
Exsolution of nanoparticles on A-site-deficient lanthanum ferrite perovskites: its effect on co-electrolysis of CO2 and H2O†
Jaesung
Kim
a,
Matthew
Ferree
a,
Seval
Gunduz
 a,
Jean-Marc M.
Millet
b,
Mimoun
Aouine
b,
Anne C.
Co
c and
Umit S.
Ozkan
*a
a,
Jean-Marc M.
Millet
b,
Mimoun
Aouine
b,
Anne C.
Co
c and
Umit S.
Ozkan
*a
aWilliam G. Lowrie Department of Chemical and Biomolecular Engineering, The Ohio State University, Columbus, OH 43210, USA. E-mail: ozkan.1@osu.edu
bInstitut de Recherche sur la Catalyse et l'Environnement de Lyon, UMR 5256, Université Claude-Bernard Lyon 1, 69626, Villeurbanne Cedex, France
cDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210, USA
First published on 22nd November 2021
Abstract
La0.7Sr0.2Ni0.2Fe0.8O3 (LSNF), having thermochemical stability, superior ionic and electronic conductivity, and structural flexibility, was investigated as a cathode in SOECs. Exsolution of nanoparticles by reduction of LSNF at elevated temperatures can modulate the characteristics of adsorption, electron transfer, and oxidation states of catalytically active atoms, consequently improving the electrocatalytic activity. The exsolution of NiFe and La2NiO4 nanoparticles to the surface of LSNF under reducing atmosphere (5% H2/N2) was verified at various temperatures (500–800 °C) by IFFT from ETEM, TPR and in situ XRD. The exsolved nanoparticles obtained uniform size distribution (4.2–9.2 nm) and dispersion (1.31 to 0.61 × 104 particle per μm2) depending on the reduction temperature (700–800 °C) and time (0–10 h). The reoxidation of the reduced LSNF (Red-LSNF) was verified by the XRD patterns, indicative of its redox ability, which allows for redistribution of the nanoparticles between the surface and the bulk. TPD-DRIFTS analysis demonstrated that Red-LSNF had superior H2O and CO2 adsorption behavior as compared to unreduced LSNF, which we attributed to the abundance of oxygen vacancy sites and the exsolved NiFe and La2NiO4 nanoparticles. After the reduction of LSNF, the decreases in the oxidation states of the catalytically active ions, Fe and Ni, were characterized on the surface by XPS as well as in the bulk by XANES. The electrochemical performance of the Red-LSNF cell was superior to that of the LSNF cell for electrolysis of H2O, CO2, and H2O/CO2.
Introduction
Carbon recycling will continue to be a major challenge for scientists in the foreseeable future as a result of the strong correlation between atmospheric CO2 levels and climate change, which is caused by the excessive use of fossil fuels.1,2 To enable fossil fuel use for safe, reliable, and affordable energy production, cost-effective processes for CO2 capture and utilization are vital. It is clear that the conversion of CO2 into valuable chemicals or fuels represents a promising strategy for reducing atmospheric CO2. A great deal of effort has been made in the past years in electrocatalytic conversion of CO2 into other chemicals.2–6 It should be noted that if renewable energy sources are available for electrolysis, directly splitting CO2 into CO and O2 using electrochemical cells is a relatively simple and effective way to promote the reduction of global CO2 concentration in the atmosphere.4,7–9 Moreover, electrochemical cells contribute to the environmentally friendly aspect of hydrogen production as opposed to current H2 production methods which heavily rely on fossil fuels, generating CO2.10 In this vision, utilization of electricity-to-chemicals through electrolysis cells is a practical method of utilizing renewable energy resources that are inherently intermittent.There have been a variety of electrolysis technologies proposed, including polymer electrolyte membrane electrolysis cells (PEMECs) with high power density, alkaline electrolysis cells (AECs) with low capital cost, and solid oxide electrolysis cells (SOECs) with high electrical efficiency and low material cost.10 Although SOEC has not yet achieved widespread commercialization and is the least developed technology compared to PEMEC and AEC, it offers greater conversion efficiency due to its ability to operate at higher temperatures, (500–1000 °C) enhancing the reaction kinetics.10–12 The electro-reduction of CO2 and H2O involves electron transfer from the SOEC cathode to adsorbed molecules, which results in the production of H2 and CO. In this process, oxygen ions are created and transported through the solid oxide electrolyte to the anode, where they are oxidized and recombined into oxygen gas molecules. The cathode and anode half-cell reactions and overall reaction are as follows:
Cathode:
| H2O + 2e− → H2 + O2− |
| CO2 + 2e− → CO + O2− |
Anode:
| 2O2− → O2 + 4e− |
Overall:
| H2O + CO2 → H2 + CO + O2 |
It is being increasingly recognized that perovskite oxides with the generic formula ABO3 are effective cathode materials for high-temperature electrolysis.4,13–17 In this chemical formula, A represents an alkali or alkaline earth metal ion coordinated to twelve oxygen atoms, B represents a transition metal ion octahedrally coordinated to six oxygen atoms, and O represents oxygen. A and/or B sites in the formula can be doped systematically to enhance the transport properties and the electrochemical activity of the material.
Based on recent research, it has been shown that the number of B-site atoms that can be mobilized to migrate onto the surface of a perovskite can be increased by subjecting the material to a controlled reducing environment at elevated temperatures. Transition metals such as nickel, cobalt, and iron migrate from the bulk to the surface during redox processes and create stable metallic nanoparticles, a process termed “redox exsolution” or “solid-state recrystallization” depending on whether it is reversible or irreversible.18–26 As opposed to physical deposition techniques, exsolution ensures that metallic nanoparticles are formed with a uniform distribution that firmly adheres to the parent perovskite surface with superior thermochemical stability.27–29 It is also possible to prepare these nanoparticles under vacuum or by applying an electric current, which offers more possibilities for an in situ modification of perovskite electrodes.24,30 There have been several ex situ investigations into the size, number density, structure and composition of perovskites with exsolved metallic nanoparticles. In situ studies using an environmental transmission electron microscopy (ETEM) and density functional theory (DFT) revealed the growth and kinetics of Ni and Co nanoparticles.31–36
Research on exsolution from perovskites has been focused mainly on metal nanoparticles and strontium titanate with its simple cubic structure. On the other hand, strontium-doped lanthanum ferrite (LSF) perovskites are characterized by a wide range of crystal structures (orthorhombic, rhombohedral, tetragonal, or cubic) dependent on the preparation method, the dopants used, and the temperatures.37,38 LSF-type perovskites also have been shown to exsolve A2BO4 phases in addition to metal nanoparticles during reduction. Moreover, strontium doped lanthanum ferrite perovskites are found to undergo a phase transition to Ruddlesden–Popper (RP) during the exsolution process.39 The structural diversity of the strontium doped lanthanum ferrite perovskites makes them an exciting category of catalysts, but also necessitates further investigation.
In this study, we prepared A-site deficient strontium- and nickel-doped lanthanum ferrite perovskite oxide (LSNF) as a cathode in SOEC. We investigated the exsolution of nanoparticles on LSNF induced by reduction and its effect on electrolysis of H2O, CO2, and H2O/CO2. A detailed examination using combined techniques such as in situ X-ray diffraction and ETEM under reducing environment revealed that structural transformations of LSNF under reducing atmosphere caused NiFe nanoparticles and La2NiO4 to be exsolved onto the parent perovskite surface. An analysis of the elemental oxidation states, the surface adsorption/desorption characteristics, and electrochemical impedance spectra of the reduced LSNF supportively showed its enhanced electrocatalytic activity on CO2/H2O electrolysis. These results provided an insight into the potential of LSNF catalysts with improved activity and greater usability as an electrode.
Results and discussion
Structural analysis of LSNF under air and inert environment
The structure of LSNF powder samples under air and inert environment was studied using transmission electron microscopy (TEM) and in situ X-ray diffraction (XRD) techniques. In terms of crystal structure shown in Fig. 1(a), the XRD pattern of LSNF powder at room temperature was in accordance with that found in LaFeO3 perovskite oxide (reference code: 00-152-6450) having orthorhombic distortion as reported in the literature.40 Further confirmation of the perovskite structure has been obtained from Rietveld refinement. Based on the XRD pattern analysis of the as-calcined LSNF powder, the lattice volume was estimated to be 0.23622 nm3 at room temperature. This value is lower than 0.24224 nm3 of LaFeO3 perovskite, which is attributed to the smaller ionic radii of Ni ion than that of Fe ion. The crystallite size of LSNF powders can be calculated using the principal peak (1 1 2) based on the Scherrer equation:41,42Here, βL represents the peak broadening (FWHM) in radians, θ denotes half of the Bragg's angle (2θ), K is a dimensionless shape factor (0.9) assuming a spherical particle shape, λ is the wavelength of Cu-Kα radiation (0.15406 nm) and L indicates the crystallite size. The calculated L value for the LSNF powder at room temperature under air was 35 nm. TEM images of the LSNF powder at room temperature are shown in Fig. 1(b) at various scales. Inverse Fast Fourier Transform (IFFT) was attempted on the region inside Fig. 1(c). The IFFT results further confirmed the LSNF's crystalline structure obtained using XRD (Table S1†).
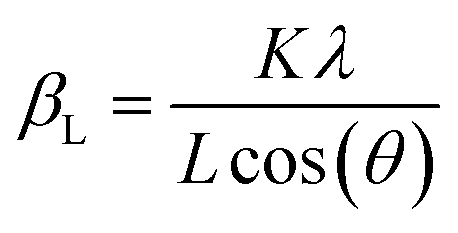
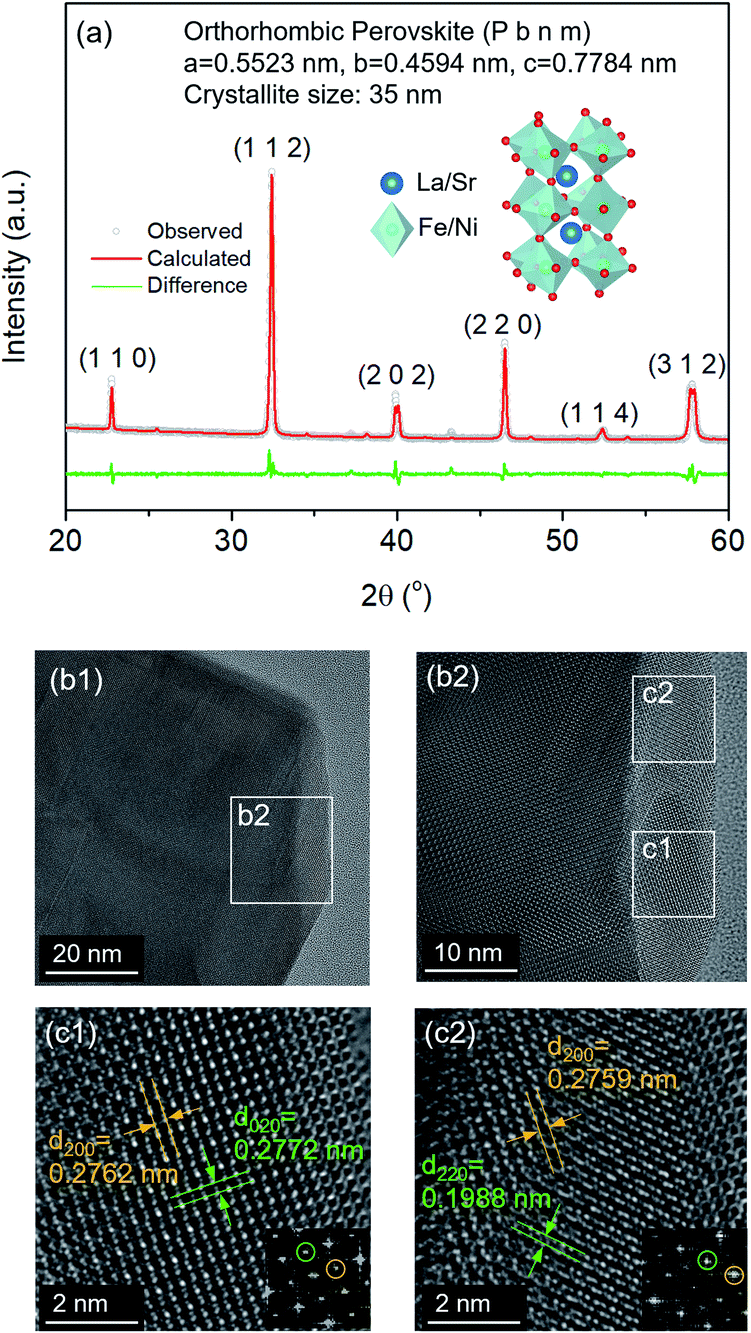 | ||
| Fig. 1 (a) XRD pattern of La0.7Sr0.2Ni0.2Fe0.8O3 perovskite (LSNF) powder at room temperature and Rietveld refinement of the XRD pattern, (b) TEM images of LSNF at room temperature, (c) enlarged TEM images of the specified area from (b) with lattice fringes and inverse fast Fourier transform (IFFT) of the selected area. | ||
The thermochemical stability of the LSNF was studied under air and helium atmospheres at 30°–800 °C using the in situ XRD technique. As shown in Fig. 2(a) and (b), LSNF exhibited perfect perovskite crystallinity under both environments at each temperature which proves its high thermochemical stability under oxidizing and inert atmospheres. Based on the assumption that LSNF powders have an orthorhombic space group (Pnma), the unit cell volume of the LSNF was calculated as a function of temperature under air and helium atmospheres as shown in Fig. 2(c). The linear average thermal expansion coefficient (TEC) of the LSNF powder under air and helium was determined to be 15 ppm °C−1 and 16 ppm °C−1, respectively, values that are compatible with gadolinium-doped ceria (GDC) buffer layers.43 Thus, LSNF is a suitable electrode that can be reliably deposited on the surface of GDC buffer layers on YSZ button cells with no significant mechanical stress arising from the heating/cooling process of the cell.
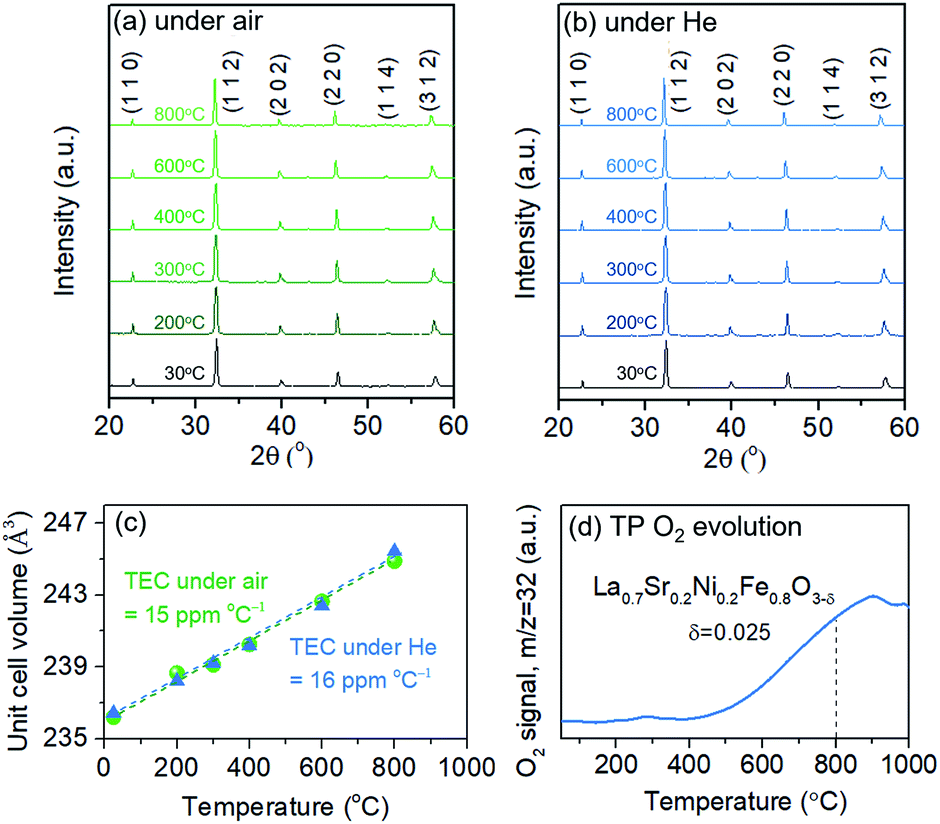 | ||
| Fig. 2 In situ XRD patterns of LSNF powders at various temperatures from 30 °C to 800 °C under (a) air and (b) helium environment, (c) thermal expansion coefficient of the LSNF powders under air and helium environment, (d) temperature-programmed O2 evolution of the LSNF powder under helium environment. δ represents the created oxygen vacancy per formula unit of LSNF from room temperature to 800 °C. | ||
The lattice oxygen, known as β-oxygen, inside LSNF structures was thermally evolved into an oxygen vacancy and molecular O2 in an inert atmosphere, which was depicted with O2 signal (m/z = 32) in Fig. 2(d).44–46 The development of oxygen vacancies plays a crucial role in LSNF functioning as an electrode, since these vacancies directly affect the ionic and electrical conductivity. The oxygen vacancy site serves as a catalytically active site where adsorption and subsequent reduction of H2O and CO2 occurs.8 The formula for the La0.7Sr0.2Ni0.2Fe0.8O3−δ perovskite was determined by measuring the amount of oxygen evolved up to 800 °C and calculating a stoichiometric value for it. The specified value of δ was estimated to be 0.025. It has been reported in literature that the LSNF perovskite exhibited a high atomic oxygen evolution, which is ascribed to the A-site deficient stoichiometry and B-site dopant Ni with weaker binding to oxygen compared to Fe.38,47 The oxygen vacancies formed during the heat-treatment reduce the valence of Fe or Ni ions as a charge compensation, resulting in increased lattice parameters. However, the estimated amount of oxygen vacancies formed during the heat-treatment in the temperature range from room-temperature to 800 °C, δ = 0.025, is 0.83% out of the stoichiometric oxygen, 3. Hence, the contributions of chemical expansion to an increase in lattice parameters would be trivial, resulting in values of TEC similar in air and in helium.
Analysis of the structural behavior of LSNF under reducing environment
Structural characteristics of LSNF perovskite under reducing environment have been examined using temperature-programmed reduction (TPR), in situ XRD, and environmental TEM (ETEM). TPR was performed under 30 ccm of 5% H2/N2 and the H2O signal (m/z = 18) was monitored as a function of temperature up to 1000 °C, as shown in Fig. 3(a). LSNF showed the first peak at around 440 °C in H2-TPR profile which is linked to the reduction of Ni3+ to Ni2+ (400 °C) and successive reduction of Ni2+ to Ni0 (460 °C). At around 650 °C, the second peak began to appear attributed to the partial reduction of Fe3+ to Fe2+ in LSNF perovskite.48 The proposed phase transformation of an ABO3-type perovskite under reducing environment is as follows:49| nABO3 → (n − 1)ABO3 + AO + BO2 |
| BO2 + H2 → B + H2O |
| ABO3 + AO → A2BO4 |
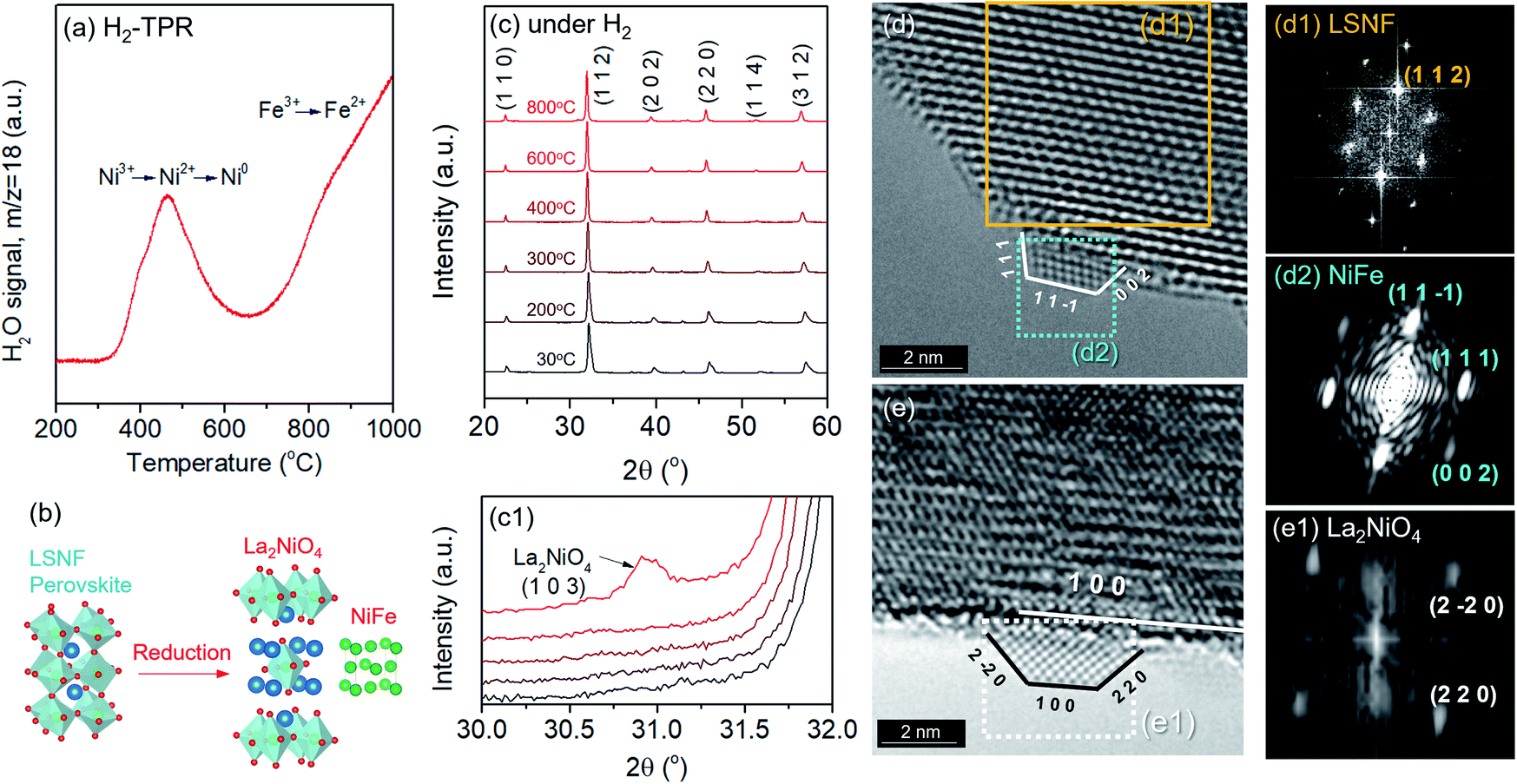 | ||
| Fig. 3 (a) Temperature-programmed reduction profile of LSNF powders under 30 ccm of 5% H2/N2 with a 20 °C min−1 ramp rate (b) illustration of the formation of zerovalent Ni and La2NiO4 in a Ruddlesden–Popper phase during the reduction and exsolution process, (c) in situ XRD patterns of LSNF powders under 5% H2/N2 atmosphere at various temperatures heating from 30 °C and 800 °C, (c1) the (1 0 3) diffraction peak of La2NiO4 in an enlarged scale, (d) in situ TEM image of LSNF under 1 mbar of H2 atmosphere at 500 °C with an exsolved NiFe nanoparticle, (d1) IFFT of the area confined for LSNF from (d), (d2) IFFT of the area selected for the exsolved NiFe nanoparticle from (d), (e) in situ TEM image of LSNF powder under 1 mbar of H2 at 700 °C with an exsolved La2NiO4 nanoparticles, (e1) IFFT of the area confined for the La2NiO4 nanoparticle from (e). | ||
Therefore, the reduction of LSNF perovskite oxide can lead to the formation of (i) a B-site metal phase, (ii) an A2BO4 oxide phase and (iii) oxygen vacancies. A2BO4 is called Ruddlesden–Popper (RP) phase. Zerovalent NiFe and La2NiO4 are accordingly responsible for B and A2BO4, respectively, as illustrated in Fig. 3(b), since the dopant Ni3+ ion is more reducible than Fe3+ ion. Because of its oxygen hyper stoichiometry, La2NiO4 is known as a mixed electronic and ionic conductor superior to the corresponding parent perovskite.38,50
In situ X-ray diffraction studies were conducted on LSNF under 5% H2/N2 at the temperatures from 30 °C to 800 °C as shown in Fig. 3(c). LSNF perovskite has demonstrated thermochemical stability at temperatures up to 700 °C under reducing conditions without exhibiting any changes to its diffraction pattern. It was found that a small peak appeared at 800 °C at approximately 31.0° 2θ in Fig. 3(c1), which is indicative of the phase transition to RP phase, La2NiO4. LSNF perovskite possibly experienced a rapid phase transition at 800 °C because the XRD peak acquisition was commenced immediately after reaching the temperature.
Phase transformation of LSNF during the reduction process has been studied by an environmental transmission electron microscopy (ETEM) carried out under hydrogen atmosphere (1 mbar) in the temperature range of 25–800 °C as shown in Fig. 3(d and e). There were no exsolved nanoparticles found on the surface of LSNF perovskites in ETEM until the temperature reached 400 °C (Fig. S1†). As the temperature rose to 500 °C, nanoparticles were formed to about 2 nm in size (Fig. 3(d)). Inverse fast Fourier transform (IFFT) analysis revealed that the nanoparticles were zerovalent NiFe monoclinic alloy (reference code: 00-901-1506). The detailed index parameters are provided in Table S2,† and EDS results further confirmed that the nanoparticles consist of Ni and Fe with 1-to-1 ratio in Fig. S2.† It was also confirmed that La2NiO4 phase had formed, and that we found it to be in the form of nanoparticles with a size of approximately 1.7 nm embedded on the surface of the parent perovskite (Fig. 3(e)). The IFFT is also depicted in the figure which illustrates the exposed planes of the particle as (1 0 0), (2 −2 0), and (2 2 0) with the particle growing in an epitaxial direction in accordance with (1 0 0) (Table S3†). Even though the exsolution of B-site-doped transition metals has been investigated extensively, we have not come across any studies investigating RP phase nanoparticles derived from exsolution. Although the number of exsolved zerovalent NiFe nanoparticles was much greater than the number of La2NiO4 nanoparticles and it is difficult to discern individual nanoparticles, the RP phase nanoparticles deserve more attention for their superior catalytic abilities.
The extent of the structural transformation in LSNF perovskite could vary with respect to the amount of time under the controlled reducing atmosphere. Fig. 4(a) shows ex situ XRD patterns of LSNF treated with 5% H2/N2 for different periods of time. The two XRD patterns on the bottom of the graph were collected from LSNF and La2NiO4 powders for comparison with treated LSNFs. La2NiO4 and zerovalent NiFe peaks appeared in the XRD pattern after 2 h of reduction. The structural transformation during the reduction seemed to be settled after 5 h because the XRD patterns were almost unchanged between 5 h and 10 h of reduction. ETEM images of LSNF powders were obtained at 700 °C, 750 °C, and 800 °C under 1 mbar H2 atmosphere (Fig. 4(b)). Images obtained during reduction were found to have stayed unchanged for an extended period implying that the extent of reduction had reached an equilibrium for each corresponding condition. Then, it may be assumed that the ETEM images obtained under 1 mbar H2 atmosphere at 800 °C correspond to the ex situ XRD patterns of LSNF powders after 5 h of reduction under 5% H2/N2. The average size and distribution could then be determined in Fig. 4(c). As the temperature increased from 700 °C to 800 °C, the number density of the nanoparticles on the surface decreased from 1.31 × 104 to 0.61 × 104 particle per μm2 and the average particle size increased from 4.5 nm to 9.2 nm. As can be seen in Fig. 4(b), NiFe nanoparticles in a narrow range of sizes were uniformly distributed over the entire surface of the perovskite, which causes the catalyst to have excellent electrochemical performance. Zerovalent NiFe nanoparticles observed in this study were in a similar size range to that reported for doped strontium titanate perovskites and are much smaller than the observed size range for Ni doped Sr2Fe1.5Mo0.5O6 double perovskites.35,51,52 It can be seen that the number densities of nanoparticles in this study are about 20 times higher than those obtained during Ni-exsolution from La0.5Ca0.4Ni0.2Ti0.8O2.95 perovskite.22 Higher density of heterogeneous phases can be obtained by exsolution from non-stoichiometric perovskites. A-site deficient materials have demonstrated migration of more B-site cations to the surface compared to their stoichiometric equivalents.20 The transformed amount of NiFe phase on the Red-LSNF after 5 h of reduction was estimated to be 6.5 wt% by Rietveld refinement in Fig. 5, which explains the massive nanoparticles on the surface.
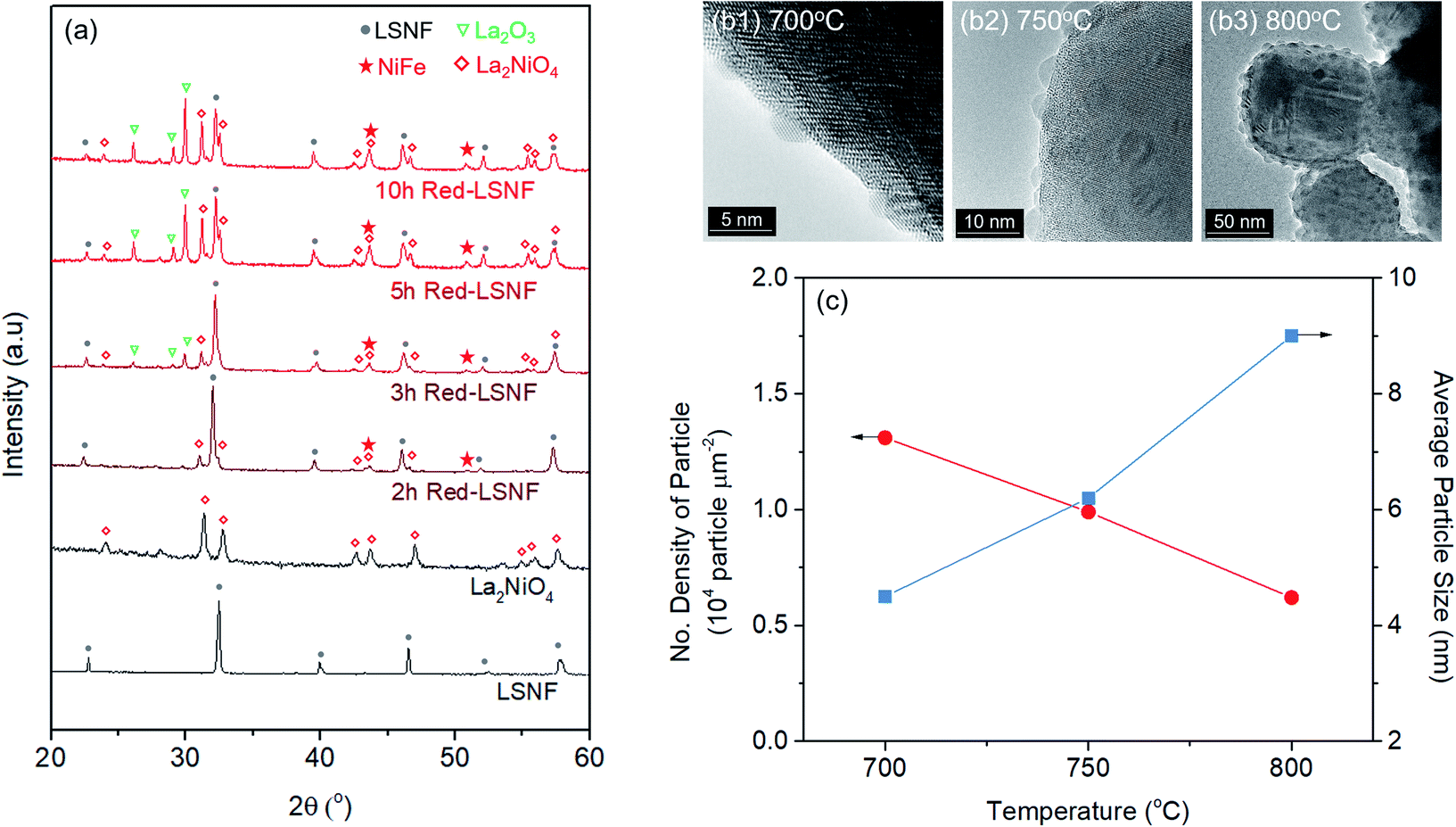 | ||
| Fig. 4 (a) XRD patterns of LSNF, La2NiO4, and LSNF treated with 5% H2/N2 atmosphere at 800 °C with different time duration, (b) in situ TEM images of LSNF powders under 1 mbar of H2 atmosphere at 700 °C, 750 °C, and 800 °C, (c) the number densities and average particles sizes of the exsolved nanoparticles on the LSNF surface at 700 °C, 750 °C, and 800 °C. | ||
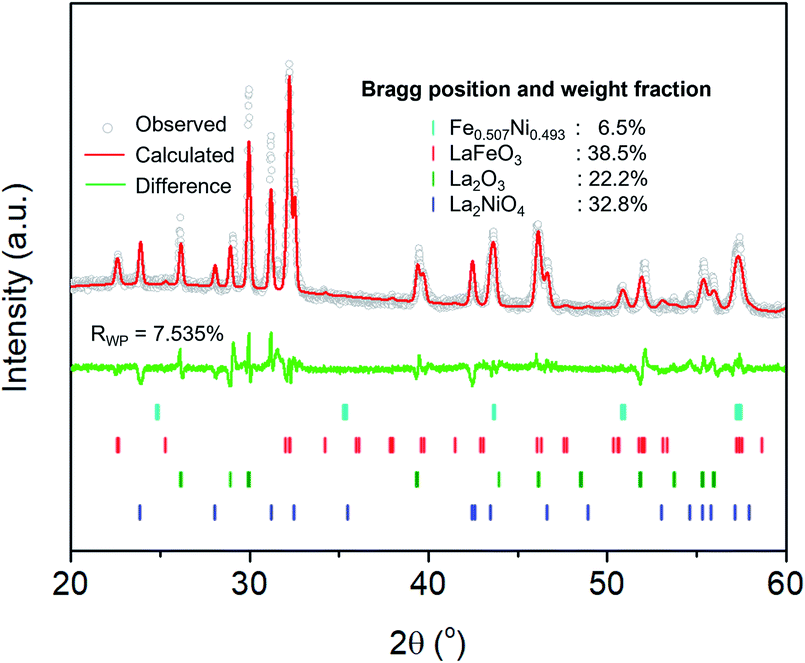 | ||
| Fig. 5 Rietveld refinement on XRD pattern of Red-LSNF after 5 h of reduction at 800 °C under 5% H2/N2. | ||
Analysis of the structural redox ability of LSNF
The redox ability of LSNF was explored by temperature-programmed oxidation (TPO) and XRD. LSNF was treated with 5% H2/N2 at 800 °C to form Red-LSNF, and TPO was carried out on the Red-LSNF to estimate the reoxidation temperature at which the exsolved nanoparticles are reversibly incorporated back into the perovskite lattice, thus restoring the original LSNF. 2% O2/He (TPO) was used to reoxidize Red-LSNF and the oxygen consumption was depicted in a plot by the difference from the level where O2 signal (m/z = 32) was stabilized at room temperature. A peak was seen for O2-TPO at 670 °C in Fig. 6(a). As compared with the result of TPR in Fig. 3(a), Red-LSNF experienced reoxidation at a temperature range of 600–800 °C while reduction began to occur at the temperature lower than 500 °C under H2.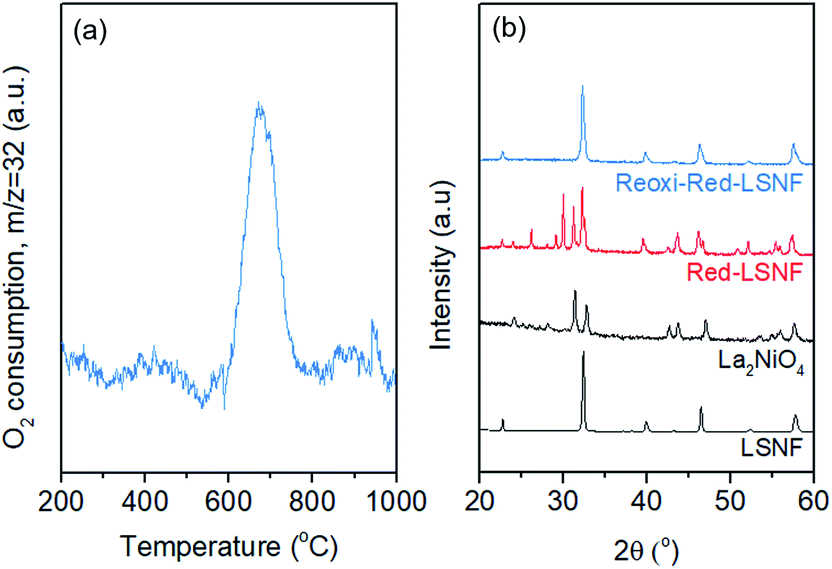 | ||
| Fig. 6 (a) Temperature-programmed oxidation profile of Red-LSNF with 30 ccm of 2% O2/He, (b) XRD patterns of LSNF, La2NiO4, Red-LSNF treated with 5% H2/N2 at 800 °C for 5 h, and re-oxidized Red-LSNF powders with 10% O2/He at 800 °C for 2 h. | ||
The structural redox ability of LSNF was also examined by XRD. Representative XRD patterns of LSNF and La2NiO4 powders are presented at the bottom in Fig. 6(b). It was found that the XRD patterns of Red-LSNF were completely reversed to that of LSNF after 2 h of reoxidation under 10% O2/He atmosphere at 800 °C. Given that 800 °C is the temperature at which co-electrolysis of CO2 and H2O is conducted, redistribution of nanoparticles can be readily achieved as needed. For conventional supported transition metal catalysts, sintering is a degradative process irreversible in nature, which restrains the number of oxidative regeneration cycles that can be applied in order to recover coked catalysts.53,54 Since LSNF retains the structural redox ability, nanoparticles, including zerovalent NiFe and La2NiO4, decorated on the perovskite surface can be repeatedly reincorporated into the parent perovskite and subsequently exsolved onto the surface preserving uniform and narrow size distribution with no loss of catalytic activity.
Investigation of the oxidation state of metal ions in LSNF and Red-LSNF
In the process of co-electrolysis of CO2 and H2O, the oxidation state and the atomic concentration of LSNF electrodes on their surfaces have substantial effects on electrochemical performance. To quantify such characteristics, X-ray photoelectron spectroscopy (XPS), a surface sensitive technique with a typical penetration depth of 5 nm, was conducted on LSNF and Red-LSNF. As illustrated in Fig. 7, the normalized XPS spectra in the Fe 2p, O 1s and Sr 3d regions were plotted as a function of binding energy for both LSNF and Red-LSNF. The graph also displays the atomic composition of the samples calculated from the respective XPS spectra.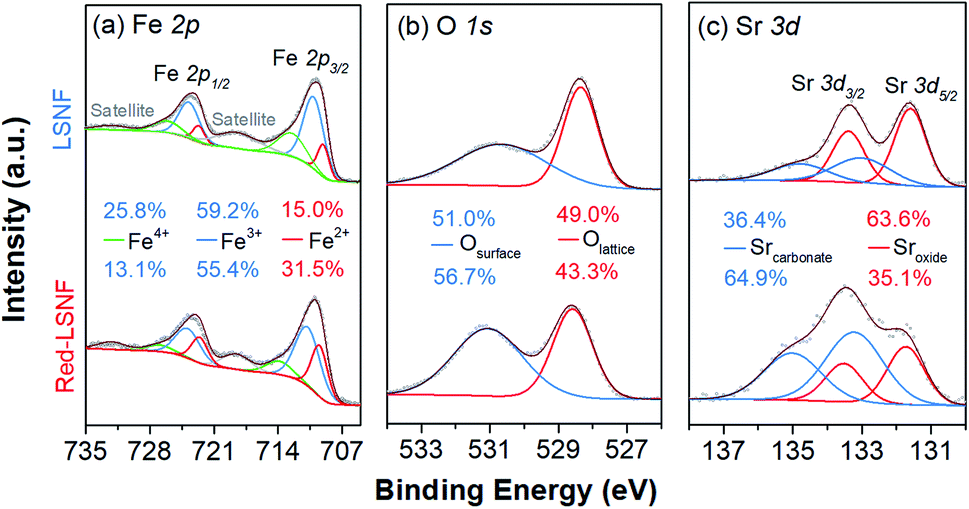 | ||
| Fig. 7 X-ray photoelectron spectra of LSNF and Red-LSNF powders: (a) Fe 2p, (b) O 1s, (c) Sr 3d. | ||
It was reported that the binding energy difference between the peak of Fe 2p3/2 and the satellite peak of Fe 2p3/2 can be used to differentiate between Fe2+ and Fe3+.55
| ΔFe 2p3/2 = BE[Sat(Fe 2p3/2)] − BE[Fe 2p3/2] |
Compounds with Fe3+ show around 8 eV of ΔFe 2p3/2, which is greater than those with Fe2+ (4.3–5.65 eV). Since ΔFe 2p3/2 was estimated to be around 8 eV for both LSNF and Red-LSNF in Fig. 7(a), this indicates that Fe3+ species are predominantly present on the surface. For Fe3+ species, the components that have binding energies of around 710.8 eV and 723.9 eV correspond to Fe 2p3/2 and Fe 2p1/2, respectively.44,56 713.9 eV and 727.0 eV are the peaks that can be attributed to Fe4+. Fe2+ peaks were observed at 709.5 eV and 722.6 eV for Fe 2p3/2 and Fe 2p5/2, respectively.57 As a result of reduction, the concentration of Fe2+ increased from 15.0% to 31.5% but Fe3+ is still dominant, 55.4%, which agrees with the analysis of ΔFe 2p3/2. It has been known that having Fe4+/Fe3+ redox couples in lanthanum ferrite perovskites (LSFs) is very crucial as they act as p-type charge carriers, therefore allowing for electronic conductivity through the catalysts.38 Based on in situ AP-XPS study, however, it has been found that the majority of electrons are located on states of O 2p character rather than Fe 3d and when electrochemically oxidizing the material with large anodic overpotentials, the Fe3+/Fe4+ state has also been observed in in situ AP-XAS study to be too far from the Fermi level.58,59
The peaks in O 1s XPS spectra were deconvoluted into two peaks at 528.3 eV for lattice oxygen and 530.7 eV for surface oxygen in Fig. 7(b).60,61 The surface oxygen includes hydroxyl group and carbonate species. The higher concentration of the surface oxygen on Red-LSNF than that on LSNF could be due to the hydroxyl group formed during the reduction under hydrogen. Moreover, the oxygen vacancies on the surface produced during reduction can be occupied by atmospheric CO2 which consequently forms carbonate.
It appears in Fig. 7(c) that Sr 3d was composed of two distinct species with an oxidation state of +2. The strontium oxide species on LSCF were corresponding to the Sr 3d5/2 peak at 131.7 eV, and the strontium carbonate species was found at 133.2 eV.62,63 Due to the migratory nature of strontium atoms and the instability of the SrO phase, they readily react with CO or CO2 to form stable strontium carbonates (SrCO3) on the surface.9 On the surface of Red-LSNF, the atomic concentration of carbonate was 64.9%, which is higher than 36.4% on LSNF. It has to do with the fact that the reduction of LSNF significantly increased the concentration of oxygen vacancies on the surface, which have been suggested to be the adsorption site for CO2.8
Since XPS is a surface sensitive technique, it is possible that the atmospheric gas reacts with species on the surface of samples when they are exposed to atmosphere, especially NiFe alloy, even though we kept the reducing environment during the cool period after reduction. However, the primary reasons why nickel could not be observed using XPS are likely its low concentration, low sensitivity, and peak overlap between Ni 2p and La 3d.
X-ray absorption near edge structure (XANES) could provide more reliable information on the oxidation state of Fe and Ni since it is a bulk-sensitive. XANES was used to investigate the relative reducibility of the B-site metal ions in LSNF. Fig. 8(a and b) illustrates the Fe K-edge and Ni K-edge XANES spectra for LSNF and Red-LSNF. The XANES spectra of Fe and Ni standards in the form of oxides as well as Fe and Ni metal foils were first obtained and shown in black and grey lines. It can be seen in Fig. 7(a) that the edge energy of Fe in LSNF is 7127.79 eV higher than that of Fe2O3 indicating that the oxidation state of Fe is higher than 3+. This is a typical characteristic found in lanthanum ferrite perovskite oxides when Sr atoms are doped on A-sites.38 After the reduction, the Fe K-edge binding energy in Red-LSNF was determined to be 7126.71 eV, which was 1.08 eV lower than that of LSNF. In the case of Ni ions, the binding energy of Ni K-edge decreased from 8349.09 eV to 8346.37 eV after the reduction of LSNF. There was a −2.72 eV difference in the Ni K-edge energy between LSNF and Red-LSNF, which was greater than −1.08 eV difference between two samples for Fe K-edge energy. In other words, Ni is more prone to reduction than Fe, which is why it is more easily exsolved onto the surface than Fe atoms.
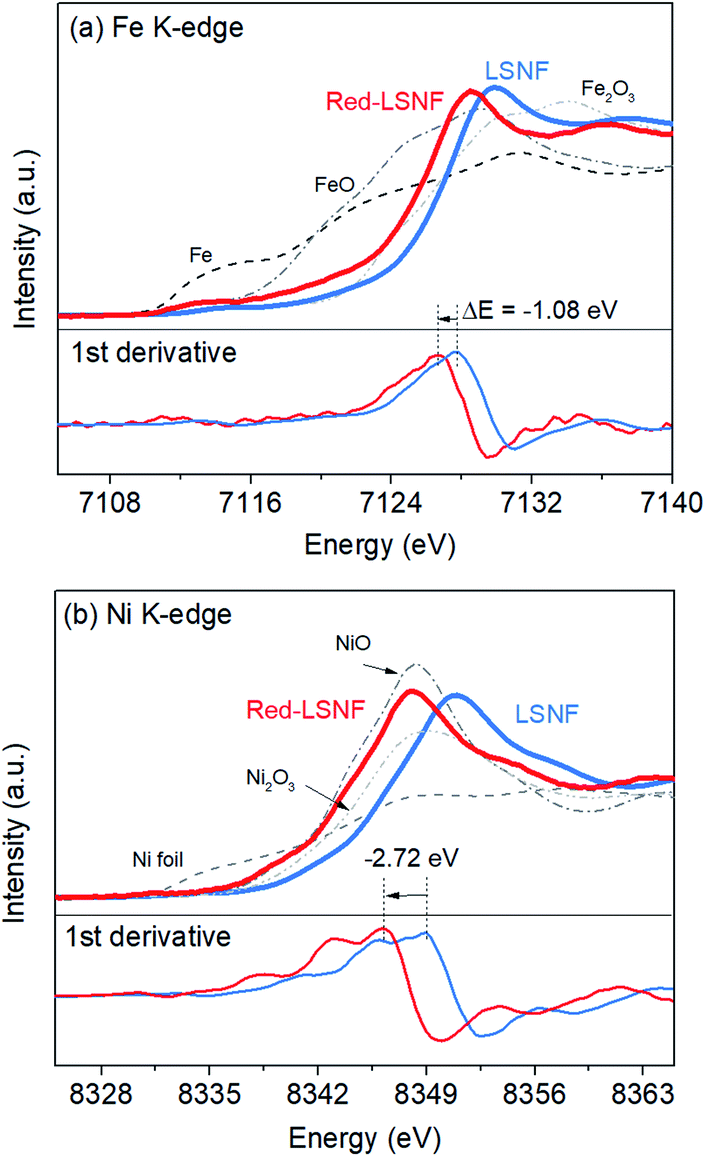 | ||
| Fig. 8 XANES of LSNF and Red-LSNF powders: (a) Fe and (b) Ni K-edge. | ||
TPD-DRIFTS on LSNF and Red-LSNF for H2O and CO2
It should be noted that during the reduction process, heterogeneous phases La2NiO4 and zerovalent Ni could form on the surface of Red-LSNF and affect its surface adsorption–desorption properties. Moreover, the oxygen vacancy sites formed during the reduction has been suggested to work as an effective adsorption site for CO2. In order to reveal any differences in adsorption–desorption performance between LSNF and Red-LSNF, TPD-DRIFTS using CO2 as the probe molecule was conducted on both samples. CO2 gas was adsorbed on LSNF and Red-LSNF powder samples at 50 °C for 20 min. As shown in Fig. 9(a), CO2 was molecularly adsorbed on both samples as evidenced with the peaks at 2380 cm−1 and 2330 cm−1, while the chemisorbed carbonate species with the adsorption peaks located at 1530 cm−1 and 1380 cm−1 were only observable on Red-LSNF.38,64,65 In both samples, physisorbed CO2 peaks gradually declined with increasing temperature. The molecular CO2 peaks were present on LSNF up to 200 °C, and then disappeared completely at 250 °C. Even though the intensity diminished, those peaks remained on Red-LSNF until 250 °C indicative of a stronger adsorption of CO2. On the other hand, the chemisorbed CO2 peaks appeared only on Red-LSNF. This observation can be attributed to the increased concentration of oxygen vacancy sites on the surface of Red-LSNF since carbonate species can be produced under CO2 atmosphere in the form of SrCO3.7 Moreover, it was reported that zerovalent NiFe nanoparticles showed a strong interaction with CO2, especially when coupled with oxygen vacancy.66 Since the chemisorbed carbonate is considered to be an intermediate for CO2 reduction to CO, Red-LSNF is expected to offer a better catalytic activity for CO2 electrolysis than LSNF.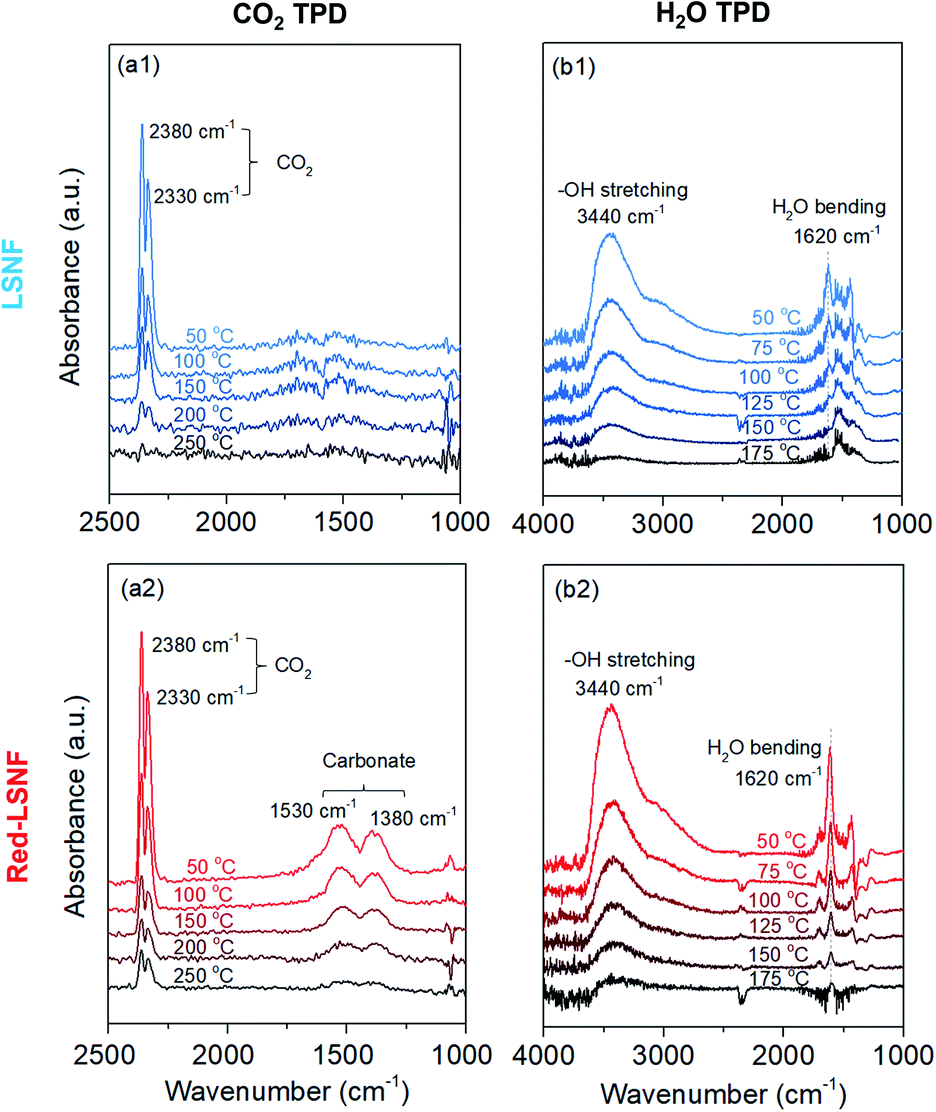 | ||
| Fig. 9 In situ TPD-DRIFTS of (a) CO2 and (b) H2O on (1) LSNF and (2) Red-LSNF. | ||
H2O-TPD was conducted separately from CO2-TPD because the absorbance of H2O bending is located in a similar range as the carbonate. Initially, the water was adsorbed at 50 °C and then the temperature increased under He atmosphere until there was no significant peak. The broad peak located at 3440 cm−1 (between 3100 cm−1 and 3500 cm−1) corresponding to the stretch modes of hydrogen-bonded OH appeared on the as-calcined and Red-LSNF in Fig. 9(b).67,68 Water molecules prefer to establish hydrogen bonds with their nearest neighbors in a tetrahedral bonding geometry. Even though the intensity of the peak was slightly higher on Red-LSNF than that on LSNF, the difference was not significant. This indicates that the hydroxide formation on the material surface is not confined to oxygen vacancy sites and exsolved nanoparticles, since LSNF is also capable of forming hydroxide on its surface when it is exposed to water vapor. There was also evidence of the OH bending vibration of residual adsorbed water at 1620 cm−1.67,69 Hydrogen bonds cause water molecules to vibrate at a frequency higher than the frequency of freely rotating water molecules with a frequency of 1595 cm−1. It corresponds to the frequency at 1620 cm−1 for dimeric water. An important observation is that the OH bending vibration peak on Red-LSNF was stronger and more thermally stable, implying a stronger interaction, compared to that on LSNF.
Electrochemical performance for co-electrolysis of CO2 and H2O
The XRD pattern of LSNF cathode on GDC showed the intact crystal structure after sintering at 1300 °C in Fig. S3.† The electrocatalytic performance of LSNF and Red-LSNF cells for electrolysis of H2O, CO2 and H2O/CO2 was compared through characteristic current–voltage (I–V) polarization curves and Nyquist plots. The resultant I–V polarization curve in Fig. 10(a) shows that the current densities on the Red-LSNF cell measured were 451, 318, and 446 mA cm−2 at 2 V (vs. OCV) for electrolysis of H2O, CO2, and H2O/CO2, respectively. It was an improvement of 66–73% over the current densities on the LSNF cell for the corresponding electrolysis. Nyquist plots under OCV are presented in Fig. 10(b) for H2O, CO2, and H2O/CO2 electrolysis on LSNF and Red-LSNF. The ohmic resistance, RS, can be attributed to the electrolyte thickness of 125 μm, the contact resistance at the interfaces, and the resistance contribution of the current collectors and contacts.70 In spite of the reduction of LSNF, there was no significant change in RS for H2O electrolysis, whereas moderate decreases were observed in RS from 2.85 Ω cm2 to 2.03 Ω cm2 for CO2 and from 2.40 Ω cm2 to 1.89 Ω cm2 for H2O/CO2 electrolysis, respectively. On the other hand, the reduction of LSNF cells led to a remarkable improvement in polarization resistances, RP. The total polarization resistance, RPt, on the LSNF cells was greater than 30 Ω cm2 for all three electrolysis reactions. RPt was, however, reduced to 6.23, 7.73, and 5.40 Ω cm2 for H2O, CO2, and H2O/CO2 electrolysis, respectively, after the reduction of the LSNF electrode. The enhanced electrocatalytic performance of Red-LSNF could be ascribed to the improved adsorption properties for H2O and CO2 and reduced oxidation states of Fe and Ni, originated from the exsolved NiFe nanoparticles and abundant oxygen vacancy sites. From the results of TPD-DRIFTS in Fig. 9, the chemisorbed CO2 was found in the carbonate form only on Red-LSNF and XPS analysis in Fig. 7 disclosed the carbonate species on the surface was increased from 36.4% to 64.9% after 5 h reduction of LSNF under H2 atmosphere. A possible reaction mechanism suggested for CO2 electrolysis was that oxygen vacancies and electrons are required to form a bidendate (CO3)˙3− adsorbate which was proposed as the decisive intermediate of CO2 activation.8 Moreover, in the heterojunction interface between NiFe nanoparticles interacting with oxygen vacancies on perovskite oxides, the NiFe nanoparticles could act synergistically to facilitate electrolysis.66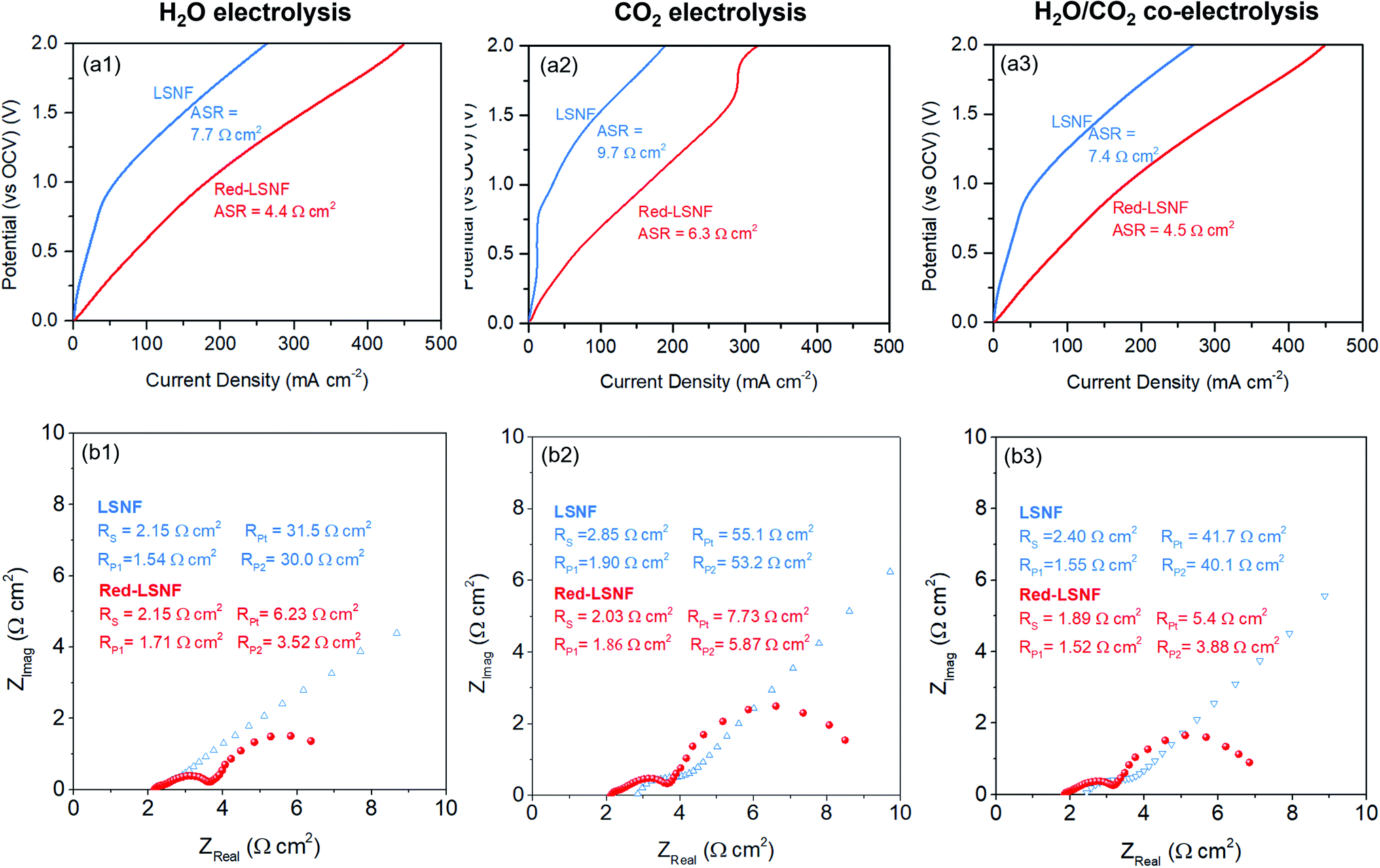 | ||
| Fig. 10 (a) Current–voltage polarization curves and (b) Nyquist plots under OCV of LSNF and Red-LSNF cells for (1) H2O, (2) CO2, (3) H2O/CO2 electrolysis. | ||
The Red-LSNF cell voltage was measured to be 0.943 V under OCV for H2O/CO2 electrolysis at 800 °C. In closed circuit, the cell voltage of the Red-LSNF for co-electrolysis of H2O/CO2 increased from 0.19 V (vs. OCV) to 0.81 V (vs. OCV) as the applied current increased from 50 mA cm−2 to 200 mA cm−2 as shown in Fig. 11(a). The area specific resistances (ASRs) derived from the polarization curve were estimated to be around 4 Ω cm2. The Red-LSNF cell was highly stable in the cell voltage under the co-electrolysis condition at all applied current densities.
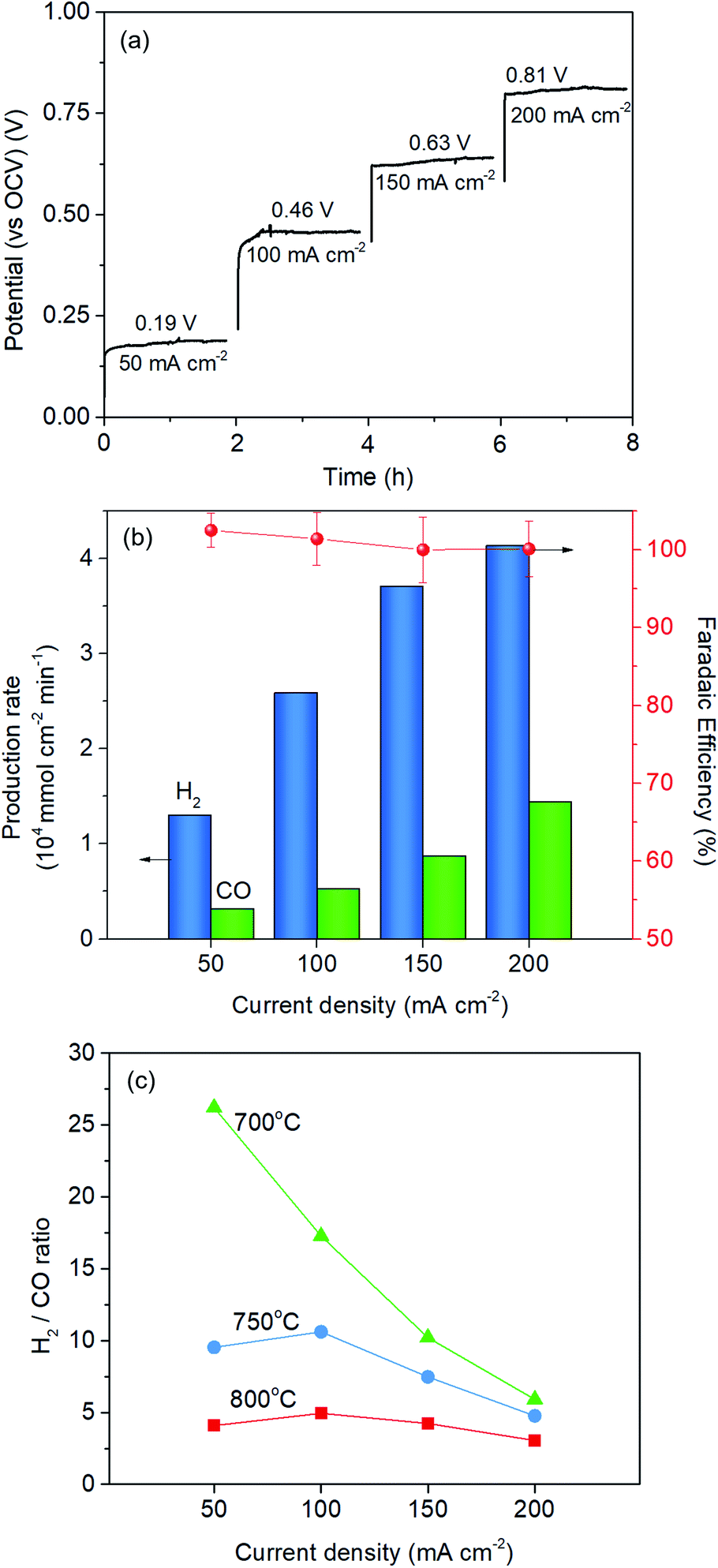 | ||
| Fig. 11 (a) Current–voltage relationship of the Red-LSNF cell, (b) normalized production rate of H2 and CO with faradaic efficiency for co-electrolysis of H2O/CO2 at 800 °C at different current densities, (c) H2/CO ratio at different temperatures and at different current densities. | ||
The normalized production rates of H2 and CO on the Red-LSNF cell during the co-electrolysis operation are displayed with faradaic efficiency at the corresponding applied current densities in Fig. 11(b). With increasing current densities, the production rates of both H2 and CO increase while faradaic efficiency remains around 100% with all current densities. When the same amount of H2O and CO2 was supplied to the system, the electrolysis of H2O was dominant over that of carbon dioxide due to the slower overall kinetics of CO2 electrolysis.5 CO2 reduction would have taken place as a result of the involvement of reverse water-gas shift reaction (RWGS).3,5 The CO production rate under OCV was 0.209 × 104 mmol cm−2 min−1, which is 51.1% for the rate at 50 mA cm−2 (0.318 × 104 mmol cm−2 min−1) and 14.5% for the rate at 200 mA cm−2 (1.442 × 104 mmol cm−2 min−1). As shown by the established model in literature, the extent of RWGS during co-electrolysis of CO2 and H2O is dependent on operating temperature, gas composition, and applied current (or voltage).71,72 An approximate identification of the role of RWGS can be made by analyzing the ASRs of the electrolysis cells operated in three different electrolysis, H2O, CO2 and H2O/CO2. From the results of the polarization curve in Fig. 10(a), ASRs for H2O and CO2 were measured to be 4.4 Ω cm2 and 6.3 Ω cm2, while the ASR for H2O/CO2 was 4.5 Ω cm2. Since the ASR for co-electrolysis H2O/CO2 was close to that for H2O electrolysis, a greater portion of the applied current was supposedly consumed by the reduction of H2O, not by CO2 reduction.
The effect of temperature on H2/CO ratio was investigated in the range of 700–800 °C. At 800 °C and 750 °C, the H2/CO ratio slightly increased with increasing the current density from 50 mA cm−2 to 100 mA cm−2 and then gradually decreased, while the ratio continuously and substantially decreased at 700 °C. This could be explained by the fact that RWGS reaction actively occurred at higher temperatures. The CO production rate through RWGS reaction under OCV was 0.209, 0.141, and 0.0775 × 104 mmol cm−2 min−1 at 800 °C, 750 °C, and 700 °C, respectively. Based on the results in Fig. 11(c), the H2/CO ratio would be readily controlled by temperature or current density.
Conclusions
Some key features of LSNF perovskites are their thermochemical stability, superior ionic and electronic conductivity, and their unique structural utility and adaptibility, making them excellent electrode catalysts in SOECs. LSNF perovskite undergoes exsolution of B-site ions, Ni and Fe, as well as structural transformation to RP phase under 5% H2/N2 atmosphere at various temperatures from 500 °C to 800 °C, as verified by different characterization techniques such as TPR, ETEM and in situ XRD. The exsolved nanoparticles obtained uniform size distribution (4.2–9.2 nm) and dispersion (1.31 to 0.61 × 104 particle per μm2) based on the reduction temperature (700–800 °C) and time (0–10 h). Further, it was observed that LSNF can be re-oxidized by treatment at 800 °C for 2 h, and this allows for reestablishing the nanoparticle distribution as needed. Based on the results of XPS and XANES, the reduction of LSNF contributed to the decreases in oxidation states of Fe and Ni ions that led to Red-LSNF being more catalytically active for co-electrolysis of H2O and CO2. This also resulted in increased amount of oxygen vacancy sites, which led to a stronger affinity toward H2O and CO2, confirmed by TPD-DRIFTS. The reduction of LSNF ultimately improved the electrochemical performance by 66–73% on ASRs for H2O, CO2, and H2O/CO2 electrolysis at 800 °C.Experimental
Preparation of catalysts
La0.7Sr0.2Ni0.2Fe0.8O3 (LSNF) was synthesized via citric acid–ethylenediaminetetraacetic acid (EDTA) complexation method as described in our earlier publication.49 Metal-nitrate salts were dissolved in 100 ml of deionized water in stoichiometric quantities. At room temperature, EDTA was added to the solution with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio compared to total metal ions. At a solution temperature of 60 °C, ammonium hydroxide was added until the pH stabilized at 6. Citric acid and ethylene glycol were added, and the solution was further heated to 90 °C while maintaining the pH at 6 using ammonium hydroxide. After evaporation, the formed gel was dried at 150 °C overnight. The dried black powder was calcined at 1000 °C for 5 h to form crystalline LSNF. For the preparation of reduced LSNF samples (Red-LSNF), the calcined LSNF powders were reduced under 5% H2/N2 at 800 °C.
:1 molar ratio compared to total metal ions. At a solution temperature of 60 °C, ammonium hydroxide was added until the pH stabilized at 6. Citric acid and ethylene glycol were added, and the solution was further heated to 90 °C while maintaining the pH at 6 using ammonium hydroxide. After evaporation, the formed gel was dried at 150 °C overnight. The dried black powder was calcined at 1000 °C for 5 h to form crystalline LSNF. For the preparation of reduced LSNF samples (Red-LSNF), the calcined LSNF powders were reduced under 5% H2/N2 at 800 °C.
Characterization
X-ray diffraction patterns of the catalysts were analyzed in a Bruker D8 Lead X-ray powder diffractometer using a Cu Kα X-ray source. The generator operated at 40 kV and 40 mA. Scans were performed within the 2θ range of 20° to 80°, and the step size was 0.02° per 0.5 s. Phase identification was done using the crystallographic open database (COD).73 Miller indices were calculated via Rietveld refinement of the XRD patterns using General Structure Analysis System-2 (GSAS-2) software.74In situ XRD required the use of an Anton Paar HTK1200 oven under 5% H2/N2 environment at various temperatures from 30 °C to 800 °C.The surface elemental oxidation state and composition were analyzed by X-ray photoelectron spectroscopy (XPS) using a Kratos Axis Ultra XPS instrument equipped with a monochromated Al Kα X-ray source (1254 eV, 12 kV, 10 mA) and a charge neutralizer at 2.05 A of element current, 1.3 V of filament bias, and 3.6 V of charge balance. Each scan was calibrated using the C 1s standard peak at 284.5 eV. The peaks for Fe 2p, O 1s, and Sr 3d were studied with high resolution scans, performing 8 sweeps for Sr 3d and O 1s and 16 sweeps for Fe 2p with a 300 ms dwell time in the narrowed binding energy ranges.
FEI Titan ETEM G2 80–300 kV instrument equipped with an objective Cs aberration corrector was used to conduct an environmental transmission electron microscopy (ETEM) characterization. ETEM study was performed at various temperatures under 1 mbar H2 atmosphere controlled by mass flow controllers and monitoring the effluent by mass spectroscopy. The concentration of hydrogen was determined by continuously analyzing the effluent leaving the microscope chamber using a mass spectrometer (Pfeiffer). Scanning transmission electron microscopy (STEM) studies were conducted to investigate Red-LSNF using a TECNAI F20 TEM operating at 200 kV equipped with an energy dispersive X-ray spectroscopy (EDS).
For temperature-programmed reduction/oxidation (TPR/TPO), sample powders were prepared by packing a quartz tube with 50 mg catalyst powder. A gas mixture was flowed through the catalyst bed, sending a portion of the effluent to an MKS Cirrus mass spectrometer. 30 ccm of 5% H2/N2 was used for TPR and 2% O2/He for TPO. The reactor was heated from room temperature to 1000 °C with a ramp rate of 20 °C min−1.
X-ray absorption near edge spectroscopy (XANES) was conducted at Sector 10-BM of the Materials Research Collaborative Access Team (MRCAT) at the Advanced Photon Source (APS, Argonne National Laboratory). The instrument was operated in transmission mode for all samples. The catalyst powders were diluted with boron nitride in a ratio of 1:5. The Fe K-edge and Ni K-edge were scanned at 7112 eV and 8333 eV, respectively, for both catalyst samples. The corresponding metal foils were used as references to calibrate the K-edge spectra. Athena software was utilized to process the collected data.75
Temperature-programmed desorption-diffuse reflectance infrared Fourier transform spectroscopy (TPD-DRIFTS) data was collected to observe the difference in affinity of CO2 and H2O towards LSNF and Red-LSNF using Thermoelectron Nicolet 6700 FTIR equipped with an MCT detector. Powder samples were diluted with potassium bromide (KBr) in a 1:20 ratio of catalyst to KBr. The sample was pretreated with helium at 450 °C to desorb any additional adsorbed species and exposed to CO2 or H2O at 50 °C.
Evaluation of electrochemical performance
Electrochemical button cells were fabricated by screen-printing on commercial yttria-stabilized zirconia (YSZ) electrolyte (25 mm diameter, 125 μm thickness, Nextech Materials). Gadolinium doped ceria interlayer was printed on the cathode side and sintered at 1400 °C for 2 h under N2 flow. LSNF was printed on the GDC layer and sintered at 1300 °C under air. LSM-YSZ was printed on the anode side and sintered at 1200 °C under air. Gold mesh was attached onto the electrodes using platinum paste. The electrode area was 0.42 cm2. Red-LSNF cathode was prepared by treating LSNF cathode under 5% H2/He for 5 h at 800 °C after sealing the button cell and 3.3% H2 was continuously flowed into the reactor during the electrochemical tests. For the electrochemical performance test, the feed gas concentrations at the cathode were (a) 3.3% H2O, (b) 3.3% CO2, and (c) 3.3% H2O and 3.3% CO2 balanced He with a total flowrate of 45 ccm. The electrochemical impedance spectra (EIS) were collected in the frequency range from 1 MHz to 0.1 Hz under open-circuit voltage (OCV) and linear sweep voltammetry (LSV) was carried out from 0 to 2 V (vs. OCV) with a 100 mV s−1 scan rate. The equivalent circuit model of RS(RP1Q1)(RP2Q2) was used consisting of an ohmic resistance RS and two contributions to polarization impedance (RP) and constant phase elements (Q) in parallel. Quantification of the gas products was performed on an on-line gas chromatograph (Shimadzu 2014) equipped with a pulse discharge ionization detector.Author contributions
Jaesung Kim: conceptualization, investigation, methodology, data curation, formal analysis, writing – original draft. Matthew Ferree: investigation, formal analysis, writing – review & editing. Seval Gunduz: investigation, formal analysis, writing – review & editing. Jean-Marc M. Millet: investigation, formal analysis, writing – review. Mimoun Aouine: investigation, formal analysis, writing – review. Anne C. Co: formal analysis, writing – review. Umit S. Ozkan: conceptualization, funding acquisition, project administration, resources, supervision, formal analysis, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We would like to gratefully acknowledge the financial support provided for this work by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under the Award Number DE-FG02-07ER15896 and the U.S. National Science Foundation, under the award number 1932638. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Part of the electron microscopy imaging was performed at the Center for Electron Microscopy and Analysis (CEMAS) at the Ohio State University.References
- E.S.R. Laboratory, Global Monitoring Laboratory, Trends in atmospheric carbon dioxide, 2019 Search PubMed.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- F. Alenazey, Y. Alyousef, O. Almisned, G. Almutairi, M. Ghouse, D. Montinaro and F. Ghigliazza, Int. J. Hydrogen Energy, 2015, 40, 10274–10280 CrossRef CAS.
- D. J. Cumming, A. R. Thompson and R. H. Rothman, J. Electrochem. Soc., 2016, 163, F3057–F3061 CrossRef CAS.
- S.-W. Kim, M. Park, H. Kim, K. J. Yoon, J.-W. Son, J.-H. Lee, B.-K. Kim, J.-H. Lee and J. Hong, Appl. Catal., B, 2017, 200, 265–273 CrossRef CAS.
- Y. Li, P. Li, B. Hu and C. Xia, J. Mater. Chem. A, 2016, 4, 9236–9243 RSC.
- D. J. Deka, J. Kim, S. Gunduz, D. Jain, Y. Shi, J. T. Miller, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2021, 283, 119642 CrossRef CAS.
- A. K. Opitz, A. Nenning, C. Rameshan, M. Kubicek, T. Gotsch, R. Blume, M. Havecker, A. Knop-Gericke, G. Rupprechter, B. Klotzer and J. Fleig, ACS Appl. Mater. Interfaces, 2017, 9, 35847–35860 CrossRef CAS PubMed.
- L. Ye, C. Pan, M. Zhang, C. Li, F. Chen, L. Gan and K. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 25350–25357 CrossRef CAS PubMed.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- X. Zhang, S. H. Chan, H. K. Ho, S.-C. Tan, M. Li, G. Li, J. Li and Z. Feng, Int. J. Hydrogen Energy, 2015, 40, 6866–6919 CrossRef CAS.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- T. Ishihara, S. Wang and K.-T. Wu, Solid State Ionics, 2017, 299, 60–63 CrossRef CAS.
- C. K. Lim, Q. Liu, J. Zhou, Q. Sun and S. H. Chan, Fuel Cells, 2017, 17, 464–472 CrossRef CAS.
- M. Maide, K. Lillmaa, G. Nurk and E. Lust, ECS Trans., 2017, 78, 3275–3281 CrossRef CAS.
- M. Torrell, S. Garcia-Rodriguez, A. Morata, G. Penelas and A. Tarancon, Faraday Discuss., 2015, 182, 241–255 RSC.
- X. Yue and J. T. S. Irvine, Solid State Ionics, 2012, 225, 131–135 CrossRef CAS.
- K. Huang, Sci. Bull., 2016, 61, 1783–1784 CrossRef.
- D. Neagu, V. Kyriakou, I. L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, ACS Nano, 2019, 13, 12996–13005 CrossRef CAS.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Menard and J. T. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- Y. Sun, J. Li, Y. Zeng, B. S. Amirkhiz, M. Wang, Y. Behnamian and J. Luo, J. Mater. Chem. A, 2015, 3, 11048–11056 RSC.
- S.-K. Otto, K. Kousi, D. Neagu, L. Bekris, J. Janek and I. S. Metcalfe, ACS Appl. Energy Mater., 2019, 2, 7288–7298 CrossRef CAS.
- S. Joo, O. Kwon, K. Kim, S. Kim, H. Kim, J. Shin, H. Y. Jeong, S. Sengodan, J. W. Han and G. Kim, Nat. Commun., 2019, 10, 697 CrossRef CAS.
- Y. R. Jo, B. Koo, M. J. Seo, J. K. Kim, S. Lee, K. Kim, J. W. Han, W. Jung and B. J. Kim, J. Am. Chem. Soc., 2019, 141, 6690–6697 CrossRef CAS PubMed.
- B. Hua, M. Li, Y. F. Sun, J. H. Li and J. L. Luo, ChemSusChem, 2017, 10, 3333–3341 CrossRef CAS PubMed.
- J. G. Lee, J. H. Myung, A. B. Naden, O. S. Jeon, Y. G. Shul and J. T. S. Irvine, Adv. Energy Mater., 2020, 10, 2070044 CrossRef CAS.
- D. Neagu, T. S. Oh, D. N. Miller, H. Menard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- S. P. Jiang, Int. J. Hydrogen Energy, 2012, 37, 449–470 CrossRef CAS.
- S. Kim, A. Jun, O. Kwon, J. Kim, S. Yoo, H. Y. Jeong, J. Shin and G. Kim, ChemSusChem, 2015, 8, 3153–3158 CrossRef CAS.
- J. H. Myung, D. Neagu, D. N. Miller and J. T. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS PubMed.
- V. Kyriakou, D. Neagu, E. I. Papaioannou, I. S. Metcalfe, M. C. M. van de Sanden and M. N. Tsampas, Appl. Catal., B, 2019, 258, 117950 CrossRef CAS.
- T. Zhu, H. E. Troiani, L. V. Mogni, M. Han and S. A. Barnett, Joule, 2018, 2, 478–496 CrossRef CAS.
- T. Wei, L. Jia, J.-L. Luo, B. Chi, J. Pu and J. Li, Appl. Surf. Sci., 2020, 506, 144699 CrossRef CAS.
- K.-Y. Lai and A. Manthiram, Chem. Mater., 2018, 30, 2838–2847 CrossRef CAS.
- Y. Wang, T. Liu, M. Li, C. Xia, B. Zhou and F. Chen, J. Mater. Chem. A, 2016, 4, 14163–14169 RSC.
- T. Liu, Y. Zhao, X. Zhang, H. Zhang, G. Jiang, W. Zhao, J. Guo, F. Chen, M. Yan, Y. Zhang and Y. Wang, J. Mater. Chem. A, 2020, 8, 582–591 RSC.
- T. Götsch, L. Schlicker, M. F. Bekheet, A. Doran, M. Grünbacher, C. Praty, M. Tada, H. Matsui, N. Ishiguro, A. Gurlo, B. Klötzer and S. Penner, RSC Adv., 2018, 8, 3120–3131 RSC.
- D. J. Deka, S. Gunduz, T. Fitzgerald, J. T. Miller, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2019, 248, 487–503 CrossRef CAS.
- Y. S. Chung, T. Kim, T. H. Shin, H. Yoon, S. Park, N. M. Sammes, W. B. Kim and J. S. Chung, J. Mater. Chem. A, 2017, 5, 6437–6446 RSC.
- L. Sangaletti, L. E. Depero, B. Allieri, P. Nunziante and E. Traversa, J. Eur. Ceram. Soc., 2001, 21, 719–726 CrossRef CAS.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- G. Xiong, Z.-L. Zhi, X. Yang, L. Lu and X. Wang, J. Mater. Sci. Lett., 1997, 16, 1064–1068 CrossRef CAS.
- R. Chockalingam, A. K. Ganguli and S. Basu, J. Power Sources, 2014, 250, 80–89 CrossRef CAS.
- W. Y. Hernández, M. N. Tsampas, C. Zhao, A. Boreave, F. Bosselet and P. Vernoux, Catal. Today, 2015, 258, 525–534 CrossRef.
- Y. Teraoka, Y. Honbe, J. Ishii, H. Furukawa and I. Moriguch, Solid State Ionics, 2002, 150–153, 681–687 CrossRef.
- N. Li, A. Boréave, J.-P. Deloume and F. Gaillard, Solid State Ionics, 2008, 179, 1396–1400 CrossRef CAS.
- M. Alifanti, J. Kirchnerova, B. Delmon and D. Klvana, Appl. Catal., A, 2004, 262, 167–176 CrossRef CAS.
- E. H. Yang, Y. S. Noh, S. S. Lim, B. S. Ahn and D. J. Moon, J. Nanosci. Nanotechnol., 2016, 16, 1938–1941 CrossRef PubMed.
- D. J. Deka, J. Kim, S. Gunduz, M. Aouine, J.-M. M. Millet, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2021, 286, 119917 CrossRef CAS.
- G. Nirala, D. Yadav and S. Upadhyay, J. Adv. Ceram., 2020, 9, 129–148 CrossRef CAS.
- Z. Du, H. Zhao, S. Yi, Q. Xia, Y. Gong, Y. Zhang, X. Cheng, Y. Li, L. Gu and K. Swierczek, ACS Nano, 2016, 10, 8660–8669 CrossRef CAS PubMed.
- T. Zhang, Y. Zhao, X. Zhang, H. Zhang, N. Yu, T. Liu and Y. Wang, ACS Sustainable Chem. Eng., 2019, 7, 17834–17844 CrossRef CAS.
- P. Steiger, M. Nachtegaal, O. Kröcher and D. Ferri, ChemCatChem, 2018, 10, 4456–4464 CrossRef CAS.
- P. Steiger, R. Delmelle, D. Foppiano, L. Holzer, A. Heel, M. Nachtegaal, O. Krocher and D. Ferri, ChemSusChem, 2017, 10, 2505–2517 CrossRef CAS PubMed.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Appl. Surf. Sci., 2000, 165, 288–302 CrossRef CAS.
- Z. Zhao, H. Dai, J. Deng, Y. Du, Y. Liu and L. Zhang, Microporous Mesoporous Mater., 2012, 163, 131–139 CrossRef CAS.
- H. Chang, E. Bjørgum, O. Mihai, J. Yang, H. L. Lein, T. Grande, S. Raaen, Y.-A. Zhu, A. Holmen and D. Chen, ACS Catal., 2020, 10, 3707–3719 CrossRef CAS.
- A. Nenning, A. K. Opitz, C. Rameshan, R. Rameshan, R. Blume, M. Havecker, A. Knop-Gericke, G. Rupprechter, B. Klotzer and J. Fleig, Journal of Physical Chemistry C: Nanomaterials and interfaces, 2016, 120, 1461–1471 CrossRef CAS PubMed.
- D. N. Mueller, M. L. Machala, H. Bluhm and W. C. Chueh, Nat. Commun., 2015, 6, 6097 CrossRef CAS PubMed.
- D. Kim, J. W. Park, M. S. Chae, I. Jeong, J. H. Park, K. J. Kim, J. J. Lee, C. Jung, C.-W. Lee, S.-T. Hong and K. T. Lee, J. Mater. Chem. A, 2021, 9, 5507–5521 RSC.
- A. Machocki, T. Ioannides, B. Stasinska, W. Gac, G. Avgouropoulos, D. Delimaris, W. Grzegorczyk and S. Pasieczna, J. Catal., 2004, 227, 282–296 CrossRef CAS.
- K. Rida, A. Benabbas, F. Bouremmad, M. A. Peña and A. Martínez-Arias, Catal. Commun., 2006, 7, 963–968 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski, Y.-L. Lee, D. Morgan, H. M. Christen, H. Bluhm and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 6081–6088 RSC.
- J. C. Lavalley, Catal. Today, 1996, 27, 377–401 CrossRef CAS.
- D. Dogu, S. Gunduz, K. E. Meyer, D. J. Deka, A. C. Co and U. S. Ozkan, Catal. Lett., 2019, 149, 1743–1752 CrossRef CAS.
- L. Ye, M. Zhang, P. Huang, G. Guo, M. Hong, C. Li, J. T. Irvine and K. Xie, Nat. Commun., 2017, 8, 14785 CrossRef CAS PubMed.
- A. Litke, Y. Su, I. Tranca, T. Weber, E. J. M. Hensen and J. P. Hofmann, Journal of Physical Chemistry C: Nanomaterials and interfaces, 2017, 121, 7514–7524 CrossRef CAS PubMed.
- Q. Du, R. Superfine, E. Freysz and Y. R. Shen, Phys. Rev. Lett., 1993, 70, 2313–2316 CrossRef CAS PubMed.
- D. J. C. Yates, J. Phys. Chem., 1961, 65, 746–753 CrossRef CAS.
- E. Hernández, F. Baiutti, A. Morata, M. Torrell and A. Tarancón, J. Mater. Chem. A, 2018, 6, 9699–9707 RSC.
- D. J. Deka, J. Kim, S. Gunduz, M. Ferree, A. C. Co and U. S. Ozkan, Appl. Catal., A, 2020, 602, 117697 CrossRef CAS.
- Y. Wang, T. Liu, L. Lei and F. Chen, Fuel Process. Technol., 2017, 161, 248–258 CrossRef CAS.
- A. Vaitkus, A. Merkysa and S. Gražulis, J. Appl. Crystallogr., 2021, 54, 661–672 CrossRef CAS PubMed.
- B. H. Toby and R. B. V. Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 537–541 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta07389c |
| This journal is © The Royal Society of Chemistry 2022 |