
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally-driven reactivity of Li0.35La0.55TiO3 solid electrolyte with LiCoO2 cathode†
Subhash
Chandra‡
 a,
Younggyu
Kim‡
a,
Daniele
Vivona
b,
Iradwikanari
Waluyo
c,
Adrian
Hunt
c,
Christoph
Schlueter
d,
Jeong Beom
Lee
e,
Yang
Shao-Horn
abf and
Bilge
Yildiz
*ag
a,
Younggyu
Kim‡
a,
Daniele
Vivona
b,
Iradwikanari
Waluyo
c,
Adrian
Hunt
c,
Christoph
Schlueter
d,
Jeong Beom
Lee
e,
Yang
Shao-Horn
abf and
Bilge
Yildiz
*ag
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: byildiz@mit.edu
bDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
cNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, New York 11973, USA
dPhoton Science, Deutsches Elektronen-Synchrotron DESY, Notkestr. 85, 22607 Hamburg, Germany
eLG Energy Solution, Ltd, LG Science Park, Magokjungang 10-ro, Gangseo-gu, Seoul 07796, Korea
fResearch Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
gDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
First published on 3rd February 2022
Abstract
A critical bottleneck for developing successful solid-state batteries is minimizing the interfacial impedances between the solid electrolyte and the active electrode materials. Advancing our understanding of the chemically and electro-chemically induced reactions of the cathode–electrolyte interface is important to achieve stable interfaces with desired charge transfer properties. The perovskite Li-solid electrolyte with the general formula Li3xLa2/3−x□1/3−2xTiO3 (LLTO) is an interesting candidate for solid-state Li-ion cells due to its superionic Li-ion conductivity. However, the chemical compatibility of LLTO with potential cathode materials is not sufficiently understood, and here we examine the LLTO interface with thin-film LiCoO2 (LCO) cathode layers. We show that elemental mixing, in particular Co diffusion into LLTO, and increased structural ordering of the LCO occurs at moderate temperatures, on-setting around 300 °C in air, even in the absence of any secondary phases. This reaction decreases the LCO|LLTO interface charge transfer resistance by almost 50% after annealing to 500 °C relative to the as-deposited state. Reduction of the interface charge transfer resistance can be attributed to the formation of a mixed electronic and ionic conducting zone near the interface upon Co diffusion into LLTO. This finding can guide the engineering of cathode–solid electrolyte interfaces with fast charge transfer kinetics.
1. Introduction
The current Li-ion technology which is based on organic liquid electrolytes has been highly successful with wide applications ranging from small portable electronics, electric vehicles (EV) to stationary energy storage.1,2 The current state-of-the-art commercial Li-ion batteries however face significant challenges of safety, and limited energy and power density concerning growing consumer demand.3 One of the main causes of safety concerns is the low flashpoint of carbonates in organic electrolytes, which causes flammability issues.4,5 Inorganic solid-state electrolytes have received increased interest as a potential solution to these safety issues because they are non-flammable. Moreover, employing solid-state electrolytes is seen as a promising strategy to enable high energy density lithium metal anodes,6 offering high energy and power densities to support applications such as electric vehicles.7,8 Although there has been considerable progress in the development of superionic solid-state Li-ion conductors (conductivity > 0.1 mS cm−1),9 the interfacial instability of electrolyte materials with anode and cathode materials presents a major challenge. The instability issues of the interfaces have been broadly discussed in recent reviews,10–14 and it is widely accepted that the high impedance arising from the cathode–electrolyte interface, due to thermally or electrochemically induced degradation of this interface, is one of the main bottlenecks for developing successful solid-state Li-ion cells. The application of coatings on cathode particles to prevent cathode–electrolyte chemical reaction is one of the strategies to address the issue,10 however, the proposed coating compositions have limited Li-ion conductivity.15 This challenge motivates us to investigate the cathode–electrolyte interface interaction and degradation mechanisms and their impact on charge transfer kinetics. A fundamental understanding of the complex phenomena occurring at the electrode–electrolyte interface could help guide the design of next generation safe and energy dense Li-ion batteries with long cycle life. In this respect, our previous works have probed the onset of thermally-driven degradation of the garnet structured Al-doped Li7La3Zr2O12 solid electrolyte with layered transition metal oxide cathode interfaces, with implications to electrochemical properties.16,17 In particular, we have found that carbon dioxide present in the environment has a detrimental effect on interface charge transfer kinetics due to the formation of lithium carbonate and other delithiated insulating secondary phases at the interface. At the same time, the local change in the structure and chemistry at the interface begins at temperatures as low as 500 °C, well-below typical ceramic sintering temperatures needed for bonding at the interface.Here we focus our attention on a promising ceramic solid electrolyte, the perovskite structured Li3xLa2/3−x□1/3−2xTiO3 (0 < x < 0.16) (LLTO). LLTO has attracted attention since its first study as a lithium-ion conductor by Inaguma et al., in the 1990s.18,19 The LLTO with composition x = 0.11 has been reported to possess the highest Li-ion bulk conductivity of 1 mS cm−1 at room temperature.20 There are few reports on the use of LLTO as a solid electrolyte20–23 or as a coating material on layered oxide cathodes24–28 and on LLTO electrochemical stability.29,30 However, to the best of our knowledge the thermal or electrochemical interaction of LLTO and cathode material at the interface and its implication on charge transfer characteristics have not been fully assessed, yet.
Lithium Cobalt Oxide (LCO) has been chosen as a cathode material in our study. LCO was been first proposed as a cathode material in 1980,31 and is still one of the most studied and widely adopted cathode material.32–34 Recent computational studies on the LLTO–Lithium Cobalt Oxide (LCO) interface based on density functional theory (DFT) have predicted low decomposition energy (−0.5 to −1 meV per atom) with the formation of Co3O4, La2Ti2O7, Li2TiO3, Li0.5CoO2 phases as reaction products.9 These computational efforts based on DFT and grand potential phase diagram construction provide incredibly valuable first insights into the thermodynamic stability and synthesizability of materials.35 However, these tools are yet far from correctly predicting the behaviour of interfaces because they do not explicitly take into account (for example) physical effects such as interfacial contact, inhomogeneities and presence of space charge layers, which result in added complexity of structure and composition of the interface. Moreover, practical consideration of the environment, such as temperature and gas species involved in the synthesis, is still difficult to simulate and can lead to divergences between computational predictions and experimental observations. For example, in our recent work on LLZO–NMC and LLZO–LCO interface,16,17 we experimentally found that decomposition products deviate from earlier computationally predicted secondary phases.35–37 Hence, experimental probing of these interfaces can not only help to elucidate fundamental mechanisms but also detect decomposition onset conditions which are important for validating and benchmarking computational predictions.
In this work, we investigated how elevated temperatures affect the Lithium Lanthanum Titanium Oxide (Li0.35La0.55TiO3) (LLTO) electrolyte and Lithium Cobalt Oxide (LiCoO2) (LCO) cathode interface from a structural, chemical, and charge transfer kinetics standpoint. The temperature effect is important because sintering at elevated temperatures can provide good interfacial contact between the ceramic cathode and ceramic electrolyte materials to reduce the interface contact resistances. However, as we see from earlier work,16,17,38 this can lead to thermally-driven interactions between the solid electrolyte and cathode and insulating secondary phases at the interface. The challenge of probing the interface was addressed by using a thin-film cathode layer deposited on top of a dense LLTO electrolyte pellet. This configuration allowed us to elucidate the onset of reactions at the interface within the detection depth of X-ray absorption spectroscopy (XAS) and hard X-ray photoelectron spectroscopy (HAXPES) at relatively low temperatures (300 °C to 700 °C). We already employed a similar setup using a thin-film cathode in our previous recent work on the garnet structured Li7La3Zr2O12 solid electrolyte.16,17 We performed synchrotron X-ray absorption near-edge spectroscopy (XAS) to probe the structural and chemical change of LCO|LLTO interface and determine the onset of reaction. Grazing incidence X-ray diffraction (GIXD) was utilized to detect the crystalline decomposition phases at the interface. The elemental diffusion profile across the interface was determined by X-ray photoelectron spectroscopy (XPS). The effect of thermal pre-treatment on the interfacial charge transfer response was observed using electrochemical impedance spectroscopy (EIS). Our results show elemental mixing, specifically Co diffusion into LLTO at moderate temperatures (300 °C to 600 °C) in addition to increased crystallinity of the LCO layer, and reduction of cobalt at higher temperatures (700 °C). The LCO layer was found to be stable up to 600 °C in air. However, at 700 °C cobalt completely migrated into the LLTO bulk, resulting in the complete loss of the LCO layer on the LLTO surface. The impedance spectra show a decrease in interfacial resistance after annealing in air up to 500 °C, by almost 50% relative to the as-deposited state. This decrease arises likely as a result of enhanced bonding of LCO and LLTO, increased crystallinity of LCO, and formation of mixed conducting zone in LLTO near the LCO|LLTO interface.
2. Experiment design and methods
Thin-film LCO cathode layers deposited onto the LLTO pellet substrates were used as a model system to understand the interfacial structure, chemistry, and charge transfer kinetics. This allowed us to keep the reaction zone within the detection depth limit of XAS. The thickness of LCO was chosen considering the mean free path of photons to be 100 nm with 1 keV energy.39 The LCO (100 nm thick) was deposited at room temperature using radio frequency (RF) sputtering on LLTO pellets (≈1 mm thick and ≈10 mm diameter) that we have synthesized with composition Li0.35La0.55TiO3. The thin film LCO deposited LLTO was annealed at 300 °C, 500 °C, 600 °C and 700 °C for 4 hours in ambient air. LLTO was synthesized by solid-state reaction following procedure reported elsewhere40 with slight modification. The average grain-size of our synthesized LLTO pellets is expected to be ∼2 to 3 μm, given the similarity in the synthesis conditions (annealed at 1150 °C during synthesis with no post-processing) reported in literature.40 The LCO layer is expected to be nano-grain, considering the thickness of the LCO layer (∼100 nm). Since the LCO layer is amorphous in the as-deposited state, the crystallite size determination is not applicable. The detailed procedure for synthesis is given in ESI.† The results from X-ray diffraction (XRD) (Fig. S1†) and electrochemical impedance spectroscopy (EIS) (Fig. S2†) for synthesized LLTO are presented in ESI.†Grazing incidence X-ray diffraction (GIXD) with Cu Kα source was utilized to assess whether and which crystalline secondary phase form at the LCO|LLTO interface. Soft X-ray absorption spectroscopy (XAS) was leveraged to understand the chemical environment and oxidation state change of the elements near the interface. Soft XAS measurements were performed at the IOS (23-ID-2) beamline at the National Synchrotron Light Source II, Brookhaven National Laboratory. Partial fluorescence yield (PFY) spectra were obtained using an energy-discriminating Vortex EM silicon drift detector. The elemental diffusion profile was obtained by X-ray photoelectron spectroscopy (XPS) using soft X-ray (Al Kα) coupled with Ar-ion sputtering over a 2 mm by 2 mm area on the sample. The sputtering setting for the Ar-ion gun was set to 1 kV and 0.78 μA. The sample was rotated while sputtering to uniformly remove the material at the spot. The sample was initially sputtered for 5 minutes to get rid of most of the gold layer on top. Scans for Au 4f, Co 2p, C 1s, Ti 2p, and O 1s was collected after every three minutes of sputtering time.
We have also used hard X-ray photoelectron spectroscopy (HAXPES) at beamline P22 at PETRA III (Hamburg, Germany)41 as a non-destructive method of probing the elemental mixing at the interface. For this experiment, 20 nm LCO film was deposited on LLTO and further annealed at 300 °C, 500 °C and 600 °C for 4 hours in ambient air. The Co 2p and La 3d spectra were collected at 45° emission angle with photon energy of 6 keV for different thermally treated samples. The integrated intensity ratio was utilized to qualitatively discuss the relative change in elemental composition near the interface of LCO|LLTO. The sample configuration used for collection is shown in ESI (Fig. S4†).
The electrochemical response of the interface was analysed using electrochemical impedance spectroscopy (EIS). For this purpose, symmetric cells (Au|LCO|LLTO|LCO|Au) were constructed comprising of LCO (100 nm thick) on both sides of the LLTO pellet (≈1 mm thick and ≈10 mm diameter) followed by annealing and finally, Au deposition by RF sputtering (60 nm thick) as the current collector.
3. Results
3.1 Chemical environment and oxidation state obtained by XAS
Here, using X-ray absorption spectroscopy, we identify that LCO|LLTO interface undergoes structural ordering upon annealing up to 600 °C and significant chemical changes take place at 700 °C given by reduction of cobalt. The XAS spectra for Co L2,3-edge, Ti L2,3-edge, and O K-edge in partial fluorescence (PFY) mode are shown in Fig. 1 for the 100 nm LCO|LLTO sample after different annealing temperatures in air. As seen in the Co L2,3-edge XAS, there is a significant change in peak shape after annealing. The Co L3 became sharper progressively by annealing till 600 °C with respect to the as-deposited state. This is indicative of an increase in local ordering of the cobalt environment.42 Additionally, the crystal field splitting feature which is given by the t2g–eg energy difference43 became evident for annealed samples. The oxidation state obtained from the Co L2,3-edge XAS for as-deposited sample and for samples annealed at 500 °C and 600 °C is Co3+ low spin (LS) as it closely matches with spectra reported elsewhere.44 Co L3-edge shifted toward the lower energy after annealing at 700 °C, which implies the reduction of Co3+ to Co2+ at 700 °C. The shape of Co L3 and Co L2 edge changed quite significantly after annealing at 700 °C. The peak at 777.4 eV (feature 1) in the L3 spectra and presence of the shoulder at around 778.6 eV (feature 2) implies the formation of Co2+ high spin (Co2+ HS), similar to the Co L-edge spectra seen in other doped LaCoO3 materials.44 This observation suggests that Co has migrated to the B-site of the perovskite LLTO structure; which will be corroborated with the diffusion profile of Co into the LLTO by XPS depth profile presented later, in Section 3.3.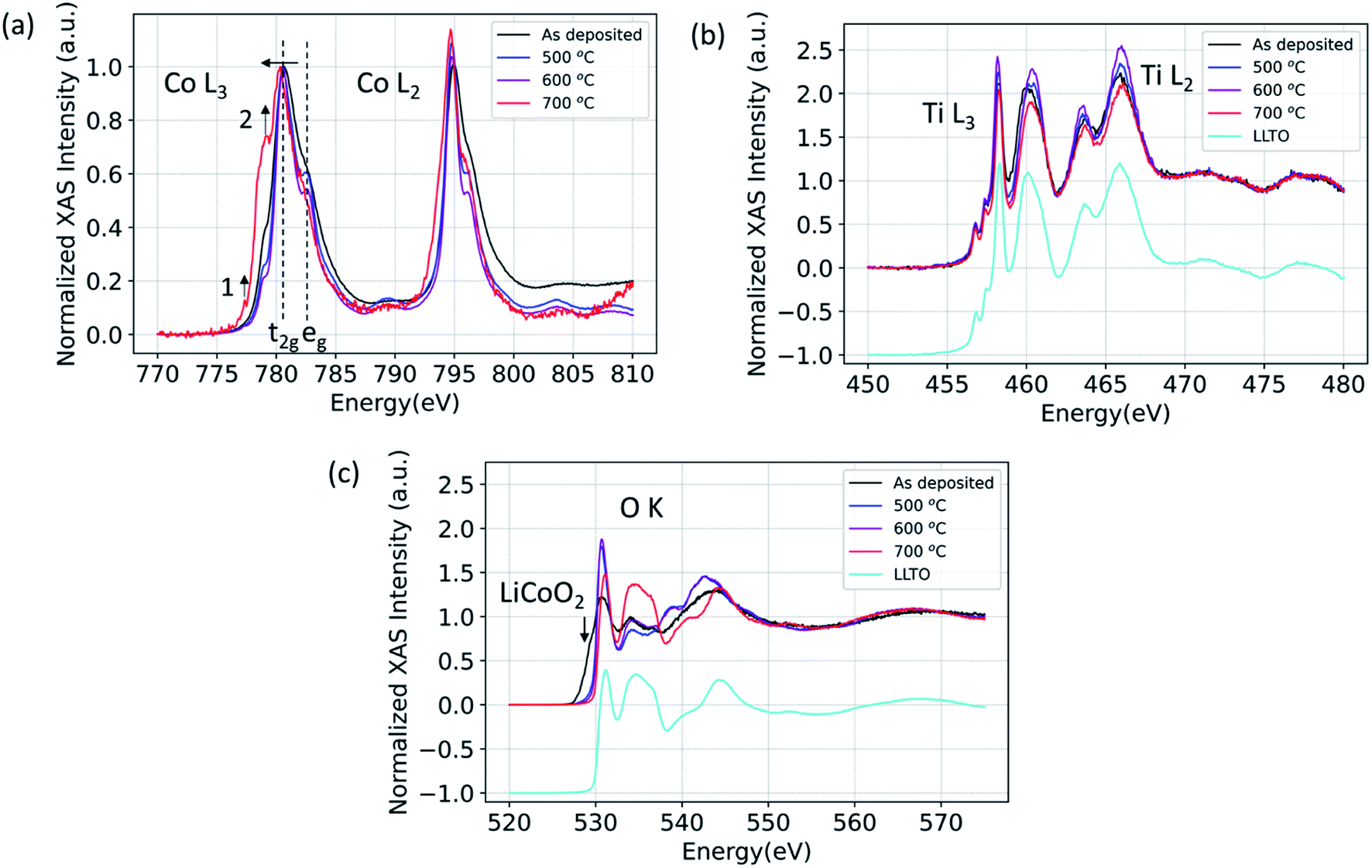 | ||
| Fig. 1 (a) Co L3 and Co L2, (b) Ti L3 and Ti L2, (c) O K X-ray absorption spectra for 100 nm LCO|LLTO for the as-deposited sample and for samples annealed at different temperatures in air. | ||
Fig. 1(b) shows the Ti L2,3-edge spectra. The position of Ti L-edge peaks for the as-deposited, 500 °C, 600 °C and 700 °C annealed samples did not change within the observable limit. This implies that the oxidation state of Ti does not change even for the 700 °C annealed sample. The O K-edge spectra for LCO|LLTO samples undergone different thermal treatment is presented along with the as-deposited LCO|LLTO sample in Fig. 1(c). The LiCoO2 pre-edge feature at about 529.5 eV (ref. 17) can be clearly identified for the as-deposited LCO|LLTO sample, and this feature abruptly decreases upon annealing at 500 °C and 600 °C. Interestingly, for the 700 °C annealed LCO|LLTO sample, the LiCoO2 feature is completely lost and the O K-edge spectra resemble the spectra collected from LLTO. Lithium carbonate (Li2CO3) which has peak at 534.1 eV (ref. 45) can be observed for the as-deposited sample and for sample annealed at 500 °C. However, it is difficult to discern Li2CO3 feature for the sample annealed at 600 °C and 700 °C. The implications for this are discussed in Section 4.
3.2 Grazing incidence X-ray diffraction
In order to resolve any phases that formed in the reaction zone of LCO|LLTO after thermal treatment, we performed the X-ray diffraction analysis with grazing incidence, with an incidence angle of 1°. As will be seen in detail below, we did not find any decomposition products in XRD irrespective of thermal treatments. Fig. 2 shows the diffraction data from samples treated with different annealing conditions. All the samples show peaks for the tetragonal (space group: P4/mmm) LLTO (ICDD: 01-070-6719),46 as we observed for the as-synthesized LLTO given in ESI (Fig. S1†). Additionally, we could identify the peaks from Au current collection layer from the top surface. The peaks from LCO from the as-deposited sample are missing because the deposited film at ambient temperature is expected to be non-crystalline. For the 300 °C annealed and 500 °C annealed samples, the annealing temperature is not sufficient to initiate long-range order in LCO on the LLTO surface, hence we did not observe diffraction peaks for these samples.47 The sample that had undergone 700 °C annealing treatment in the air also did not show the LiCoO2 diffraction peaks because of the loss of LCO layer on the LLTO electrolyte, as seen from the O K-edge spectrum and also as we discuss in the XPS depth profile analysis later. Interestingly, we did not observe any secondary phase formation in the X-ray diffraction. This observation means that the reaction zone either remained amorphous, or the secondary phase crystallites formed in the reaction zone are too low in quantity to be observed by lab XRD, or that secondary phases did not form. The stability of the LCO|LLTO mixed powders was investigated by Liao et al., and the structure was found stable till 700 °C without secondary phase formation, and at 800 °C β-LLTO was formed as a secondary phase.48 In another study, Co3O4 and La2Ti2O7 were found as secondary phases at 800 °C in system comprising of impregnated LCO in honeycomb LLTO.49 In this work, the important point to highlight is that, even if we do not observe formation of any decomposition products in XRD as a result of the thermal treatment, the LCO|LLTO interface has already interacted significantly due to diffusion of Co into LLTO.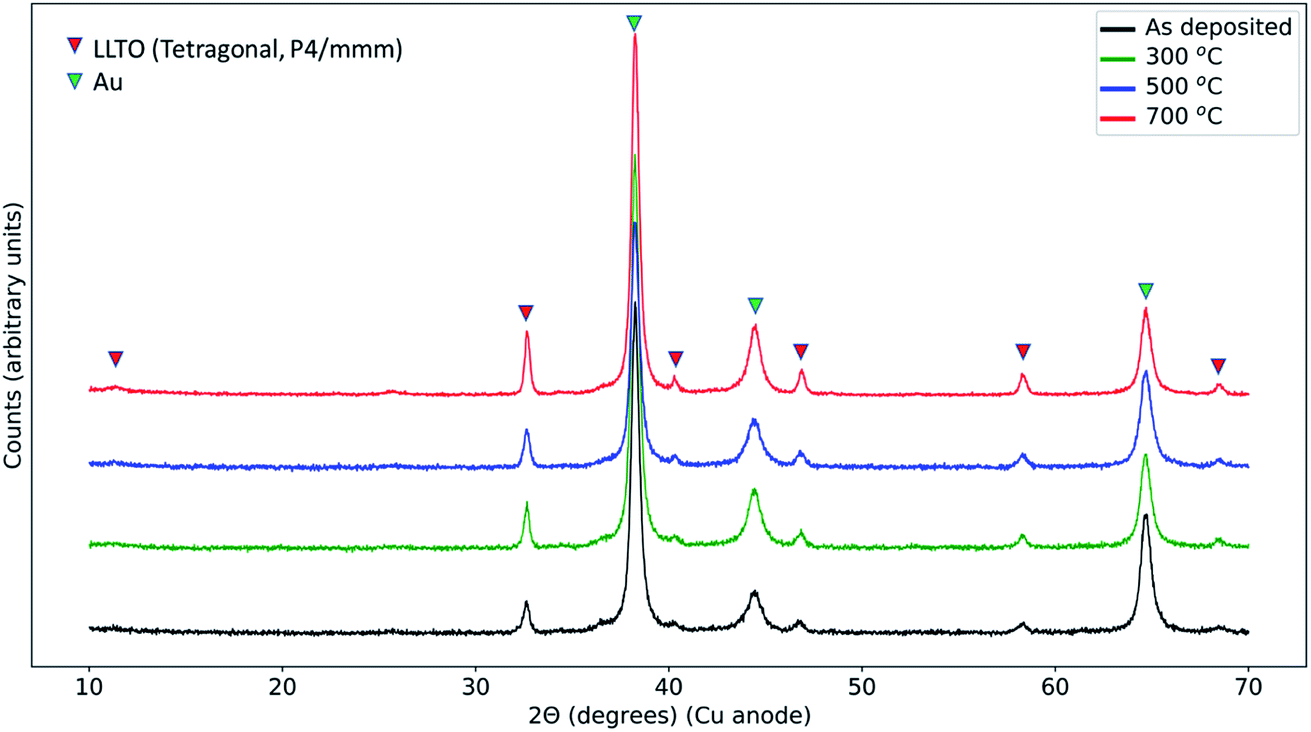 | ||
| Fig. 2 Grazing incidence X-ray diffraction (GIXD) pattern form Au|LCO|LLTO|LCO|Au samples for the as-deposited state, and after annealing at 300 °C, 500 °C, and 700 °C. Gold (Au) was deposited after the annealing as a current collector. | ||
3.3 Elemental depth profile by X-ray photoelectron spectroscopy
The objective in using XPS with depth profiling was to quantitatively resolve the elemental intermixing tendencies due to ion inter-diffusion as a function of annealing temperature. Here, X-ray photoelectron spectroscopy with both soft X-rays and hard X-rays revealed that elemental intermixing, in particular diffusion of Co into LLTO, occurs at the LCO|LLTO interface, onsetting at as low a temperature as 300 °C. Annealing at 700 °C leads to complete migration of Co into the LLTO resulting in loss of the LCO layer on LLTO.The same Au|LCO|LLTO|LCO|Au symmetric cell samples prepared for electrochemical impedance spectroscopy (EIS) measurement were utilized for elemental depth profile with soft X-rays (Al Kα). Fig. 3 shows the elemental composition as a function of sputtering time for the as-deposited, 500 °C, and 700 °C annealed samples. The top gold layer, followed by the LCO layer can be distinctively identified by observing the gold and cobalt composition profile for the as-deposited and 500 °C annealed samples. The zone where cobalt composition starts to deplete and titanium start to grow, i.e. at around 100 minutes of sputtering time, marks the LCO|LLTO interface. Based on this marker point, we estimate the Ar-ion sputtering rate to be approximately 14 Å min−1. A careful comparison of cobalt composition profile shows that after annealing at 500 °C the cobalt region broadened relative to the as-deposited sample. This profile indicates cobalt diffusion into LLTO at 500 °C. It is also important to note that the cobalt profile never completely decays in the shown sputtering time regime (Fig. 3(a and b)). We believe this is an artefact due to the destructive nature of the ion-milling, which also introduces inter-mixing in addition to sputtering off the surface layers. The sample annealed at 700 °C shows a drastic change in the Co profile. The LCO layer, which is clearly observed from the Co composition profile for the as-deposited and 500 °C annealed samples, is completely absent for the 700 °C annealed sample. However, we did observe an almost constant and very low Co concentration profile throughout the sputtering sequence. The observations revealed that LCO disappeared and Co completely diffused into the LLTO pellet as a consequence of annealing at 700 °C in air. The Li depth profile could not be obtained quantitatively because the signal from Ti 3s (59 eV) and Co 3p (60 eV) is overlapping partly with Li 1s (56 eV).
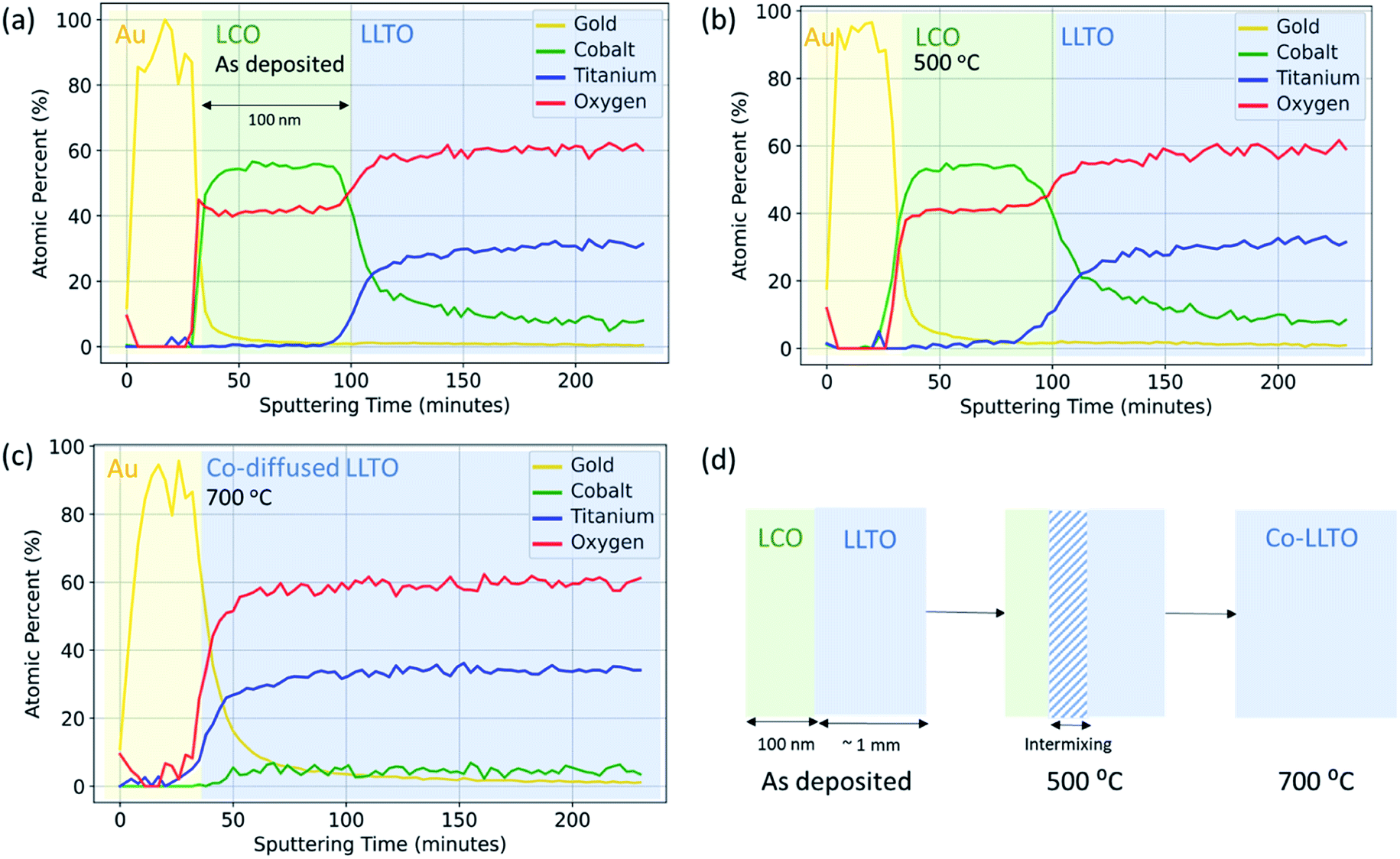 | ||
| Fig. 3 Depth profile for Au, Co, Ti and O for Au|LCO|LLTO with 100 nm LCO thickness obtained by X-ray photoelectron spectroscopy (XPS) with soft X-ray (Al Kα) and Ar-ion sputtering in the (a) as-deposited, (b) after annealing at 500 °C and (c) after annealing at 700 °C. The spectra for Au 4f, Co 2p, C 1s, Ti 2p, and O 1s were collected after every 3 minutes of sputtering interval. (d) Schematic of reaction between LCO and LLTO up to 700 °C in air. | ||
Fig. S5 in ESI† shows the C 1s spectra collected as a function of sputtering time for different thermally treated samples to determine if Li2CO3 formed at the LCO|LLTO interface. We did not observe carbon in any form in the XPS depth profile beyond the surface. We discuss the implication of this result in Section 3.
We can next compare the results from non-destructive hard X-ray photoelectron spectroscopy (HAXPES) to those obtained above by XPS depth profiling with ion milling. Fig. 4 shows the Co/La ratio obtained by HAXPES for the as-deposited sample and for samples annealed at 300 °C, 500 °C and 600 °C. The Co/La ratio progressively decreases on increasing annealing temperature. Similar to the results above, this decrease of Co/La ratio also indicates that Co diffuses into the LLTO with increasing temperature. As observed in the XPS depth profile, Co completely diffused into the LLTO resulting in loss of the LCO layer for the sample annealed at 700 °C (Fig. 3(c)). Hence, due to poor signal quality of Co during HAXPES measurement, the data for Co/La at 700 °C ratio is not shown in Fig. 4. The consistency between the non-destructive HAXPES and the XPS depth profiling is reassuring that indeed there is Co diffusion from LCO into LLTO.
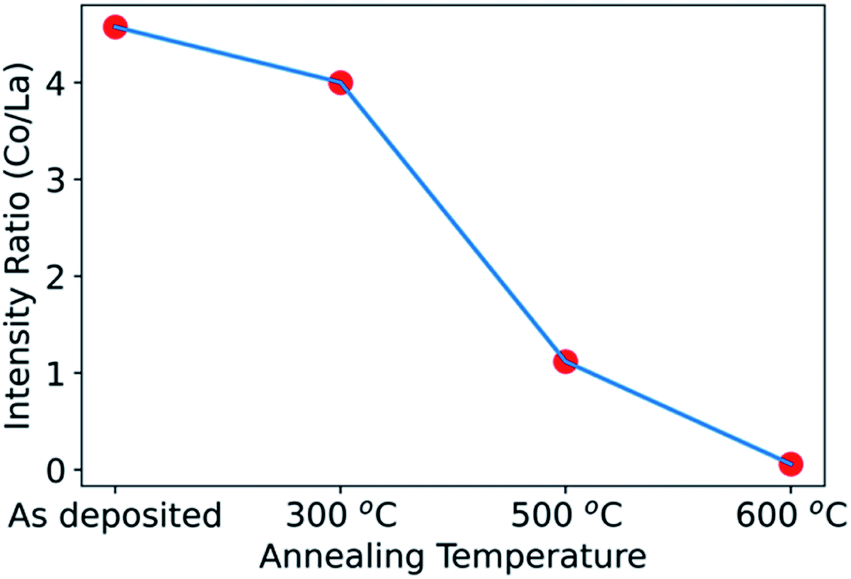 | ||
| Fig. 4 Co/La ratio obtained by Co 2p and La 3d hard X-ray photoelectron spectroscopy at 45° emission angle for the LCO|LLTO samples with a 20 nm deposited LCO layer on LLTO, in the as-deposited state and after annealing at 300 °C, 500 °C and 600 °C. The Co/La ratio was obtained by integrating intensity for Co 2p and La 3d. | ||
3.4 Interfacial impedance as a result of annealing in air
Electrochemical impedance spectroscopy revealed that the interfacial charge transfer at the LCO|LLTO interface decreased progressively upon annealing at higher temperatures. The interface impedance was no longer present in the limit of a totally depleted LCO layer after annealing at 700 °C. Fig. 5 shows the electrochemical impedance spectra obtained from Au|LCO|LLTO|LCO|Au symmetric cells for different annealing treatments. Fig. 5(b and c) show the corresponding equivalent circuit used to model the obtained impedance spectra for the cells. A resistor (Rbulk) in parallel with a capacitor (Cbulk) (i.e. Rbulk∥Cbulk) was used to model the bulk lithium-ion conductivity in LLTO. Secondly, resistor Rboundary in parallel with constant phase element (CPEboundary) (i.e. Rboundary∥CPEboundary) was used to model the domain boundary lithium-ion conductivity in LLTO. The combination of a resistor (R) in parallel with a capacitor or a constant phase element (CPE) is common to model bulk and domain boundary contribution for Li-ion conduction in LLTO.18,50–52 The use of CPE allows quantification of different polarization contributions in electrochemical systems as suggested by authors describing separation of bulk and boundary conduction in solid electrolytes.50 To model the interfacial impedance feature, a resistor Rinterface in parallel with constant phase element (CPEinterface) (i.e. Rinterface∥CPEinterface) was utilized. The approach for modelling the interfacial feature was adopted from previous studies.16,53,54 Finally, the tail was modelled by using another constant phase element (CPEtail) in series with the above circuit elements, as reported elsewhere to model Li-blocking Au electrode.51,52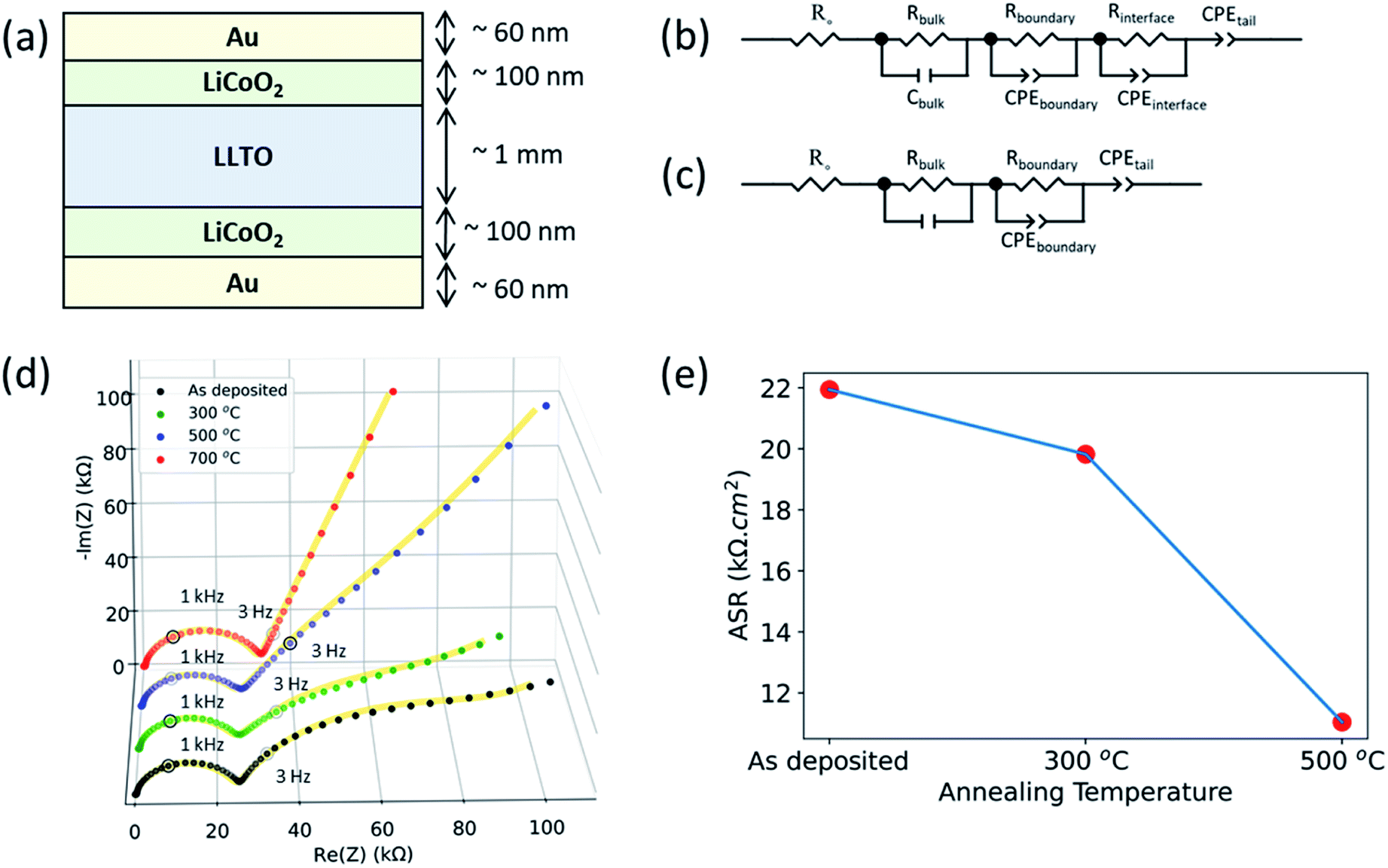 | ||
| Fig. 5 Electrochemical impedance spectroscopy (EIS) for Au|LCO|LLTO|LCO|Au symmetric cells. (a) Schematic of the cell configuration. Equivalent circuit models for EIS spectra, (b) for the as-deposited sample and the samples annealed at 300 °C and 500 °C, and (c) for the samples annealed at 700 °C. (d) Electrochemical impedance spectra of samples annealed at different temperatures. Solid line for each spectra corresponds to data obtained from equivalent circuit model after fitting the spectra with the models shown in (b) and (c). (e) Area-specific resistance (ASR) for a single LCO|LLTO interface. | ||
Fig. 5(d) show the full EIS spectra. The magnified EIS spectra in high frequency region can be found in ESI: Fig. S4(a–d).† All the samples show the feature arising from bulk grain-interior Li-ion conductivity, and grain boundary Li-ion conductivity from the LLTO pellets, at around 2 MHz and 1 kHz respectively. The above two semicircles observed for these cells for different annealing treatment are consistent with the observed EIS spectra from the bare LLTO pellets presented in ESI (Fig. S2†). The frequency region over which these two features appear51 and the observed bulk Li-ion conductivity (1.28 mS cm−1) agree with earlier studies of Li-ion conduction in LLTO.55
As seen in Fig. 5(d), the as-deposited, 300 °C annealed and 500 °C annealed samples additionally show a third arc in the low-frequency region at around 3 Hz which was not observed for LLTO pellet. The EIS spectra for LLTO pellet is shown in ESI (Fig. S2†). Hence, we attribute this feature at around 3 Hz to the interfacial charge transfer impedance of the LCO|LLTO interface. The 700 °C annealed sample did not show the third arc for the interface impedance at the low frequency. This is consistent with the loss of the LCO layer upon annealing at 700 °C as seen by the XAS and XPS results. The tail at the low-frequency region is observed in all the samples, arising from the Li-blocking nature of the Au electrode.
Fig. 5(e) shows the area-specific charge transfer resistance (ASR) of the LCO|LLTO interface obtained from EIS after equivalent circuit modelling. Rinterface element in the equivalent circuit construction corresponds to ASR. Since this comes from the two equivalent LCO|LLTO interfaces due to symmetry of the cell as shown in Fig. 5(a), Rinterface was divided by two to obtain the interface charge transfer resistance. It is interesting to note that the interfacial resistance reduces by almost 50% after annealing to 500 °C relative to the as-deposited sample, i.e. from 21.94 kΩ cm2 to 11.04 kΩ cm2. The ASR from the 700 °C annealed sample could not be deduced as LCO layer was completely lost after annealing at 700 °C.
4. Discussion
The XAS results from Co L-edge prove that there is no oxidation state change of cobalt from the as-deposited state to annealing up to 600 °C, but increased ordering of cobalt local environment, increased crystallinity, became evident. This can be seen from the Co L2,3-edge spectra with the appearance of crystal field splitting (t2g–eg split) and narrowing of peak after the annealing treatment till 600 °C. The Ti L2,3-edge XAS showed us no change in oxidation state of Ti even after annealing till 700 °C. These results rule out the possibility of formation of any decomposition phase that has a different oxidation state of Co (than in LCO) and of Ti (than in LLTO) till 600 °C.Although we have no evidence of secondary phase formation at the LCO|LLTO interface at temperatures below 700 °C, there is clear evidence from XPS and HAXPES that there is elemental mixing onsetting at relatively low temperatures. The XPS depth profile has shown that Co from LCO migrates into the LLTO when subjected to annealing in air, and has completely diffused into LLTO by 700 °C. The complete loss of the LCO layer after annealing at 700 °C resulted in the loss of the LCO|LLTO interface, and hence we could not observe interfacial features in the EIS spectra. The XPS depth profile, however, cannot say what the nature of Co is when diffused into LLTO. The Co L-edge XAS spectra for the 700 °C annealing condition provides hints as to where the Co goes. The observed spectra closely resemble the Co L-edge in doped lanthanum cobalt oxide perovskite (LaCoO3) measured by electron energy loss spectroscopy (EELS), showing partial reduction of Co3+ to Co2+ HS44 when doping is introduced. We also know Co can easily substitute into the perovskite lattice, forming Co-doped LLTO.56 The Co composition profile together with the Co oxidation state indicate strongly that the Co diffuses into LLTO upon annealing at temperatures as low as 300 °C and higher. While Co–Ti exchange has been also shown at the LCO|LATP interface,57 the reaction mechanism that we have found at the LCO|LLTO interface is different. At the LCO|LATP interface, an intermixed layer comprising Co and Ti forms after annealing57 due to thermodynamically favorable Co–Ti exchange at the interface.58 For the LLTO|LCO case however, the compositional depth profile showed that Co diffused significantly into the bulk LLTO pellet at 500 °C. However, we didn't detect Ti diffusion into the LCO layer. This makes us to believe that the reaction mechanism does not involve Co–Ti exchange, but rather Co dissolution from LCO into LLTO. The reduction of Co in LLTO can take place either due to the easier reducibility of Co compared to Ti upon annealing as can be expected from the standard reduction potentials,59 or due to Co and Li co-diffusion into LLTO where Li+ is then charge compensated by reduced Co ions. We could not resolve whether the latter mechanism is in place due to overlap of the Ti 3s (59 eV) and Co 3p (60 eV) peaks partly with the Li 1s (56 eV) peak in XPS.
Since the XPS depth profile shows the presence of the LCO layer on the LLTO pellet up to 500 °C, we can interpret the role of Co inter-diffusion on the interface impedance. The ASR reduces upon annealing to 500 °C. Other than relative structural ordering, the LCO|LLTO interface has not undergone any detectable secondary phase formation. The decrease in the interface ASR likely arises from better crystallinity of LCO and increased bonding between LCO and LLTO up to 500 °C. In addition, the diffusion of Co from the LCO layer into LLTO should form a mixed electronic-ionic conductor phase (Co-doped LLTO) near this interface. Co-doping in LLTO enhances the electronic conductivity by almost 3 orders of magnitude (10−9 to 10−6 S cm−1).56 Presence of such a mixed conducting phase can improve charge transfer kinetics60 between LLTO and LCO. For example, introducing mixed electronic-ionic conductor at cathode|solid electrolyte interface improved charge transfer properties in All-solid-state Li-ion Batteries based on Li7La3Zr2O12 solid electrolyte.61 All these effects can assist charge transfer and reduce the interface ASR of LCO|LLTO interface upon annealing to 500 °C.
We did not observe formation of Li2CO3 at the interface from the XRD and XPS depth profile for the LCO|LLTO system. This is of high significance as we know from previous studies that Li2CO3 is a detrimental secondary phase when formed at the solid electrolyte surface. For example, on the widely studied garnet solid electrolyte surface (for example the Al-substituted Li7La3Zr2O12), formation of Li2CO3 detrimentally increases the impedance at the interface with both the Li anode and the cathode.16,17,45 Our results on LCO|LLTO which showcased decreased interfacial impedance upon thermal treatment is an important finding on guiding engineering principles for designing desired cathode–electrolyte interfaces.
The as-deposited LCO thin film is amorphous, since we deposit at room temperature, in order not to cause any thermal interaction with the electrolyte during the deposition of the LCO film. Then upon annealing, we expect crystallization of the LCO layer as we have seen in our earlier papers,16,17 however, here before crystallization with detectable grain sizes, there is also Co-diffusion into LLTO and loss of LCO. We believe this is why we are not easily seeing LCO in the XRD patterns. The chemical environment of LCO is already known from XAS (oxidation state, Fig. 1) and XPS (elemental content, Fig. 3), respectively.
Lastly, it is interesting to note that the XRD results revealed no decomposition products in the reaction zone of the LCO|LLTO interface within the resolution limit. This is in contrast to the computational predictions that LCO|LLTO forms Co3O4, La2Ti2O7, Li2TiO3, and Li0.5CoO2 decomposition products.9 The apparent discrepancy between our experimental finding and prior theoretical predictions could arise from the fact that the thermodynamic driver for the LLTO–LCO reactions was predicted to be small, and could be overcome by other factors including interface energy of thin films and high nucleation barriers. According to DFT calculations, the reaction energy between delithiated LCO and LLTO is zero, and the reaction energy between fully lithiated LCO and LLTO is very small (−0.5 meV per atom).9 Small deviations in the Li stoichiometry could reduce this reaction energy even more. Therefore, regardless of lithium content in the LCO, DFT calculations predict a relatively stable interface, with only a small driving force at fully lithiated conditions. This small driving force may be overcome by other factors in the ultrathin film structure, such as the amorphous state or the interface energy contribution to the thermodynamics of secondary phase formation,62,63 and by high energy barriers for these phases to nucleate and grow at the interface. For example, it has been shown that the elements in the ultrathin layer can diffuse into the bulk material at the interface, before nucleation of the most thermodynamically stable phase.64 Hence, it is possible that what we find here, as Co-diffusion into LLTO and loss of LCO in the absence of any other secondary phases, is due to such a metastable path, instead of formation of the phases that were predicted by DFT to be thermodynamically stable. It is also possible that, even if such secondary phases formed, they were in too small amounts or not well crystallized, that we could not detect them with XRD up to 600 °C. Our results from XRD are in agreement with the observed stability of the LCO|LLTO mixed powders by Liao et al., who also did not observe secondary phase formation till 700 °C.48 Importantly, our findings in this work show that the LCO|LLTO system undergoes significant interaction, especially in the form of elemental mixing (of Co in particular) upon thermal treatment, even if no secondary phases were detectable by XRD or XAS up to 700 °C.
5. Conclusion
We have investigated the interface of lithium cobalt oxide (LiCoO2) and Li0.35La0.55TiO3 (LLTO) to understand the thermally driven interaction between the solid electrolyte and cathode material, as relevant to sintering and bonding of these components. The results showed that the LCO|LLTO system undergoes elemental mixing at the interface that leads to the formation of a mixed ionic-electronic conducting phase due to Co substitution at B-site in LLTO crystal lattice. Although there is no secondary phases that were detected in XRD, the interaction between LCO and LLTO does take place and onsets at relatively low temperatures around 300 °C as detected by XPS, HAXPES and XAS. The implication of this is evident from the interfacial electrochemical response from LCO|LLTO interface. The interfacial impedance decreases progressively upon increasing the annealing temperature until the thin LCO layer is completely lost at 700 °C. The mixed conducting phase formed due to Co substituted LLTO could be the reason for this, and motivate composite cathodes with such mixed ionic electronic doped LLTO components in solid-state Li-ion batteries. This finding has the potential to replace or limit the use of electronic and ionic conductive additive coating layers that is generally incorporated in cathode–electrolyte interface fabrication.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the support of LG Chem for this project through the Global Innovation Contest program. This work made use of the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-1419807. Deposition by RF sputtering was carried out through the use of MIT.nano's facilities. XAS data were acquired at IOS (23-ID-2) beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. We thank Andrei Gloskovskii for valuable technical assistance at the P22 beamline of DESY. Funding for the HAXPES instrument at beamline P22 is provided by the Federal Ministry of Education and Research (BMBF) under contracts 05KS7UM1 and 05K10UMA with Universität Mainz; 05KS7WW3, 05K10WW1 and 05K13WW1 with Universität Würzburg. We thank Georgios Dimitrakopoulos for his guidance in making the LLTO pellets, Jiayue Wang and Dongha Kim for their guidance with the XPS analysis.References
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Electrochem., 2017, 21, 1939–1964 CrossRef CAS
.
- D. Andre, S. J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed
- K. Takada, Acta Mater., 2013, 61, 759–770 CrossRef CAS
- X. B. Cheng, C. Z. Zhao, Y. X. Yao, H. Liu and Q. Zhang, Chem, 2019, 5, 74–96 CAS
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 Search PubMed
- Y. Horowitz, C. Schmidt, D. Hwan Yoon, L. M. Riegger, L. Katzenmeier, G. M. Bosch, M. Noked, Y. Ein-Eli, J. Janek, W. G. Zeier, C. E. Diesendruck and D. Golodnitsky, Energy Technol., 2020, 8, 2000580 CrossRef CAS
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC
- A. Banerjee, X. Wang, C. Fang, E. A. Wu and Y. S. Meng, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS PubMed
- A. C. Luntz, J. Voss and K. Reuter, J. Phys. Chem. Lett., 2015, 6, 4599–4604 CrossRef CAS PubMed
- C. Yu, S. Ganapathy, E. R. H. V. Eck, H. Wang, S. Basak, Z. Li and M. Wagemaker, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed
- Y. Xiao, Y. Wang, S. H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS
- J. Dai, C. Yang, C. Wang, G. Pastel and L. Hu, Adv. Mater., 2018, 30, 1802068 CrossRef PubMed
- S. Xu, R. M. Jacobs, H. M. Nguyen, S. Hao, M. Mahanthappa, C. Wolverton and D. Morgan, J. Mater. Chem. A, 2015, 3, 17248–17272 RSC
- Y. Kim, D. Kim, R. Bliem, G. Vardar, I. Waluyo, A. Hunt, J. T. Wright, J. P. Katsoudas and B. Yildiz, Chem. Mater., 2020, 32, 9531–9541 CrossRef CAS
- G. Vardar, W. J. Bowman, Q. Lu, J. Wang, R. J. Chater, A. Aguadero, R. Seibert, J. Terry, A. Hunt, I. Waluyo, D. D. Fong, A. Jarry, E. J. Crumlin, S. L. Hellstrom, Y. M. Chiang and B. Yildiz, Chem. Mater., 2018, 30, 6259–6276 CrossRef CAS
- Y. Inaguma, C. Liquan, M. Itoh, T. Nakamura, T. Uchida, H. Ikuta and M. Wakihara, Solid State Commun., 1993, 86, 689–693 CrossRef CAS
- Y. Inaguma, L. Chen, M. Itoh and T. Nakamura, Solid State Ionics, 1994, 70–71, 196–202 CrossRef CAS
- S. Stramare, V. Thangadurai and W. Weppner, Chem. Mater., 2003, 15, 3974–3990 CrossRef CAS
- F. Aguesse, V. Roddatis, J. Roqueta, P. García, D. Pergolesi, J. Santiso and J. A. Kilner, Solid State Ionics, 2015, 272, 1–8 CrossRef CAS
- F. Aguesse, J. M. López Del Amo, V. Roddatis, A. Aguadero and J. A. Kilner, Adv. Mater. Interfaces, 2014, 1, 1–9 Search PubMed
- A. R. Symington, J. Purton, J. Statham, M. Molinari, M. S. Islam and S. C. Parker, J. Mater. Chem. A, 2020, 8, 19603–19611 RSC
- D. Qian, B. Xu, H. M. Cho, T. Hatsukade, K. J. Carroll and Y. S. Meng, Chem. Mater., 2012, 24, 2744–2751 CrossRef CAS
- S. Song, X. Peng, K. Huang, H. Zhang, F. Wu, Y. Xiang and X. Zhang, Nanoscale Res. Lett., 2020, 15, 110 CrossRef CAS PubMed
- J. Yi, P. He, H. Liu, H. Ni, Z. Bai and L. Z. Fan, J. Energy Chem., 2021, 52, 202–209 CrossRef
- M. Prabu, M. V. Reddy, S. Selvasekarapandian, S. Admas, K. P. Loh, G. V. S. Rao and B. V. R. Chowdari, J. Electrochem. Soc., 2013, 160, A3144–A3147 CrossRef CAS
- H. G. Song, J. Y. Kim and Y. J. Park, Electrochim. Acta, 2011, 56, 6896–6905 CrossRef CAS
- X. Hu, G. Yan, X. Cheng, J. Malzbender, W. Qiang and B. Huang, J. Am. Ceram. Soc., 2019, 102, 1953–1960 CAS
- K. Kishida, N. Wada, H. Adachi, K. Tanaka, H. Inui, C. Yada, Y. Iriyama and Z. Ogumi, Acta Mater., 2007, 55, 4713–4722 CrossRef CAS
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS
- L. Wang, B. Chen, J. Ma, G. Cui and L. Chen, Chem. Soc. Rev., 2018, 47, 6505–6602 RSC
- Y. Lyu, X. Wu, K. Wang, Z. Feng, T. Cheng, Y. Liu, M. Wang, R. Chen, L. Xu, J. Zhou, Y. Lu and B. Guo, Adv. Energy Mater., 2021, 11, 2000982 CrossRef CAS
- A. Manthiram and J. B. Goodenough, Nat. Energy, 2021, 6, 323 CrossRef CAS
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 CrossRef CAS
- Y. Xiao, L. J. Miara, Y. Wang and G. Ceder, Joule, 2019, 3, 1252–1275 CrossRef CAS
- L. J. Miara, W. D. Richards, Y. E. Wang and G. Ceder, Chem. Mater., 2015, 27, 4040–4047 CrossRef CAS
- K. Park, B. C. Yu, J. W. Jung, Y. Li, W. Zhou, H. Gao, S. Son and J. B. Goodenough, Chem. Mater., 2016, 28, 8051–8059 CrossRef CAS
-
J. Stöhr, NEXAFS Spectroscopy, Springer Science & Business Media, 1992, vol. 25 Search PubMed
- T. Polczyk, W. Zając, M. Ziąbka and K. Świerczek, J. Mater. Sci., 2021, 56, 2435–2450 CrossRef CAS
-
C. Schlueter, A. Gloskovskii, K. Ederer, I. Schostak, S. Piec, I. Sarkar, Y. Matveyev, P. Lömker, M. Sing, R. Claessen, C. Wiemann, C. M. Schneider, K. Medjanik, G. Schönhense, P. Amann, A. Nilsson and W. Drube, in AIP Conference Proceedings, AIP Publishing LLC, 2019, vol. 2054, p. 040010 Search PubMed
- J. P. Strachan, J. Joshua Yang, R. Münstermann, A. Scholl, G. Medeiros-Ribeiro, D. R. Stewart and R. Stanley Williams, Nanotechnology, 2009, 20, 485701 CrossRef PubMed
- H. Ikeno, I. Tanaka, Y. Koyama, T. Mizoguchi and K. Ogasawara, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 75123 CrossRef
- M. Merz, P. Nagel, C. Pinta, A. Samartsev, H. V. Löhneysen, M. Wissinger, S. Uebe, A. Assmann, D. Fuchs and S. Schuppler, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 174416 CrossRef
- L. Cheng, E. J. Crumlin, W. Chen, R. Qiao, H. Hou, S. Franz Lux, V. Zorba, R. Russo, R. Kostecki, Z. Liu, K. Persson, W. Yang, J. Cabana, T. Richardson, G. Chen and M. Doeff, Phys. Chem. Chem. Phys., 2014, 16, 18294–18300 RSC
- J. Ibarra, A. Várez, C. León, J. Santamaría, L. M. Torres-Martínez and J. Sanz, Solid State Ionics, 2000, 134, 219–228 CrossRef CAS
- B. Wang, J. B. Bates, F. X. Hart, B. C. Sales, R. A. Zuhr and J. D. Robertson, J. Electrochem. Soc., 1996, 143, 3203–3213 CrossRef CAS
- C. L. Liao, C. H. Wen and K. Z. Fung, J. Alloys Compd., 2007, 432, L22–L25 CrossRef CAS
- M. Kotobuki, Y. Suzuki, H. Munakata, K. Kanamura, Y. Sato, K. Yamamoto and T. Yoshida, J. Power Sources, 2010, 195, 5784–5788 CrossRef CAS
- P. Braun, C. Uhlmann, A. Weber, H. Störmer, D. Gerthsen and E. Ivers-Tiffée, J. Electroceram., 2017, 38, 157–167 CrossRef CAS
- W. J. Kwon, H. Kim, K. N. Jung, W. Cho, S. H. Kim, J. W. Lee and M. S. Park, J. Mater. Chem. A, 2017, 5, 6257–6262 RSC
- A. Mei, X. L. Wang, J. Le Lan, Y. C. Feng, H. X. Geng, Y. H. Lin and C. W. Nan, Electrochim. Acta, 2010, 55, 2958–2963 CrossRef CAS
- T. Kobayashi, A. Yamada and R. Kanno, Electrochim. Acta, 2008, 53, 5045–5050 CrossRef CAS
- W. Zhang, F.
H. Richter, S. P. Culver, T. Leichtweiss, J. G. Lozano, C. Dietrich, P. G. Bruce, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 22226–22236 CrossRef CAS PubMed
- Y. Harada, T. Ishigaki, H. Kawai and J. Kuwano, Solid State Ionics, 1998, 108, 407–413 CrossRef CAS
- X. Cheng, J. Wang, W. Qiang and B. Huang, J. Am. Ceram. Soc., 2020, 103, 3698–3705 CrossRef CAS
- Y. Liu, Q. Sun, J. Liu, M. Norouzi Banis, Y. Zhao, B. Wang, K. Adair, Y. Hu, Q. Xiao, C. Zhang, L. Zhang, S. Lu, H. Huang, X. Song and X. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 2293–2298 CrossRef CAS PubMed
- H. K. Tian, R. Jalem, B. Gao, Y. Yamamoto, S. Muto, M. Sakakura, Y. Iriyama and Y. Tateyama, ACS Appl. Mater. Interfaces, 2020, 12, 54752–54762 CrossRef CAS PubMed
-
P. Vanysek, Electrochemical Series, in CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 96th edn, 2015 Search PubMed
- M. J. Wang, J. B. Wolfenstine and J. Sakamoto, Adv. Funct. Mater., 2020, 30, 1909140 CrossRef CAS
- C. Wang, L. Zhang, H. Xie, G. Pastel, J. Dai, Y. Gong, B. Liu, E. D. Wachsman and L. Hu, Nano Energy, 2018, 50, 393–400 CrossRef CAS
- M. Wagemaker, F. M. Mulder and A. Van Der Ven, Adv. Mater., 2009, 21, 2703–2709 CrossRef CAS
- A. Navrotsky, ChemPhysChem, 2011, 12, 2207–2215 CrossRef CAS PubMed
- D. L. M. Cordova and D. C. Johnson, ChemPhysChem, 2020, 21, 1345–1368 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08853j |
| ‡ Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2022 |