
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme-activated probes in optical imaging: a focus on atherosclerosis
Edward R. H.
Walter
 ab,
Saul M.
Cooper
ab,
Joseph J.
Boyle
b and
Nicholas J.
Long
*a
ab,
Saul M.
Cooper
ab,
Joseph J.
Boyle
b and
Nicholas J.
Long
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: n.long@imperial.ac.uk
bNational Heart and Lung Institute, Imperial College London, London, W12 0NN, UK
First published on 27th September 2021
Abstract
Enzyme-activated probes enable complex biological processes to be studied in real-time. A wide range of enzymes are modulated in diseases, including cancer, inflammatory diseases and cardiovascular disease, and have the potential to act as vital diagnostic and prognostic biomarkers to monitor and report on disease progression. In this perspective article, we discuss suitable design characteristics of enzyme-activated fluorescent probes for ex vivo and in vivo optical imaging applications. With a particular focus on atherosclerosis imaging, we highlight recent approaches to report on the activity of cathepsins (K and B), matrix metalloproteinases (MMP-2 and MMP-9), thrombin, heme oxygenase-1 (HO-1) and myeloperoxidase (MPO).
1. Introduction
Optical Imaging (OI) is a powerful, highly sensitive and cost effective non-invasive diagnostic technique routinely used in chemical biology.1 ‘Activity’ or ‘smart’ fluorescent probes are highly advantageous for use in OI, and typically display a rapid and selective fluorescence activation mechanism resulting in a high signal-to-noise ratio against the background signal. Additionally, such probes have the advantage of visualising direct molecular processes and abnormalities, compared to Molecular Resonance Imaging (MRI) and Ultrasound (US) which provide much more detailed anatomical information.Over the years, a wide range of activity-based probes have been designed and developed to detect a number of metal ions,2,3 pH fluctuations4 and biologically relevant enzymes5–7 in real-time. In particular, enzymes represent important biomarkers for diagnosis and monitoring the progression of a number of chronic diseases such as Parkinson's disease,8 arthritis9 and many types of cancer.10 Therefore, enzyme-activated fluorescent probes are an important area in this field.
One growing area of interest is utilising enzyme-activated fluorescent probes for the OI of atherosclerosis. Acting as a prerequisite to Cardiovascular disease (CVD) and stroke – two leading causes of death worldwide,11 atherosclerosis is a chronic and often asymptomatic condition that is characterised by the narrowing of arteries following a steady build-up of plaque over a prolonged period of time.12 Atherosclerotic plaque broadly takes two phenotypes, named stable and vulnerable. One of the main challenges in this field is differentiating between these two dissimilar forms, particularly in the planning of medical interventions.
For example, vulnerable plaque is characterised by a high burden of inflammatory macrophages and a thin fibrous cap that is prone to rupture. One of the key processes in disruption is intraplaque hemorrhage (IPH), a bleed within the plaque itself, and strongly associated with more complete plaque disruption causing thrombosis and blockage of the lumen.13,14 Therefore, atherosclerosis is accelerated by IPH, and often results in the onset of ischemic stroke and myocardial infarction.15 In contrast, stable plaques display the opposite features and are significantly less prone to rupture.
In this perspective, we summarise the design aims and activation mechanisms that may guide the future development of enzyme-activated probes for disease diagnosis and prognosis. In particular, we focus on recently developed enzyme-activatable fluorescent probes for imaging atherosclerosis, and future directions that could be taken in this important area of fluorescence medical imaging. Chemical probes for other biological targets secreted by macrophages, such as cytokines, will not be discussed.
2. Enzyme-activated probes in OI
Enzyme-activated probes are highly advantageous for OI applications, allowing molecular processes to be detected in real-time. Probes of this nature typically contain an enzyme-specific functionality and a fluorescent reporter moiety to provide the signal output. During the design and development process, a number of different factors must be considered, including the photophysical properties of the fluorophore and biological considerations of the probe prior to, and following activation. Moreover, these design requirements for enzyme-activatable fluorescent probes are inherently generalisable so would also apply to other medical OI probes in various biochemical contexts.2.1 Considerations in probe design
As with all fluorescent probes used in OI applications, the signal-transduction and photophysical properties of enzyme-activated fluorogenic probes must be carefully considered. The fluorescence readout observed following activation is most commonly intensity-based via a ‘turn-on/off’ mechanism. However, in some instances, the comparison of two or more wavelengths enables a ratiometric measurement. Such a ratiometric response is highly advantageous, due to enablement of self-calibration, eliminating artefacts resulting from the degree of cellular uptake or intracellular quenching during imaging.16 This facilitates much more precise assessment.In a biological setting a low-energy excitation (long wavelength) is extremely desirable for two reasons. The first is reduced absorbance by endogenous biomolecules such as hemoglobin,17 allowing an increased depth of tissue penetration. To this end, a range of fluorophores with a NIR-1 (700–900 nm)18 and NIR-2 (1000–1700 nm)19 spectral window have been developed in recent years for in vivo imaging applications. Additionally, probes that enable two-photon excitation are also extremely sought-after, due to the use of the lower energy excitation of two individual NIR photons.20
It is also important to consider the relative energy difference between the absorbance and emission of light of a particular fluorophore – termed the Stokes’ shift. Fluorophores that display a large Stokes’ shift are preferred and prevent the self-absorption of emitted light by molecules in the ground state.21 Finally, the fluorescent reporter must also be photostable with a high quantum yield (ϕ) and large extinction coefficient (ε).
In addition to the photophysical properties of the fluorophore, a number of biological factors must also be considered. The probe must respond selectively to the necessary enzymatic target in a complex biological environment at physiological pH.22 Additionally, the probe, and the products generated following enzyme activation, must be non-cytotoxic in order to prevent intracellular damage during in vivo imaging applications.
In many instances, it is also desirable to incorporate biological targeting functionalities into probe design to ensure localisation in a specific region of the body or cellular compartment.23 The unique properties of functionalised nanoparticles are often used to this end, as a strategy for the delivery of small molecule-based probes. One such approach, used in several probes designed for activation by proteases, is the use of a peptide-appended pegylated graft co-polymer to aid delivery through enhanced circulation of the agent.24–26 Additionally, functionalised liposomal nanoparticles can encapsulate an OI probe for delivery to a specific vascular site.27 In a very interesting approach, the endogenous protective nanoparticle High-Density Lipoprotein (HDL) has been reverse engineered as a potential carrier for therapeutic and imaging reagents.28,29 Furthermore, inorganic nanoparticles (composed of iron oxide,30,31 gold32 or silica33) and solid lipid nanoparticles34 have been developed as a carrier for an array of imaging techniques.
A further approach, used to good effect in the application of lipophilic cation-functionalised probes to facilitate mitochondrial localisation, includes a small targeting group on the probe scaffold itself.35,36 An example by Urano and co-workers uses the specificity provided by antibodies to their cellular targets, in the application of activatable probe–antibody conjugates to cancer cells.37
The nature of the fluorophore itself can strongly influence the pharmacokinetic profile of the imaging agent, particularly in the case of extended systems for signal generation in high molecular weight probes. Therefore, an optimal balance between the desired NIR properties of a probe, and the undesirable perturbation it induces in the pharmacokinetics of a delivery system needs to be established. As such, strategies to improve localisation of an OI probe at the site of interest, through additional targeting moieties, encapsulation, or extended circulation times, are of great contemporary interest.38
2.2 Fluorescence activation mechanisms
A wide range of different fluorescence activation mechanisms in OI have been reported to date, in part reflecting a variety of different applications and enzymatic targets. Here, we summarise three common fluorescence activation mechanisms, with recently developed examples from the literature. The first two examples described in this section are particularly widespread and are characterised by an enzyme-specific cleavage that is accompanied by a fluorescence response. Such a cleavage is typically either a linker between two fluorophores (e.g. a specific, short peptide sequence) (Fig. 1A), or a functional group cleavage on a single fluorophore (e.g. an ester/thioester) (Fig. 1B).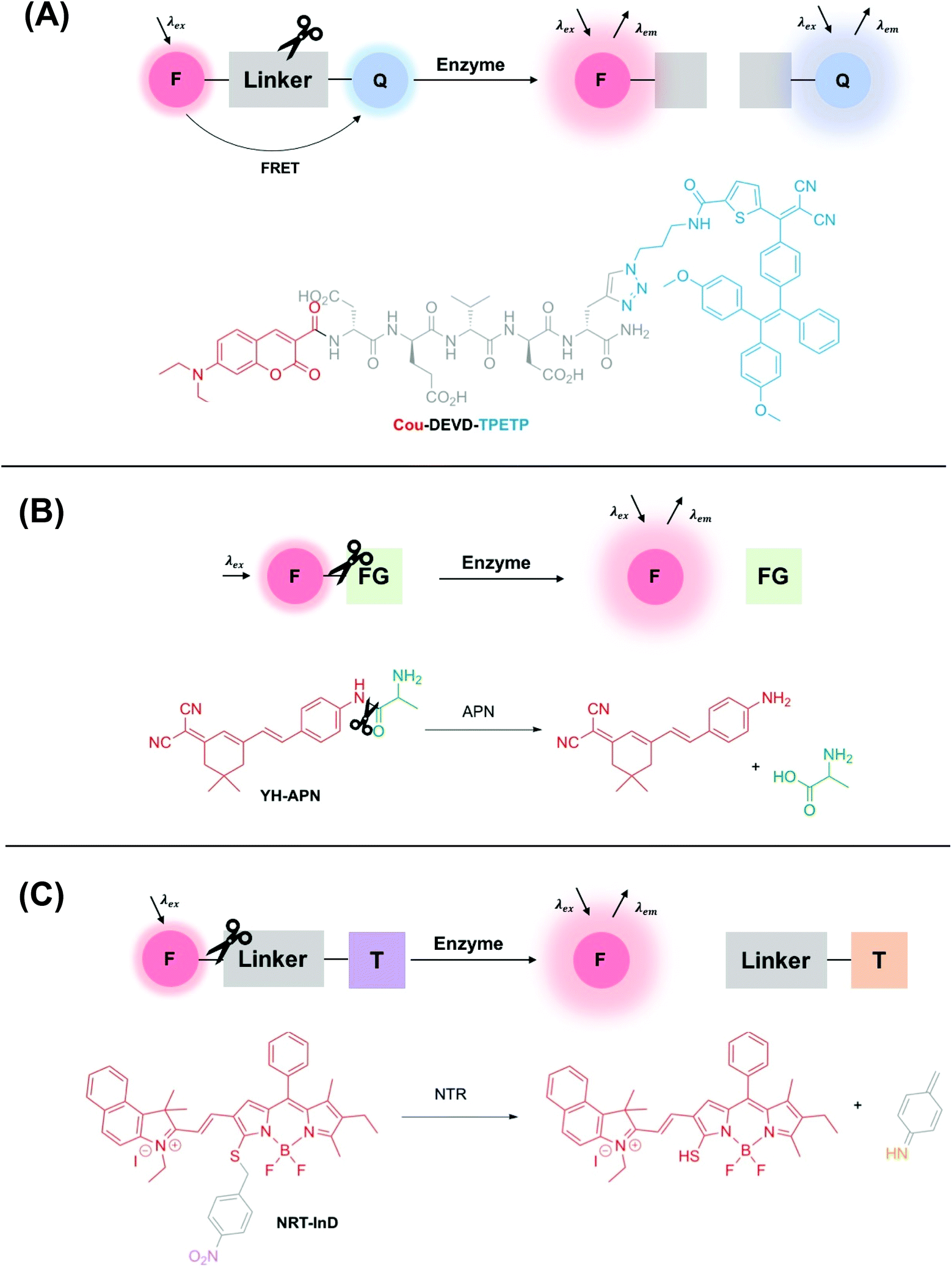 | ||
| Fig. 1 Schematic showing three common examples of enzyme-activity probes in the literature. (A) A ‘break-apart’ probe with a cleavable linker between two fluorophores e.g.Cou-DEVD-TPETP from Liu and co-workers.39 Another common approach utilises self-quenching of two fluorophores linked by an enzyme cleavable peptide sequence. (B) An enzyme-specific functional group cleavage e.g.YH-APN developed by Peng and Yoon,40 and (C) a self-immolative probe for nitroreductase, NRT-InD developed by Zhao and co-workers.41 F = fluorophore, Q = FRET pair/fluorescent quencher, FG = enzyme-selective functional group, T = trigger group. | ||
Additionally, another common class of enzyme-activated fluorogenic probes involve the use of a self-immolative linker that masks the fluorescence of a fluorophore prior to enzymatic activation in the cellular environment. In this case, following enzyme activation, a chemical trigger group initiates a cascade of chemical reactions, initiating an increase in a fluorescence output (Fig. 1C).
Of course, other activation mechanisms are entirely possible too, but are not within the scope of this perspective. We point to two informative recent reviews in this area for a more detailed literature evaluation of a number of disease-relevant enzymes, particularly in oncology.42,43
The incorporation of a short and specific enzyme-cleavable peptide sequence that also acts as a linker between a pair of fluorophores is frequently the method of choice for monitoring the activity of a range of different proteases, a hydrolase enzyme, in real-time.44–46 Following selective enzymatic scission, a change in fluorescence response is observed, often due to the perturbation of Fluorescence Resonance Energy Transfer (FRET)† between two fluorophores (Fig. 1A). Typically, to minimise the interference of a background signal, the acceptor fluorophore is deployed as a fluorescent quencher.
Liu and co-workers used this approach to design a dual-signal ‘turn-on’ probe for caspase-3 – an enzyme responsible for the initiation and activation of cell apoptosis. Probe Cou-DEVD-TPETP contains a coumarin donor fluorophore and a tetraphenylethenethiophene (TPETP) acceptor acting as a fluorescent quencher (Fig. 1A).39 Following caspase-3 initiated cleavage of the DEVD peptide, FRET was inhibited and accompanied by a ‘turn-on’ in coumarin fluorescence. Additionally, self-validation of caspase-3 activity was provided by TPETP, displaying a red fluorescence ‘turn-on’ due to aggregation of the released TPETP acceptor via aggregation-induced emission (AIE). Due to this dual-signal approach, Cou-DEVD-TPET was used successfully to self-validate caspase-3 activity in live fluorescence imaging of HeLa and MDA-MB-231 cells.39
It is also important to note that in some cases, fluorescence quenching can occur by photoinduced electron transfer (PET)‡ prior to activation.47,48 A self-quenching mechanism can also be utilised to good effect with two fluorophores in close proximity, quenching the fluorescence prior to activation. Probes with these design features are collectively named enzyme “break-apart” probes, with enzymatic cleavage accompanied by a fluorogenic response.
Probes for other types of hydrolase enzymes, such as esterases and thioesterases, contain enzyme-cleavable ester and thioester functional groups respectively.49–51 A similar functional group cleavage activation mechanism is observed for aminopeptidases, and this approach was used by Peng and Yoon to develop a CD13/aminopeptidases (APN) N-activatable fluorescent probe for tracking metastatic cancer (Fig. 1B). Probe YH-APN comprises a dicyanoisophorone fluorophore and an APN selective recognition site, L-alanine. Following a selective enzymatic activation by APN, YH-APN displayed a large Stokes’ shift of 205 nm and a detection limit of 0.13 ng mL−1.40 Crucially, from cell morphology, YH-APN could distinguish cells in a mixed cultivation of normal (LO2) and cancerous cells (Hep-G2), an experiment designed to simulate the environment of a tumour.52 To achieve the mixed cultivation, cell planting and culture was achieved in a step-wise manner, within the same confocal dish over a 48 h period. A fluorescence signal of 655–755 nm was detected in the mixed culture and attributed to enzyme activation of YH-APN in Hep-G2 cells only. Furthermore, in the study, cancerous tissues were successfully identified by in situ spraying, outlining a high tumour-to-normal tissue ratio. Lesions smaller than 1 mm could be imaged precisely, superior to other diagnostic techniques used in clinical practice such as MRI and US. Such approaches, therefore, show a great deal of promise for use in image-guided surgery in the future.40
Finally, ‘turn-on’ fluorescent probes incorporating a self-immolative linker are increasingly being developed to detect disease-relevant enzymes. For example, in 2019 Zhao and co-workers developed a series of BODIPY-based activity probes for nitroreductase (NTR) and NAD(P)H:quinone oxidoreductase isozyme 1 (NQO1) for imaging hypoxia in cancer cells (Fig. 1C).41 All probes in this study contained a self-immolative benzyl thioether linker, that resulted in a bathochromic-shift in the absorbance and emission wavelength maxima following activation by the enzymatic target. For example, following detection of NTR (in DMSO/Tris-HCl, v/v 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8), probe NTR-InD displayed a significant decrease in fluorescence at 612 nm (λex = 535 nm) and a 12-fold ‘turn-on’ in NIR fluorescence at 900 nm (λex = 730 nm). A highly favourable Stokes’ shift of 170 nm was displayed. Such a significant bathochromic-shift in fluorescence following enzyme activation enabled NTR-InD to be analysed in A549 cells by two-channel ratiometric fluorescence imaging. Additionally, promising results were displayed in a murine in vivo model where a time-dependent NIR fluorescence was displayed specifically in the tumour.41
:8), probe NTR-InD displayed a significant decrease in fluorescence at 612 nm (λex = 535 nm) and a 12-fold ‘turn-on’ in NIR fluorescence at 900 nm (λex = 730 nm). A highly favourable Stokes’ shift of 170 nm was displayed. Such a significant bathochromic-shift in fluorescence following enzyme activation enabled NTR-InD to be analysed in A549 cells by two-channel ratiometric fluorescence imaging. Additionally, promising results were displayed in a murine in vivo model where a time-dependent NIR fluorescence was displayed specifically in the tumour.41
3. Enzyme-activated probes for atherosclerosis imaging
Over the years an array of invasive and non-invasive medical imaging techniques has been used to detect the pathogenesis of atherosclerosis. Non-invasive imaging modalities utilising an assortment of biological targeting moieties for use in MRI,31,53,54 OI,55 Photoacoustic Imaging (PAI)56 and Positron Emission Tomography (PET)57,58 have been developed, and summarised in an informative and recent review by Gao and co-workers.59Here, in contrast, we summarise recent efforts to develop activatable fluorescent probes for enzymatic biomarkers associated with the diagnosis and pathogenesis of atherosclerosis. The primary challenge in this field is accurately differentiating between stable and vulnerable plaque, the latter of which is significantly more prone to rupture, due in part to the high burden of inflammatory macrophages.
Macrophages are responsible for the secretion of enzymes, reactive oxygen species (ROS) and cytokines as the primary response of a host to disease.60,61 Consequently, the development of chemical probes to monitor the enzymatic activity of macrophages is a popular approach to image plaque instability in atherosclerotic lesions.62 These include recent approaches to report on the activity of proteases (cathepsins, matrix metalloproteinases (MMPs), and thrombin), heme oxygenase-1 (HO-1) and myeloperoxidase (MPO). Due to commonality of molecular disease mechanisms, the enzymatic biomarkers developed for atherosclerosis may also be useful markers of other diseases, characterised by perturbation of the same enzyme activity.42,43,61
3.1 Monitoring the enzyme-activity of proteases
As described in section 2, the design of enzyme-activatable probes for proteases predominantly involves the incorporation of a specific enzyme-cleavable peptide sequence as a linker between two fluorophores. Proteases (e.g. cathepsins, MMPs and thrombin) are a class of enzymes that are primarily responsible for the digestion of the extracellular matrix (ECM), the weakening of the fibrous cap, and subsequently leading to plaque destabilisation and rupture from this vulnerable state.63,64 Additionally, proteases play a major role in vascular remodelling, and over time can lead to the development of more advanced atherosclerotic plaques, and possible thrombus formation.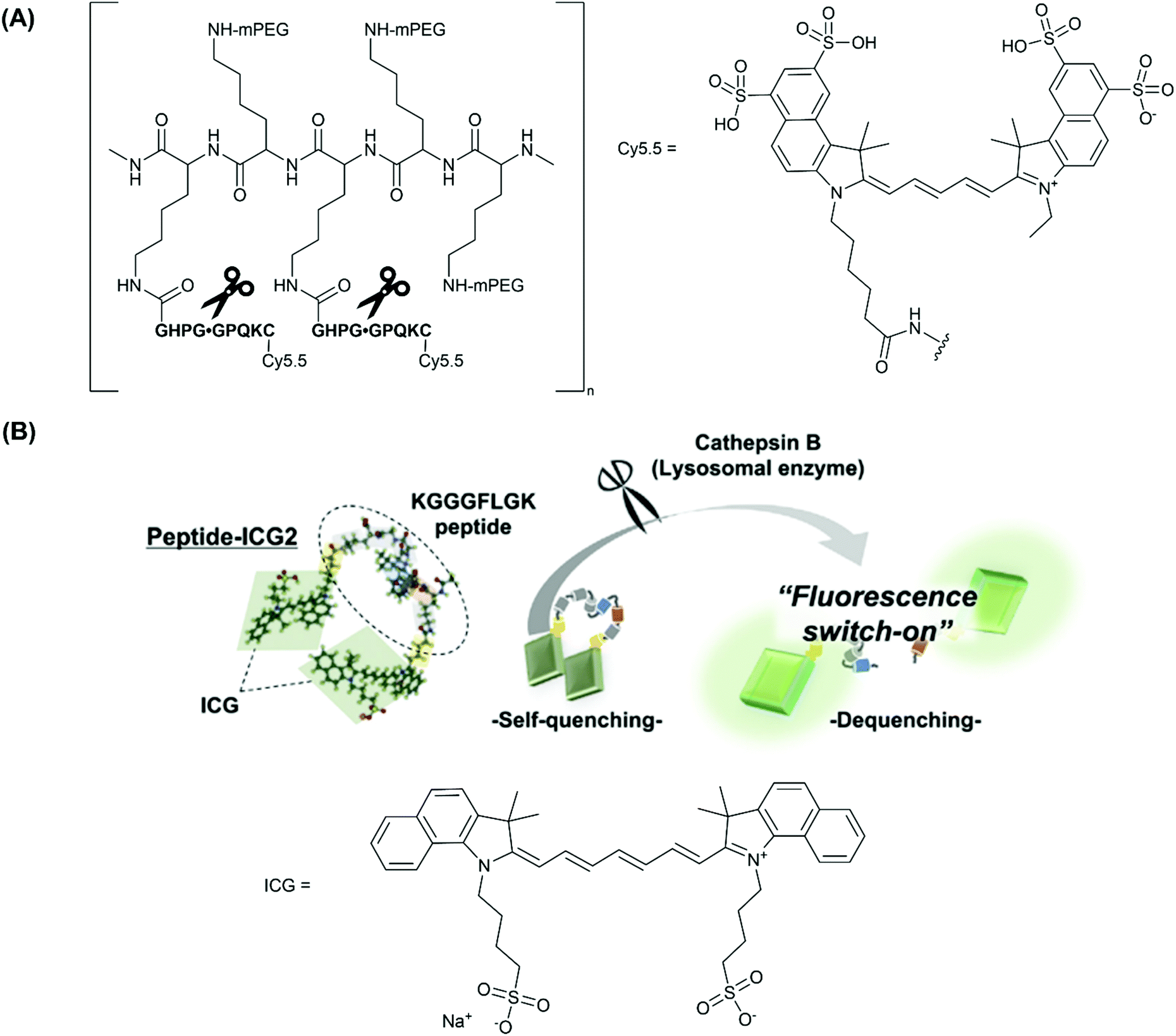 | ||
| Fig. 2 (A) The structure of CatK-FITC-PGC-Cy5.5, emphasising the three modular components; the Cy.5.5 unit, a CatK-specific GHPGGPQKC peptide substrate (cleaved at the glycyl-glycyl bond), and partially pegylated polylysine graft co-polymer to aid delivery of the agent, reported by Weissleder and co-workers.26 (B) The fluorescence ‘turn-on’ mechanism following enzymatic cleavage by cathepsin-B and the elimination of self-quenching utilised in peptide-ICG2, reported by Ogawa and co-workers.27 Reprinted with permission from ref. 27. Copyright (2019) Elsevier B.V. | ||
Earlier studies have also investigated a cathepsin B (CatB)-activated analogue in a murine atherosclerosis model.24,25 Indeed this probe, ProSense750/VM110, was used in the first demonstration of real-time intravascular Near-Infrared Fluorescence (NIRF) catheter-based imaging, by Jaffer and co-workers in 2008.66 Ultimately, two forms of commercially-available cathepsin-activated NIRF probes have been developed, based on the scaffold outlined above; ProSense 750/VM110, activated by cathepsins B, L and S, and ProSense680, activated by cathepsins B, L, S, K, V and D.67
More recently, Ogawa and co-workers employed another fluorescent ‘turn-on’ probe selective for CatB, designed to target macrophages in vulnerable atherosclerotic plaque, whilst avoiding the use of large co-polymer scaffold.27 Rather, phosphatidylserine (PS)-functionalised liposomes were used as a macrophage-specific delivery mechanism. In this study, indocyanine green (ICG, Fig. 2B), an FDA (U.S. Food and Drug Administration) approved NIR-1 cyanine dye, was used as the fluorophore of choice.
Probe peptide-ICG2 contains two ICG fluorophores linked via a small peptide sequence (KGGGFLGK) that can be selectively cleaved by cathepsin B. Initially, the NIR emission of peptide-ICG2 was quenched 15-fold compared to ICG, assumed by the authors to be due to π-stacking between the two ICG moieties (Fig. 2B). Following addition of cathepsin B, a time-dependent increase in the fluorescence intensity at 845 nm was observed in sodium acetate buffer (20 mM, pH = 5). Such an increase was attributed to the elimination of ICG self-quenching, and verification of cathepsin B-dependent peptide cleavage was confirmed following addition of leupeptin, a classical cathepsin B inhibitor.68
For in vitro and ex vivo imaging applications, peptide-ICG2 was encapsulated in a (PS)-functionalised liposome to target macrophages that had infiltrated vulnerable atherosclerotic plaque.69 For NIR-1 dyes, encapsulation in an nanocarrier platform can be highly advantageous as it dramatically increases the circulation time of the probe, allowing substantial accumulation in a biological target of choice. It is also thought that the stability of ICG and other cyanine dyes can increase considerably following encapsulation.70
NIR ex vivo fluorescence imaging of the aortae of a murine imaging model (Apolipoprotein E knockout (ApoE−/−)) confirmed that the CatB-induced fluorescence signal observed was located in atherosclerotic plaque macrophages. Analogous results have been displayed in work by Kim and co-workers.71 In the latter, a weaker fluorescence intensity was displayed in mouse aortas with a high-fat diet following addition of atorvastatin, a known statin medication widely characterised to prevent cardiovascular disease by reducing plaque.71 Furthermore, it was suggested by the authors that a weaker fluorescence intensity correlates with more stable atherosclerotic plaque, an outcome that could not be predicted from a visual inspection.71 However, without any internal calibration or ratiometric response, this must be expressed with some caution as intensity-based probes can have systematic errors such as differing cell uptake, potentially affecting the intensity of the fluorescence output observed. One major limitation of self-quenching fluorophores is this inherent difficulty in obtaining ratiometric measurements. Such a response is achieved more readily when a donor–acceptor FRET quenching mechanism is employed.
Taken together these results are still very encouraging and demonstrate that OI probes for cathepsins are excellent candidates for imaging plaque vulnerability.
In a notable example, Deguchi and co-workers developed a NIR Cy5.5-based self-quenching fluorogenic probe incorporating two Cy5.5 fluorophores linked via a GGPRQITAG peptide sequence on a pegylated polylysine backbone.75 The NIR probe was based on work by Huang and co-workers,76 and demonstrated a 200-fold increase in fluorescence intensity following cleavage by MMP-2 and MMP-9.75Ex vivo fluorescence imaging of atherosclerotic aortas of a ApoE−/− murine model showed an MMP-2/MMP-9 specific increase in NIR fluorescence (Fig. 3). Furthermore, histological validation confirmed that NIR fluorescence was localised in macrophage-rich regions of the aorta. In vivo analysis revealed an enhanced MMP activity in atherosclerotic lesions, indicating that related probes could help distinguish stable and vulnerable plaques.75 However, as with peptide-ICG2 (described in section 3.1.1), the non-ratiometric behaviour of these probes would limit precision of in vivo imaging applications.
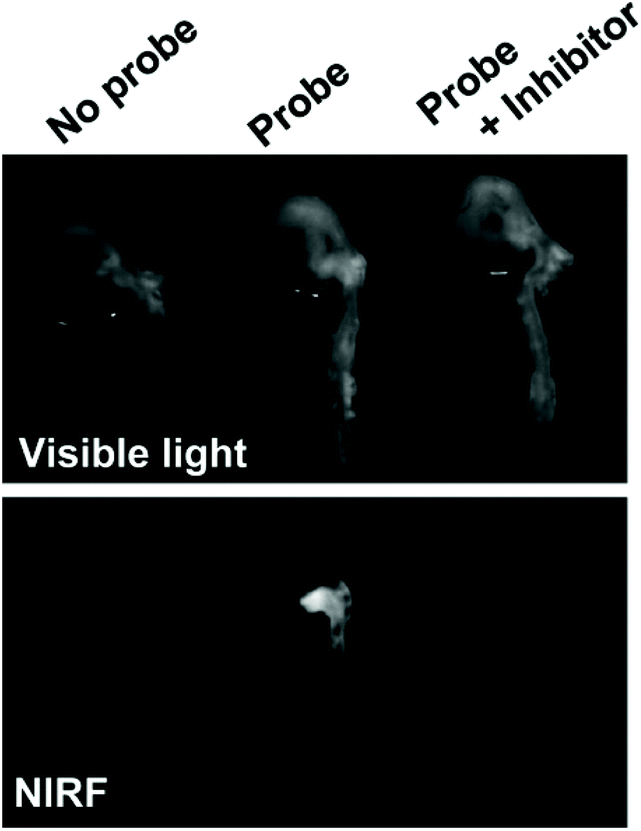 | ||
| Fig. 3 Ex vivo analysis of MMP-2 and MMP-9 activity in atherosclerotic aortas in a ApoE−/− murine model by Deguchi and co-workers.75 Fluorogenic selectivity was confirmed following addition of MMP-2/MMP-9 inhibitor III, resulting in no significant increase in NIR fluorescence versus the control. Reprinted with permission from ref. 75. Copyright (2006), Wolters Kluwer Health. | ||
Another probe developed by Tung and co-workers bears a NIR Cy5.5-labelled peptide, containing a thrombin-activated cleavable peptide substrate, appended to a partially pegylated polylysine grafted co-polymer as a delivery vector. An average of 23 fluorophores were attached to each polymeric carrier molecule. A high specificity for thrombin was demonstrated in PBS buffer, with no NIR fluorescence observed without thrombin (control) or following addition of CatD or CatB (Fig. 4A). Additionally, such a probe was shown to exhibit an 18-fold increase in fluorescence intensity in vitro, using both exogenous thrombin in human blood (Fig. 4B) and in a mouse intravascular thrombosis model.79 In the latter case, the probe could be detected in the loci of the induced thrombi. Whereas these studies have shown the possibility of significant uptake of this probe in fresh thrombi, it has not been investigated in the context of older thrombi. Nevertheless, likewise to cathepsin and MMP-activated agents, thrombin-activated probes for OI are expected to be relevant in a wide range of disease contexts.
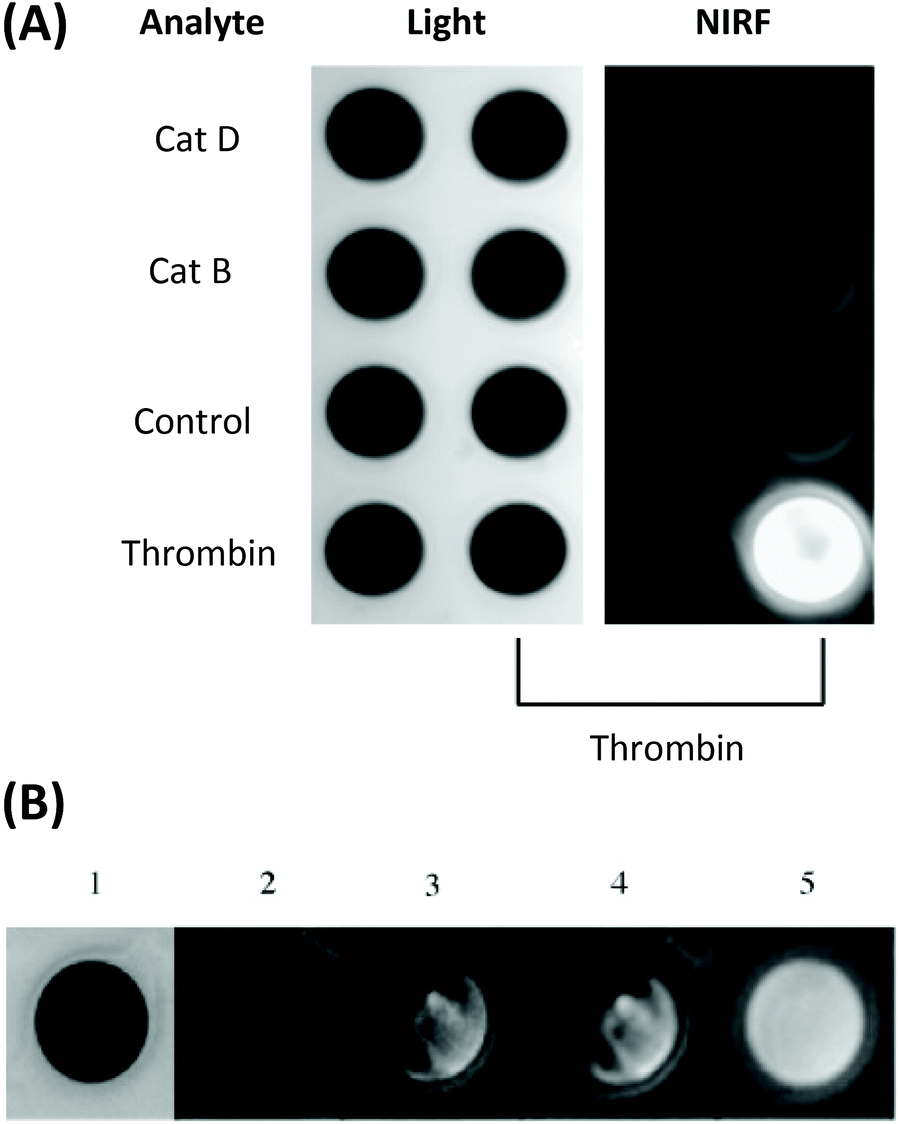 | ||
| Fig. 4 (A) The specificity of the thrombin-activated NIR fluorescent probe in PBS buffer (probe concentration = 0.5 μM). (B) Time-dependent increase in NIR fluorescence in whole blood with exogenous thrombin. 1 = light, 2 = NIR fluorescence image before the addition of thrombin, 3–5 = images at 3 min, 60 min, and 24 h respectively.78 Reprinted with permission from ref. 78. Copyright (2002) WILEY-VCH Verlag GmbH. | ||
3.2 Heme oxygenase-1 (HO-1)
The vast majority of probes described in section 3.1 make use of a short, cleavable peptide sequence to facilitate enzymatic cleavage at sites of interest. However, a focus on different enzyme targets can facilitate activatable NIR probes based on alternative molecular scaffolds. Such scaffolds have the potential to be synthetically more accessible than high molecular weight bioconjugates based on co-polymers, peptides or proteins.Heme oxygenase-1 (HO-1) plays a crucial role in vascular health, playing a protective role in hemorrhage and hematoma resolution.80 HO-1 is induced during the IPH of vulnerable plaque, and has somewhat been neglected as a biomarker in atherosclerosis imaging to date.
HO-1 catalytically degrades ‘free’ (or unbound) heme, with regioselectivity for the α-meso-position, from which it releases carbon monoxide (CO) (Fig. 5).81 Whilst heme is an essential prosthetic group in numerous enzymes and hemoglobin, in the ‘free’ form it is cytotoxic due to the generation of highly reactive oxygen intermediates. During this three-step process bilirubin is formed, and ferrous iron is directed to safe storage in ferritin, preventing the generation of reactive oxygen species via the Fenton reaction.82 Each of the degradation products of heme/HO-1 (bilirubin, CO and Fe-ferritin) are well-known to display antioxidant and antiapoptotic properties.83,84
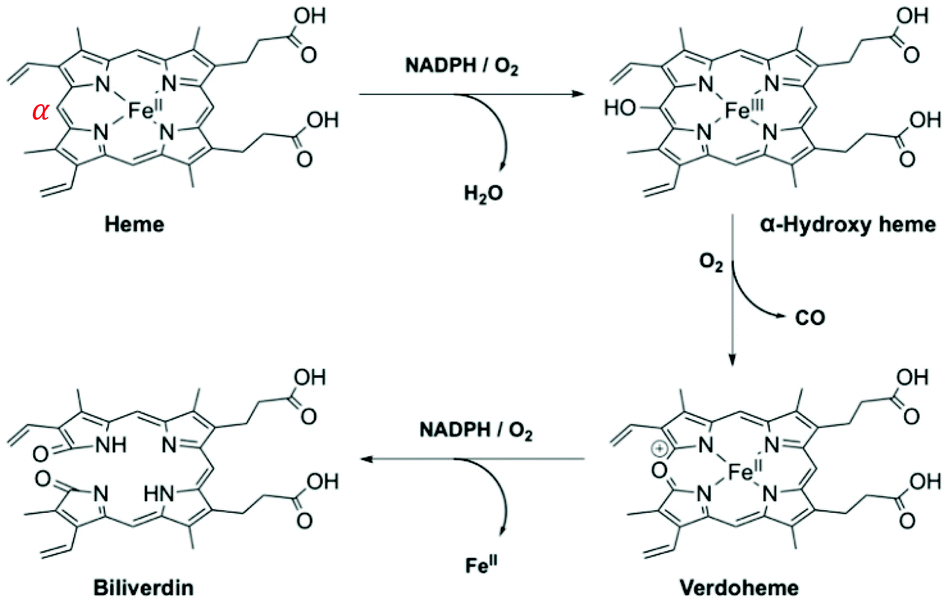 | ||
| Fig. 5 HO-1 catabolism of ‘free’ heme.85,86 | ||
To the best of our knowledge, there have been no known chemical probes to report on the activity of HO-1, holding back this field of research considerably. Our group has recently reported the design, development, photophysical and biological characterisation of the first known chemical probe to report on the activity of HO-1.85 Probe Fe–L1 (Fig. 6A) is based on a symmetrical analogue of heme, known to display an activity for HO-1.87 The α-carbon atom was functionalised with a hydroxycoumarin donor fluorophore, and displayed an efficient FRET to the porphyrin acceptor moiety prior to HO-1 catabolism.85
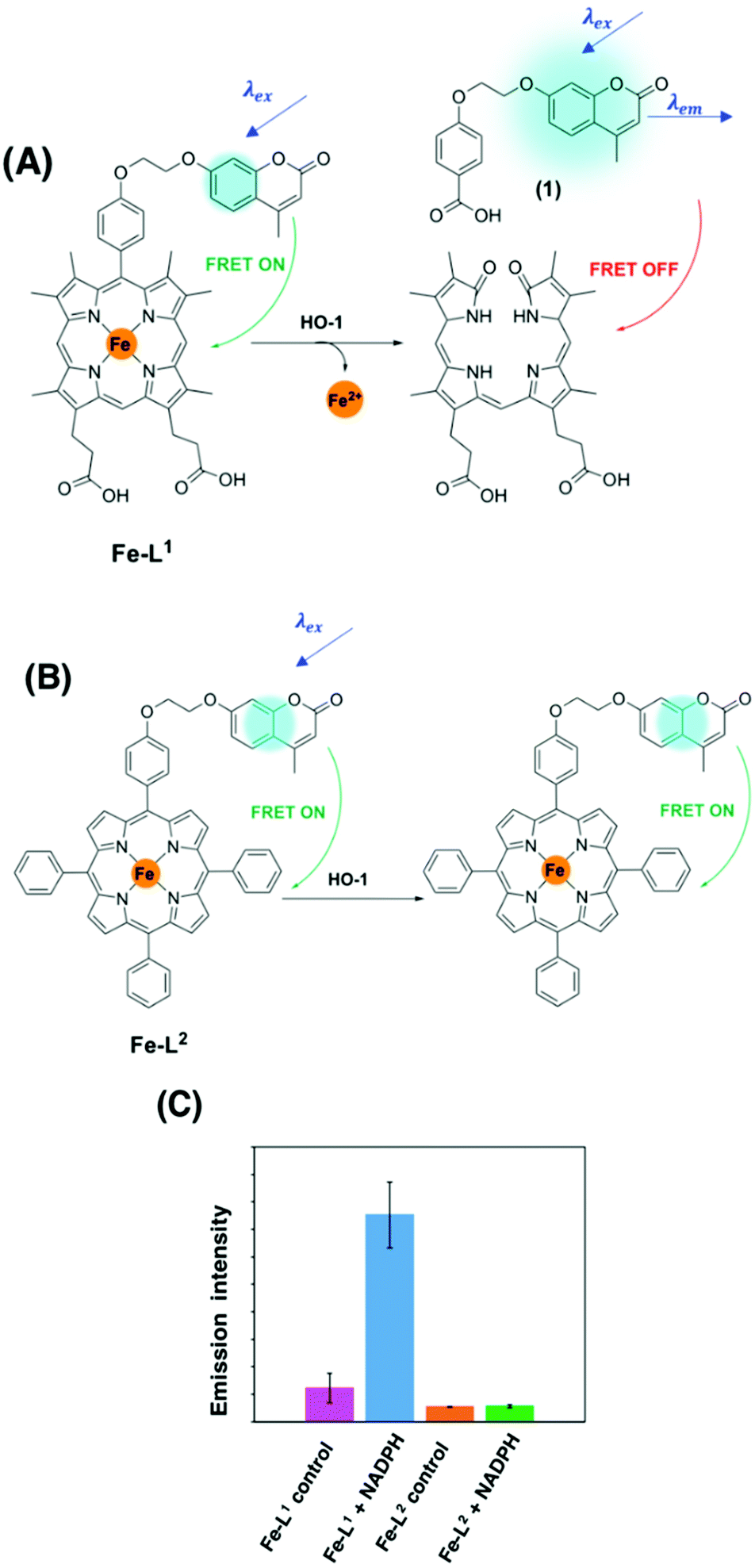 | ||
| Fig. 6 (A) HO-1 catalysed Regioselective α-cleavage of Fe–L1. (B) Diad Fe–L2 and (C) the change in coumarin fluorescence at 383 nm of Fe–L1 and Fe–L2 in E. coli lysates with and without incubation with NADPH.85 Reprinted with permission from ref. 85, https://pubs.acs.org/doi/abs/10.1021/jacs.0c12864. Further permissions related to the material excerpted should be directed to the ACS. | ||
HO-1 activity was quantified in Escherichia coli (E. coli) lysates overexpressing human HO-1 (hHO-1) in a proof-of-concept study. Following addition and incubation with NADPH, a 6-fold increase in coumarin fluorescence intensity versus the control was reported. Mass spectrometry analysis (LC-MS and MALDI-MS) determined that porphyrin cleavage was regiospecific at the α-position, forming compound 1, and perturbing the efficient FRET process from the coumarin donor to the porphyrin acceptor moiety (Fig. 6). Diad Fe–L2 (Fig. 6B) was not a substrate for HO-1 and was subsequently used as a control.85
The development of a FRET break-apart probe for HO-1 is a significant step forward towards the translation of such an approach into a real-time imaging agent. Such an advancement would be a valuable addition to this field and could allow the detection of IPH and plaque vulnerability, as well as other internal hemorrhages caused by aneurysm or stroke. One key area to improve on in the near future is the development of new analogues with red-shifted excitation and emission wavelengths.
Currently, Fe–L1 displays photophysical properties within the ultraviolet (UV) region of the electromagnetic spectrum. However, high energy excitation is undesirable in OI due to a poor biological tissue penetration and can trigger intracellular damage as a consequence of the formation of ROS. Work in our research group is currently on-going to develop analogues of Fe–L1 with red-shifted absorbance and emission wavelengths for live-fluorescence imaging applications. For this purpose, probes with a high FRET efficiency remain essential, enabling a high signal-to-noise ratio and maximising the ‘turn-on’ increase in fluorescence observed following detection of HO-1 activity.
3.3 Myeloperoxidase (MPO)
Myeloperoxidase (MPO) is a member of the heme peroxidase family that catalyses the formation of a number of reactive oxidant species including hypochlorous acid and peroxynitrite. MPO utilises hydrogen peroxide (H2O2) and a chloride anion (Cl−) as substrates to form hypochlorous acid (HOCl).88 At physiological pH, approximately half of all HOCl exists as a hypochlorite anion (OCl−), an extremely strong natural oxidant capable of causing tissue damage during inflammation.MPO is secreted by neutrophils and macrophages in human atherosclerotic lesions.89,90 Therefore, MPO provides another interesting enzymatic biomarker for the diagnosis and pathogenesis of atherosclerosis, through the detection of HOCl/OCl− activity.
In 2007, Libby and co-workers developed a self-immolative xanthene-based probe SNAPF, for selective detection of HOCl in MPO+ macrophages in atherosclerotic lesions. Probe SNAPF displayed an 8-fold increase in fluorescence intensity in PBS buffer at physiological pH (λex = 625 nm, λem = 676 nm), with a high specificity over other reactive oxygen and nitrogen species (Fig. 7A).91 Additionally, SNAPF was able to detect the generation of HOCl in a murine in vivo model and in frozen tissue sections of human atherosclerotic plaque. Such a study showed great promise and was one of the first of its kind.91
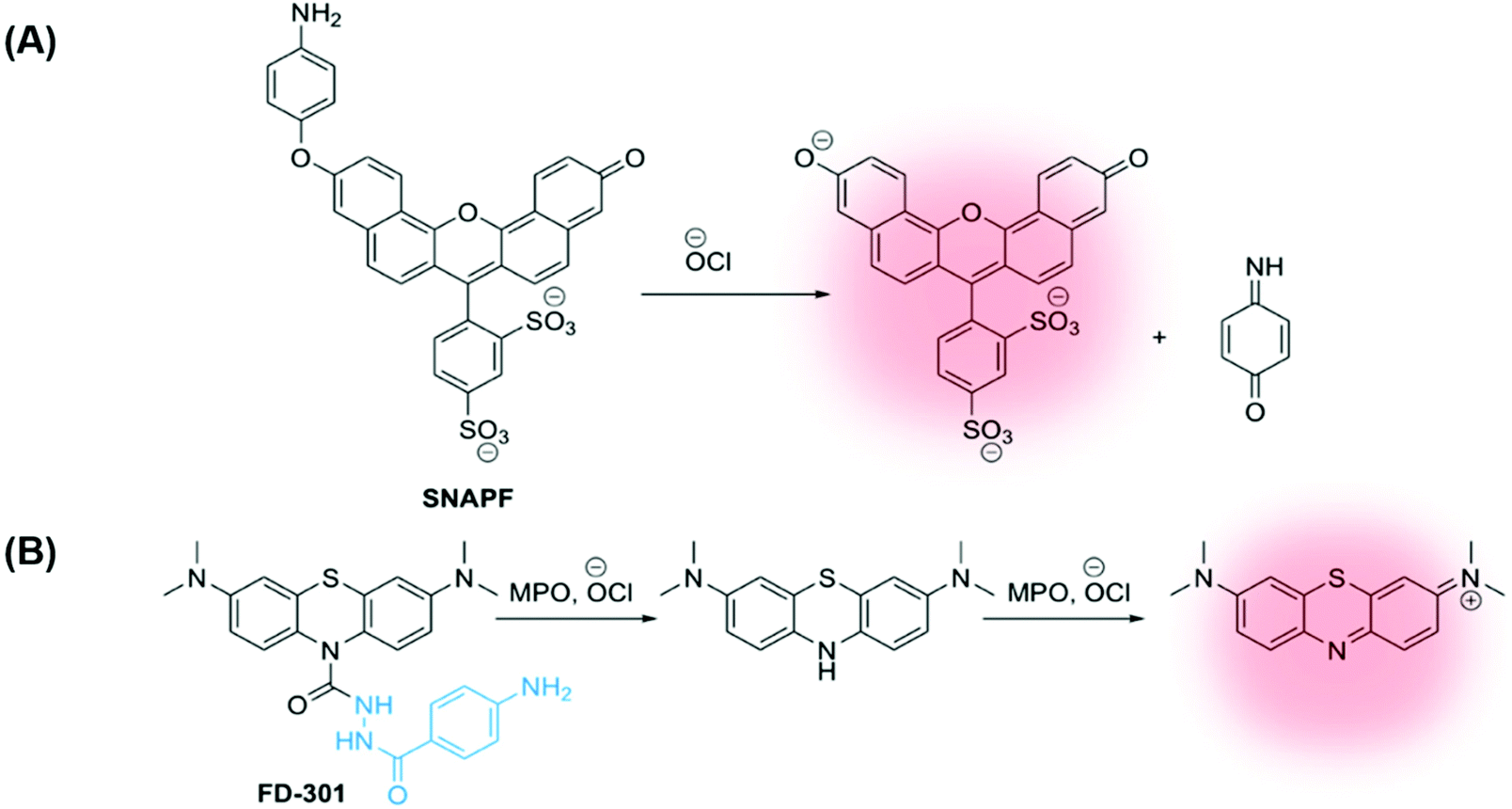 | ||
| Fig. 7 (A) Xanthene-based probe SNAPF for the detection of HOCl in atherosclerotic-associated macrophages, developed by Libby and co-workers.91 (B) A methylene blue (MB)-based NIR emissive ‘turn-on’ probe, FD-301, to detect basal MPO activity. MPO inhibitor 4-aminobenzoic acid hydrazide is highlighted in blue.92 | ||
More recently, Yi and Wei developed a NIR emissive ‘turn-on’ probe to detect basal MPO activity, FD-301 (Fig. 7B).92 4-Aminobenzoic acid hydrazide, a known MPO inhibitor,93 has been incorporated into the design of FD-301, facilitating binding to the MPO active site. However, from molecular docking and in vitro experiments, it was determined by the authors that the enzyme activity in FD-301 is not inhibited. This is likely due to the steric hindrance resulting from the presence of the methylene blue (MB) fluorophore in the structure of the probe. Such an approach is advantageous enabling the MB fluorophore to be in close proximity to the MPO active site, reducing complications arising from the short half-life of HOCl. Following formation of HOCl, the hydrazine bond in FD-301 is cleaved and a ‘turn-on’ increase in MB fluorescence is observed.92
The study by Yi and Wei shows promising data for the selective detection and quantification of HOCl and, by implication MPO activity in vivo. In this case, FD-301 was studied in vitro and in vivo in murine models for arthritis and ulcerative colitis.92 However, using a similar probe design and imaging approach in the future could accurately detect the overexpression of MPO in macrophages found in human atherosclerotic lesions.
4. Summary and perspective
Using recent approaches from the literature, we have summarised the desirable design considerations and activation mechanisms for enzyme-activatable probes in OI applications. With a particular focus on atherosclerosis imaging, and the detection of vulnerable atherosclerotic plague, an array of relevant enzymatic biomarkers has been evaluated, encompassing proteases (cathepsins, MMPs and thrombin), HO-1 and MPO.Differentiating between vulnerable and stable plaque remains the greatest challenge in this field. Whilst approaches described here are promising, they predominantly involve a “turn-on” activation mechanism, rather than a ratiometric behaviour. It remains key that new probes are developed in the future to ensure vascular diseases are detected earlier and more efficiently, before the onset of severe complications. The enzymes that are overexpressed in atherosclerosis also play key roles in a range of other chronic disorders, marked by inflammation and wound healing. Conversely, much information could be gained by repurposing enzyme-activated probes developed for other diseases, such as cancer. Such a strategy would improve the hugely challenging synthetic complexity which is often characteristic of enzyme-activatable probes. Ultimately, however, the development of new fluorogenic biomarkers for atherosclerosis is really required to significantly increase our understanding of this disease.
In particular, enzyme-activated fluorescent probes to image atherosclerotic lesions offer extremely promising applications in catheter-based OI. However, further clinical application of such probes is still dependent on the availability of a clinically approved catheter.67 In any case, the development of new fluorescent probes for less invasive in vivo imaging applications is also highly desirable. New NIR probes with high quantum yields, high stability, a high Stokes’ shift and low toxicity are required to this end. Furthermore, probes utilising two-photon excitation are also becoming increasingly desirable to facilitate greater depth penetration.
There are several challenges in developing OI agents applicable to atherosclerosis. These include a major trade-off between accessibility and wavelength. Longer wavelengths are better for tissue penetration and minimise tissue damage. For example, UV-excitable probes would be unacceptable. Yet, broadly, the longer the wavelength, the larger the dye molecule. Large fluorophores present challenges with entry in and out of the cells and tissue and may well present significant issues with steric hindrance of the enzyme site. Toxicity is a further consideration, particularly given the tissue is itself diseased. Appropriate instrumentation and route of delivery still remain challenges, although minimally invasive intracoronary approaches are now commonplace.
Other techniques, not discussed in detail over the course of this perspective, such as Photoacoustic Imaging (PAI) and luciferase-induced Bioluminescence Imaging (BLI), are appealing alternatives to OI. For example, PAI combines the advantages of OI and US imaging modalities, and possesses a higher intravascular resolution compared to MRI and CT,94 as well as a superior tissue penetration over OI.95 PAI is being validated at preclinical studies, where it allows relatively high-resolution imaging of vessels. Additionally, the technique is being incorporated into multimodality coronary imaging. Enzyme-activatable PAI probes have been developed for MMP-2,56,96 and will form a vital part of imaging toolkit to improve the diagnosis of vascular disease moving forward.
Luciferase-based imaging is already widely used in preclinical application, albeit predominantly based on genetic modifications, of a sort that would be challenging to translate clinically. For example, it has been widely used to image gene activation, based on insertion of artificial constructs comprised of gene regulatory sequence and an exogenous luciferase gene from firefly or jellyfish. A further evolution of this approach has been Bioluminescence Resonance Energy Transfer (BRET) which has primarily been used in the imaging of cell signalling.
Dual-modal imaging techniques are highly advantageous and will be further explored in the future. Such approaches have the potential to utilise an enzyme-activatable fluorophore to directly report on molecular abnormalities, in combination with a modality to report in detail on anatomical abnormalities. The combination of OI with MRI, for example, is one possibility due to the high spatial resolution and depth penetration that is characteristic of MRI, but both significantly lacking in OI. Research to this end is currently of major interest,97,98 and is likely to be studied further in the near future to develop the next generation of chemical probes for atherosclerosis imaging.
It is hoped that this perspective will provide an insight into the enzymatic targets for OI imaging of vulnerable atherosclerotic plaques, representing an exciting and emerging field within medical imaging. There are a number of directions to explore in the future, that could improve the diagnosis of vascular disease.
Conflicts of interest
The authors have applied for IP protection for a novel class of HO-1 fluorescence probes.Acknowledgements
We thank the British Heart Foundation (Project Grants PG/17/71/33242 and PG/21/10422) (EHRW, SMC, NJL and JJB) and EPSRC Centre for Doctoral Training in Medical Imaging (EP/L015226/1) (SMC) for funding. NJL is grateful for a Royal Society Wolfson Research Merit Award.References
- K. L. Wong, C. S. Lee and G. L. Law, ChemPlusChem, 2020, 85, 1093–1094 CrossRef PubMed.
- A. T. Aron, M. O. Loehr, J. Bogena and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 14338–14346 CrossRef PubMed.
- C. Y. S. Chung, J. M. Posimo, S. Lee, T. Tsang, J. M. Davis, D. C. Brady and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18285–18294 CrossRef PubMed.
- Y. Koide, R. Kojima, K. Hanaoka, K. Numasawa, T. Komatsu, T. Nagano, H. Kobayashi and Y. Urano, Commun. Chem., 2019, 2, 1–7 CrossRef.
- A. J. Lee, A. A. Ensign, T. D. Krauss and K. L. Bren, J. Am. Chem. Soc., 2010, 132, 1752–1753 CrossRef CAS PubMed.
- X. Jia, Q. Chen, Y. Yang, Y. Tang, R. Wang, Y. Xu, W. Zhu and X. Qian, J. Am. Chem. Soc., 2016, 138, 10778–10781 CrossRef PubMed.
- M. Rehm, H. Düßmann, R. U. Jänicke, J. M. Tavaré, D. Kögel and J. H. M. Prehn, J. Biol. Chem., 2002, 277, 24506–24514 CrossRef PubMed.
- J. DiFrancisco-Donoghue, E. Rabin, E. M. Lamberg and W. G. Werner, Mov. Disord. Clin. Pract., 2014, 1, 348–353 CrossRef PubMed.
- F. Sabeh, D. Fox and S. J. Weiss, J. Immunol., 2010, 184, 6396–6406 CrossRef PubMed.
- M. H. Baig, M. Adil, R. Khan, S. Dhadi, K. Ahmad, G. Rabbani, T. Bashir, M. A. Imran, F. M. Husain, E. J. Lee, M. A. Kamal and I. Choi, Semin. Cancer Biol., 2019, 56, 1–11 CrossRef PubMed.
- World Health Organisation (WHO): The top ten causes of death, http://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
- J. Frostegård, BMC Med., 2013, 11, 1–8 CrossRef PubMed.
- C. Silvestre-Roig, M. P. De Winther, C. Weber, M. J. Daemen, E. Lutgens and O. Soehnlein, Circ. Res., 2014, 114, 214–226 CrossRef PubMed.
- L. Parma, F. Baganha, P. H. A. Quax and M. R. de Vries, Eur. J. Pharmacol., 2017, 816, 107–115 CrossRef PubMed.
- J. J. Boyle, M. Johns, J. Lo, A. Chiodini, N. Ambrose, P. C. Evans, J. C. Mason and D. O. Haskard, Arterioscler. Thromb. Vasc. Biol., 2011, 31, 2685–2691 CrossRef PubMed.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- L. Y. Huang, S. Zhu, R. Cui and M. Zhang, Anal. Chem., 2020, 92, 535–542 CrossRef CAS.
- F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932–940 CrossRef.
- E. R. H. Walter, C. Hogg, D. Parker and J. A. G. Williams, Coord. Chem. Rev., 2021, 428, 213622 CrossRef.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef PubMed.
- B. M. Luby, D. M. Charron, C. M. MacLaughlin and G. Zheng, Adv. Drug Delivery Rev., 2017, 113, 97–121 CrossRef PubMed.
- R. Weissleder, C. H. Tung, U. Mahmood and A. Bogdanov, Nat. Biotechnol., 1999, 17, 375–378 CrossRef PubMed.
- J. Chen, C. H. Tung, U. Mahmood, V. Ntziachristos, R. Gyurko, M. C. Fishman, P. L. Huang and R. Weissleder, Circulation, 2002, 105, 2766–2771 CrossRef PubMed.
- F. A. Jaffer, D. E. Kim, L. Quinti, C. H. Tung, E. Aikawa, A. N. Pande, R. H. Kohler, G. P. Shi, P. Libby and R. Weissleder, Circulation, 2007, 115, 2292–2298 CrossRef PubMed.
- Y. Narita, K. Shimizu, K. Ikemoto, R. Uchino, M. Kosugi, M. B. Maess, Y. Magata, N. Oku and M. Ogawa, J. Controlled Release, 2019, 302, 105–115 CrossRef PubMed.
- T. Skajaa, D. P. Cormode, E. Falk, W. J. M. Mulder, E. A. Fisher and Z. A. Fayad, Arterioscler. Thromb. Vasc. Biol., 2010, 30, 169–176 CrossRef PubMed.
- A. Bernal, C. Calcagno, W. J. M. Mulder and C. Pérez-Medina, Curr. Opin. Chem. Biol., 2021, 63, 78–85 CrossRef CAS PubMed.
- H. Qiao, Y. Wang, R. Zhang, Q. Gao, X. Liang, L. Gao, Z. Jiang, R. Qiao, D. Han, Y. Zhang, Y. Qiu, J. Tian, M. Gao and F. Cao, Biomaterials, 2017, 112, 336–345 CrossRef.
- R. J. Evans, B. Lavin, A. Phinikaridou, K. Y. Chooi, Z. Mohri, E. Wong, J. J. Boyle, R. Krams, R. Botnar and N. J. Long, Nanotheranostics, 2020, 4, 184–194 CrossRef PubMed.
- A. V. Ramsey, A. J. Bischoff and M. B. Francis, J. Am. Chem. Soc., 2021, 143, 7342–7350 CrossRef PubMed.
- M. Wu, X. Li, Q. Guo, J. Li, G. Xu, G. Li, J. Wang and X. Zhang, Nanomedicine, 2021, 32, 102330 CrossRef PubMed.
- K. Oumzil, M. A. Ramin, C. Lorenzato, A. Hémadou, J. Laroche, M. J. Jacobin-Valat, S. Mornet, C. E. Roy, T. Kauss, K. Gaudin, G. Clofent-Sanchez and P. Barthélémy, Bioconjugate Chem., 2016, 27, 569–575 CrossRef PubMed.
- A. J. Smith, B. E. Osborne, G. P. Keeling, P. J. Blower, R. Southworth and N. J. Long, Dalton Trans., 2020, 49, 1097–1106 RSC.
- Y. Wei, Y. Liu, Y. He and Y. Wang, J. Mater. Chem. B, 2021, 9, 908–920 RSC.
- Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104–109 CrossRef PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef PubMed.
- Y. Yuan, R. Zhang, X. Cheng, S. Xu and B. Liu, Chem. Sci., 2016, 7, 4245–4250 RSC.
- H. Li, Q. Yao, W. Sun, K. Shao, Y. Lu, J. Chung, D. Kim, J. Fan, S. Long, J. Du, Y. Li, J. Wang, J. Yoon and X. Peng, J. Am. Chem. Soc., 2020, 142, 6381–6389 CrossRef CAS PubMed.
- R. Wang, J. Chen, J. Gao, J. A. Chen, G. Xu, T. Zhu, X. Gu, Z. Guo, W. H. Zhu and C. Zhao, Chem. Sci., 2019, 10, 7222–7227 RSC.
- W. Chyan and R. T. Raines, ACS Chem. Biol., 2018, 13, 1810–1823 CrossRef PubMed.
- J. Zhang, X. Chai, X. P. He, H. J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683–722 RSC.
- S. Y. Li, H. Cheng, B. R. Xie, W. X. Qiu, L. L. Song, R. X. Zhuo and X. Z. Zhang, Biomaterials, 2016, 104, 297–309 CrossRef PubMed.
- R. Tian, M. Li, J. Wang, M. Yu, X. Kong, Y. Feng, Z. Chen, Y. Li, W. Huang, W. Wu and Z. Hong, Org. Biomol. Chem., 2014, 12, 5365–5374 RSC.
- H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef PubMed.
- S. U. Hettiarachchi, B. Prasai and R. L. McCarley, J. Am. Chem. Soc., 2014, 136, 7575–7578 CrossRef PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef PubMed.
- W. Chyan, H. R. Kilgore, B. Gold and R. T. Raines, J. Org. Chem., 2017, 82, 4297–4304 CrossRef PubMed.
- T. Shen, S. Zang, W. Shu, L. Nie, J. Jing and X. Zhang, Dyes Pigm., 2020, 178, 108345 CrossRef CAS.
- R. S. Kathayat, Y. Cao, P. D. Elvira, P. A. Sandoz, M. E. Zaballa, M. Z. Springer, L. E. Drake, K. F. Macleod, F. G. Van Der Goot and B. C. Dickinson, Nat. Commun., 2018, 9, 334 CrossRef PubMed.
- H. Li, Q. Yao, F. Xu, N. Xu, R. Duan, S. Long, J. Fan, J. Du, J. Wang and X. Peng, Biomaterials, 2018, 179, 1–14 CrossRef PubMed.
- H. Xing, S. Zhang, W. Bu, X. Zheng, L. Wang, Q. Xiao, D. Ni, J. Zhang, L. Zhou, W. Peng, K. Zhao, Y. Hua and J. Shi, Adv. Mater., 2014, 26, 3867–3872 CrossRef PubMed.
- A. Phinikaridou, F. L. Ruberg, K. J. Hallock, Y. Qiao, N. Hua, J. Viereck and J. A. Hamilton, Circ. Cardiovasc. Imaging, 2010, 3, 323–332 CrossRef PubMed.
- D. Peters, M. Kastantin, V. R. Kotamraju, P. P. Karmali, K. Gujraty, M. Tirrell and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9815–9819 CrossRef PubMed.
- H. Qin, Y. Zhao, J. Zhang, X. Pan, S. Yang and D. Xing, Nanomedicine, 2016, 12, 1765–1774 CrossRef PubMed.
- H. Orbay, H. Hong, Y. Zhang and W. Cai, Theranostics, 2013, 3, 894–902 CrossRef PubMed.
- J. H. F. Rudd, E. A. Warburton, T. D. Fryer, H. A. Jones, J. C. Clark, N. Antoun, P. Johnström, A. P. Davenport, P. J. Kirkpatrick, B. N. Arch, J. D. Pickard and P. L. Weissberg, Circulation, 2002, 105, 2708–2711 CrossRef PubMed.
- R. Qiao, X. Huang, Y. Qin, Y. Li, T. P. Davis, C. E. Hagemeyer and M. Gao, Nanoscale, 2020, 12, 8040–8064 RSC.
- P. J. Murray and T. A. Wynn, Nat. Rev. Immunol., 2011, 11, 723–737 CrossRef CAS PubMed.
- A. Fernández and M. Vendrell, Chem. Soc. Rev., 2016, 45, 1182–1196 RSC.
- G. W. van Lammeren, F. L. Moll, G. Jan De Borst, D. P. V. de Kleijn, J.-P. P. M. de Vries and G. Pasterkamp, Curr. Cardiol. Rev., 2012, 7, 22–27 CrossRef PubMed.
- A. Zia, Y. Wu, T. Nguyen, X. Wang, K. Peter and H. T. Ta, Cardiovasc. Res., 2020, 116, 2055–2068 CrossRef PubMed.
- Z. S. Galis, G. K. Sukhova, M. W. Lark and P. Libby, J. Clin. Invest., 1994, 94, 2493–2503 CrossRef PubMed.
- C. F. Zhao and D. M. Herrington, Am. J. Cardiovasc. Dis., 2016, 6, 163–170 CAS.
- F. A. Jaffer, C. Vinegoni, M. C. John, E. Aikawa, H. K. Gold, A. V. Finn, V. Ntziachristos, P. Libby and R. Weissleder, Circulation, 2008, 118, 1802–1809 CrossRef PubMed.
- H. Khraishah and F. A. Jaffer, Curr. Cardiovasc. Imaging Rep., 2020, 13, 8 CrossRef.
- A. Baici and M. Gyger-Marazzi, Eur. J. Biochem., 1982, 129, 33–41 CrossRef CAS PubMed.
- M. Ogawa, I. O. Umeda, M. Kosugi, A. Kawai, Y. Hamaya, M. Takashima, H. Yin, T. Kudoh, M. Seno and Y. Magata, J. Nucl. Med., 2014, 55, 115–120 CrossRef CAS PubMed.
- Z. Qu, J. Shen, Q. Li, F. Xu, F. Wang, X. Zhang and C. Fan, Theranostics, 2020, 10, 2631–2644 CrossRef CAS PubMed.
- D. E. Kim, J. Y. Kim, D. Schellingerhout, S. M. Shon, S. W. Jeong, E. J. Kim and W. K. Kim, Mol. Imaging, 2009, 8, 291–301 CrossRef CAS PubMed.
- T. Myochin, K. Hanaoka, S. Iwaki, T. Ueno, T. Komatsu, T. Terai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 4759–4765 CrossRef CAS PubMed.
- B. Mills, D. Norberg, K. Dhaliwal, A. R. Akram, M. Bradley and A. Megia-Fernandez, Chem. Commun., 2020, 56, 9962–9965 RSC.
- W. Pham, Y. Choi, R. Weissleder and C. H. Tung, Bioconjugate Chem., 2004, 15, 1403–1407 CrossRef PubMed.
- J. O. Deguchi, M. Aikawa, C. H. Tung, E. Aikawa, D. E. Kim, V. Ntziachristos, R. Weissleder and P. Libby, Circulation, 2006, 114, 55–62 CrossRef PubMed.
- J. Chen, C. H. Tung, J. R. Allport, S. Chen, R. Weissleder and P. L. Huang, Circulation, 2005, 111, 1800–1805 CrossRef PubMed.
- N. Jaberi, A. Soleimani, M. Pashirzad, H. Abdeahad, F. Mohammadi, M. Khoshakhlagh, M. Khazaei, G. A. Ferns, A. Avan and S. M. Hassanian, J. Cell. Biochem., 2019, 120, 4757–4765 CrossRef PubMed.
- C. H. Tung, R. E. Gerszten, F. A. Jaffer and R. Weissleder, ChemBioChem, 2002, 3, 207–211 CrossRef PubMed.
- F. A. Jaffer, C. H. Tung, R. E. Gerszten and R. Weissleder, Arterioscler. Thromb. Vasc. Biol., 2002, 22, 1929–1935 CrossRef PubMed.
- A. Seneviratne, Y. Han, E. Wong, E. R. H. Walter, L. Jiang, L. Cave, N. Long, D. Carling, J. Mason, D. Haskard and J. Boyle, Circ. Res., 2020, 127, 928–944 CrossRef PubMed.
- P. R. Ortiz De Montellano, Curr. Opin. Chem. Biol., 2000, 4, 221–227 CrossRef.
- S. M. H. Sadrzadeh, E. Graf, S. S. Panter, P. E. Hallaway and J. W. Eaton, J. Biol. Chem., 1984, 259, 14354–14356 CrossRef CAS.
- S. M. Kapetanaki, M. J. Burton, J. Basran, C. Uragami, P. C. E. Moody, J. S. Mitcheson, R. Schmid, N. W. Davies, P. Dorlet, M. H. Vos, N. M. Storey and E. Raven, Nat. Commun., 2018, 9, 907 CrossRef.
- S. W. Ryter, J. Alam and A. M. K. Choi, Physiol. Rev., 2006, 86, 583–650 CrossRef CAS.
- E. R. H. Walter, Y. Ge, J. C. Mason, J. J. Boyle and N. J. Long, J. Am. Chem. Soc., 2021, 143, 6460–6469 CrossRef CAS PubMed.
- M. Unno, T. Matsui and M. Ikeda-Saito, Nat. Prod. Rep., 2007, 24, 553–570 RSC.
- R. B. Frydman, M. L. Tomara, G. Buldain, J. Awruch, L. Diaz and B. Frydman, Biochemistry, 1981, 20, 5177–5182 CrossRef CAS PubMed.
- S. J. Weiss, R. Klein, A. Slivka and M. Wei, J. Clin. Invest., 1982, 70, 598–607 CrossRef CAS.
- A. Daugherty, J. L. Dunn, D. L. Rateri and J. W. Heinecke, J. Clin. Invest., 1994, 94, 437–444 CrossRef PubMed.
- S. J. Nicholls and S. L. Hazen, Arterioscler. Thromb. Vasc. Biol., 2005, 25, 1102–1111 CrossRef PubMed.
- J. Shepher, S. A. Hilderbrand, P. Waterman, J. W. Heinecke, R. Weissleder and P. Libby, Chem. Biol., 2007, 14, 1221–1231 CrossRef PubMed.
- L. Liu, P. Wei, W. Yuan, Z. Liu, F. Xue, X. Zhang and T. Yi, Anal. Chem., 2020, 92, 10971–10978 CrossRef PubMed.
- A. J. Kettle, C. A. Gedye and C. C. Winterbourn, 1996, 508, 1–6.
- X. Bai, X. Gong, W. Hau, R. Lin, J. Zheng, C. Liu, C. Zeng, X. Zou, H. Zheng and L. Song, PLoS One, 2014, 9, 1–6 Search PubMed.
- L. V. Wang and S. Hu, Science, 2012, 335, 1458–1462 CrossRef CAS PubMed.
- K. Yang, L. Zhu, L. Nie, X. Sun, L. Cheng, C. Wu, G. Niu, X. Chen and Z. Liu, Theranostics, 2014, 4, 134–141 CrossRef CAS PubMed.
- L. E. Jennings and N. J. Long, Chem. Commun., 2009, 3511–3524 RSC.
- M. A. Bruckman, K. Jiang, E. J. Simpson, L. N. Randolph, L. G. Luyt, X. Yu and N. F. Steinmetz, Nano Lett., 2014, 14, 1551–1558 CrossRef CAS PubMed.
Footnotes |
| † FRET is a distance–dependent interaction that involves the donation of energy from a donor to an acceptor fluorophore proving that there is a good spectral overlap, and there are in close proximity. |
| ‡ PET is a non-radiative decay process that quenches luminescence. It is characterised by an electron transfer from the highest occupied molecular orbital (HOMO) of a receptor (electron donor) to the HOMO of a lumiphore in its excited state. |
| This journal is © The Royal Society of Chemistry 2021 |