
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced electrocatalytic hydrogen evolution by molybdenum disulfide nanodots anchored on MXene under alkaline conditions†
Xiaoyu
Wang‡
a,
Wenbin
You‡
b,
Liting
Yang
b,
Guanyu
Chen
b,
Zhengchen
Wu
b,
Chang
Zhang
b,
Qianjin
Chen
 *a and
Renchao
Che
*bc
*a and
Renchao
Che
*bc
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, 201620, P. R. China. E-mail: qianjinchen@dhu.edu.cn
bLaboratory of Advanced Materials, Shanghai Key Lab of Molecular Catalysis and Innovative Materials, Department of Materials Science, Fudan University, Shanghai 200438, P. R. China. E-mail: rcche@fudan.edu.cn
cJoint-Research Center for Computational Materials, Zhejiang Laboratory, Hangzhou 311100, P. R. China
First published on 14th July 2022
Abstract
Efficient hydrogen production through electrocatalysis represents a promising path for the future clean energy. Molybdenum disulfide (MoS2) is a good substitute for platinum-based catalysts, due to its low cost and high activity. However, the limited active sites and low electrical conductivity of MoS2 hinder its large-scale industrial application under alkaline conditions. Herein, we constructed MoS2 nanodots anchored on an MXene/nickel foam (MoS2 NDs/MXene/NF) heterostructure by a cascade polymerization synthesis and in situ vulcanization. The prepared heterostructure displays an ultralow overpotential of 94 mV at a current density of 10 mA cm−2 with a Tafel slope of only 59 mV dec−1 in alkaline (1 M KOH) hydrogen evolution reaction (HER), and is better than conventional MoS2 electrocatalysts reported so far. Fine structural analysis indicates that MoS2 NDs are dispersed uniformly on the surface of the heterostructure with consistent orientation, leading to the improvement of MoS2 conductivity with more paths for electron transfer. Moreover, the orientation of the synthesized MoS2 NDs was verified to expose the more (002) crystal plane, which exhibits higher activity than other planes. Our results demonstrate that MoS2 NDs with heterostructure design and preferential growth can serve as high-efficiency noble-metal free electrocatalysts for the HER in alkaline solution.
Introduction
Hydrogen energy is deemed a promising alternative to fossil fuels, due to its features of high energy capacity and environmental friendliness.1–3 The efficient and economical way of hydrogen production from electrochemical water splitting is a long-standing goal for the energy crisis solution.4–6 Despite the HER being easier under acidic conditions, accompanying corrosion of electrolytic equipment and destruction of catalysts become a huge bottleneck for its industrial application.7 Instead, alkaline electrolysis, due to the long durability of the electrolyzer, the high reliability of the production process, and the long life of the catalyst, has attracted tremendous attention in the past decade.8 Platinum and its derivatives till now are still the most common HER catalysts, both under acidic and alkaline conditions.9,10 Considering the scarcity of noble-metal catalysts, the development of sustainable catalysts with low cost and abundance has been the focus of intensive research efforts.11The concentration of hydrogen ions in alkaline medium is ∼8 to 14 orders of magnitude lower than that in acidic medium, resulting in the drawbacks of sluggish water adsorption and poor dissociation dynamics during water splitting. Thus, greater efforts on increasing the accessibility of active sites and surface modification with water adsorption components have been adopted to develop high-performance alkaline HER electrocatalysts.11–14 Recently, non-precious Mo-based materials have been proved experimentally to replace platinum and its derivatives,11,12 because of the lower Gibbs-energy of hydrogen adsorption analogous to platinum. Typically, MoS2 with a two-dimensional layered nanostructure, recognized as the most important one, shows catalytic sites at its layer edges during the Volmer reaction.15–19 The vertical growth of MoS2 on a flat substrate or nanowire surfaces can expose more edge sites.20,21 Meanwhile, MoS2 nanosheets, even ultrasmall nanodots, are preferable because the accompanying higher specific surface area and more edge atoms strikingly boost the HER catalytic activity.22–24 Worse still, the nanostructures tend to agglomerate after being formed into electrodes, which hinders sustainable electrocatalytic activity, thus far from being satisfactory for practical electrolysis.25,26 As for surface modification to enhance the catalyst hydrophilicity and cleave the HO–H bond, nano-assembly hierarchical structures, such as MoS2-Ni/Co compounds with porous media,14 have been constructed and used as highly efficient electrocatalysts. However, the even worse conductivity of Ni/Co compounds, such as hydroxide, sulfide, and phosphide, causes more energy waste in hydrogen production.
The electrical conductivity enhancement of electrocatalysts can simultaneously accelerate the reaction rate and reduce overpotentials. Due to the semiconducting properties of molybdenum disulfide, a minimized bandgap means less difficulty for carrier migration. Nevertheless, the bulk form of MoS2 tends to have a smaller bandgap (1.2 eV) than that of few-layer MoS2 (1.9 eV for monolayer MoS2). In another word, the MoS2 nanosheets or nanodots possess poor electrical conductivity. Thus, defect engineering strategies, such as introducing holes, cracks, and crimps, have been extensively developed and utilized to minimize the bandgap, and achieve higher active site exposure22,27 at the same time. However, most defects influence slightly the intrinsic property regulation of MoS2. In this context, introducing a highly conductive substrate could simultaneously optimize the HER performance of MoS2 nanosheets or nanodots. Of all substrates, the Ti3C2Tx MXene shows great promise due to its good hydrophilicity and metallic conductivity (up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 S cm−1), having been applied in supercapacitors, water treatment, microwave absorption,28 and electromagnetic interference shielding.10 The structure design between MXene and MoS2 is assumed to improve the conductivity of the electrocatalyst and deliver enough activity toward the HER, while the remaining challenges of uniform dispersion of catalytically active sites and effective protection of the MXene conductive substrate from surface oxidization and anti-aggregation should be overcome first.16,20,29
000 S cm−1), having been applied in supercapacitors, water treatment, microwave absorption,28 and electromagnetic interference shielding.10 The structure design between MXene and MoS2 is assumed to improve the conductivity of the electrocatalyst and deliver enough activity toward the HER, while the remaining challenges of uniform dispersion of catalytically active sites and effective protection of the MXene conductive substrate from surface oxidization and anti-aggregation should be overcome first.16,20,29
Motivated by the above considerations, we developed an MXene/NF substrate with preferential growth MoS2 nanodots (MoS2/MXene/NF), of which the more active edge exposure enhances the HER performance. Notably, the chelated molybdate ions and Ti3C2Tx MXene flakes encapsulated by polydopamine (PDA), used as a precursor, provide uniformly dispersed MoS2 nanodots anchored on MXene with ultra-small size and inhibit surface oxidation of MXenes during annealing and sulfurization treatment.30 The synthesized MoS2/MXene heterostructure, confirmed by morphology, structure, phase, and elemental chemical valence characterization, displays a synergistic effect of active MoS2 nanodots and conductive Ti3C2Tx MXene flakes for fast charge transfer. The optimal MoS2/MXene/NF yields remarkable HER activities with a Tafel slope of 59 mV dec−1 and an overpotential of 94 mV at 10 mA cm−2. Due to its excellent stability over 2000 cycles of cyclic voltammetry tests in an alkaline electrolyte, MoS2/MXene/NF has been certified superior to most non-noble metal catalysts in alkaline solution. This work sheds new insight into nanostructure construction for Mo-based catalysts to enhance their capability of catalytic water decomposition.
Results and discussion
Fig. 1 shows the schematic illustration of a novel strategy for constructing MoS2 NDs/MXene with abundant exposed sites grown on a 3D self-supporting electrode. First, ultrathin MXenes were obtained by selectively etching the Al element in the MAX phase (Ti3AlC2) with hydrofluoric acid and the subsequent exfoliation procedure (experimental section in the ESI†). During the in situ polymerization process of PDA under weakly alkaline conditions, the mixture of few-layer MXenes and molybdate ions was well encapsulated on the NF substrate surface. The molybdate ions were chelated in PDA (Mo-PDA) as the precursor of MoO3, and few-layer MXenes were fully wraped with Mo-PDA. Then, a heterostructure of Ti3C2Tx MXene sheets decorated with MoO3 NDs was obtained by annealing Mo-PDA/Ti3C2Tx/NF at 650 °C under a N2 atmosphere, which inhibits the agglomeration of MoO3 NDs and oxidation of Ti3C2Tx due to the confinement effect of PDA. Finally, as-synthesized MoO3 NDs/MXene was controllably sulfurized to MoS2 NDs/MXene at sulfurization temperatures from 500 to 800 °C in a N2 atmosphere.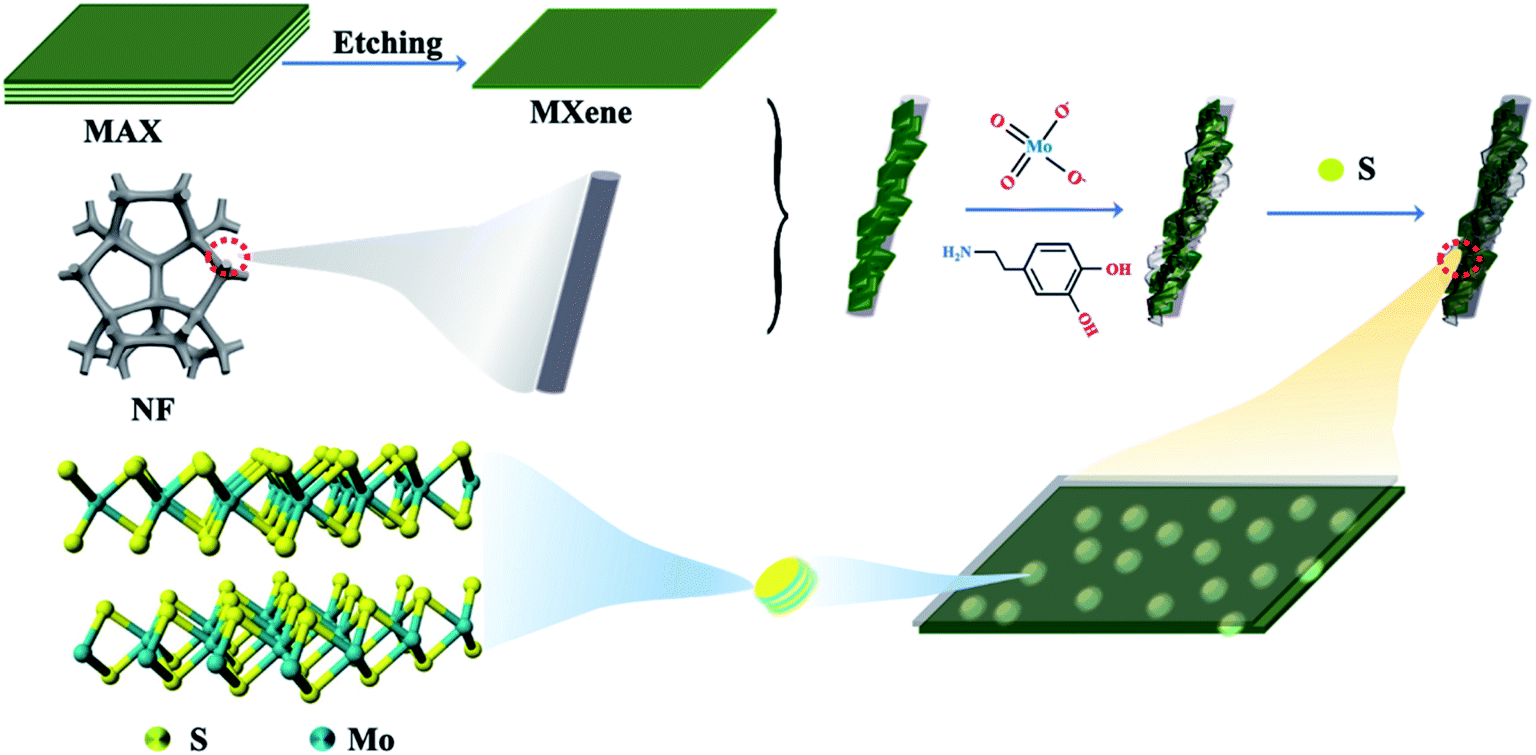 | ||
| Fig. 1 Schematic diagram of the MoS2 NDs/MXene/NF synthetic route. | ||
The products of the schematic step are characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) to verify the synthetic process. The as-prepared MXene shows a layered structure in the SEM image (Fig. S1b†).31 The clear few-layered structure of 2D MXene is further confirmed by the TEM image (Fig. S2a†). By in situ polymerization of dopamine, the relatively smooth surface of NF with micropores (Fig. 2a) is wrapped by a uniformly layered structure network combined with 2D MXene and Mo-PDA (Fig. S1d†). After annealing at 650 °C, the Mo-PDA transforms into MoO3 NDs dispersed in the MXene, while the MoO3/MXene/NF (Fig. 2b) shows a similar morphology to the Mo-PDA/MXene/NF precursor (Fig. S1d†). Moreover, the well-defined ND decorated 2D MXene structure was maintained standing on NF after the sulfurization process (Fig. 2c). The uniform distributions of MoO3 NDs and MoS2 NDs in carbide are more clearly seen in the TEM images (Fig. 2d and e). As for Mo-PDA/NF and its derivative MoS2/NF (Fig. S3a–c†), a flower-like structure with about 400 nm diameter was formed on the NF surface, which is similar to the folded surface in Mo-PDA/MXene/NF (Fig. S1d†). This revealed that the Ti3C2Tx with oxygen- or fluorine-containing terminations was identified as a charged substrate to adsorb dopamine hydrochloride chelated Mo precursors, realizing well dispersed nanostructured Mo compounds.32–34
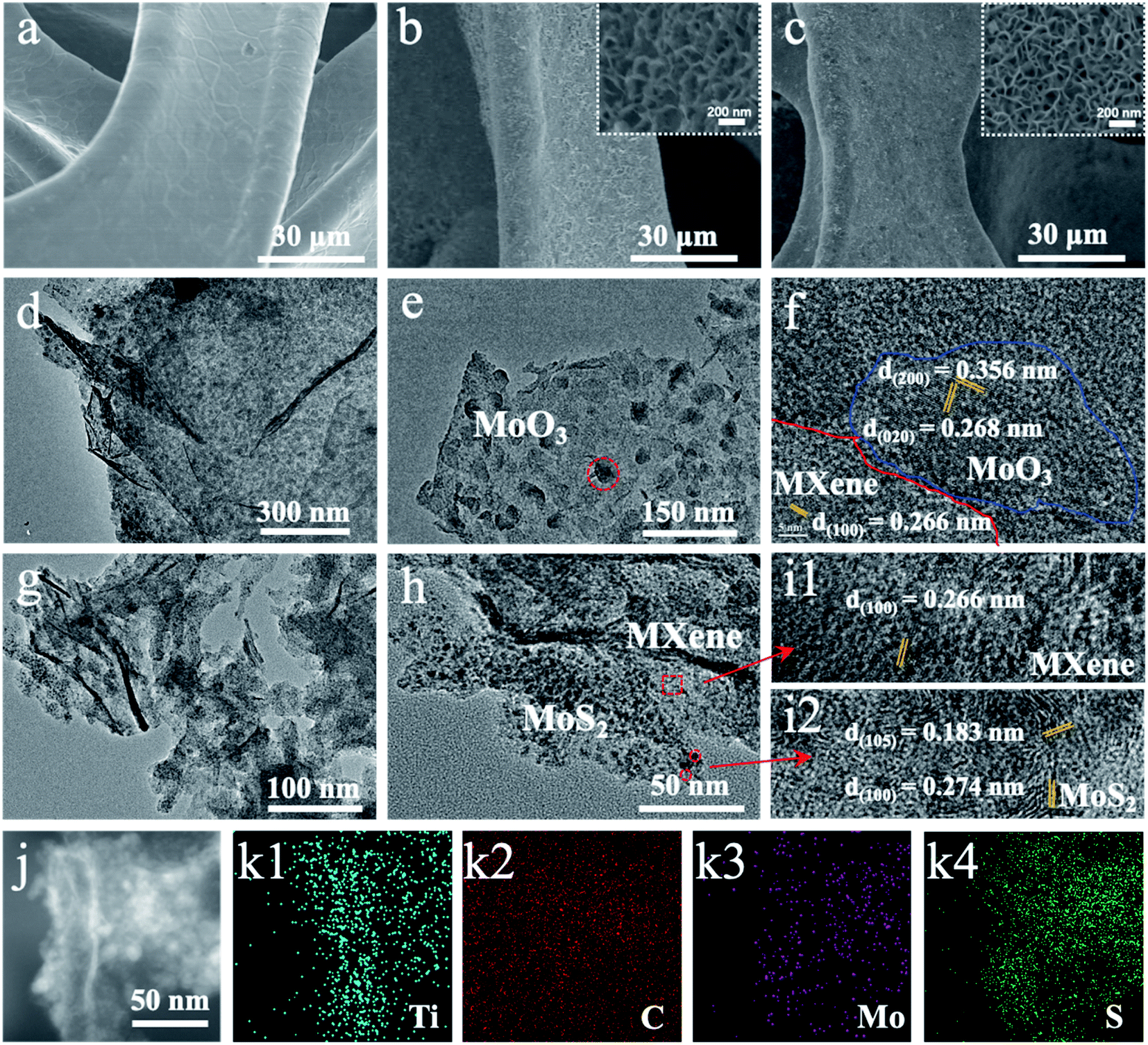 | ||
| Fig. 2 SEM images of (a) NF, (b) MoO3/MXene/NF, and (c) MoS2/MXene/NF. The insets of (b and c) are images at high magnification. (d and e) TEM images, and (f) HRTEM image of MoO3/MXene removed from MoO3/MXene/NF. (g and h) TEM images, (i) HRTEM image, (j) HAADF image, and (k) EDX elemental distributions of MoS2/MXene removed from MoS2/MXene/NF. | ||
The structures of MXene and its composites were further investigated by X-ray diffraction (XRD) and high-resolution TEM (HRTEM). The XRD pattern of MXenes (Fig. S2c†) shows a strong diffraction peak at 6.9°, corresponding to the typical (002) plane of Ti3C2Tx.33,35 Meanwhile, the HRTEM image (Fig. S2b†) can also index the lattice fringes of 2D MXene. As for NF (Fig. S4†), three strong peaks located at 44.7, 52.0, and 76.5 can be well identified as Ni (JCPDS no. 04-0850). The diffraction peaks of NF didn't shift or change during the processes of in situ polymerization and high-temperature treatment. In addition, the (002) Ti3C2Tx diffraction peak disappeared in the XRD pattern of MoO3/MXene/NF and MoS2/MXene/NF (Fig. S4†), which should be attributed to the PDA decomposition and carbon layer restacking on the surface of MXene flakes.30,36 What's more, no obvious crystal diffraction peaks of MoO3 and MoS2 were found in XRD patterns with NF due to the ultra-small size of NDs and the extremely strong diffraction peak of NF.24 The enlarged TEM image (Fig. 2e) exhibits ultrasmall MoO3 nanodots uniformly decorated on the surface of MXene. Lattice fringes of both MoO3 and MXene can be found in the HRTEM image (Fig. 2f). The carbon-coated Ti3C2Tx MXene derived from PDA/Ti3C2Tx retains the flat surface without the presence of TiO2 species, which demonstrated that tightly covered PDA on the MXene surface could inhibit surface oxidation during the annealing process, to maintain excellent electrical conductivity. The morphologies of the Ti3C2Tx MXene and molybdenum derivative are slightly changed after high-temperature sulfurization (Fig. 2g). Even smaller MoS2 nanodots were observed on the MXene surface (Fig. 2h) after the MoO3 vulcanization process, meaning the existence of multiple reaction points resulting in fission of MoO3 dots at the end of the vulcanization. The corresponding HRTEM image revealed lattice fringes of 0.62 nm on the nanodot matching with the (002) plane of 2H-MoS2 (Fig. 2i). The lattice of MoS2 has an obvious distortion compared with MoO3 due to the unavoidable vacancies appearing during high-temperature treatment and the two-dimensional microstructure wrinkling in MoS2. Further analysis revealed that most of the sulfurized MoS2 preferentially grows in the vertical direction of Ti3C2Tx MXene flakes with (002) crystal plane exposure. The high-angle annular dark field (HAADF) image under scanning TEM mode (Fig. 2j) indicates abundant bright dots anchored on the MXene flakes. The corresponding elemental distribution images under energy-dispersive X-ray spectroscopy (EDX) show the distributions of Ti, C, Mo, and S elements (Fig. 2k), further confirming the uniform MoS2 NDs/MXene heterostructure.
Given that EDX and HRTEM only provide foundational composition information, the X-ray photoelectron spectroscopy (XPS) analysis is further used to elucidate the electronic valence of compositions and coordination structures. Fig. S5a† shows the full spectrum of MoO3/MXene removed from MoO3/MXene/NF, which evidences the existence of Ti, C, Mo, O, and N. The peak of N may stem from the carbonized PDA. The signal attenuation caused by the active material package is scanned under high-resolution. By calibrating the carbon peak C (284.8 eV), +0.7 eV peak shifted, the high-resolution XPS spectrum of Mo 3d is obtained in Fig. S5b.† The peaks at 229.9, 233.2, and 236.3 eV, which can be attributed to the Mo4+ 3d5/2, Mo6+ 3d5/2, and Mo6+ 3d3/2, are assigned to the existence of MoO3 after 600 °C high-temperature treatment. In addition, the deconvolution of the Ti 2p XPS spectrum (Fig. S5c†) can be fitted with TiO2 (Ti4+) Ti–X (Ti2+, X = OH), Ti–C (Ti+), and Ti–X (Ti2+, X = F) at the peaks of 455.6, 458.4, 460.9, and 464.3 eV, respectively. Compared to the brand-new Ti3C2Tx MXene after etching, the binding energy of Ti–C and Ti–X positively shifts,37 revealing the obvious electron transfer from Ti3C2Tx to MoO3 nanodots in the heterostructure.
After further vulcanization to obtain MoS2/Ti3C2Tx, the signal peak of sulfur can be found initially in the full spectrum peak (Fig. 3a). In the corresponding Mo 3d spectrum (Fig. 3b), the distinct peaks at 229.6 and 232.7 eV are assigned to the Mo4+ 3d5/2 and Mo4+ 3d3/2 in MoS2, whereas the signals at 226.9 and 234.4 eV can be attributed to the S and Mo–O (MO6+), respectively. The S 2p convolutional high-resolution spectrum (Fig. 3c) is divided into two peaks of S 2p3/2 and S 2p1/2 at 162.1 and 163.5 eV, revealing the −2 oxidation state of S in MoS2. These phenomena are consistent with previous studies.22,39 As for the Ti 2p XPS spectrum after vulcanization (Fig. 3d), similar to that of MoO3/Ti3C2Tx, signals appear at 455.7, 459.1, 461.2, and 464.8 eV, stemming from TiO2 (Ti4+), Ti–X (Ti2+, X = OH), Ti–C (Ti+), and, Ti–X (Ti2+, X = F). Compared to the MoO3 nanodots on Ti3C2Tx, the Ti–C and Ti–X binding energies of Ti3C2Tx in the MoS2/Ti3C2Tx heterostructure positively shift about 0.3 eV and 0.7 eV, while the Mo4+ 3d5/2 peak shifts negatively from 229.9 to 229.6 eV in Mo–S. The decreasing Mo 3d binding energy and increasing Ti 2p binding energy indicate that more electrons were transferred from Ti3C2Tx to MoS2 nanodots. The above XPS results reveal that MoS2 nanodots are strongly adhered to the Ti3C2Tx MXene surface, accompanied by strong interfacial interactions existing at the Ti3C2Tx–MoS2 heterogeneous interfaces.
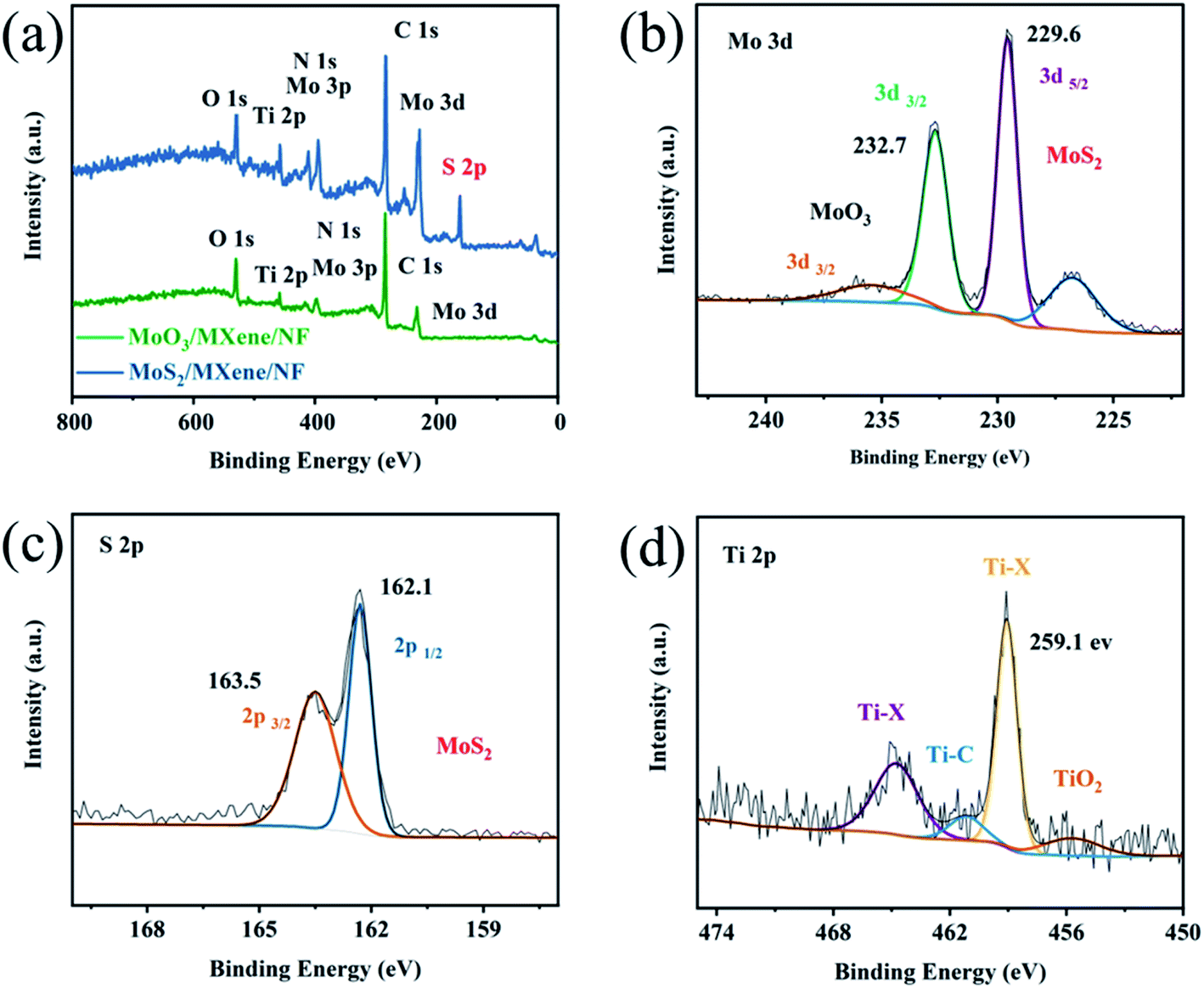 | ||
| Fig. 3 (a) Full spectra of MoO3/MXene and MoS2/MXene removed from MoO3/MXene/NF and MoS2/MXene/NF. High-resolution XPS (b) Mo 3d (c) S 2p, and (d) Ti 2p spectra of MoS2/MXene. | ||
To evaluate the electrocatalytic activity of MoS2/MXene/NF towards water splitting, HER performance was investigated in a traditional three-electrode system, in which a N2-saturated 1 M KOH solution was employed as the electrolyte. For comparison, NF, MXene/NF, and MoS2/NF MoO3/MXene/NF were also tested as control samples. Fig. 4a shows the steady-state linear sweep voltammograms (LSV) of the above electrodes, whose activity trend is as follows: NF < MXene/NF < MoO3/MXene/NF < MoS2/MXene/NF. The MXene/NF and NF show relatively worse HER activity, suggesting that both MXene and NF are not very suitable for the HER. The MoS2/MXene/NF exhibits a low onset overpotential of 94 mV at 10 mA cm−2 (η10) vs. reversible hydrogen electrode (RHE), which is much better than those of MoS2/NF (η10 = 164 mV, Fig. S6a†), MoO3/MXene/NF (η10 = 204 mV) and MXene/NF (η10 = 189 mV) (Fig. 4c). The Tafel slopes, as a pivotal kinetic parameter to elucidate the HER mechanism, were calculated using the Tafel equation. Here, the Tafel slope of MoS2/MXene/NF is 59 mV dec−1 (Fig. 4b), and the smallest among all the compared samples.
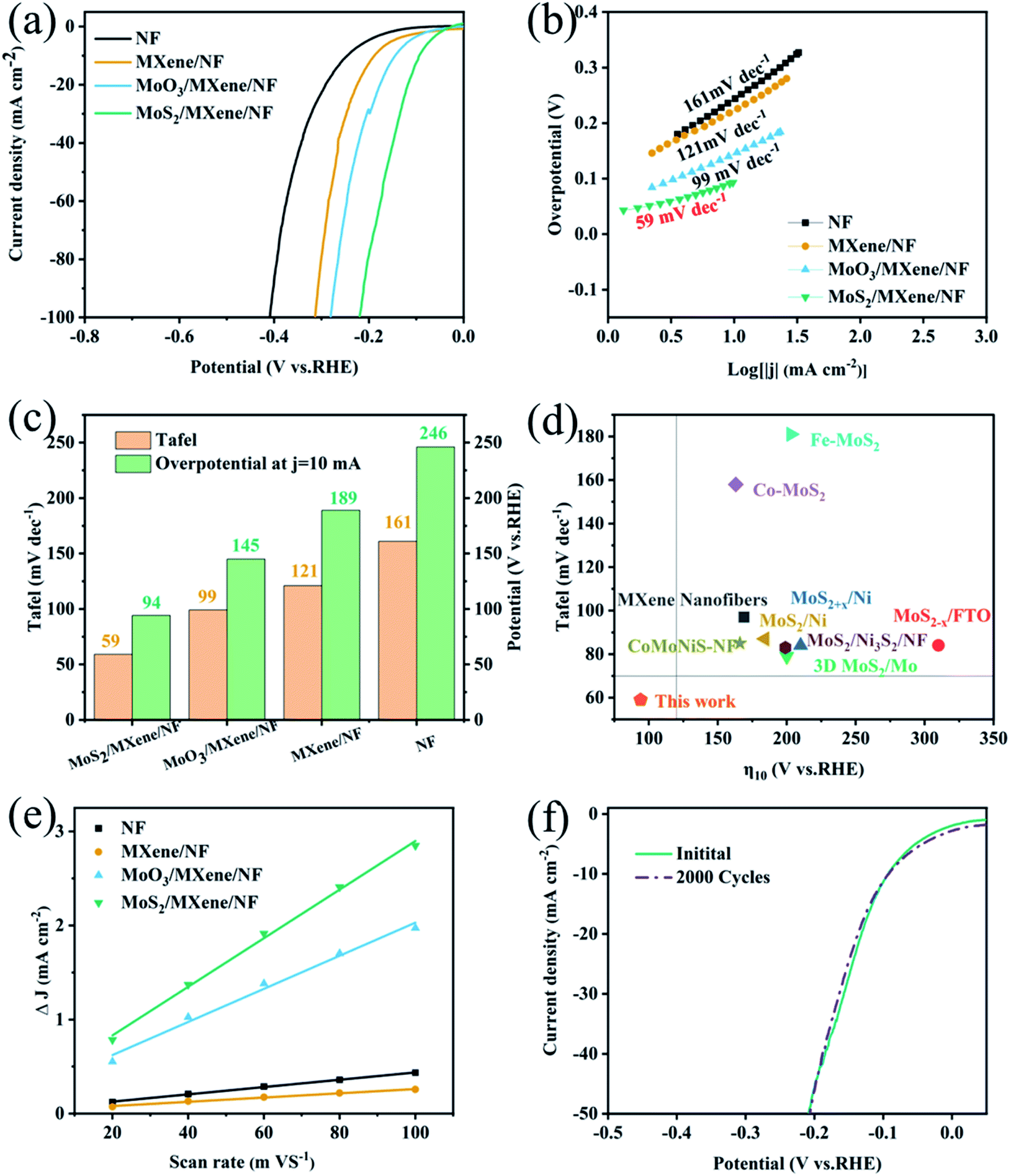 | ||
| Fig. 4 Electrochemical HER performance. (a) Polarization curves and (b) corresponding Tafel plots of MoS2/MXene/NF, MoO3/MXene/NF, MXene/NF, NF in 1 M KOH. (c) Comparison of overpotentials at 10 mA cm−2 (η10) and Tafel slopes for prepared samples. (d) Electrocatalytic activity comparison of MoS2 based materials.14,23,29,35,38,39 (e) LSV curves of MoS2/MXene/NF before and after 2000 cyclic potential scans. (f) Linear fits of half capacitive currents vs. scan rates for the extraction of Cdl. | ||
The Tafel slope curves indicate that MoS2/MXene/NF has extremely fast reaction kinetics. As shown in Fig. 4d and Table S1,† the comparison of HER performance between MoS2/MXene/NF and other non-noble-metal electrocatalysts from the literature is presented.14,23,29,35,38,39 Most of them require larger overpotentials (η10 over 100 mV) and higher Tafel slopes (over 60 mV dec−1), which are far worse than the catalytic performance of Pt. Some non-noble metal catalysts may have a low overpotential (η10 < 50 mV), while their poor kinetic conditions (higher Tafel slopes exceeding 50 mV dec−1) also make the performance inexhaustible. Therefore, MoS2/MXene/NF with an ultra-low overpotential (η10 = 94 mV) and Tafel slope (59 mV dec−1) have great potential to replace Pt in industrial applications. Meanwhile, the stability of the electrocatalyst, which is a critical indicator for catalyst practical application, has also been investigated through cyclic voltammetry (CV) cycles over 200 mV vs. RHE (Fig. 4f) and the chronoamperometry (CP) test at a constant overpotential (Fig. S7d†). The LSV curve of MoS2/MXene/NF shows a negligible negative shift after 2000 cycles in current density compared to the initial curve. Meanwhile, the MoS2/MXene/NF exhibits greater long-term stability, only a slight decrease over 60000 s. SEM and XRD characterization studies of MoS2/MXene/NF (Fig. S7a–c†) were further conducted to confirm the outstanding durability of its morphology, structure, and phase after the stability test. Similar to MoS2/NF after electrochemical tests (Fig. S3d†), the SEM image shows that only the folded surface of the PDA carbonation layer is destroyed, while the main skeleton structure and phase of MoS2/MXene/NF remain unchanged. The excellent durability of MoS2/MXene/NF can be attributed to the confinement effect of carbonated PDA, which protects the ion-exchange interface during the electrocatalytic process.
The above results imply that the HER activity of both Ti3C2Tx and MoS2 is unsatisfactory under alkaline conditions, whereas modulating the heterogeneous structure can result in an optimized HER activity. To better understand the function of the MoS2/Ti3C2Tx heterostructure during the HER process, the electrochemical impedance spectroscopy (EIS) measurement was performed at 200 mV vs. RHE from 106 Hz to 0.01 Hz. Fig. S8† displays the Nyquist plots of the above electrodes, in which the charge transfer resistance (Rct) is related to the electrocatalytic kinetics at the interfaces. The MoS2/MXene/NF delivers a much smaller Rct than MoO3/MXene/NF and MXene/NF, evidencing faster electron transfer between the MoS2–Ti3C2Tx heterostructure. Both the Ti3C2Tx flake and MoS2 nanodot contribute to the higher electrical conductivity and faster HER kinetics. Generally, the effective electrochemically active area (ECSA) is related to the electrocatalyst surface area and the number of active sites, which is estimated from the double-layer capacitance (Cdl) by performing cyclic voltammetry with different sweep speeds under the voltage range of the non-Faradic current (electrochemical tests). The Cdl value was calculated by the slope between the half capacitive current density at the median of the applied potential and the scan rates. The measured Cdl value for MoS2/MXene/NF is 0.23 F cm−2 (Fig. 4e and S8†), considerably better than those of MoO3/MXene/NF (0.39 F cm−2), MXene/NF (1.8 F cm−2), and NF (2.6 F cm−2). As the MXene/NF displays the worst ECSA, the HER active sites are assumed from the MoO3 and MoS2 nanodots in the heterostructure. Significantly, the ECSA of MoS2/MXene/NF is larger than that of MoO3/MXene/NF, indicating the extension of the electrochemical active area or the more catalytic active sites obtained after high-temperature sulfurization. Experimental studies and theoretical calculations have certified that MoS2 active sites are mainly concentrated on the edge.15–21 Therefore, the fixed orientation for MoS2 NDs preferential growth on MXene flake exposes more (002) crystal planes, which exhibit more active sites for excellent HER than other planes.
Obviously, the HER performance is associated with the varied ratio of MoS2/MXene and the construction of MoS2–MXenes interfaces. The different ratios of MoS2/MXene can be adjusted by c the usage of ammonium molybdate and MXene in the precursor reaction process. Though more Mo active sites are beneficial for HER performance enhancement, excessive Mo will result in larger MoS2 dots, which have lower conductivity and a smaller percentage of active sites. What's more, a larger ratio of MoS2 and MXene may be accompanied by the formation of MoS2 microspheres (Fig. S3†), of which the HER performance is very poor (Fig. S6†). As for the role of the MoS2–MXenes interface, it was known that Ti3C2Tx itself shows negligible HER activity. So, the function of Ti-based MXenes at the interfaces is to provide a better hydrophilicity and conductivity, which will promote more efficient and direct electron transfer (Fig. 3) to improve the electrocatalytic performance. Meanwhile, the exposures of MoS2 edges in the outside of interfaces provide active sites for the generation of H*.
The effect of sulfurization temperatures (500 °C and 800 °C) on HER activity was evaluated. The SEM image (Fig. 5a) of MoS2/MXene/NF powder obtained after sulfurization treatment at 500 °C displays negligible change in morphology, and structure, while obvious particles appear in MXene flakes (Fig. 5b), identified as TiO2 (Fig. S10,† JCPDS no. 21-1271) when increasing the sulfurization temperature to 800 °C. As shown in Fig. 5c, the overpotentials for achieving 10 mA cm−2 for samples treated at 500 °C and 800 °C are 151 and 128 mV, respectively. Meanwhile, the corresponding Tafel slopes are 128 and 126 mV dec−1 (Fig. 5d), respectively. Obviously, the MoS2/MXene/NF treated at 650 °C exhibits the best HER activity among the three samples. The deteriorating HER performance of the MoS2/MXene/NF treated at 800 °C should be attributed to the high temperature induced oxidation of MXene which would reduce the MoS2/MXene conductivity and block the electron transfer channel. The MoS2/MXene/NF treated at low temperature (500 °C) has even poorer intrinsic activity than initial MoO3/MXene/NF (Fig. 4a and b), which may be related to the nanostructure destruction of MoO3 and MoS2 under insufficient sulfurization. According to the literature,32,39,40 temperature can control the degree of sulfurization, and molybdenum disulfide is continuously formed on the surface of molybdenum oxide at high temperatures. For the MoO3/MXene/NF electrode at low temperature, few active sites on the molybdenum disulfide edge can be exposed. And molybdenum oxide is destroyed in the sulfurization process at the same time, so the active sites of the catalyst are greatly reduced. What's more, the alkaline HER process includes two microscopic steps:41,42 (a) H2O + e− → H* + OH− (Volmer reaction); (2) H* + H2O + e− → H2 + OH− (Heyrovsky reaction) or H* + H* → H2 (Tafel reaction). The Tafel slope of MoS2/MXene/NF (500 °C) is 128 mV dec−1 (>120 mV), indicating that the Volmer reaction is the rate-limiting step of the HER due to the deficiency of active sites. The Tafel slope of MoO3/MXene/NF and MoS2/MXene/NF (650 °C) declined to 99 and 59 mV dec−1, respectively, implying that the Heyrovsky reaction or Tafel reaction has transited to the rate-determining step. A comparison experiment further indicated that both MXene flakes with higher conductivity and MoS2 nanodots with more active site exposure are significant to MoS2/MXene/NF with extraordinary HER performance.
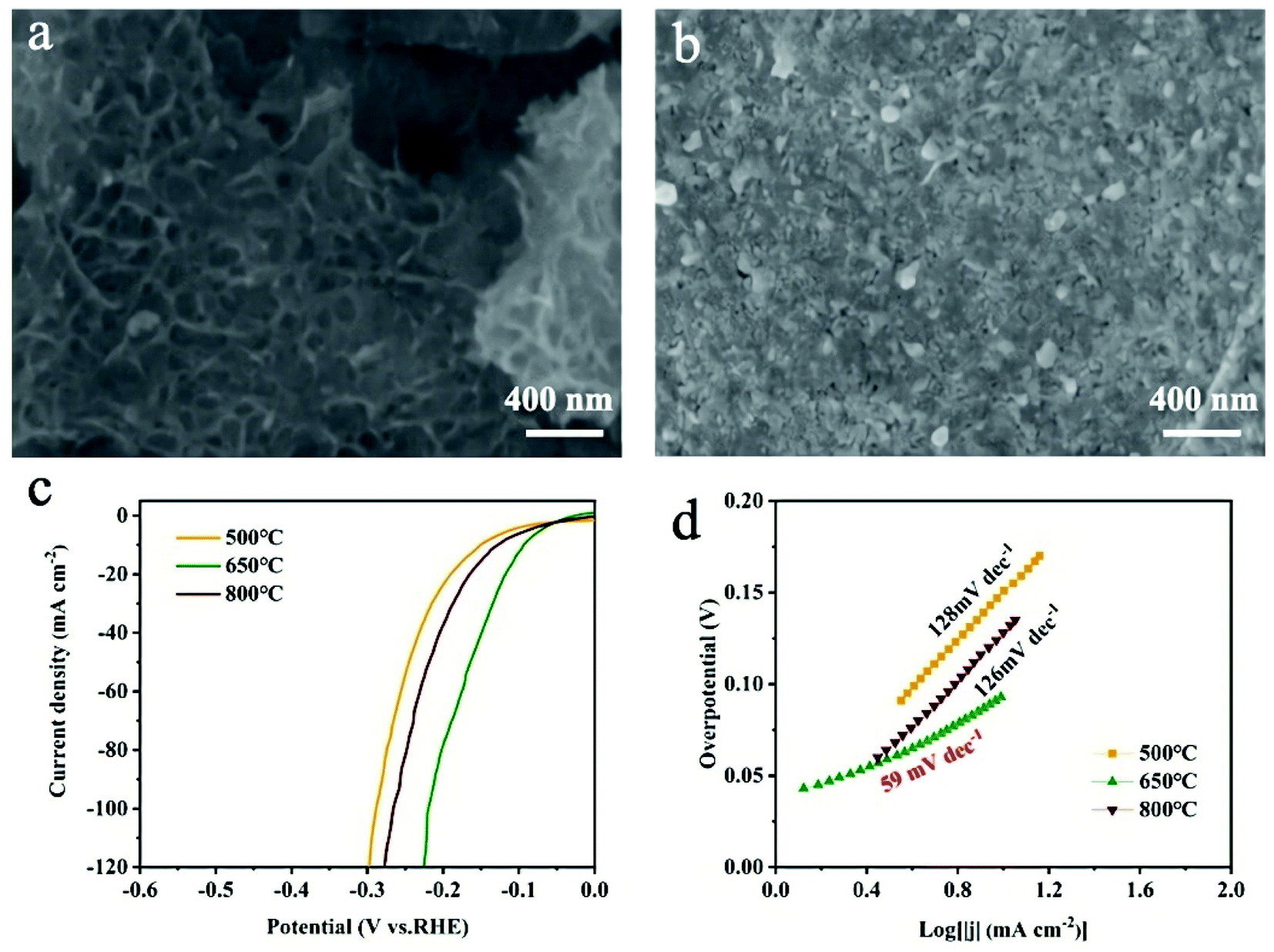 | ||
| Fig. 5 SEM images of MoS2/MXene/NF after sulfurization at (a) 500 °C and (b) 800 °C. (c) Polarization curves and (d) corresponding Tafel plots of MoS2/MXene/NF sulfurized at different temperatures. | ||
Based on the aforementioned analysis, the remarkable HER activity of MoS2/MXene/NF (650 °C) is assumed to originate from the following factors: (1) in situ polymerized PDA with chelated molybdate ions plays a key role in avoiding Ti3C2Tx aggregation and protecting the structure; (2) PDA chelated MoO42− makes the molybdenum compound highly disperse ultrasmall nanostructures on the surface of the MXene flakes; (3) the special heterostructures of MoS2 nanodots and Ti3C2Tx flakes guarantee excellent HER performance and long-term stability; (4) after sulfurization at an appropriate temperature, MoS2 nanodots grow vertically on Ti3C2Tx MXene flakes, accompanied by the high accessibility of active sites for the HER at the MoS2 nanodot edge; (5) the intense synergistic effect of the MoS2–Ti3C2Tx heterostructure provides a resistance-less channel for faster electron transfer when the active sites of MoS2 generate H*.
Conclusion
In summary, a high-activity and high-stability MoS2/MXene/NF catalyst based on non-precious metal was developed through an in situ polymerization strategy and high-temperature sulfurization treatment. The HAADF image and EDX element distribution showed the synthetic MoS2 anchored on Ti3C2Tx MXene flakes with uniformly dispersed ultrasmall nanodots. Electronic microscopies indicated that the constructed MoS2/MXene heterostructure displayed excellent stability, preventing the aggregation of both the Ti3C2Tx flakes and molybdenum compound nanodots, thus providing high accessibility exposure of active sites for the HER. The Ti3C2Tx flakes between MoS2 and NF have multiple functions, which can protect the NF substrate from the sulfurization reaction, increase the stability of the substrate, and enhance the conductivity of electron transport. The MoS2 nanodots sulfurized from MoO3 achieve an exposure of high-activity edges on the (002) plane, providing better HER performance in an alkaline solution. Compared with NF, MXenes, MoO3, and MoS2, the prepared MoS2/MXene/NF exhibited a Tafel slope of 59 mV dec−1 and an overpotential of 94 mV at 10 mA cm−2, which is better than those of most of the non-noble metal catalysts in alkaline solution. These findings and insights encourage further heterostructure design and coupling effect investigation of HER catalysts, and point to a new pathway for industrial alkaline catalysis for water splitting.Author contributions
Xiaoyu Wang: experiments and data collection; Wenbin You: characterization and analysis; Liting Yang, Guanyu Chen, Zhengchen Wu, and Chang Zhang: characterization; Qianjin Chen and Renchao Che: experimental design and project lead. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (2232020A-09 and 2232021G-04), National Natural Science Foundation of China (21804018, 51725101, 11727807, 51672050, 61790581, and 22088101), the Ministry of Science and Technology of China (973 Project No. 2018YFA0209102 and 2021YFA1200600), Infrastructure and Facility Construction Project of Zhejiang Laboratory, and the National Science Foundation of Shanghai (19ZR1470800) and was sponsored by the Shanghai Sailing Program (21YF1401800).References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355 Search PubMed.
- F. Dawood, M. Anda and G. M. Shafiullah, Int. J. Hydrogen Energy, 2020, 45, 3847–3869 CrossRef CAS.
- K. T. Moller, T. R. Jensen, E. Akiba and H.-w. Li, Prog. Nat. Sci.: Mater. Int., 2017, 27, 34–40 CrossRef.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- M. Mitov, E. Chorbadzhiyska, R. Rashkov and Y. Hubenova, Int. J. Hydrogen Energy, 2012, 37, 16522–16526 CrossRef CAS.
- Y. Zheng, Y. Jiao, A. Vasileff and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed.
- S. Ghasemi, S. R. Hosseini and S. Nabipour, J. Iran. Chem. Soc., 2019, 16, 101–109 CrossRef CAS.
- Z. Wu, Z. Yang, C. Jin, Y. Zhao and R. Che, ACS Appl. Mater. Interfaces, 2021, 13, 5866–5876 CrossRef CAS PubMed.
- Q. Ding, B. Song, P. Xu and S. Jin, Chem, 2016, 1, 699–726 CAS.
- X. Geng, W. Wu, N. Li, W. Sun, J. Armstrong, A. Al-hilo, M. Brozak, J. Cui and T.-p. Chen, Adv. Funct. Mater., 2014, 24, 6123–6129 CrossRef CAS.
- M. I. A. A. Maksoud, A. G. Bedir, M. Bekhit, M. M. Abouelela, R. A. Fahim, A. S. Awed, S. Y. Attia, S. M. Kassem, M. Abd Elkodous, G. S. El-Sayyad, S. G. Mohamed, A. I. Osman, A. a. H. Al-Muhtaseb and D. W. Rooney, Environ. Chem. Lett., 2021, 19, 3645–3681 CrossRef.
- Y. Yang, H. Yao, Z. Yu, S. M. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao, G. Sun, H. Li, S. Ma, P. Zapol and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 10417–10430 CrossRef CAS PubMed.
- H. Wang, X. Xiao, S. Liu, C.-L. Chiang, X. Kuai, C.-K. Peng, Y.-C. Lin, X. Meng, J. Zhao, J. Choi, Y.-G. Lin, J.-M. Lee and L. Gao, J. Am. Chem. Soc., 2019, 141, 18578–18584 CrossRef CAS PubMed.
- J. Hu, C. Zhang, L. Jiang, H. Lin, Y. An, D. Zhou, M. K. H. Leung and S. Yang, Joule, 2017, 1, 383–393 CrossRef CAS.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jorgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Norskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- D. Voiry, R. Fullon, J. Yang, C. d. C. Castro e Silva, R. Kappera, I. Bozkurt, D. Kaplan, M. J. Lagos, P. E. Batson, G. Gupta, A. D. Mohite, L. Dong, D. Er, V. B. Shenoy, T. Asefa and M. Chhowalla, Nat. Mater., 2016, 15, 1003–1009 CrossRef CAS PubMed.
- B. Zhang, J. Liu, J. Wang, Y. Ruan, X. Ji, K. Xu, C. Chen, H. Wan, L. Miao and J. Jiang, Nano Energy, 2017, 37, 74–80 CrossRef CAS.
- G. Zhao, P. Li, K. Rui, Y. Chen, S. X. Dou and W. Sun, Chem.–Eur. J., 2018, 24, 11158–11165 CrossRef CAS PubMed.
- G. Ou, P. Fan, X. Ke, Y. Xu, K. Huang, H. Wei, W. Yu, H. Zhang, M. Zhong, H. Wu and Y. Li, Nano Res., 2018, 11, 751–761 CrossRef CAS.
- J. Zhang, T. Wang, P. Liu, S. Liu, R. Dong, X. Zhuang, M. Chen and X. Feng, Energy Environ. Sci., 2016, 9, 2789–2793 RSC.
- S. Xu, D. Li and P. Wu, Adv. Funct. Mater., 2015, 25, 1127–1136 CrossRef CAS.
- S. Anantharaj, S. Noda, V. R. Jothi, S. Yi, M. Driess and P. W. Menezes, Angew. Chem., Int. Ed., 2021, 60, 18981–19006 CrossRef CAS PubMed.
- X. Wang, Y. Zheng, W. Sheng, Z. J. Xu, M. Jaroniec and S.-Z. Qiao, Mater. Today, 2020, 36, 125–138 CrossRef CAS.
- J. Lin, P. Wang, H. Wang, C. Li, X. Si, J. Qi, J. Cao, Z. Zhong, W. Fei and J. Feng, Adv. Sci., 2019, 6, 1900246 CrossRef PubMed.
- X. Li, W. You, L. Wang, J. Liu, Z. Wu, K. Pei, Y. Li and R. Che, ACS Appl. Mater. Interfaces, 2019, 11, 44536–44544 CrossRef CAS PubMed.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS PubMed.
- H. Huang, J. Cui, G. Liu, R. Bi and L. Zhang, ACS Nano, 2019, 13, 3448–3456 CrossRef CAS PubMed.
- Y. Wei, R. A. Soomro, X. Xie and B. Xu, J. Energy Chem., 2021, 55, 244–255 CrossRef.
- H. Wang, Y. Lin, S. Liu, J. Li, L. Bu, J. Chen, X. Xiao, J.-H. Choi, L. Gao and J.-M. Lee, J. Mater. Chem. A, 2020, 8, 7109–7116 RSC.
- X. Li, X. Lv, X. Sun, C. Yang, Y.-Z. Zheng, L. Yang, S. Li and X. Tao, Appl. Catal., B, 2021, 284, 119708 CrossRef CAS.
- J. Liu, Y. Liu, D. Xu, Y. Zhu, W. Peng, Y. Li, F. Zhang and X. Fan, Appl. Catal., B, 2019, 241, 89–94 CrossRef CAS.
- W. Yuan, L. Cheng, Y. An, H. Wu, N. Yao, X. Fan and X. Guo, ACS Sustainable Chem. Eng., 2018, 6, 8976–8982 CrossRef CAS.
- X. Zhan, C. Si, J. Zhou and Z. Sun, Nanoscale Horiz., 2020, 5, 235–258 RSC.
- S. Seyedin, E. R. S. Yanza and J. M. Razal, J. Mater. Chem. A, 2017, 5, 24076–24082 RSC.
- C. G. Morales-Guio, L. Liardet, M. T. Mayer, S. D. Tilley, M. Graetzel and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 664–667 CAS.
- B. He, L. Chen, M. Jing, M. Zhou, Z. Hou and X. Chen, Electrochim. Acta, 2018, 283, 357–365 CrossRef CAS.
- H. F. Liu, S. L. Wong and D. Z. Chi, Chem. Vap. Deposition, 2015, 21, 241–259 CrossRef CAS.
- Q. Hu, Z. Wang, X. Huang, Y. Qin, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Energy Environ. Sci., 2020, 13, 5097–5103 RSC.
- X. Liu, Q. Hu, B. Zhu, G. Li, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, Small, 2018, 14, 1802755 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00376g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |