
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast and reliable generation of [18F]triflyl fluoride, a gaseous [18F]fluoride source†
A.
Pees
 *a,
C.
Sewing
a,
M. J. W. D.
Vosjan
b,
V.
Tadino
c,
J. D. M.
Herscheid
a,
A. D.
Windhorst
a and
D. J.
Vugts
a
*a,
C.
Sewing
a,
M. J. W. D.
Vosjan
b,
V.
Tadino
c,
J. D. M.
Herscheid
a,
A. D.
Windhorst
a and
D. J.
Vugts
a
aDepartment of Radiology & Nuclear Medicine, Location Radionuclidecenter, De Boelelaan 1085C, 1081 HV Amsterdam, The Netherlands. E-mail: a.pees@vumc.nl
bBV Cyclotron VU, De Boelelaan 1081, 1081 HV Amsterdam, The Netherlands
cORA Neptis, Rue de la Gendarmerie 50/B, 5600 Philippeville, Belgium
First published on 23rd August 2018
Abstract
A novel strategy for the production of reactive [18F]fluoride has been developed, omitting time consuming azeotropic drying procedures. Gaseous [18F]triflyl fluoride is formed instantaneously at room temperature from hydrated [18F]fluoride, followed by distillation in less than 5 minutes into a dry aprotic solvent, in which dry [18F]fluoride is released in presence of base with >90% radiochemical yield. The reactivity of the [18F]fluoride has been confirmed by reaction with several model compounds and by the synthesis of the PET tracers [18F]fluoroestradiol ([18F]FES) and O-2-[18F]fluoroethyl-L-tyrosine ([18F]FET), providing good isolated radiochemical yields and molar activities of up to 123 GBq μmol−1.
Fluorine-18, a popular radionuclide used for Positron Emission Tomography (PET), can be produced via the nuclear reaction 18O(p,n)18F by irradiation of an oxygen-18 enriched water target and is obtained as strongly hydrated [18F]fluoride, which is unreactive towards nucleophilic substitution reactions. To enhance the reactivity of [18F]fluoride and recover expensive enriched target water, the radionuclide is processed before further reaction. Most commonly, [18F]fluoride is trapped on an anion-exchange resin, eluted with a mixture of MeCN and water containing a complex of inorganic base and cryptand (typically K2CO3/kryptofix-K2.2.2.) and dried by repeated azeotropic distillation with MeCN.1,2
However, this procedure has several drawbacks: (i) it is time-consuming and complicated to miniaturise, e.g. for the use in microfluidic devices; (ii) significant amounts of radioactivity can be lost during azeotropic drying due to decay and unspecific adsorption to the walls of the reaction vessel; (iii) most importantly substantial amounts of base are needed to isolate [18F]fluoride from the cartridge thereby limiting the subsequent reaction scope, since the base cannot be removed.3 As a consequence, alternative methods have been developed to circumvent the drying procedure. Firstly, the eluent for eluting [18F]fluoride from the anion-exchange resin has been varied: the use of strong organic bases in MeCN,3 tetraethylammonium hydrogen carbonate in polar aprotic solvents,4 protic solvents in combination with cryptand and potassium salt,5 solutions of sulfonyl derivatives6 as well as alcoholic solutions of precursors containing a quaternary ammonium, diaryliodonium or triarylsulfonium functionality7 have been reported. Secondly, modified anion exchange resins have been used, containing e.g. macroporous copolymers loaded with long alkyl chain quaternary ammonium salts2 or the phosphonium borane [(Ph2MeP)C6H4(BMes2)]+.8 Thirdly, the use of additives in the labelling solution has been described, which make azeotropic drying of the eluate unnecessary. Sergeev et al. reported the use of titanium dioxide nanoparticles as catalyst and water adsorbent allowing radiofluorination in aqueous mixtures with up to 25 vol% of water.9 Finally, electrodeposition has been extensively studied, especially with regard to its use in microfluidic devices: after anodic deposition of [18F]fluoride from the water target, it is recovered under reversed voltage in aprotic solvent containing a phase transfer catalyst.10–14 In conclusion, all these alternative and improved methods solve one or more of the aforementioned issues.
We present a novel strategy which allows for fast generation of dry [18F]fluoride which can be broadly applied in radiofluorination reactions. In a first step, [18F]fluoride is trapped on an anion-exchange column to recover the target water, followed by elution from the column with an aqueous potassium salt solution and conversion to volatile [18F]triflyl fluoride (b.p. −25 °C)15 using bistriflate 1. After distillation [18F]triflyl fluoride is trapped in a dry aprotic solvent and dry [18F]fluoride is released in the presence of base and kryptofix-K2.2.2. (see Scheme 1). The type and amount of base are variable and thus can be adapted with regard to the subsequent radiofluorination reaction. This method provides dry [18F]fluoride in a very short time and allows for subsequent radiofluorination under mild conditions. Furthermore, gaseous [18F]triflyl fluoride 2 can be transported over longer distance, as e.g. is also done with [11C]CO2. During the course of our work a patent application outlining the role of sulfonyl fluorides in volatilising fluoride for PET was published, although exemplification did not extend to triflyl fluoride.6b
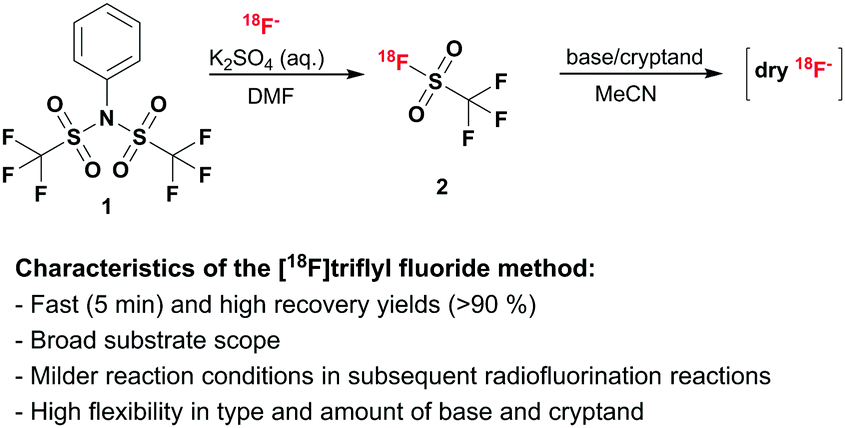 | ||
| Scheme 1 [18F]Triflyl fluoride formation and reaction to dry [18F]fluoride. | ||
Initial studies showed, that [18F]fluoride can be eluted from an anion-exchange cartridge using a 0.1 M potassium sulfate solution without the need for a cryptand, such as kryptofix. In reactions of aliquots of this aqueous [18F]fluoride-containing solution with bistriflate 1 in polar aprotic solvent, volatile [18F]triflyl fluoride 2 was formed at room temperature. Furthermore, the addition of base, e.g. KHCO3 or K2CO3 used for the standard elution procedure, led to decomposition of the product and formation of free [18F]fluoride. This fuelled our idea to use [18F]triflyl fluoride as carrier to transfer [18F]fluoride from an aqueous to a polar aprotic medium. Hence, the following reaction setup was developed: [18F]fluoride was eluted from the cartridge using a 0.1 M potassium sulfate solution. An aliquot or the total volume was taken and added to a solution of bistriflate 1 in DMF. Instantaneously formed [18F]triflyl fluoride 2 was distilled at room temperature under a gentle stream of helium (10 mL min−1) to a second reaction vessel, where it was trapped in a solution of KHCO3 and kryptofix in MeCN. Hereinafter, the optimisation process of the whole setup is described.
To investigate the efficiency of the elution of [18F]fluoride from the anion exchange resin (Chromafix® 30-PS-HCO3) with 0.1 M potassium sulfate solution, [18F]fluoride was eluted in small steps of 100 μL and the eluted amount of radioactivity was measured (n = 4). Fig. 1 shows the percentage of eluted radioactivity relative to the starting amount of radioactivity on the cartridge for volumes up to 500 μL. Compared to the standard eluent, the elution efficiency was much higher: with only 500 μL of 0.1 M potassium sulfate solution, ≥99% of the [18F]fluoride was eluted from the cartridge, whereas commonly 1 mL of K2CO3/kryptofix-K2.2.2. solution is needed for complete elution. However, it was noticed that this high elution efficiency was only realised with the stepwise elution. When eluting with 500 μL 0.1 M potassium sulfate at once, only 85 ± 5% of the radioactivity was recovered (n = 6). Because of the small volume of the eluent it came to significant losses of radioactivity in the tubing. By flushing the column and the tubes with 850 μL DMF after the elution with 0.1 M potassium sulfate, the efficiency could be increased to 97 ± 3% (n = 8).
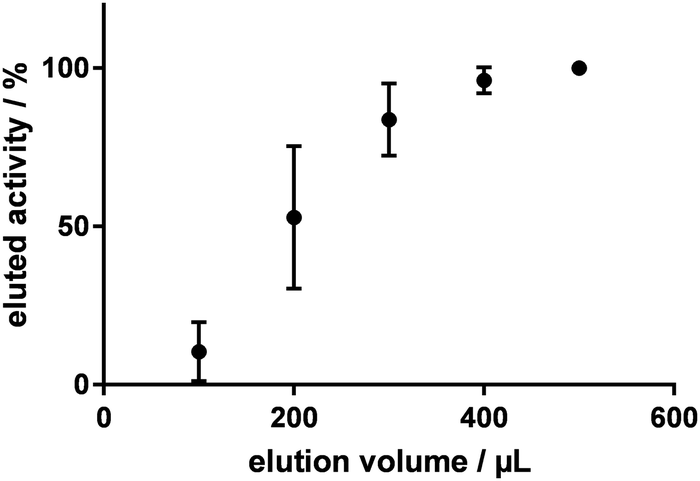 | ||
| Fig. 1 Elution efficiency of 0.1 M K2SO4 in water. | ||
The conversion of the eluted [18F]fluoride to [18F]triflyl fluoride was performed in different solvents to determine which of them provided the best radiochemical yield. Reactions in DMF, MeCN, THF and dioxane were studied. MeCN and DMF both resulted in radiochemical yields of over 90%, while reaction in dioxane and THF provided much lower radiochemical yields (see Table S2 of the ESI†). Because DMF proved to be slightly superior to MeCN in the overall radiochemical yield, it was used for all further experiments.
Next, the amount of water that is allowed in the [18F]triflyl fluoride formation was investigated. When precursor 1 was reacted for 5 minutes at room temperature in DMF in the presence of different amounts of water, ∼5% of water led to optimal radiochemical yields (>90%, determined by HPLC) for all examined precursor concentrations, being 5, 10, 50 and 100 mM. With increased water content, the radiochemical yields dropped and became less reproducible (see Table S3 of the ESI†). However, when the temperature was raised from 20 to 40 °C, up to 33% of water was allowed and >90% isolated radiochemical yield could be obtained again.
To promote dryness of the distilled radioactivity in the second reaction vessel for subsequent radiofluorination, [18F]triflyl fluoride was distilled over a drying column. Different materials were examined (see Table 1). Whereas calcium sulfate trapped a notable amount of radioactivity during the distillation, only low amounts of radioactivity (≤1%) remained on the drying column when using copper(II)sulfate, magnesium sulfate, sodium sulfate or phosphorus pentoxide (sicapent). P2O5 showed 0.2 ± 0.1% (n = 2) retained radioactivity and high radiochemical conversions of the distilled [18F]triflyl fluoride in the subsequent reactions.
| Drying column | Remaining activity on drying column (%) |
|---|---|
| CaSO4 | 15.0 ± 1.0 |
| CuSO4 | 0.6 ± 0.6 |
| MgSO4 | 1.0 ± 0.0 |
| Na2SO4 | 0.7 ± 0.2 |
| P2O5 | 0.2 ± 0.1 |
Finally, the trapping efficiency of [18F]triflyl fluoride was investigated, which proved to be rather difficult. Only at temperatures of −100 °C, satisfactory trapping of [18F]triflyl fluoride in THF with radiochemical yields of up to 95% could be achieved, but the yields were strongly varying. A more reproducible trapping efficiency was obtained at higher temperatures, when the [18F]triflyl fluoride was converted instantaneously in the trapping solution into free [18F]fluoride and trapped as such in a solution of KHCO3 and kryptofix-K2.2.2.. 94 ± 1% (n = 3) radiochemical yield (percentage of distilled and trapped radioactivity relative to the eluted amount of [18F]fluoride) was obtained by trapping the [18F]triflyl fluoride by quantitative conversion to [18F]fluoride in a 26.6 μM KHCO3/kryptofix-K2.2.2. solution in MeCN at room temperature. The distillate was used in a model reaction to test the reactivity of the [18F]fluoride. 1,4-Dinitrobenzene 3 was converted to 4-[18F]fluoronitrobenzene at 50 °C within 5 minutes with a radiochemical yield of >95% (determined by HPLC) (see Scheme 2). To assess the suitability of other solvents for trapping at room temperature, solutions of KHCO3 and kryptofix-K2.2.2. in DMF, DMA, THF and DMSO were tested. Trapping in DMF and DMA resulted in comparable trapping efficiencies as MeCN providing the [18F]fluoride in radiochemical yields of 95 ± 1% and 95 ± 3% (n = 2), respectively. Also in THF a good trapping efficiency was achieved, however radiochemical yields were slightly lower than with DMF and DMA as solvent (90 ± 4%; n = 2). In DMSO, the trapping was least efficient and not very reproducible: dry [18F]fluoride was obtained in radiochemical yields of 62 ± 19% (n = 2). Besides KHCO3, the use of other bases has been investigated (see Table S5 of the ESI†). With a solution of K2CO3 and kryptofix in MeCN, a base-cryptand combination often used in radiofluorination reactions, a bit lower trapping efficiencies of 72–92% were obtained. Similarly efficient trapping (77–92% radiochemical yield) was observed, when dibenzo-18-crown-6/K2CO3 or (n-Bu)4NH4SO4/Na2CO3 was employed. Only very low trapping efficiencies of 1–15% were observed when using DABCO and kryptofix-K2.2.2. without any base at room temperature. A further possibility, which may be convenient in the automated synthesis of a tracer, is immediate reaction of the trapped radioactivity with the precursor. Thus, [18F]triflyl fluoride was directly distilled into a solution of 1,4-dinitrobenzene, KHCO3 and kryptofix-K2.2.2. in MeCN heated at 50 °C and reacted for 5 minutes. 82 ± 3% of the radioactivity of the [18F]triflyl fluoride reaction (n = 2) was trapped in the 1,4-dinitrobenzene solution by near quantitative conversion to [18F]fluoronitrobenzene. Thus, this method had a high trapping efficiency as well as a high conversion to the final radiofluorinated product. Additionally, the procedure was time-saving as distillation and reaction were performed at once.
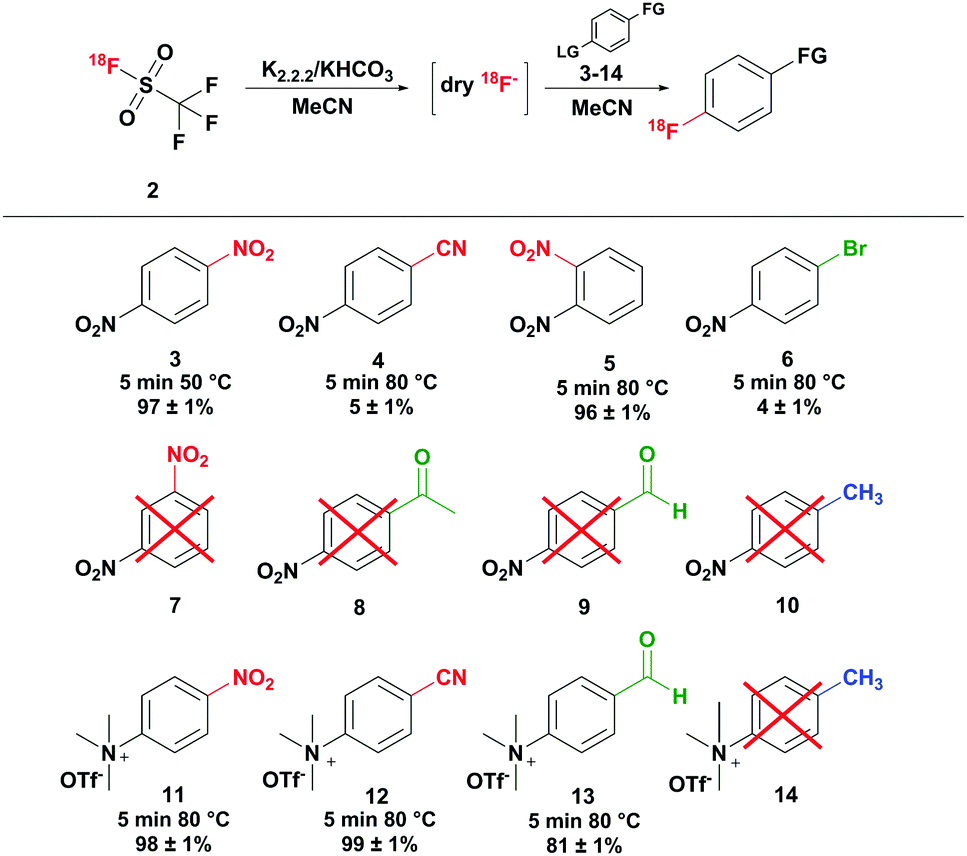 | ||
Scheme 2 Aromatic [18F]fluorination of different model compounds. *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Red: strong electron withdrawing; green: weak electron withdrawing; blue: electron donating; non-isolated and non-decay corrected, n = 2. Red: strong electron withdrawing; green: weak electron withdrawing; blue: electron donating; non-isolated and non-decay corrected, n = 2. | ||
Next to the radiofluorination of 1,4-dinitrobenzene, radiofluorination of other compounds has been carried out to investigate the performance of the [18F]triflyl fluoride derived [18F]fluoride in radiofluorination reactions. Aromatic molecules with strong electron withdrawing groups, weak electron withdrawing groups and electron donating groups (see Scheme 2) as well as aliphatic molecules with different leaving groups (see Scheme 3) were chosen as model compounds. The radiochemical yields were determined by HPLC for all model reactions (n = 2). Of the nitrobenzenes, para-dinitrobenzene 3 and ortho-dinitrobenzene 5 could be radiofluorinated in MeCN at 50 °C in 97 ± 1% and 96 ± 1% yield, respectively, whereas meta-dinitrobenzene 7 could not be converted to the desired product under these conditions. Compared to conventional syntheses described in literature, shorter reaction times and lower temperatures could be applied.16,17 For example the radiofluorination of ortho-dinitrobenzene 5 resulted after 5 minutes at 50 °C in 96 ± 1% yield, while according to literature temperatures of 130 °C and reaction times of 10 minutes are needed.16 The radiofluorination yield of 4-nitrobenzonitrile 4 was low as expected (5 ± 1%). Also para-substituted nitrobenzenes carrying a methyl, bromo, aldehyde or methyl ester group showed no or minor conversion to the radiofluorinated product because the aromatic rings were not sufficiently activated by the substituents for radiofluorination (see Scheme 2). Besides nitro precursors, also para-substituted N,N,N-trimethylammonium benzene precursors have been explored. With strong electron withdrawing groups such as the nitro and nitrile group high radiochemical yields close to quantitative were obtained. Analogous to the nitro precursor, no conversion to the [18F]fluorinated product was observed with the electron donating methyl group. With the weak electron withdrawing aldehyde group good yields of 81 ± 1% were obtained, which is comparable to literature results.18
 | ||
| Scheme 3 Aliphatic [18F]fluorination of different model compounds; non-isolated and non-decay corrected, n = 2. | ||
As aliphatic model compounds, a series of 3-substituted phenyl propanes with different halogens and sulfonates as leaving groups has been studied (see Scheme 3). With all leaving groups, high radiochemical yields of over 90% were obtained in the [18F]fluorination reaction. However, reaction conditions giving the optimal yields were differing depending on the leaving group. Whereas 1-phenyl-3-tosyl propane 15 was [18F]fluorinated in 5 minutes at 80 °C, radiofluorination of the corresponding mesylate 16 required higher temperatures of 120 °C and quantitative [18F]fluorination of the halides only occurred after 15 minutes at 80 °C. Next to the phenyl propanes, two different 3-substituted tosyl propanes were radiofluorinated. 1-[18F]Fluoro-3-tosyl propane was obtained from the ditosylate precursor 19 in good radiochemical yields (71 ± 3%) at moderate temperature (50 °C). In literature, higher reaction temperatures (90 °C) were reported,19 indicating that [18F]fluoride obtained by the [18F]triflyl fluoride method enables labelling under milder reaction conditions. Labelling of the corresponding tosyl azide at 80 °C yielded 89 ± 7% of 1-azido-3-[18F]fluoro propane after 5 minutes reaction time.
In addition to the model compounds, two tracers were synthesized from [18F]triflyl fluoride derived [18F]fluoride to prove the applicability of this method in PET tracer synthesis (see Scheme 4). [18F]Fluoroestradiol ([18F]FES) 22, which targets the estrogen receptor and is used for breast cancer imaging, was successfully synthesized with isolated radiochemical yields of 17 and 57%. The amino acid analogue O-2-[18F]fluoroethyl-L-tyrosine ([18F]FET) 23 was synthesized from the ((2S)-O-(2-tosyloxyethyl))-N-trityl-tyrosine-tert-butyl ester precursor and was likewise obtained with moderate to good radiochemical yields (10 and 55%). The molar activity of the formulated products ranged from 34 to 123 GBq μmol−1 and a radiochemical purity of >95% was obtained. A comparable range in molar activity and similar yields were observed using the standard elution and drying procedure.
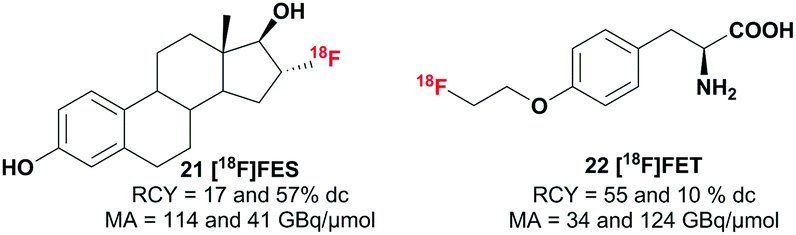 | ||
| Scheme 4 PET tracers synthesized with the [18F]triflyl fluoride method (n = 2). | ||
To conclude, we developed a valuable new elution and drying procedure for [18F]fluoride with several advantages over already existing drying procedures: (1) generation of dry [18F]fluoride via [18F]triflyl fluoride is fast and reliable. Dry [18F]fluoride can be obtained within 5 minutes and with recovery yields of >90%, whereas the conventional drying procedure can take 10–30 minutes and have losses of 15–50%; (2) [18F]triflyl fluoride derived [18F]fluoride has a broad substrate scope, unlike many other alternative drying strategies where the labelling solution contains too much water, base or other additives; (3) this methods allows in some instances for labelling under milder conditions than conventionally dried [18F]fluoride; (4) the type and amount of base and cryptand can be varied freely and therefore can be tailored to the need of the subsequent radiofluorination reaction which is in contrast to conventional labelling conditions and many of the alternatives summarized in this manuscript.
The project is financially supported by The Netherlands Organization of Scientific Research (NWO) grant no. 731.015.413 and BV Cyclotron VU.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Hamacher, H. H. Coenen and G. Stocklin, J. Nucl. Med., 1986, 27, 235–239 Search PubMed.
- J. Aerts, S. Voccia, C. Lemaire, F. Giacomelli, D. Goblet, D. Thonon, A. Plenevaux, G. Warnock and A. Luxen, Tetrahedron Lett., 2010, 51, 64–66 CrossRef.
- C. F. Lemaire, J. J. Aerts, S. Voccia, L. C. Libert, F. Mercier, D. Goblet, A. R. Plenevaux and A. J. Luxen, Angew. Chem., Int. Ed., 2010, 49, 3161–3164 CrossRef PubMed.
- L. Brichard and F. I. Aigbirhio, Eur. J. Org. Chem., 2014, 6145–6149 CrossRef.
- S. Lindner, C. Rensch, S. Neubaur, M. Neumeier, R. Salvamoser, V. Samper and P. Bartenstein, Chem. Commun., 2016, 52, 729–732 RSC.
- (a) D. Zhou, W. Chu and J. Katzenellenbogen, J. Nucl. Med., 2017, 58, 403 CrossRef PubMed; (b) D. Zhou and W. Chu, US Pat., 0197912, 2017 Search PubMed.
- R. Richarz, P. Krapf, F. Zarrad, E. A. Urusova, B. Neumaier and B. D. Zlatopolskiy, Org. Biomol. Chem., 2014, 12, 8094–8099 RSC.
- C. Perrio, S. Schmitt, D. Pla, F. P. Gabbaï, K. Chansaenpak, B. Mestre-Voegtle and E. Gras, Chem. Commun., 2017, 53, 340–343 RSC.
- M. E. Sergeev, F. Morgia, M. Lazari, C. Wang and R. M. Van Dam, J. Am. Chem. Soc., 2015, 137, 5686–5694 CrossRef PubMed.
- K. Hamacher, T. Hirschfelder and H. H. Coenen, Appl. Radiat. Isot., 2002, 56, 519–523 CrossRef PubMed.
- H. Saiki, R. Iwata, H. Nakanishi, R. Wong, Y. Ishikawa, S. Furumoto, R. Yamahara, K. Sakamoto and E. Ozeki, Appl. Radiat. Isot., 2010, 68, 1703–1708 CrossRef PubMed.
- F. Kügler, D. Roehrens, M. Stumpf, C. Drerup, J. Ermert, K. Hamacher and H. H. Coenen, Appl. Radiat. Isot., 2014, 91, 1–7 CrossRef PubMed.
- R. Wong, R. Iwata, H. Saiki, S. Furumoto, Y. Ishikawa and E. Ozeki, Appl. Radiat. Isot., 2012, 70, 193–199 CrossRef PubMed.
- S. Sadeghi, V. Liang, S. Cheung, S. Woo, C. Wu, J. Ly, Y. Deng, M. Eddings and R. M. van Dam, Appl. Radiat. Isot., 2013, 75, 85–94 CrossRef PubMed.
- M. Kobayashi, T. Inoguchi, T. Iida, T. Tanioka, H. Kumase and Y. Fukai, J. Fluorine Chem., 2003, 120, 105–110 CrossRef.
- H. F. VanBrocklin, J. P. O’Neil, D. L. Hom and A. R. Gibbs, J. Labelled Compd. Radiopharm., 2001, 1, 880–882 CrossRef.
- M. Constantinou, F. Shah and V. W. Pike, J. Labelled Compd. Radiopharm., 2001, 1, 889–891 CrossRef.
- C. Lemaire, L. Libert, A. Plenevaux, J. Aerts, X. Franci and A. Luxen, J. Fluorine Chem., 2012, 138, 48–55 CrossRef.
- C.-Y. C.-Y. Shiue, G. G. Shiue, A. A. Alavi, S. Jones and M. A. Zasloff, J. Labelled Compd. Radiopharm., 2001, 1, 391–392 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of reference compounds and experimental details. See DOI: 10.1039/c8cc03206h |
| This journal is © The Royal Society of Chemistry 2018 |