
Self-assembled peptide–inorganic nanoparticle superstructures: from component design to applications
Claudia
Pigliacelli
 ,
Rosalía
Sánchez-Fernández
,
Marcos D.
García
,
Carlos
Peinador
and
Elena
Pazos
*
,
Rosalía
Sánchez-Fernández
,
Marcos D.
García
,
Carlos
Peinador
and
Elena
Pazos
*
Departamento de Química, Facultade de Ciencias and Centro de Investigacións Científicas Avanzadas (CICA), Universidade da Coruña, 15071 A Coruña, Spain. E-mail: elena.pazos@udc.es
First published on 4th June 2020
Abstract
Peptides have become excellent platforms for the design of peptide–nanoparticle hybrid superstructures, owing to their self-assembly and binding/recognition capabilities. Morover, peptide sequences can be encoded and modified to finely tune the structure of the hybrid systems and pursue functionalities that hold promise in an array of high-end applications. This feature article summarizes the different methodologies that have been developed to obtain self-assembled peptide–inorganic nanoparticle hybrid architectures, and discusses how the proper encoding of the peptide sequences can be used for tailoring the architecture and/or functionality of the final systems. We also describe the applications of these hybrid superstructures in different fields, with a brief look at future possibilities towards the development of new functional hybrid materials.
 Claudia Pigliacelli | Claudia Pigliacelli received her PhD in Pharmacy in 2015 from the University of East Anglia (UK). She has been working as a research assistant (2013–2015) and postdoctoral fellow (2015–2016) in the SupraBioNano Lab at the Politecnico di Milano (Italy), and in the Molecular Materials group in the Applied Physics Department of Aalto University (Finland, 2016–2019). In 2020 she joined the group of Elena Pazos at the Advanced Scientific Research Center (CICA). Her research interests include nanoparticle design and self-assembly, peptide and peptide–nanoparticle systems and in general nanomaterials for biomedical applications. |
 Rosalía Sánchez-Fernández | Rosalía Sánchez-Fernández obtained her BSc degree in Biotechnology from the Universidad de Oviedo in 2018. The following year, she received her MSc degree in Supramolecular Chemistry from the Universidad de Salamanca. In 2020, Rosalía joined the Universidade da Coruña as a PhD student, where she is working under the supervision of Dr Elena Pazos and Prof. Carlos Peinador. Her research is focused on the development of metallopeptides to control the formation of nanostructures. |
 Marcos D. García | Marcos D. García studied Chemistry at the Universidade de Santiago de Compostela, where he completed his PhD in 2005 under the supervision of Prof. F. Fernández and Prof. O. Caamaño. After post-doctoral studies at the University of Leicester with Prof. Paul R. Jenkins, in 2007 he moved back to Spain, joining the Universidade da Coruña as an Isidro Parga Pondal Fellow. Marcos currently holds a position at the UDC as Associate Professor, focusing his research interests on the field of molecular self-assembly, trying to find new ways to make heterocyclic compounds obey his will. |
 Carlos Peinador | Carlos Peinador studied chemistry at Universidade de Santiago de Compostela, receiving his PhD degree from the Universidade da Coruña where he has been working as Associate Professor from 2000 to 2018 and as Full Professor since 2018. His current research interests are oriented towards the field of Supramolecular Chemistry, with a particular focus on the use of 4,4′-bipyridinium-based cations for the self-assembly of catenanes and inclusion complexes and their implementation into stimuli-responsive supramolecular systems. He is the author of more than 100 research articles in peer-reviewed journals, and has written several Reviews and book chapters. |
 Elena Pazos | Elena Pazos obtained her PhD in 2012 from the Universidade de Santiago de Compostela under the supervision of Prof. José L. Mascareñas and Prof. M. Eugenio Vázquez. In 2012 she received the Fundación Barrié Postdoctoral fellowship and joined Prof. Samuel I. Stupp at Northwestern University. She then joined Medcom Advance in 2014, and from December 2015 worked in the group of Prof. Ramón A. Álvarez-Puebla at Centre Tecnològic de la Química. In July 2017 she joined the Universidade da Coruña as an InTalent researcher, where her group is working on the development of new peptide-based materials and biosensors. |
Introduction
In chemistry, self-assembly refers to the process by which discrete constituents, encoded with appropriate information, spontaneously organize into highly ordered entities.1,2 This concept was promptly implemented in the field of nanotechnology, with nanoparticle (NP) research expanding its main focus from NP synthesis, morphological control, and colloidal stability, towards hierarchical organization into superstructures.3–7 Such interest arose from the fact that NP properties not only depend on their shape, size, and composition, but also on their spatial distribution.8 Given the attention gained by these superstructures, several approaches have been proposed for programming and tuning NP self-assembly,3–5 ranging from the use of weak intermolecular forces,9–15 to the application of external inputs, such as electric or magnetic fields.16,17 Moreover, different biomolecules, including DNA, proteins, lipids, etc., have been used as linkers or templates for the design of a large variety of hybrid superstructures.3,4,18,19 Notably, biomolecule-templated routes offer the possibility of achieving controlled periodicity of the assembled NPs, and even the programmed assembly of hierarchical structures, via the introduction and controlled arrangement of specific reactive sites for the nucleation and/or binding of NPs.In this context, peptide-driven inorganic nanoparticle superstructures, which combine the features of inorganic nanoscale particles with the inherent recognition properties of both naturally occurring and synthetic peptides, have gained a prominent role in the last two decades.20–26 Peptides are essential structural and signalling molecules in biological systems and, thanks to their self-assembly and molecular recognition capabilities, have turned into excellent platforms for the development of many supramolecular self-assembled nanostructures.27–31 Taking in consideration the intrinsic biological activity and biocompatibility of peptides, as well as their synthetic and chemical versatility, the resulting materials have been exploited in many different applications, ranging from biomedicine to even electronics.29,32–34 Furthermore, the possibility of encoding different self-assembling peptide motifs with selective binding or reactive sites for a chosen inorganic material gives access to a comprehensive toolbox of templates for spatially-controlled NP synthesis and assembly into tailored peptide–NP hybrid superstructures.35–38 Besides, the self-assembling peptide sequences can be further functionalized to endow these hybrid systems with specific interaction/binding sites and/or stimuli responsiveness. Therefore, the combination of peptides’ versatility together with the unique optical and chemical properties of nanoscale particles grants the possibility to devise functional hybrid superstructures, having tuned architectures and properties, exploitable in a wide range of high-end applications (Fig. 1).
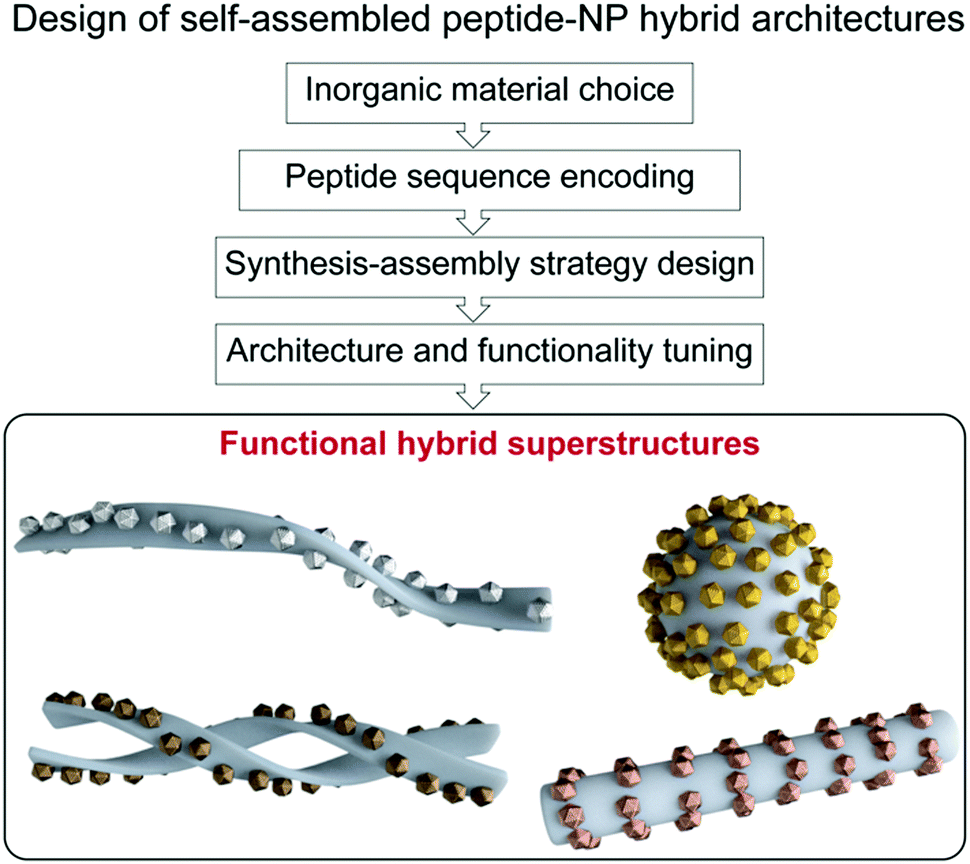 | ||
| Fig. 1 Schematic representation of the self-assembled peptide–NP superstructure design process. | ||
These types of hybrid superstructures have been reviewed in the last few years.29,39–41 However, these review papers are mainly focused on the description of peptide–NP hybrid systems and their applications in different fields, while in this feature article we summarize the different synthetic strategies that have been developed to obtain self-assembled peptide–NP hybrid architectures, and discuss how the proper encoding of the peptide sequences modulates their structure–function relationship, enabling innovative and/or collective properties. We also describe the applications of these peptide-based hybrid superstructures in different fields, foregrounding their potential role in advancing nanomaterials research and technologies.
Synthetic strategies for obtaining self-assembled peptide–nanoparticle hybrid architectures
In living systems many inorganic materials, such as bones, teeth, or shells, are produced under mild aqueous conditions. For their synthesis, organisms use peptides and proteins to interact with the precursor inorganic species and assemble them into ordered structures. These biomineralization processes have served as inspiration for chemists, who have made use of peptides as scaffolds for the preparation of hybrid self-assembled peptide–NP superstructures.38,42In this scenario, peptides can be seen as multifunctional reagents for the synthesis of inorganic NPs, since they can display a variety of reactive functional groups in their amino acid side chains that can work both as reducing and capping/stabilizing agents.43 Moreover, thanks to their synthetic simplicity, peptides can be further modified to include non-natural functionalities within their sequences and structures. Considering this chemical versatility of peptides, and the possible combination with external reducing agents or additional orthogonal intermolecular interactions, several approaches have been described for the production of custom superstructures. In this review, we have classified the different synthetic strategies reported in the literature according to the level of preorganization of the peptide and inorganic NP building blocks employed to obtain the hybrid architectures, namely one-pot synthesis and self-assembly, peptide nanostructure-templated synthesis, and self-assembly of peptide nanostructures and NPs. A summary of the different tactics presented for each synthetic strategy is shown in Table 1.
| Synthetic strategy | Tactic | Inorganic material | Ref. |
|---|---|---|---|
| One-pot synthesis and self-assembly | Peptide-induced (AYSSGAPPMPPF) | Au | 44, 47 and 48 |
| Iodide-induced | Au | 37 | |
| Peptide nanostructure-templated synthesis | Metal ion binding and reduction | Au, Ag, Cu, Ni, Pd, Pt | 49 and 51–58 |
| Tollens’ reaction | Ag | 36 | |
| Self-assembly of peptide nanostructures and NPs | Hydrogen-bond formation | Au | 59 and 60 |
| Thiol-based immobilization | Au | 55, 61 and 62 | |
| Electrostatic interaction | Au, Pt | 63–65 |
One-pot synthesis and self-assembly
In 2008, the group of Rosi reported a new direct approach that coupled both peptide self-assembly and NP synthesis into a synchronous process, demonstrating that the design of specific peptide conjugates can produce homogeneous hybrid systems with precise arrangement of the NPs within the superstructure.44 The designed peptide conjugates included the AYSSGAPPMPPF sequence (PEPAu), which was identified by phage-display45 and was already known to produce homogenous gold NPs (AuNPs) in HEPES buffer.46 In addition, a C12 hydrophobic chain was attached to the N-terminus of the sequence (Fig. 2a). The obtained conjugates nicely self-assembled into uniform left-handed twisted-nanoribbons in HEPES buffer, driven by the hydrophobic effect and the formation of parallel β-sheets between the AYSS amino acids (Fig. 2b and c). The self-assembly into nanoribbons was favoured over the formation of condensed nanofibers due to the presence of bulky amino acids over their surface (a phenylalanine and four proline residues). Consequently, left-handed double helices decorated with monodisperse AuNPs were obtained when HAuCl4 and C12-PEPAu solutions were mixed, without the need for an external reducing agent (Fig. 2e and f). In this case, the synthesis of the AuNPs was aided by the tyrosine residue (Y) on the peptide and the HEPES buffer. Interestingly, the hybrid superstructures appeared highly regular and maintained the structural properties of the self-assembled peptides, i.e. chirality and pitch. Therefore, the peptide conjugates could control the formation of hybrid double helices and interact with AuNPs without impacting their self-assembly pattern, demonstrating the potential of this methodology in obtaining highly regular superstructures in a synchronous orthogonal fashion.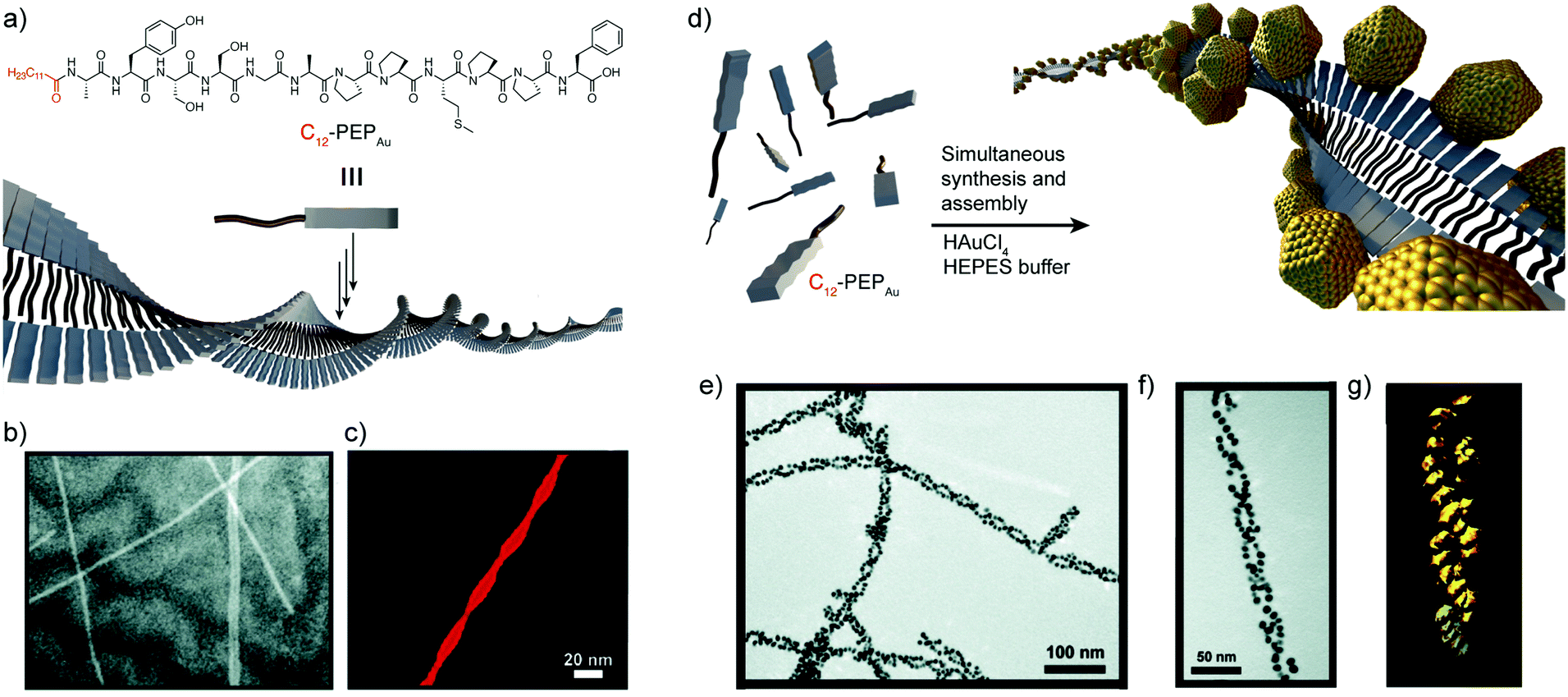 | ||
| Fig. 2 (a) C12-PEPAu chemical structure and representation of its self-assembly into twisted-nanoribbons. (b) TEM and (c) AFM images of C12-PEPAu nanoribbons. (d) Schematic representation of the AuNP double helices. (e and f) TEM images, and (g) electron tomography reconstruction of the peptide–AuNP superstructures. Adapted with permission from ref. 44. Copyright 2008 American Chemical Society. | ||
Later, the same research group expanded this methodology to obtain size-controlled hollow spherical peptide–AuNP superstructures.47,48 First, they showed that small modifications in the peptide conjugates could modify the superstructure topology, leading to spherical instead of monodimensional superstructures. Indeed, the substitution of the C12 hydrophobic chain by a C6 tail, together with the inclusion of two alanine residues (AA) at the N-terminus of the PEPAu sequence, favoured the formation of well-defined and monodisperse hollow spherical superstructures (diameter ≈ 52 nm), showing the versatility of the one-pot methodology and the possibility of programming the shape of the superstructures by encoding the peptide sequences.47 One year later, the authors described how the substitution of the C6 tail by a biphenyl unit (BP) in the peptide sequence (BP-AA-PEPAu) allowed the tuning of the spherical assembly size, with the formation of larger hollow vesicular superstructures (diameter ≈ 60–270 nm). Moreover, upon incorporation of hydrophobic residues at the N-terminus of the sequence (i.e. BP-AAA-PEPAu), further changes were observed in the self-assembled structures, yielding homogeneous spherical superstructures (diameter ≈ 29 nm) and emphasising the rich structural diversity that can be reached using this synthetic methodology.48
Recently, Pigliacelli et al. have developed a new one-pot methodology for the preparation of hybrid peptide–AuNP superstructures (Fig. 3).37 In this case, they made use of the reducing and self-assembly capabilities of iodophenylalanine residues (F(I)), introduced in the amyloidal peptide sequence DF(I)NKF, to simultaneously synthesize and self-assemble peptide–gold hybrid superstructures in aqueous conditions, without the need for supporting reducing agents. The proposed mechanism for the formation of these superstructures relies on the self-assembly of the peptides into spherical structures covered with Au(III) ions, after the incubation of the reagent mixture at 60 °C. Then, the Au(III) ions are reduced to Au(I) by the amino groups present in the peptide sequences. Subsequently, Au(I) activates the C–I bond of iodophenylalanine to produce iodide ions, which can further reduce more Au(III) ions to Au(I). In addition, the Au(I) ions can function as a source of metallic gold by disproportionation, leading to the formation of AuNPs over the surface of the spheres. This mechanism was corroborated by the addition of 2,2′-bipyridine, known to complex Au(I) in solution, which inhibited the formation of AuNPs. Furthermore, the research group also studied the potential of this methodology to obtain hybrid superstructures with other topologies, observing that the introduction of two iodophenylalanine residues in the KLVF(I)F(I) sequence could lead to the formation of ribbon-like superstructures decorated with AuNPs along their surface (Fig. 3e and f). Overall, this example further demonstrated the generality of the one-pot approach to obtain hybrid peptide–AuNP superstructures with different morphologies in aqueous media.
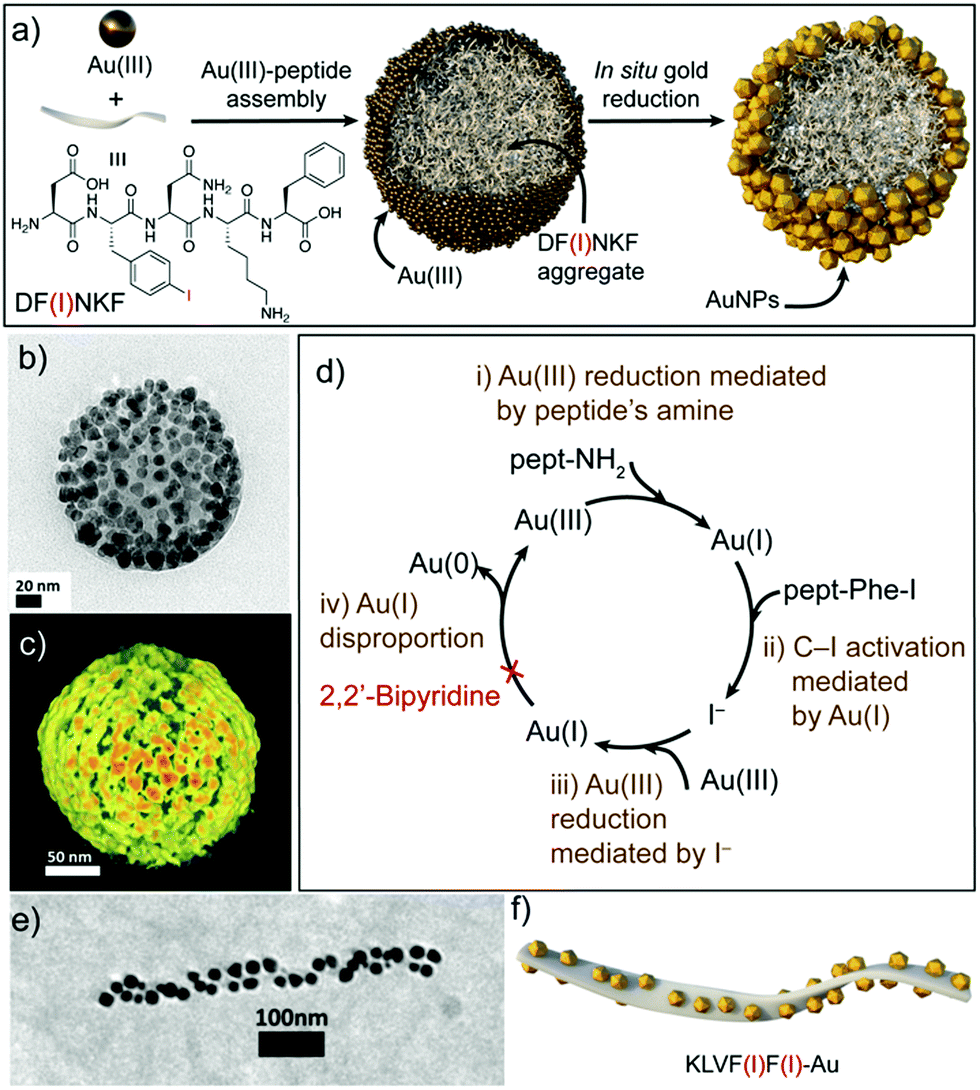 | ||
| Fig. 3 (a) Schematic representation of the formation of spherical DF(I)NKF–Au hybrid superstructures. (b) TEM image and (c) electron tomography reconstruction of DF(I)NKF–Au superstructures. (d) Representation of the gold reduction mechanism. (e) TEM image and (f) schematic representation of helical KLVF(I)F(I)–Au superstructures. Adapted with permission from ref. 37. | ||
These systems clearly exemplify the advantages of the one-pot strategy, which gives in situ access to well-defined hybrid superstructures under environmentally friendly conditions and requires fewer synthetic steps than other bottom-up approaches. However, until now this approach has been largely restricted to the preparation of Au-based architectures.
Peptide nanostructure-templated synthesis
Matsui and co-workers pioneered the use of pre-assembled peptide nanostructures as templates for the synthesis of Au hybrid superstructures.49 Their first example relied on the use of glycine based bolaamphiphiles, which self-assemble into nanotubes and incorporate high-affinity binding sites for peptides. These extra binding sites were used to immobilize histidine-rich peptides over the nanotube surface. The incubation of these nanostructures with a gold salt, and the subsequent addition of NaBH4, resulted in the formation of hybrid superstructures decorated with AuNPs over their surface. In later work, the same research group showed that, by controlling the pH of the solutions, they could finely modulate the density of Au binding sites within the peptide nanostructures and, therefore, tune the number of nucleation sites and the AuNP packing density.50 In addition, they also showed that, by modifying the peptide sequence immobilized over the nanostructures, this strategy could be extended for the preparation of other hybrid superstructures decorated with Ag, Cu and Ni NPs.51–53Other examples described in the literature report on the use of different peptide-based self-assembled nanostructures for the formation of hybrid architectures. In these cases, the peptide sequences include amino acids that bind different metal ions, which are then reduced, yielding Au, Ag, Pd or Pt hybrid superstructures.54–58
A related strategy was followed by Pazos et al., who described the use of aldehyde-modified peptide-amphiphiles for the development of peptide–silver NP (AgNP) hybrid nanofibres.36 In this case, the peptide-amphiphiles were modified to include an aldehyde moiety that, upon peptide self-assembly, was exposed on the surface of the nanofibres. This functional group was oxidized into a carboxylic acid via the Tollens’ reaction, consequently reducing Ag(I) ions to Ag2 clusters, without the need for any external reducing agent or additives to control the nucleation (Fig. 4b). Different experiments suggested that the rearrangement of the peptide–amphiphile molecules within the supramolecular fibre aided the clustering of the Ag atoms into larger and monodisperse particles, which were spatially ordered along the nanofibres (Fig. 4c). The monodispersity arose from different interactions, such as cohesive forces and electrostatic repulsion between the peptide monomers, as well as interactions between the carboxylic groups of the monomers and the surface of the NPs. In turn, the ordered distribution of the AgNPs within the superstructures was due to the minimization of the electrostatic repulsion between neighbouring AgNP–peptide-amphiphile aggregates.
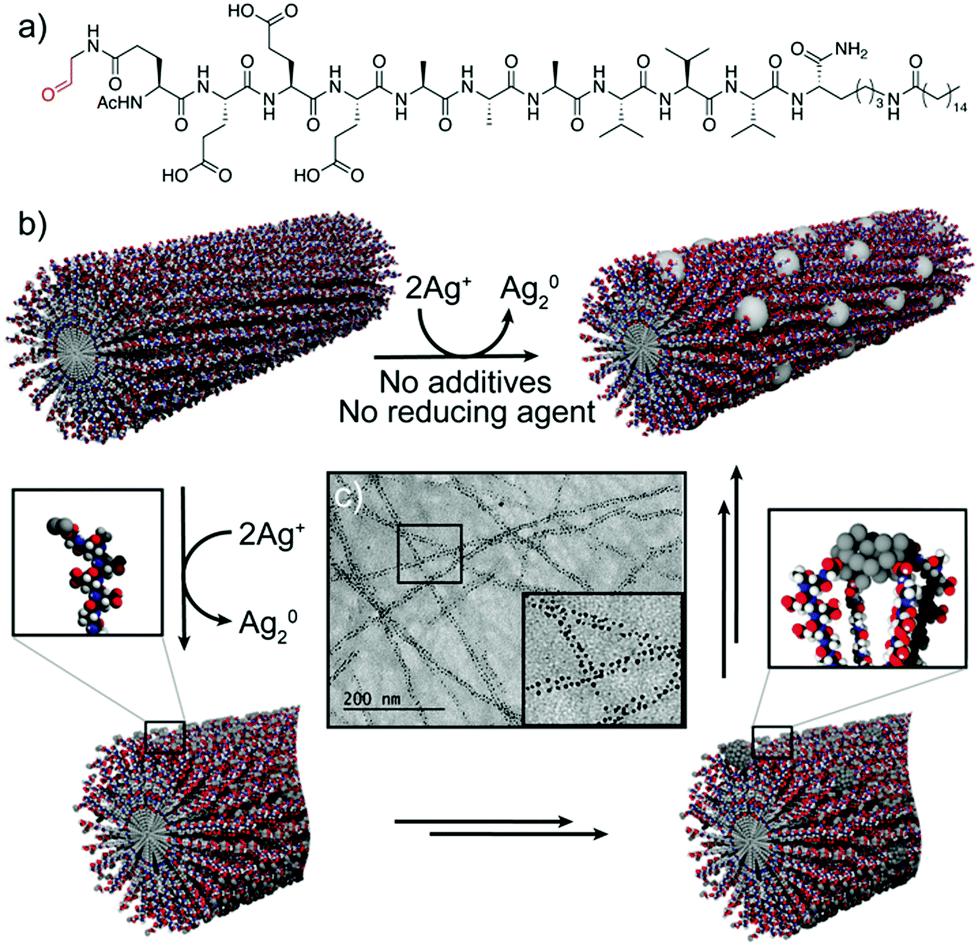 | ||
| Fig. 4 (a) Chemical structure of the peptide–amphiphile. (b) Schematic representation of the proposed mechanism for the formation of peptide–AgNP hybrid nanofibres. (c) TEM image of the hybrid nanofibres. Adapted with permission from ref. 36. | ||
Although this strategy involves more steps than the one-pot methodology and in many cases requires the addition of reducing agents, it has been successfully exploited for the preparation of hybrid systems in aqueous media with other transition metals than Au. Moreover, this strategy allows to modulate the NP density over the peptide nanostructure or the nanocrystal shape by controlling the reaction conditions.
Self-assembly of peptide nanostructures and nanoparticles
Several types of interactions have been exploited to obtain hybrid superstructures from the combination of self-assembled peptide-based nanostructures and pre-synthesized metal NPs. In 2001, the group of Matsui described the use of glycine-based bolaamphiphile nanotubules to which AuNPs, capped with 11-mercaptoundecanoic acid, were attached by means of hydrogen bonding between the carboxylic acids of the AuNPs and the free amide bonds present on the surface of the tubules.59 Later, Stupp and co-workers developed self-assembled peptide-amphiphile nanofibres decorated on their surface with thymine moieties, which were introduced by co-assembling these peptide-amphiphile monomers with non-modified counterparts. These thymine scaffolds were used to immobilize the AuNPs, capped with diaminopyridine, over the surface of the nanofibers through the formation of hydrogen bonds between both units.60An alternative approach involved the addition of thiol moieties (usually incorporating Cys residues within the peptide sequences) that were then exposed over the surfaces of the self-assembled peptide nanostructures. This functional group, which serves as a gold-binding ligand, allowed the immobilization of AuNPs over a variety of structures, such as nanotubules, coaxial nanowires or 2D plates, to obtain the corresponding superstructures.55,61,62
Other strategies exploited electrostatic interactions to drive the self-assembly of oppositely charged peptide nanostructures and metal NPs.63–65 Interestingly, Pochan and co-workers used this type of interactions to obtain fibril-like hybrid architectures that display characteristic arrays of laterally-spaced NPs.64 To achieve this, the authors designed an amphiphilic peptide able to self-assemble into β-sheet filaments, driven by the hydrophobic collapse of the apolar valine residues from two β-sheets, with the positively charged lysine residues exposed at the outer faces. As a result, these filaments stacked to yield fibril laminates consisting of alternating layers of apolar and positively charged residues, in which the layers of lysine residues are laterally spaced with a periodicity of 2.5 nm. The addition of 2 nm negatively charged AuNPs to these fibrils resulted in the formation of hybrid superstructures, in which the distribution of the AuNPs followed linear arrays with a highly conserved spacing between them (Fig. 5).
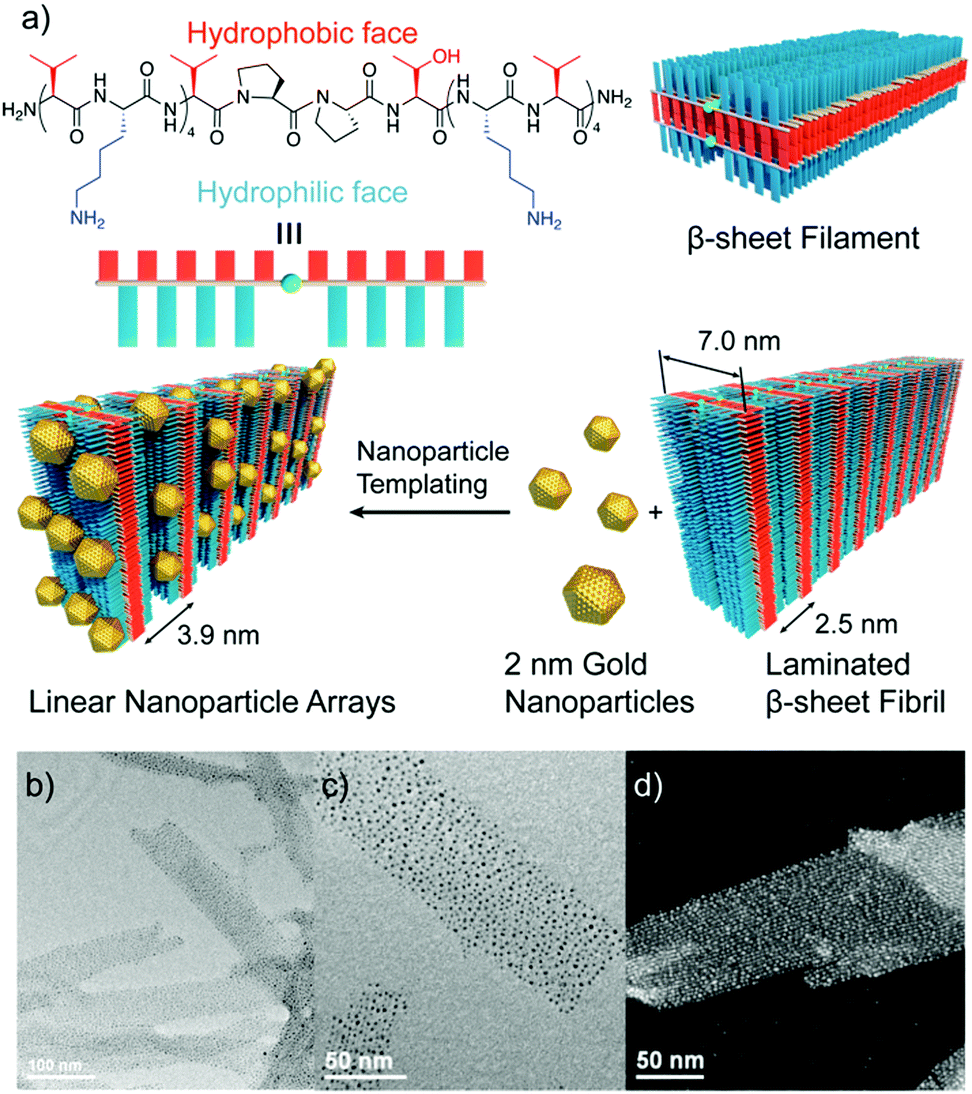 | ||
| Fig. 5 (a) Chemical structure of the amphiphilic peptide and schematic representation of the formation of the hybrid architectures. (b and c) TEM images, and (d) high resolution TEM image showing the conserved spacing between AuNP arrays. Adapted with permission from ref. 64. | ||
Clearly, this strategy represents the most tedious one in terms of the number of synthetic steps; however, it provides a higher level of control of the peptide–NP interaction, which is mediated by specific functional groups introduced for this purpose on the NPs. On the other hand, this approach would allow greater control over the final superstructures’ morphology, because the metal NPs are prepared and stabilized before their assembly in the final superstructure, making it possible to assemble NPs with other shapes than the classical spherical NPs.
Structure–function relationship
It is well-known that the sequence of a peptide defines its self-assembly pattern, its interaction ability and the possible functionality arising from these properties. Thus, proper encoding of the amino acid sequence can lead to the formation of tailored functional peptide-based structures.66 Many different peptide motifs associated with specific functions and/or properties have been identified in natural proteins, and integrated in artificial peptides or conjugated to target molecules/materials to endow them with desired functions.67 Similarly, peptide sequences employed in the design of hybrid peptide–NP systems can be encoded to confer to the superstructure a chosen functionality, which is strongly related to the designed architecture and to the NP arrangement.Chiroptical activity and chiral plasmonics
A seminal example of peptides’ ability to endow nanoscale hybrid materials with innovative properties has been shown in the work by Slocik et al. that reported how the functionalization of AuNPs with random coil and α-helix peptides could impart chirality to the AuNPs, changing their spectroscopic behaviour. Indeed, the circular dichroism (CD) profile of the chiral peptide–NP complex showed the activation of a signal at the AuNP surface plasmon resonance (SPR) frequency, proving the formation of chiroptically-active AuNPs.68 Moreover, the arrangement of inorganic NPs on helical peptide scaffolds represents one of the most exploited strategies to develop chiral plasmonic structures. A peptide sequence can be programmed to self-assemble into chiral architectures and also designed to bind or associate with NP surfaces. If the spatial distribution of NPs along the peptide fibre is uniform, and the interparticle gap is properly tailored, a collective plasmonic mode can be formed along the handed peptide structure, yielding a bisignate CD signal in the visible area of the electromagnetic spectrum.69Rosi's research group pioneered the topic, exploiting the self-assembly propensity of the peptide conjugate C12-PEPAu to drive the synthesis and assembly of AuNPs along the peptide nanoribbons.44 Notably, the left-handed helicity of these fibres arose from the chirality of the L-amino acids within the C12-L-PEPAu conjugates. By replacing the L-amino acids with the corresponding D-configured ones, the formation of right-handed peptide–AuNP superstructures could also be achieved (Fig. 6a and b). The change in handedness was reflected in the CD spectra of the superstructures, which showed vertically mirrored CD signals at 562 nm (Fig. 6c), proving the possibility of tailoring the superstructure's CD response by properly encoding the PEPAu sequence.35 In further studies reported by the same research group, the oxidation of the C18-(PEPAu)2 methionine residue to the corresponding sulfoxide (C18-(PEPAuM_ox)2) led to a change in the AuNP shape and self-assembly profile, with the formation of rod-like AuNPs along single left-handed helix superstructures (Fig. 6e). The growth of the rod-like AuNPs over the helical structure resulted in a strong bisignate CD signal, centred at approximately 600 nm, near the collective plasmonic extinction band for the assemblies (Fig. 6g). Notably, the unoxidized C18-(PEPAu)2 could only lead to the formation of double helices, highlighting the role of the methionine residue in both defining the superstructure morphology and controlling the particle shape and dimensions. Therefore, chemical modifications of a motif or an amino acid residue in the peptide sequence can be employed to modulate the final superstructure architecture and, consequently, its spectroscopic behaviour.70 Moreover, the position of the methionine residue in the peptide sequence, and of its oxidized counterparts, was shown to play a key role in determining the size and position of AuNPs, while possible changes in the motif driving the peptide self-assembly are envisaged to impact more on the final architecture of the superstructure.71
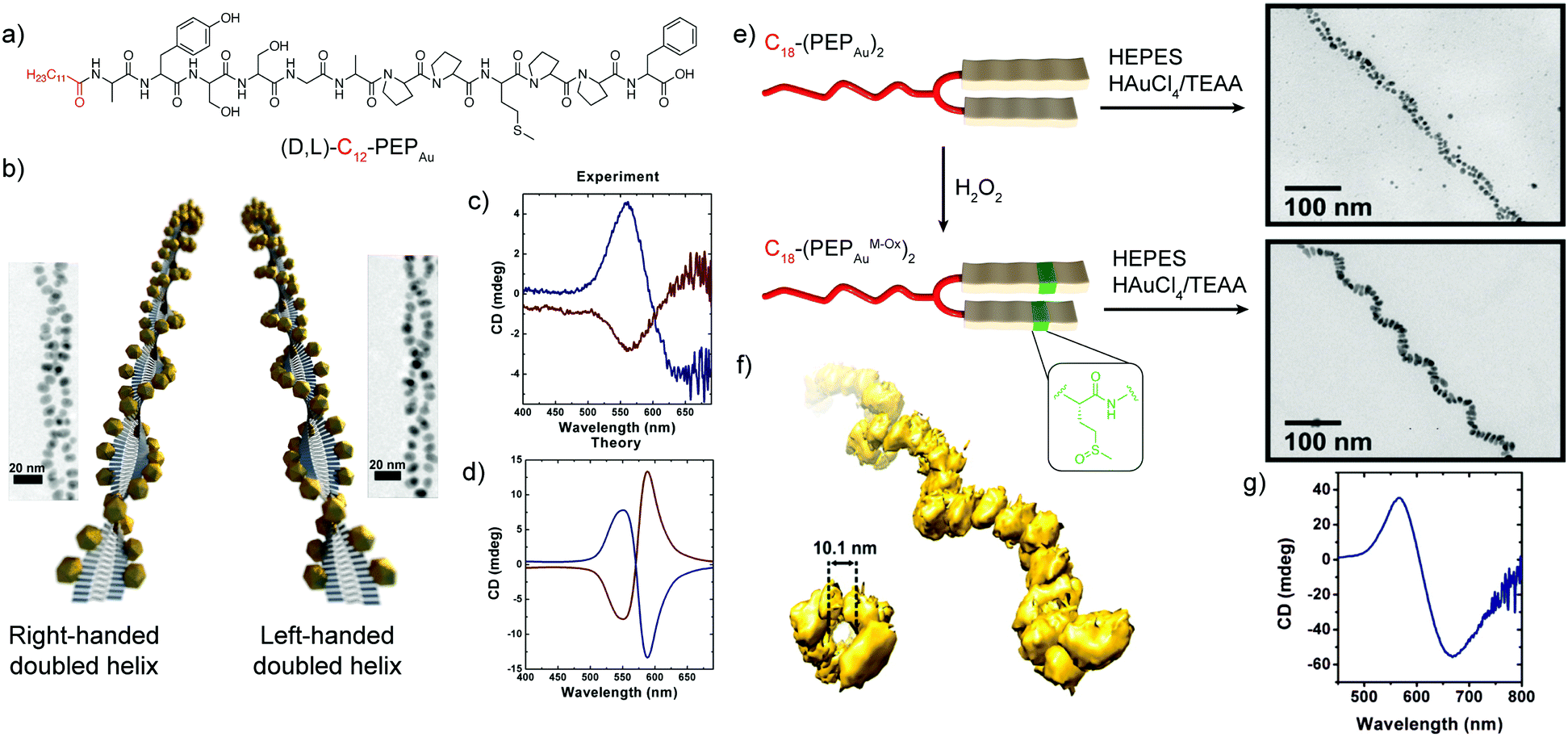 | ||
| Fig. 6 (a) Chemical structure of (D,L)-C12-PEPAu peptide conjugates. (b) Schematic representation of the left- and right-handed superstructures and their corresponding TEM images. (c) Experimental and (d) theoretical CD spectra of the left- and right-handed superstructures. Adapted with permission from ref. 35. Copyright 2013 American Chemical Society. (e) Schematic representation of C18-(PEPAu)2 and C18-(PEPAuM_ox)2 peptide conjugates and TEM images of their corresponding hybrid superstructures. (f) Electron tomography reconstruction of the C18-(PEPAuM_ox)2 single helical superstructures, and (g) their CD spectrum. Adapted with permission from ref. 70. Copyright 2016 American Chemical Society. | ||
Catalytic activity
Besides defining optical properties, the structure characterizing peptide–NP systems has been shown to strongly affect their potential catalytic abilities.72 Several studies revealed the relation between the observed catalytic activity and peptide arrangement along the NP surface, confirming the need to properly tune the peptide architecture and its binding to the inorganic moiety of the hybrid system.73 Mikolajczak et al. designed the peptides E3H15 and K3, based on the heterodimeric IAAL-E3/IAAL-K3 coiled coil system, to study the role of the peptide's conformation in controlling the catalytic activity of peptide–AuNP systems. While E3H15 was esterolytically active, its complementary counterpart K3 was catalytically inactive. E3H15@AuNPs were shown to effectively accelerate ester hydrolysis, thanks to the histidine imidazole moiety, which works as a catalytically active charge-relay site. Such functionality was proved to be disrupted upon heterodimerization of E3H15@AuNPs with K3 and the formation of the corresponding coiled coil structures, which hindered the E3H15@AuNP interaction with the substrate, lowering the overall catalytic activity.74Structure–function relationship studies on peptide–AuNP systems were performed also by Bedford et al., who employed a series of Au binding peptides (PEPCANs) to investigate sequence-dependent structural and functional changes in catalytically active peptide–AuNP systems.75 The conformational freedom of the peptide at the biotic/abiotic interface was identified as one of the main factors influencing the catalytic activity, in which peptide sequences without strongly pinned terminal or central loop segments showed lower activation energies. Therefore, the catalytic activity was found to be favoured by high dynamic exposure of reactive sites on the peptide–AuNP interface and limited by conformational constraints.
Notably, peptide–AuNP hybrid superstructures have been shown to significantly enhance the catalytic activity in the reduction of 4-nitrophenol, when compared to template-free AuNPs.54 Indeed, using a fragment of the amyloid-β peptide (Aβ25–35), several self-assembled peptide–AuNP architectures were obtained by changing the reaction conditions, i.e. reaction time and solvent. The obtained structures efficiently worked as catalysts for the reduction of 4-nitrophenol, showing activation energies strongly reduced with respect to template-free AuNPs, and higher conversions towards 4-aminophenol. Individually dispersed peptide–AuNPs (4.5 nm in diameter) yielded the highest catalytic activity thanks to their high surface area, which provided an increased number of interaction sites for the substrate and, consequently, a higher reaction rate. Upon self-assembly of the AuNPs into peptide–Au nanoribbons, the surface area was significantly lowered, so proper diffusion of the substrate through the peptide assembled layer was needed for the reaction to occur. This led to a decreased reaction rate and higher activation energy in comparison to the single peptide–AuNPs, but the overall catalytic activity was still significantly improved with respect to template free AuNPs. Finally, peptide–AuNP nanofibers with a more densely packed framework of small sized AuNPs could drive the 4-nitrophenol conversion at a faster rate, providing comparable catalytic activity to the colloidal peptide–AuNPs.
Stimuli-responsiveness
Peptide sequences can also be programmed to respond to different stimuli, and consequently impart specific stimuli-responsiveness to the final peptide–AuNP hybrid material.76 Such a design strategy has been widely employed to develop innovative biosensing platforms, in which the NP optical properties and peptide recognition and responsiveness are combined in a unique nanostructure.77 Protease-triggered disassembly of peptide–AuNP aggregates was devised by Laromaine et al., who reported on a short peptide-functionalized AuNP system whose self-organization was controlled by the protease thermolysin.78 The formation of the aggregates was driven by the tripeptide Fmoc-Gly-Phe-Cys-NH2, which was encoded to have as key features: a thiol containing residue able to bind Au, cysteine; a protease cleavable portion, the amide bond between glycine and phenylalanine; and a self-assembly actuator, in this case 9-fluorenylmethoxycarbonyl (Fmoc), known to induce self-assembly through π-stacking interactions. Thermolysin could catalyse the hydrolysis of the amide bond between the glycine and phenylalanine residues, triggering the disassembly of the system and the subsequent dispersion of individual peptide–AuNPs (Fig. 7). Crucially, the process causes an optical response, with a blue-shift of the SPR signal in the UV-vis spectrum translated into a visible colour change in the solution, features that render the system a possible platform for optical and colorimetric sensing applications.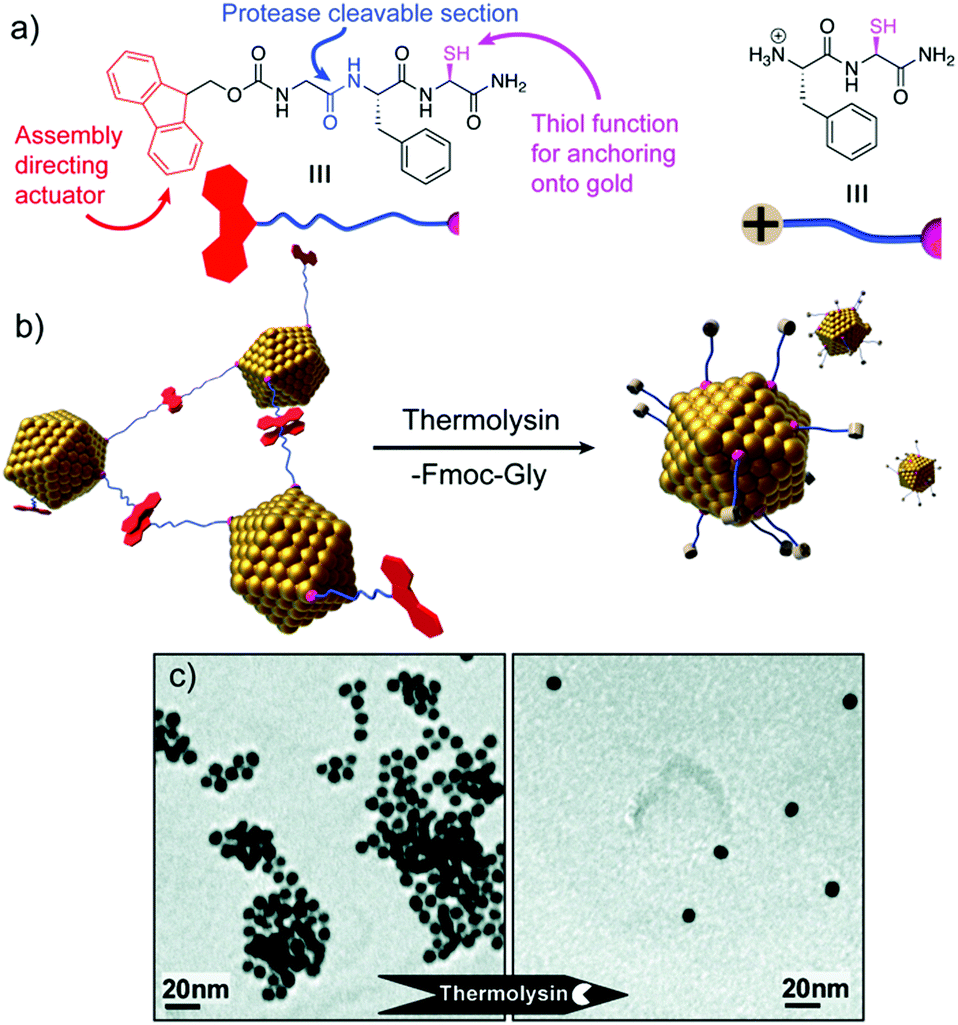 | ||
| Fig. 7 (a) Chemical structure and schematic representation of the peptide fragments. (b) Schematic representation of the disassembly mechanism. (c) TEM images of peptide–AuNP systems before and after exposure to protease. Adapted with permission from ref. 78. Copyright 2007 American Chemical Society. | ||
Similarly, peptide–AuNP hybrid systems could also be employed for the colorimetric detection of blood coagulation Factor XIII.79 To achieve responsiveness to its activity, AuNPs were functionalized with a Factor XIII reactive peptide domain, having glutamine or lysine residues. Upon exposure of peptide–AuNPs to the targeted enzyme, the activity of the blood coagulation factor resulted in the formation of covalent crosslinking between the peptide chains, via ε-(γ-glutamyl)-lysine isopeptide bonds, leading to AuNP aggregation. As a consequence of the particles’ assembly, the SPR signal was strongly red-shifted, with a rapid change in the sample colour from purple to blue being observed, confirming the system's potential application as a colorimetric sensing tool.
Assembly and disassembly of peptide–NP systems can also be triggered by pH changes in the sample medium. Indeed, electrostatic and hydrogen bond interactions contributing to the self-organization of peptide–NP nanostructures can be finely tuned by proper pH adjustments, causing protonation or deprotonation of the functional groups involved in the interactions, triggering aggregate rearrangement.80
Challenges in the structure–function relationship and functionality design
As described in the previous sections, several hybrid nanomaterials, endowed with specific tuneable properties, have been produced by peptide-sequence rational design. Although still being a challenge for materials scientists, in depth control of the designed self-assembled hybrid structures is pivotal for the development of tailored functional nanoscale materials. In this scenario, the combination of experimental and computational approaches has been shown to provide key information about the structure of the biotic/abiotic interface of peptide–NP systems, peptide assembly and template efficiency, peptide interaction with potential analytes and responsiveness to different stimuli.81 Still, the acquisition of structural data for peptide-enabled hybrid materials remains highly challenging and the elucidation of their structure at the atomic and molecular level is frequently limited. For instance, the structural resolution of single molecular components by NMR spectroscopy can be strongly hindered by their arrangement in the self-assembled system and X-ray diffraction is not well-suited for studying peptides at interfaces.82 Moreover, although CD spectroscopy can probe the presence of peptides’ secondary structures, it can be uninformative when used to study the occurrence of peptides’ conformational and orientational changes upon binding to NP surfaces.81 Recently, the use of 2D NMR techniques and of sum-frequency generation (SFG) vibrational spectroscopy has allowed significant progress for the study of peptide and protein structures at the solid/liquid interface. Specifically, SFG has emerged as an exceptional tool to probe peptides’ folding, binding, orientation, hydration, and dynamics even after their self-assembly on metal surfaces.82 Although the rational design of a peptide sequence represents the first step towards a specific functional hybrid system, the acquisition of structural information on the self-assembled systems is essential to further predict and tune the targeted functionalities.83,84 Indeed, the elucidation of the relationship between the structural data and the properties of peptide-enabled hybrid materials could open new avenues for the design of custom functional nanostructures, advancing technologies based on self-organized materials for plasmonics, catalysis, biosensing and nanomedicine.Applications of self-assembled peptide–inorganic nanoparticle hybrid systems
Biomedical applications
| Peptide sequence | NPs | Superstructure morphology | Application | Ref. |
|---|---|---|---|---|
| Fmoc-FFECG | AgNPs | Fibres | Antimicrobial (B. subtilis and E. coli) | 56 |
| Ac-E(ald)E3A3V3K(C16)-NH2 | AgNPs | Fibres | Antimicrobial (E. coli) | 36 |
| FF | AuNPs | Spheres | H2O2 sensing | 98 |
| CD44 biomarker detection | 100 | |||
| Cholesterol detection | 101 | |||
| PdNPs | Fibres | Catalysis (Suzuki and hydrogenation react.) | 107 | |
| VIAGASLWWSEKLVIA | AgNPs | Fibres | H2O2 sensing | 97 |
| CCYAEAKAEAKAEAKAEAKRGD | AuNPs | Fibres | H2O2 and Hg2+ detection | 103 |
| RGDAEAKAEAKCCYYCCAEAKAEAKRGD | AuNCs | Fibres | Temperature sensing and cell imaging | 102 |
| PC10ARGD (full sequence in ref. 99) | AuNPs | Spheres | Cell imaging | 99 |
| Octreotide | AgNPs | Cubes | Photothermal therapy | 105 |
| C12-VVAGHH-NH2 | PdNPs | Fibres | Catalysis (Suzuki react.) | 57 |
| Oligo-Arg | PdNPs | Fibres | Catalysis (C–S coupling react.) | 108 |
| Aha-(Nce)6(N4-Clpe)6-NH2 | PdNPs | Fibres | Catalysis (olefin hydrogenation) | 109 |
| I3K | PtNPs | Fibres | Catalysis (H2 and MeOH electro-oxidation) | 58 |
| Aniline-GGAAKLVFF | PtNPs | Fibres | Catalysis (O2 reduction) | 65 |
| Ac-FFACD | AgNPs | Fibres | Catalysis (4-nitrophenol reduction) | 110 |
| GSNKGAIIGLM | AuNPs | Fibres | Catalysis (4-nitrophenol reduction) | 54 |
More recently, Pazos et al. demonstrated that supramolecular peptide–amphiphile nanofibres decorated with AgNPs of 3 nm on their surface had a similar bacteriostatic effect against E. coli to AgNO3 (Table 2).36 Moreover, when tested with eukaryotic cells (C2C12, mouse myoblast cell line), these metallized nanofibres were more than 30 times less toxic towards eukaryotic cells than to bacteria and, importantly, less cytotoxic than AgNO3 alone. In addition, the study showed that the metallized nanofibres could form gels when pipetted into a CaCl2 solution. The obtained gels retained the antimicrobial properties against E. coli cultures, demonstrating that is possible to prepare hydrogels or surface films of hybrid peptide–AgNP superstructures with effective antimicrobial properties employable for medicine applications.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 inclusion complexes with two diphenylalanine peptide sequences, present both in the hybrid superstructures and in the peptides bound to the gold electrode. Therefore, this example demonstrates that the peptide-based hybrid structures can have a dual function, as a scaffold for the self-assembly of the NPs as well as recognition elements. The research group of Li extended the strategy developed by Yang and co-workers for the detection of cholesterol (Table 2).101 In this case, the surface of the superstructures is modified with cholesterol oxidase to obtain the sensing electrode, thus probing the versatility of hybrid superstructure-modified electrodes for the development of new biosensors.
:2 inclusion complexes with two diphenylalanine peptide sequences, present both in the hybrid superstructures and in the peptides bound to the gold electrode. Therefore, this example demonstrates that the peptide-based hybrid structures can have a dual function, as a scaffold for the self-assembly of the NPs as well as recognition elements. The research group of Li extended the strategy developed by Yang and co-workers for the detection of cholesterol (Table 2).101 In this case, the surface of the superstructures is modified with cholesterol oxidase to obtain the sensing electrode, thus probing the versatility of hybrid superstructure-modified electrodes for the development of new biosensors.
In 2014, Su and Wei developed self-assembled peptide nanofibre–AgNP hybrid superstructures that were subsequently assembled electrostatically into graphene nanosheets (Table 2).97 These superstructure–graphene nanohybrids exhibited high sensitivity and selectivity for the sensing of H2O2, without the need for incorporating enzymes, providing an alternative to overcome the potential limitations of enzyme-based sensors (e.g. susceptibility to pH and temperature or inherent instability). More recently, this research group has reported a self-assembled peptide nanofibre–AuNP hybrid that can work both as a H2O2 electrochemical sensor, when deposited over a carbon electrode, and as a colorimetric sensor in aqueous media with specificity towards Hg2+ (Table 2).103
Besides the sensing capabilities of peptide–inorganic nanoparticle superstructures, Shang and co-workers have developed self-assembled peptide nanofibers with embedded gold nanoclusters (AuNCs) that can be applied for temperature sensing and cellular imaging.102 The designed peptides, in addition to being encoded with the self-assembling sequence and the reactive/binding site that yields the final hybrid structures, included the RGD sequence, known to have high binding affinity for tumour cells that overexpress integrin receptors (Table 2 and Fig. 8). The resulting hybrid nanofibres showed a 70-fold increase in luminescence intensity, when compared with the peptide–AuNCs before self-assembling into nanofibres. Interestingly, their luminescent intensity and lifetimes decreased with temperature, being not the case for the non-assembled peptide–AuNCs, making these hybrid superstructures good systems for thermometry applications. Furthermore, the hybrid nanofibres, which are expected to target integrin-rich cancer cells and thus label them, are effectively internalized by HeLa cells without affecting their morphology and viability, proving that they are promising probes for imaging cellular processes.
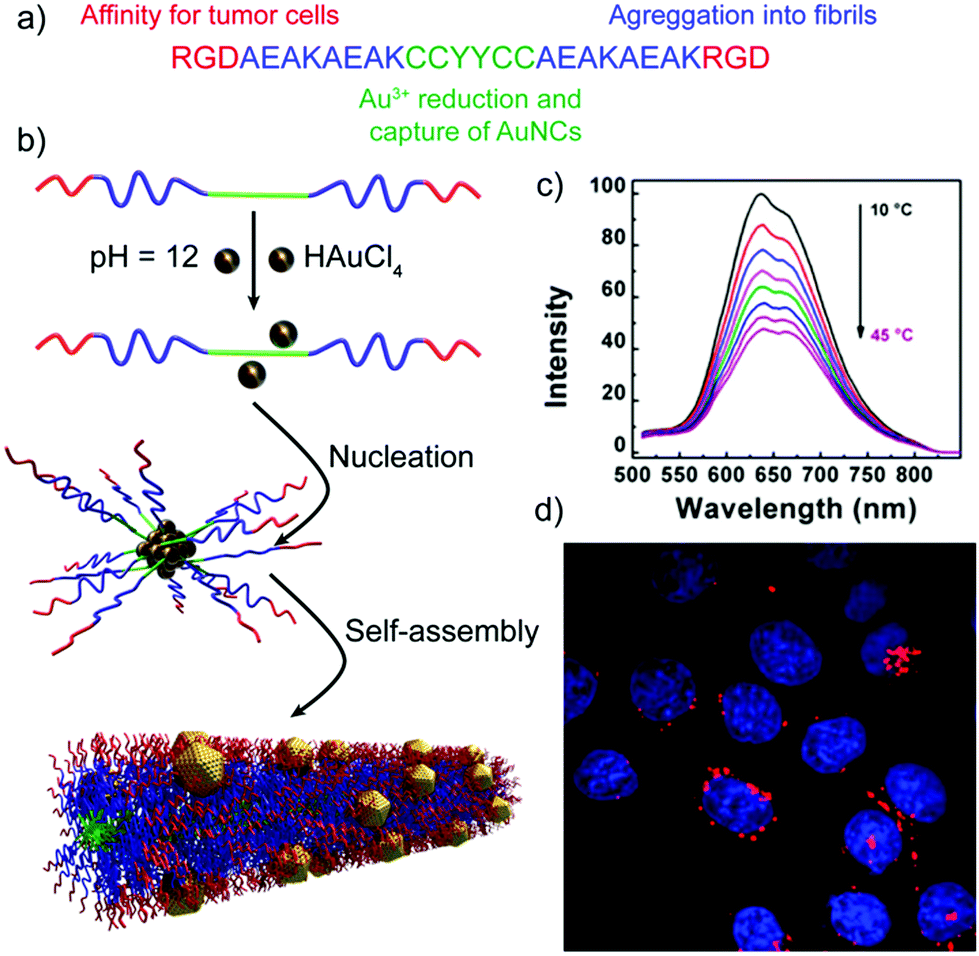 | ||
| Fig. 8 (a) Peptide sequence. (b) Schematic representation of the formation mechanism of peptide nanofibers with embedded AuNCs. (c) Luminescence emission spectra of the hybrid structures at different temperatures. (d) Confocal image of HeLa cells upon internalization of the peptide–AuNC hybrids (red dots) with nuclear staining (in blue). Adapted with permission from ref. 102. | ||
An alternative imaging strategy has been developed by Liu and co-workers, who described self-assembled peptide nanogel–AuNP hybrid spheres that can be used for targeted photoacoustic imaging (Table 2).99 This imaging modality consists in acoustic signals that are generated by transient temperature variations caused by the absorbed light, and offers high contrast and spatial resolution, and deep penetration. The photoacoustic signal produced by these hybrid structures, which incorporate the integrin binding sequence RGD, proved their higher internalization by HeLa cells, which overexpress integrin receptors, than by MCF-7 cells, with lower expression of these receptors, converting these hybrid structures into potential contrast agents for targeted photoacoustic imaging of tumour cells. Furthermore, the authors demonstrated that these hybrid nanogels can encapsulate and release drugs, such as doxorubicin, in vitro, thus having the potential to work as theragnostic agents.
Catalytically active superstructures
Metal-catalysed reactions are commonly used by organic chemists for the preparation of many important compounds. Therefore, transition-metal NPs are good candidates for heterogenous catalysis due to their high surface area. However, the metal NPs need to be stable to avoid aggregation under reaction conditions. As has been previously mentioned, peptide-templates can control parameters such as the nanoparticle size and shape, or adjust their surface chemistry, ultimately improving the catalytic properties of NPs.106For instance, diphenylalanine fibres have been vertically self-assembled and immobilised in the reaction zone of a microfluidic device by Kim and co-workers, and PdNPs were subsequently synthesised over their surface, giving access under mild conditions to an aligned array of hybrid superstructures in the microchannel (Table 2).107 The catalytic performance in hydrogenation and Suzuki reactions of this modified microfluidic device was compared with that of a control device having immobilised PdNPs on the surface of the microchannels. For both reactions, the microfluidic device modified with the hybrid superstructures showed higher conversion yields, demonstrating the utility of these modified devices for heterogeneous catalysis.
A similar approach, developed by the group of Guler, used self-assembled peptide-amphiphile nanofibers coated with PdNPs as catalysts in Suzuki reactions (Table 2 and Fig. 9).57 They tested different iodo- and bromo-aryl substrates, with both electron donating and withdrawing groups. Interestingly, the authors observed that most of the reactions were completed in almost 2 h with high conversions, and also that the reactions of aryl bromides could be carried out in water at room temperature with high yields. Furthermore, the catalyst could be isolated and reused at least five times without notable loss in activity or structural integrity, proving its potential application in industrial catalytic processes.
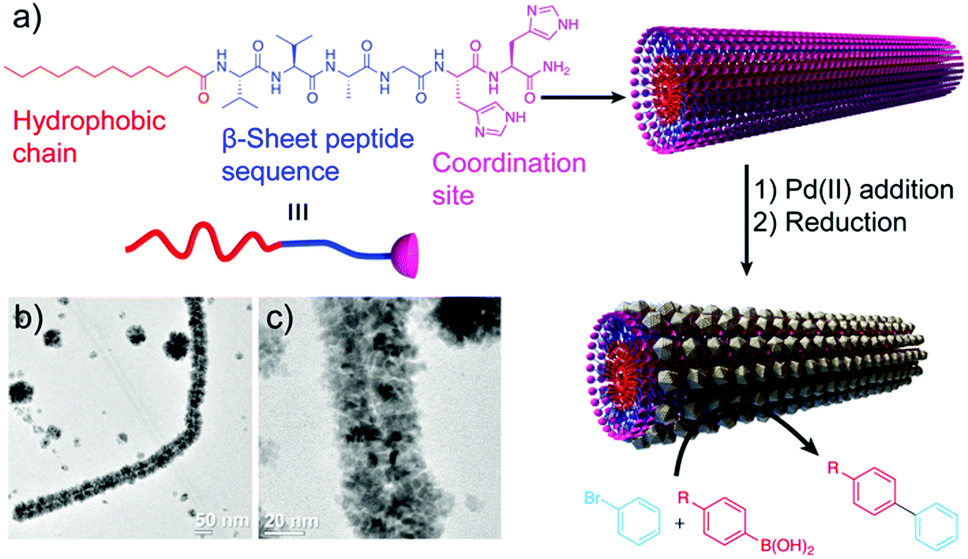 | ||
| Fig. 9 (a) Peptide chemical structure and schematic representation of the formation mechanism of the hybrid structures, which work as catalysts in Suzuki reactions. (b) and (c) TEM images of the resulting superstructures. Adapted from ref. 57 with permission from the Royal Society of Chemistry. | ||
Self-assembled peptide–PdNP architectures have also been tested as catalysts in C–S coupling reactions for the synthesis of alkyl/aryl sulphides (Table 2).108 For example, Ghorbani-Choghamarani et al. proved their use in reactions between aryl halides and thiourea or 2-mercaptobenzothiazol, and demonstrated that for the synthesis of diphenyl sulphide their Pd-hybrid catalyst provided better yields and shorter reaction times when compared with other nanoparticle and metal catalysts. More recently, Knecht and co-workers reported that the morphology of self-assembled peptoid–PdNP hybrid superstructures has an effect on their catalytic properties (Table 2).109 In this case, peptoids self-assembled into 1D fibres and coated with PdNPs displayed enhanced catalytic activity in olefin hydrogenation than 2D assemblies, corroborating that the morphology of the superstructures plays an important role in controlling the material properties.
Besides PdNP-based hybrid superstructures, self-assembled peptide nanofibre–PtNP superstructures have also been described in the literature and tested as electrocatalysts.58,65 Lu and co-workers have shown that superstructures coated with a continuous layer of PtNPs had better efficiency in the electro-oxidation of hydrogen and methanol with respect to superstructures bearing more dispersed PtNPs over their surface, and even when compared with colloidal PtNPs (Table 2).58 In the same year, Liu et al. published another example of self-assembled peptide nanofibres coated with PtNPs, further corroborating their better performance as electrocatalysts in oxygen reduction than other Pt nanomaterials (Table 2).65 Both examples verify the potential of Pt-based superstructures as electrocatalysts for fuel cells.
Finally, self-assembled peptide nanofibres have also been used to prepare Au and AgNP superstructures with catalytic properties.54,110 Song and co-workers developed self-assembled peptide–AgNP hybrid superstructures that showed good catalytic activity towards 4-nitrofenol reduction, and that were also much more efficient than colloidal AgNPs for the degradation of rhodamine B (Table 2).110 More recently, the group of Liu described how the morphology of self-assembled peptide nanostructures, i.e. ribbons or fibres, affects both the spatial distribution of the AuNPs synthesized over their surface and the catalytic activity of the hybrid superstructures for the reduction of 4-nitrophenol,54 in agreement with the well-known fact that the properties of metal NPs are dependent on their spatial distribution (Table 2).
Outlook and future perspectives
Combining the unique properties of nanoscale objects and the versatility of supramolecular systems, peptide–inorganic NP superstructures have already become promising in several high-end applications, showing their potential key role in the progress of nanotechnology. Still, the engineering of these hybrid systems is quite recent and holds great promise in multiple fields, in which the application of peptide–NP hybrids is currently remote or in its beginning.While the use of peptides as templates to arrange NPs in a chiral manner is well consolidated, their possible ability to impart chiral geometry to inorganic objects is still quite unexplored. In this direction, Lee et al. devised an elegant strategy to tune the growth of pre-synthesized AuNPs into helicoidal cubes, driven by cysteine or cysteine-based peptides, having chiral conformations. The obtained morphology was the result of the different growth rates characterizing certain gold facets, in the presence of the peptide or amino acid. Notably, the conformation of the peptide controlled the handedness of the resulting nanostructures, with opposite configurations formed when using L- or D-amino acids. The obtained helicoidal structures were endowed with chiral plasmonic optical activity, and the resulting CD response followed the handedness of the system. Moreover, by changing the molecules driving the seed growth (e.g. from Cys to glutathione) or the initial shape of the seed particles, the nanostructure chiral morphology and optical response could be finely manipulated.111 The use of peptides to control the chiral growth of inorganic and hybrid structures opens up new possibilities towards the development of metamaterials with chiral selectivity, light-manipulating capabilities or polarization control. These properties are essential features for the exploitation of nanotools in several fields, such as chiral sensing, colour displays, holography and reconfigurable switching. An illustrative example of a chiral sensing application was reported by Ma et al., who developed DNA connected side-by-side gold nanorod (AuNR) assemblies with strong polarization rotation, able to detect DNA markers at attomolar concentration.112 The chiroptical activity of side-by-side assemblies obtained employing different DNA templates could be used as a probe for the detection of oligonucleotides, as the amplitude of the CD bisignate signal of the obtained assemblies demonstrated exceptional linear correlation with the amount of DNA linker acting as a target. This work represents an excellent example of the possible use of chiroptical metamaterials in biosensing applications, proving the advantages of the higher sensitivity of the chiral plasmonic method in comparison to other techniques and its unique ability in detecting analytes larger than 2–5 nm.113
Given the promise shown by biomolecule–NP chiral systems, it can be envisaged that peptide–NP hybrid structures can gain a primary role towards the development of valuable chiroptical sensing tools, employable for the diagnosis of different diseases, as well as for forensics and environmental analysis.114,115 In this context, the interaction between AuNPs and amyloid peptides has attracted significant research interest. We have previously discussed the ability of amyloidal and β-sheet forming peptides to interact with Au ions and drive the formation and self-assembly of AuNPs. Additionally, AuNPs have been reported for their impact on amyloidal peptide and protein self-assembly behaviour, with both inhibition and promotion effects described for different systems and conditions.116,117 Amyloids are known to self-assemble into β-sheet-rich fibrillar nanostructures and are associated with several neurodegenerative diseases, being, in particular, essential constituents of Alzheimer's disease plaques.118–120 The interaction between AuNPs and amyloidal fibrils can potentially lead to the formation of fibril–AuNP hybrid systems that, upon chiral arrangement of the AuNPs along the fibrils, could generate chiral plasmonic responses. This strategy was employed by Kumar et al. to detect α-synuclein amyloid fibrils at low concentration, exploiting the helical alignment of plasmonic AuNRs on the amyloid fibrils. Notably, the AuNRs did not interact with α-synuclein monomers, proving the high selectivity of the method. In contrast, addition of nanomolar concentrations of assembled fibrils to AuNR samples led to spectral changes in both UV-vis absorption, with a red-shift of the SPR signal in agreement with the AuNR assembly, and in the CD spectrum, with the activation of a bisignate signal at the longitudinal surface plasmon wavelength (Fig. 10). Importantly, the technique could be successfully employed to identify the presence of amyloidal fibrils in brain homogenates from patients affected by Parkinson's disease and infectious amyloids formed by prion proteins, confirming the suitability of this innovative method for in vitro detection and, particularly, the wide potential of the technique in neurodegenerative disease diagnosis.121
 | ||
| Fig. 10 (a) TEM image of α-synuclein fibrils. (b and c) TEM images of α-synuclein fibrils in the presence of AuNRs at 2 and 0.5 nM concentrations, respectively. (d) Cryo-TEM tomography reconstruction image showing a 3D chiral arrangement of the AuNRs. Reproduced with permission from ref. 121. | ||
Inspired by these results, it can be foreseen that the interaction and assembly of inorganic NPs with peptides and proteins could be used as a diagnostic and therapeutic tool in protein-misfolding related diseases. Zhang et al. recently described the application of chiral L/D-FexCuySe NPs in inhibiting and reversing the formation of amyloid-β peptide 42 (Aβ42) fibrils, by interfering with the Aβ42 self-assembly under near-infrared irradiation, resulting in a decrease of neurotoxicity in vivo. Isothermal titration calorimetry studies proved an excellent binding of the NPs with the KLVFF peptide, the hydrophobic portion of Aβ42, suggesting a key role of the NP–peptide interaction in the disassembly of amyloidal fibrils.122 Similarly, AuNPs were shown to effectively inhibit Aβ40 fibrillization, and to induce the rearrangement of mature fibrils into smaller AuNP–peptide fibrillar structures or amorphous aggregates.123
Beyond the potential biomedical applications of peptide–NP hybrid structures, the use of these systems could also be envisioned in other research fields. For instance, since peptide–NP hybrid superstructures have been successfully applied as catalysts in aqueous media54,57,109,110 and internalized in cells,99,102,105 taking advantage of the intrinsic biocompatibility of peptides, the use of these hybrid catalysts could be extended to living cells, mimicking the action mode of enzymes. In a different context, several peptide–NP hybrid materials have shown conductive properties.124–126 For example, the research group of Guler used amyloid-like peptides to obtain monodimensional superstructures decorated with AuNPs over their surface.125 These hybrid systems showed high conductivity, due to the presence of a substantial number of gaps between the gold domains, and as dry films showed tunnelling-dominated conductance and resistive switching. Taking into consideration these properties, peptide–NP hybrid superstructures could hold potential in the development of devices that require optically transparent and conductive coatings, such as solar cells, touch-interface devices or flexible displays.
In summary, the organization of inorganic NPs and peptides into hybrid self-assembled systems offers a diversified toolbox to fabricate functional structures. The choice of the inorganic material, the encoding of the peptide sequence, and the fine-tuning of the self-assembly process provide multiple opportunities to materials scientists for tailoring the architecture and functionality of the final system. Given the unique combination of properties and versatility offered by peptide–NP hybrids, it is foreseeable that their application in high-end fields might rapidly take place. Nevertheless, many challenges remain unmet, including the scaling up of peptide–NP superstructure production, the acquisition of structural data, the fine manipulation of the self-assembly processes and precise control over the envisioned optical properties. It is expected that unforeseen structures and synthetic strategies could pave the way to meet these challenges, and to further consolidate the role of peptide–inorganic NP superstructures in optics, sensing, nanomedicine and other never-imagined fields. With this review we provided an overview of the current state of the art of peptide–inorganic nanoparticle superstructures, with a glimpse of future possibilities towards innovative functional systems that could meet current and future technological needs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the funding received from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 851179), the Ministerio de Economía y Competitividad and Fondo Europeo de Desarrollo Regional (FEDER) (CTQ2016-75629-P), the Agencia Estatal de Investigación and FEDER (CTQ2017-89166-R) and the Consellería de Educación, Universidade e Formación Profesional, Xunta de Galicia (ED431C 2018/39). E. P. thanks the UDC-Inditex InTalent Programme for her research contract and funding and the Xunta de Galicia for the Oportunius Programme.Notes and references
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- L. Xu, W. Ma, L. Wang, C. Xu, H. Kuang and N. A. Kotov, Chem. Soc. Rev., 2013, 42, 3114–3126 RSC.
- L. Wang, L. Xu, H. Kuang, C. Xu and N. A. Kotov, Acc. Chem. Res., 2012, 45, 1916–1926 CrossRef CAS PubMed.
- Y. Xia and Z. Tang, Chem. Commun., 2012, 48, 6320–6336 RSC.
- M. R. Jones, K. D. Osberg, R. J. MacFarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS PubMed.
- Z. Tang and N. A. Kotov, Adv. Mater., 2005, 17, 951–962 CrossRef CAS.
- A. B. Descalzo, R. Martínez-Máñez, F. Sancenón, K. Hoffmann and K. Rurack, Angew. Chem., Int. Ed., 2006, 45, 5924–5948 CrossRef CAS PubMed.
- A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS PubMed.
- M. A. Olson, A. Coskun, R. Klajn, L. Fang, S. K. Dey, K. P. Browne, B. A. Grzybowski and J. F. Stoddart, Nano Lett., 2009, 9, 3185–3190 CrossRef CAS PubMed.
- R. Klajn, M. A. Olson, P. J. Wesson, L. Fang, A. Coskun, A. Trabolsi, S. Soh, J. F. Stoddart and B. A. Grzybowski, Nat. Chem., 2009, 1, 733–738 CrossRef CAS PubMed.
- Nonappa, J. S. Haataja, J. V. I. Timonen, S. Malola, P. Engelhardt, N. Houbenov, M. Lahtinen, H. Häkkinen and O. Ikkala, Angew. Chem., Int. Ed., 2017, 56, 6473–6477 CrossRef CAS PubMed.
- D. Maiolo, C. Pigliacelli, P. Sánchez Moreno, M. B. Violatto, L. Talamini, I. Tirotta, R. Piccirillo, M. Zucchetti, L. Morosi, R. Frapolli, G. Candiani, P. Bigini, P. Metrangolo and F. Baldelli Bombelli, ACS Nano, 2017, 11, 9413–9423 CrossRef CAS PubMed.
- C. Pigliacelli, D. Maiolo, Nonappa, J. S. Haataja, H. Amenitsch, C. Michelet, P. Sánchez Moreno, I. Tirotta, P. Metrangolo and F. Baldelli-Bombelli, Angew. Chem., Int. Ed., 2017, 56, 16186–16190 CrossRef CAS PubMed.
- K. D. Hermanson, S. O. Lumsdon, J. P. Williams, E. W. Kaler and O. D. Velev, Science, 2001, 294, 1082–1086 CrossRef CAS PubMed.
- S. L. Tripp, R. E. Dunin-Borkowski and A. Wei, Angew. Chem., Int. Ed., 2003, 42, 5591–5593 CrossRef CAS PubMed.
- B. Li, N. You, Y. Liang, Q. Zhang, W. Zhang, M. Chen and X. Pang, Energy Environ. Mater., 2019, 2, 38–54 CrossRef CAS.
- X. Wu, C. Hao, J. Kumar, H. Kuang, N. A. Kotov, L. M. Liz-Marzán and C. Xu, Chem. Soc. Rev., 2018, 47, 4677–4696 RSC.
- M. G. Ryadnov, B. Ceyhan, C. M. Niemeyer and D. N. Woolfson, J. Am. Chem. Soc., 2003, 125, 9388–9394 CrossRef CAS PubMed.
- M. M. Stevens, N. T. Flynn, C. Wang, D. A. Tirrell and R. Langer, Adv. Mater., 2004, 16, 915–918 CrossRef CAS.
- S. Si, M. Raula, T. K. Paira and T. K. Mandal, ChemPhysChem, 2008, 9, 1578–1584 CrossRef CAS PubMed.
- D. Aili, K. Enander, J. Rydberg, I. Nesterenko, F. Björefors, L. Baltzer and B. Liedberg, J. Am. Chem. Soc., 2008, 130, 5780–5788 CrossRef CAS PubMed.
- D. Aili, K. Enander, L. Baltzer and B. Liedberg, Nano Lett., 2008, 8, 2473–2478 CrossRef CAS PubMed.
- O. K. Zahr and A. S. Blum, Nano Lett., 2012, 12, 629–633 CrossRef CAS PubMed.
- A. Schreiber, M. C. Huber, H. Cölfen and S. M. Schiller, Nat. Commun., 2015, 6, 6705 CrossRef CAS PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- E. De Santis and M. G. Ryadnov, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- R. V. Ulijn and R. Jerala, Chem. Soc. Rev., 2018, 47, 3391–3394 RSC.
- K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Chem. Soc. Rev., 2018, 47, 7539–7551 RSC.
- S. S. Panda, H. E. Katz and J. D. Tovar, Chem. Soc. Rev., 2018, 47, 3640–3658 RSC.
- M. Amit, S. Yuran, E. Gazit, M. Reches and N. Ashkenasy, Adv. Mater., 2018, 30, 1707083 CrossRef PubMed.
- C. Song, M. G. Blaber, G. Zhao, P. Zhang, H. C. Fry, G. C. Schatz and N. L. Rosi, Nano Lett., 2013, 13, 3256–3261 CrossRef CAS PubMed.
- E. Pazos, E. Sleep, C. M. Rubert Pérez, S. S. Lee, F. Tantakitti and S. I. Stupp, J. Am. Chem. Soc., 2016, 138, 5507–5510 CrossRef CAS PubMed.
- C. Pigliacelli, K. B. Sanjeeva, Nonappa, A. Pizzi, A. Gori, F. B. Bombelli and P. Metrangolo, ACS Nano, 2019, 13, 2158–2166 CAS.
- C. L. Chen and N. L. Rosi, Angew. Chem., Int. Ed., 2010, 49, 1924–1942 CrossRef CAS PubMed.
- X. Yu, Z. Wang, Z. Su and G. Wei, J. Mater. Chem. B, 2017, 5, 1130–1142 RSC.
- T. R. Walsh and M. R. Knecht, Chem. Rev., 2017, 117, 12641–12704 CrossRef CAS PubMed.
- S. Corra, M. S. Shoshan and H. Wennemers, Curr. Opin. Chem. Biol., 2017, 40, 138–144 CrossRef CAS PubMed.
- G. Zan and Q. Wu, Adv. Mater., 2016, 28, 2099–2147 CrossRef CAS PubMed.
- Y. N. Tan, J. Y. Lee and D. I. C. Wang, J. Am. Chem. Soc., 2010, 132, 5677–5686 CrossRef CAS PubMed.
- C.-L. Chen, P. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2008, 130, 13555–13557 CrossRef CAS PubMed.
- R. R. Naik, S. J. Stringer, G. Agarwal, S. E. Jones and M. O. Stone, Nat. Mater., 2002, 1, 169–172 CrossRef CAS PubMed.
- J. M. Slocik, M. O. Stone and R. R. Naik, Small, 2005, 1, 1048–1052 CrossRef CAS PubMed.
- C. Song, G. Zhao, P. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 14033–14035 CrossRef CAS PubMed.
- L. Hwang, G. Zhao, P. Zhang and N. L. Rosi, Small, 2011, 7, 1939–1942 CrossRef CAS PubMed.
- R. Djalali, Y. F. Chen and H. Matsui, J. Am. Chem. Soc., 2002, 124, 13660–13661 CrossRef CAS PubMed.
- R. Djalali, Y. F. Chen and H. Matsui, J. Am. Chem. Soc., 2003, 125, 5873–5879 CrossRef CAS PubMed.
- L. Yu, I. A. Banerjee and H. Matsui, J. Am. Chem. Soc., 2003, 125, 14837–14840 CrossRef CAS PubMed.
- I. A. Banerjee, L. Yu and H. Matsui, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14678–14682 CrossRef CAS PubMed.
- L. Yu, I. A. Banerjee, M. Shima, K. Rajan and H. Matsui, Adv. Mater., 2004, 16, 709–712 CrossRef CAS.
- Y. Feng, H. Wang, J. Zhang, Y. Song, M. Meng, J. Mi, H. Yin and L. Liu, Biomacromolecules, 2018, 19, 2432–2442 CrossRef CAS PubMed.
- Y. Tian, H. V. Zhang, K. L. Kiick, J. G. Saven and D. J. Pochan, Chem. Mater., 2018, 30, 8510–8520 CrossRef CAS.
- Y. Wang, L. Cao, S. Guan, G. Shi, Q. Luo, L. Miao, I. Thistlethwaite, Z. Huang, J. Xu and J. Liu, J. Mater. Chem., 2012, 22, 2575–2581 RSC.
- M. A. Khalily, O. Ustahuseyin, R. Garifullin, R. Genc and M. O. Guler, Chem. Commun., 2012, 48, 11358–11360 RSC.
- K. Tao, J. Wang, Y. Li, D. Xia, H. Shan, H. Xu and J. R. Lu, Sci. Rep., 2013, 3, 2565 CrossRef PubMed.
- H. Matsui, S. Pan and G. E. Douberly, J. Phys. Chem. B, 2001, 105, 1683–1686 CrossRef CAS.
- L. S. Li and S. I. Stupp, Angew. Chem., Int. Ed., 2005, 44, 1833–1836 CrossRef CAS PubMed.
- I. A. Banerjee, L. Yu and H. Matsui, Nano Lett., 2003, 3, 283–287 CrossRef CAS.
- O. Carny, D. E. Shalev and E. Gazit, Nano Lett., 2006, 6, 1594–1597 CrossRef CAS PubMed.
- N. Sharma, A. Top, K. L. Kiick and D. J. Pochan, Angew. Chem., Int. Ed., 2009, 48, 7078–7082 CrossRef CAS PubMed.
- M. S. Lamm, N. Sharma, K. Rajagopal, F. L. Beyer, J. P. Schneider and D. J. Pochan, Adv. Mater., 2008, 20, 447–451 CrossRef CAS.
- B. Zhou, Z. Sun, D. Li, T. Zhang, L. Deng and Y.-N. Liu, Nanoscale, 2013, 5, 2669–2673 RSC.
- S. Abb, L. Harnau, R. Gutzler, S. Rauschenbach and K. Kern, Nat. Commun., 2016, 7, 10335 CrossRef CAS PubMed.
- K. Shiba, Chem. Soc. Rev., 2010, 39, 117–126 RSC.
- J. M. Slocik, A. O. Govorov and R. R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS PubMed.
- M. Hentschel, M. Schäferling, X. Duan, H. Giessen and N. Liu, Sci. Adv., 2017, 3, e1602735 CrossRef PubMed.
- A. D. Merg, J. C. Boatz, A. Mandal, G. Zhao, S. Mokashi-Punekar, C. Liu, X. Wang, P. Zhang, P. C. A. van der Wel and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 13655–13663 CrossRef CAS PubMed.
- S. Mokashi-Punekar, T. R. Walsh and N. L. Rosi, J. Am. Chem. Soc., 2019, 141, 15710–15716 CrossRef CAS PubMed.
- B. D. Briggs, Y. Li, M. T. Swihart and M. R. Knecht, ACS Appl. Mater. Interfaces, 2015, 7, 8843–8851 CrossRef CAS PubMed.
- D. J. Mikolajczak, A. A. Berger and B. Koksch, Angew. Chem., Int. Ed., 2020, 59, 8776–8785 CrossRef CAS PubMed.
- D. J. Mikolajczak, J. L. Heier, B. Schade and B. Koksch, Biomacromolecules, 2017, 18, 3557–3562 CrossRef CAS PubMed.
- N. M. Bedford, Z. E. Hughes, Z. Tang, Y. Li, B. D. Briggs, Y. Ren, M. T. Swihart, V. G. Petkov, R. R. Naik, M. R. Knecht and T. R. Walsh, J. Am. Chem. Soc., 2016, 138, 540–548 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed.
- A. Laromaine, L. Koh, M. Murugesan, R. V. Ulijn and M. M. Stevens, J. Am. Chem. Soc., 2007, 129, 4156–4157 CrossRef CAS PubMed.
- R. Chandrawati and M. M. Stevens, Chem. Commun., 2014, 50, 5431–5434 RSC.
- S. Si and T. K. Mandal, Langmuir, 2007, 23, 190–195 CrossRef CAS PubMed.
- T. R. Walsh, Acc. Chem. Res., 2017, 50, 1617–1624 CrossRef CAS PubMed.
- S. Hosseinpour, S. J. Roeters, M. Bonn, W. Peukert, S. Woutersen and T. Weidner, Chem. Rev., 2020, 120, 3420–3465 CrossRef CAS PubMed.
- P. A. Mirau, R. R. Naik and P. Gehring, J. Am. Chem. Soc., 2011, 133, 18243–18248 CrossRef CAS PubMed.
- B. Ding, J. Jasensky, Y. Li and Z. Chen, Acc. Chem. Res., 2016, 49, 1149–1157 CrossRef CAS PubMed.
- D. Campoccia, L. Montanaro and C. R. Arciola, Biomaterials, 2013, 34, 8533–8554 CrossRef CAS PubMed.
- M. Rai, A. Yadav and A. Gade, Biotechnol. Adv., 2009, 27, 76–83 CrossRef CAS PubMed.
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS PubMed.
- L. Rizzello and P. P. Pompa, Chem. Soc. Rev., 2014, 43, 1501–1518 RSC.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, Coord. Chem. Rev., 2018, 357, 1–17 CrossRef CAS.
- I. Kim, H.-H. Jeong, Y.-J. Kim, N.-E. Lee, K. Huh, C.-S. Lee, G. H. Kim and E. Lee, J. Mater. Chem. B, 2014, 2, 6478–6486 RSC.
- L. Polavarapu, J. Pérez-Juste, Q. H. Xu and L. M. Liz-Marzán, J. Mater. Chem. C, 2014, 2, 7460–7476 RSC.
- J. Yao, M. Yang and Y. Duan, Chem. Rev., 2014, 114, 6130–6178 CrossRef CAS PubMed.
- A. Ghasemi, N. Rabiee, S. Ahmadi, S. Hashemzadeh, F. Lolasi, M. Bozorgomid, A. Kalbasi, B. Nasseri, A. Shiralizadeh Dezfuli, A. R. Aref, M. Karimi and M. R. Hamblin, Analyst, 2018, 143, 3249–3283 RSC.
- D. Aili and M. M. Stevens, Chem. Soc. Rev., 2010, 39, 3358–3370 RSC.
- K. Welser, R. Adsley, B. M. Moore, W. C. Chan and J. W. Aylott, Analyst, 2011, 136, 29–41 RSC.
- J. Zong, S. L. Cobb and N. R. Cameron, Biomater. Sci., 2017, 5, 872–886 RSC.
- J. Wang, X. Zhao, J. Li, X. Kuang, Y. Fan, G. Wei and Z. Su, ACS Macro Lett., 2014, 3, 529–533 CrossRef CAS.
- Y. Gong, X. Chen, Y. Lu and W. Yang, Biosens. Bioelectron., 2015, 66, 392–398 CrossRef CAS PubMed.
- R. M. Jin, M. H. Yao, J. Yang, D. H. Zhao, Y. Di Zhao and B. Liu, ACS Sustainable Chem. Eng., 2017, 5, 9841–9847 CrossRef CAS.
- J. Zhao, Y. Tang, Y. Cao, T. Chen, X. Chen, X. Mao, Y. Yin and G. Chen, Electrochim. Acta, 2018, 283, 1072–1078 CrossRef CAS.
- K. Wang, Z. Li, C. Wang, S. Zhang, W. Cui, Y. Xu, J. Zhao, H. Xue and J. Li, J. Colloid Interface Sci., 2019, 557, 628–634 CrossRef CAS PubMed.
- W. Zhang, D. Lin, H. Wang, J. Li, G. U. Nienhaus, Z. Su, G. Wei and L. Shang, Bioconjugate Chem., 2017, 28, 2224–2229 CrossRef CAS PubMed.
- W. Zhang, J. Xi, Y. Zhang, Z. Su and G. Wei, Arab. J. Chem., 2020, 13, 1406–1414 CrossRef CAS.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- K. Bian, X. Zhang, K. Liu, T. Yin, H. Liu, K. Niu, W. Cao and D. Gao, ACS Sustainable Chem. Eng., 2018, 6, 7574–7588 CrossRef CAS.
- W. Wang, C. F. Anderson, Z. Wang, W. Wu, H. Cui and C.-J. Liu, Chem. Sci., 2017, 8, 3310–3324 RSC.
- H. I. Ryoo, J. S. Lee, C. B. Park and D. P. Kim, Lab Chip, 2011, 11, 378–380 RSC.
- A. Ghorbani-Choghamarani and Z. Taherinia, RSC Adv., 2016, 6, 59410–59421 RSC.
- N. A. Merrill, F. Yan, H. Jin, P. Mu, C. L. Chen and M. R. Knecht, Nanoscale, 2018, 10, 12445–12452 RSC.
- W. Xu, Y. Hong, Y. Hu, J. Hao and A. Song, ChemPhysChem, 2016, 17, 2157–2163 CrossRef CAS PubMed.
- H.-E. Lee, H.-Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–365 CrossRef CAS PubMed.
- W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. A. Kotov, Nat. Commun., 2013, 4, 2689 CrossRef PubMed.
- C. Hao, L. Xu, H. Kuang and C. Xu, Adv. Mater., 2019, 1802075 CrossRef PubMed.
- G. González-Rubio and L. M. Liz-Marzán, Nature, 2018, 556, 313–314 CrossRef PubMed.
- J. M. Slocik, F. Tam, N. J. Halas and R. R. Naik, Nano Lett., 2007, 7, 1054–1058 CrossRef CAS PubMed.
- T. John, A. Gladytz, C. Kubeil, L. L. Martin, H. J. Risselada and B. Abel, Nanoscale, 2018, 10, 20894–20913 RSC.
- L. Bellucci, A. Ardèvol, M. Parrinello, H. Lutz, H. Lu, T. Weidner and S. Corni, Nanoscale, 2016, 8, 8737–8748 RSC.
- I. Cherny and E. Gazit, Angew. Chem., Int. Ed., 2008, 47, 4062–4069 CrossRef CAS PubMed.
- M. R. Elkins, T. Wang, M. Nick, H. Jo, T. Lemmin, S. B. Prusiner, W. F. DeGrado, J. Stöhr and M. Hong, J. Am. Chem. Soc., 2016, 138, 9840–9852 CrossRef CAS PubMed.
- J. D. Pham, N. Chim, C. W. Goulding and J. S. Nowick, J. Am. Chem. Soc., 2013, 135, 12460–12467 CrossRef CAS PubMed.
- J. Kumar, H. Eraña, E. López-Martínez, N. Claes, V. F. Martín, D. M. Solís, S. Bals, A. L. Cortajarena, J. Castilla and L. M. Liz-Marzán, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3225–3230 CrossRef CAS PubMed.
- H. Zhang, C. Hao, A. Qu, M. Sun, L. Xu, C. Xu and H. Kuang, Angew. Chem., Int. Ed., 2020, 59, 7131–7138 CrossRef CAS PubMed.
- Y.-H. Liao, Y.-J. Chang, Y. Yoshiike, Y.-C. Chang and Y.-R. Chen, Small, 2012, 8, 3631–3639 CrossRef CAS PubMed.
- T. Scheibel, R. Parthasarathy, G. Sawicki, X. M. Lin, H. Jaeger and S. L. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4527–4532 CrossRef CAS PubMed.
- H. Acar, R. Genc, M. Urel, T. S. Erkal, A. Dana and M. O. Guler, Langmuir, 2012, 28, 16347–16354 CrossRef CAS PubMed.
- M. Rubio-Martínez, J. Puigmartí-Luis, I. Imaz, P. S. Dittrich and D. Maspoch, Small, 2013, 9, 4160–4167 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |