ed., 2018, 57, 16511–16515 CrossRef CAS PubMed.
- L. Najafi, S. Bellani, R. OropesaNuñez, M. Prato, B. MartínGarcía, R. Brescia and F. Bonaccorso, ACS Nano, 2019, 13, 3162–3176 CrossRef CAS PubMed.
- I. S. Filimonenkov, C. Bouillet, G. Kéranguéven, P. A. Simonov, G. A. Tsirlina and E. R. Savinova, Electrochim. Acta, 2019, 321, 134657 CrossRef CAS.
- A. MartínezSéptimo, M. A. Valenzuela, P. Del Angel and R. D. G. GonzálezHuerta, Int. J. Hydrogen Energy, 2021, 46, 25918–25928 CrossRef.
- L. Wang, F. Song, G. Ozouf, D. Geiger, T. Morawietz, M. Handl, P. Gazdzicki, C. Beauger, U. Kaiser, R. Hiesgen, A. S. Gago and K. A. Friedrich, J. Mater. Chem. A, 2017, 5, 3172–3178 RSC.
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS.
- X. Kang and M. Zhu, Small, 2019, 15, 1902703 CrossRef CAS PubMed.
- L. Zhang, J. Liang, L. Yue, K. Dong, J. Li, D. Zhao, Z. Li, S. Sun, Y. Luo, Q. Liu, G. Cui, A. Ali Alshehri, X. Guo and X. Sun, Nano Res. Energy, 2022, 1, 9120028 CrossRef.
- L. Zhuang, F. Xu, K. Wang, J. Li, C. Liang, W. Zhou, Z. Xu, Z. Shao and Z. Zhu, Small, 2021, 17, 2100121 CrossRef CAS PubMed.
- K. Zhang, W. Mai, J. Li, H. Wang, G. Li and W. Hu, J. Mater. Sci., 2020, 55, 3507–3520 CrossRef CAS.
- Y. Yao, S. Hu, W. Chen, Z. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- P. Li, X. Duan, Y. Kuang and X. Sun, Small, 2021, 17, 2102078 CrossRef CAS PubMed.
- K. Sun, Y. Zhao, J. Yin, J. Jin, H. Liu and P. Xi, Acta Phys.-Chim. Sin., 2021, 38, 2107005 Search PubMed.
- A. L. Kozlovskiy and M. V. Zdorovets, Composites, Part B, 2020, 191, 107968 CrossRef CAS.
- T. Kwon, A. Yu, S. J. Kim, M. H. Kim, C. Lee and Y. Lee, Appl. Surf. Sci., 2021, 563, 150293 CrossRef CAS.
- Z. Liu, Y. Liu, S. Liu, S. Liu, Y. Tang and Z. Tang, Sustainable Energy Fuels, 2022, 6, 3649–3657 RSC.
- M. Li and L. Feng, Chin. J. Struct. Chem., 2022, 41, 2201019–2201024 CAS.
- W. Qiao, X. Yang, M. Li and L. Feng, Nanoscale, 2021, 13, 6884–6889 RSC.
- Y. Li, Y. Wang, J. Lu, B. Yang, X. San and Z. Wu, Nano Energy, 2020, 78, 105185 CrossRef CAS.
- X. Li, Q. Hu, H. Yang, T. Ma, X. Chai and C. He, Chin. Chem. Lett., 2022, 33, 3657–3671 CrossRef CAS.
- B. Jiang, Y. Guo, J. Kim, A. E. Whitten, K. Wood, K. Kani, A. E. Rowan, J. Henzie and Y. Yamauchi, J. Am. Chem. Soc., 2018, 140, 12434–12441 CrossRef CAS PubMed.
- L. Yang, K. Zhang, H. Chen, L. Shi, X. Liang, X. Wang, Y. Liu, Q. Feng, M. Liu and X. Zou, J. Energy Chem., 2022, 66, 619–627 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- Y. Pi, N. Zhang, S. Guo, J. Guo and X. Huang, Nano Lett., 2016, 16, 4424–4430 CrossRef CAS PubMed.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef PubMed.
- X. Lin, Y. Hu, K. Hu, X. Lin, G. Xie, X. Liu, K. M. Reddy and H.-J. Qiu, ACS Mater. Lett., 2022, 4, 978–986 CrossRef CAS.
- J. Xie, X. Zhang, N. Yu, R. Luan, D. Zhang, J. Zeng, Y. Chai and B. Dong, Mater. Today Phys., 2022, 27, 100778 CrossRef CAS.
- S. Yan, M. Zhong, W. Zhu, W. Li, X. Chen, M. Li, C. Wang and X. Lu, Inorg. Chem. Front., 2022, 9, 6225–6236 RSC.
- J. Yan, F. Ye, Q. Dai, X. Ma, Z. Fang, L. Dai and C. Hu, Nano Res. Energy, 2023, 2, e9120047 CrossRef.
- W. Hu, H. Zhong, W. Liang and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 12729–12736 CrossRef CAS PubMed.
- S. Chatterjee, S. Intikhab, L. Profitt, Y. Li, V. Natu, R. Gawas and J. Snyder, J. Catal., 2021, 393, 303–312 CrossRef CAS.
- W. Zhao, D. Huang, Q. Yuan and X. Wang, Nano Res., 2016, 9, 3066–3074 CrossRef CAS.
- C. Shang, Y. Guo and E. Wang, Nano Res., 2018, 11, 4348–4355 CrossRef CAS.
- W. Zhong, Z. Lin, S. Feng, D. Wang, S. Shen, Q. Zhang, L. Gu, Z. Wang and B. Fang, Nanoscale, 2019, 11, 4407–4413 RSC.
- M. Sung and J. Kim, Int. J. Hydrogen Energy, 2018, 43, 2130–2138 CrossRef CAS.
- Q. Li, F. Sun, D. Zhang, H. Sun, Q. Wang, J. Qi, H. Wang, Z. Li, Z. Hu and B. Wang, Chem. Eng. J., 2023, 452, 139232 CrossRef CAS.
- Y. Z. Wang, M. Yang, Y. Ding, N. Li and L. Yu, Adv. Funct. Mater., 2022, 32, 2108681 CrossRef CAS.
- G. Meng, W. Sun, A. A. Mon, X. Wu, L. Xia, A. Han, Y. Wang, Z. Zhuang, J. Liu, D. Wang and Y. Li, Adv. Mater., 2019, 31, 1903616 CrossRef PubMed.
- Y. Zheng, F. Zhang, G. Wang, D. Lai, L. Zou, Q. Cheng, J. Li, Z. Zou and H. Yang, J. Power Sources, 2022, 528, 231189 CrossRef CAS.
- J. Zhu, M. Xie, Z. Chen, Z. Lyu, M. Chi, W. Jin and Y. Xia, Adv. Energy Mater., 2020, 10, 1904114 CrossRef CAS.
- T. Kwon, M. Jun and K. Lee, Adv. Mater., 2020, 32, 2001345 CrossRef CAS PubMed.
- L. Zhang, H. Jang, Z. Li, H. Liu, M. G. Kim, X. Liu and J. Cho, Chem. Eng. J., 2021, 419, 129604 CrossRef CAS.
- I. Boshnakova, E. Lefterova and E. Slavcheva, Int. J. Hydrogen Energy, 2018, 43, 16897–16904 CrossRef CAS.
- M. Fathi Tovini, A. M. Damjanovic, H. A. El-Sayed, J. Speder, C. Eickes, J. Suchsland, A. Ghielmi and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 064521 CrossRef.
- I. G. Kim, A. Lim, J. H. Jang, K. Y. Lee, I. W. Nah and S. Park, J. Power Sources, 2021, 501, 230002 CrossRef CAS.
- J. Xu, J. Li, Z. Lian, A. Araujo, Y. Li, B. Wei, Z. Yu, O. Bondarchuk, I. Amorim, V. Tileli, B. Li and L. Liu, ACS Catal., 2021, 11, 3402–3413 CrossRef CAS.
- Z. Shi, J. Li, J. Jiang, Y. Wang, X. Wang, Y. Li, L. Yang, Y. Chu, J. Bai, J. Yang, J. Ni, Y. Wang, L. Zhang, Z. Jiang, C. Liu, J. Ge and W. Xing, Angew. Chem., Int. Ed., 2022, 61, e202212341 CAS.
- S. W. Lee, C. Baik, D. Kim and C. Pak, J. Power Sources, 2021, 493, 229689 CrossRef CAS.
- H. S. Oh, H. N. Nong, T. Reier, M. Gliech and P. Strasser, Chem. Sci., 2015, 6, 3321–3328 RSC.
| This journal is © The Royal Society of Chemistry 2023 |