
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of naturally occurring isoquinoline alkaloids: (S)-(−)-trolline and (R)-(+)-oleracein E†
Weilong
Lin
a and
Shengming
Ma
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn
bDepartment of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P.R. China
First published on 6th March 2017
Abstract
The highly efficient asymmetric syntheses of a pair of enantiomers, (S)-(−)-trolline and (R)-(+)-oleracein E, from commercially available 2-methyl-3-butyn-2-ol, benzaldehyde and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline have been achieved in five steps with the catalytic enantioselective C1-alkynylation of 6,7-dimethoxy-3,4-dihydroisoquinoline as a key step, providing an efficient general approach for this type of naturally occurring chiral isoquinoline alkaloids.
Introduction
Naturally occurring isoquinoline alkaloids have attracted organic and medicinal chemists’ interest because of their important biological activities.1–6 For example, a pair of enantiomers, (S)-(−)-trolline and (R)-(+)-oleracein E, have a tetrahydroisoquinoline-containing tricyclic core structure with various useful activities (Fig. 1): (S)-(−)-trolline, isolated from Trollius chinensis, represents a potent antibacterial activity against respiratory bacteria such as Staphylococcus aureus, Streptococcus pneumoniae, and Klebsiella pneumoniae, as well as antiviral activity against influenza viruses A and B.4 On the other hand, (R)-(+)-oleracein E was isolated from Portulaca oleracea L. and it shows DPPH radical scavenging activity.5,6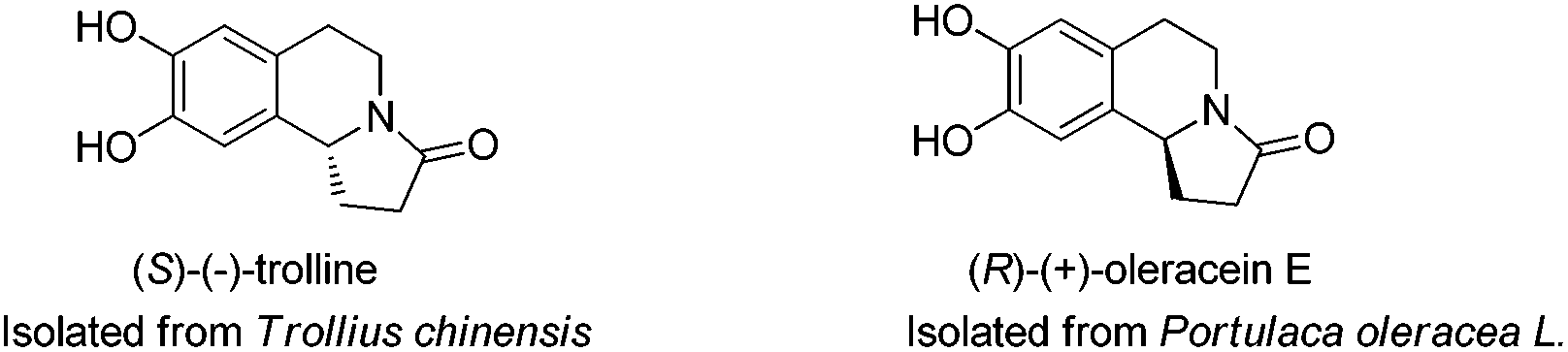 | ||
| Fig. 1 Tetrahydroisoquinoline alkaloids. | ||
The reported synthesis of optically active (S)-(−)-trolline and (R)-(+)-oleracein E takes 15 steps from veratraldehyde, with 88% ee and 86% ee to construct the chiral tetrahydroisoquinoline skeleton, respectively;8a (S)-(−)-trolline was also reported to be synthesized from 6,7-dimethoxy-3,4-dihydroisoquinoline using the toxic hydrogen cyanide.8b Thus, the development of general and efficient approaches for the highly enantioselective synthesis of these two enantiomers remains important. Previously, we have developed the highly enantioselective synthesis of C1 substituted chiral tetrahydroisoquinolines with readily available terminal alkynes and benzaldehyde under Cu(I) catalysis.9–11 Herein, we wish to demonstrate a successful application to the highly asymmetric synthesis of (S)-(−)-trolline and (R)-(+)-oleracein E (Scheme 1).
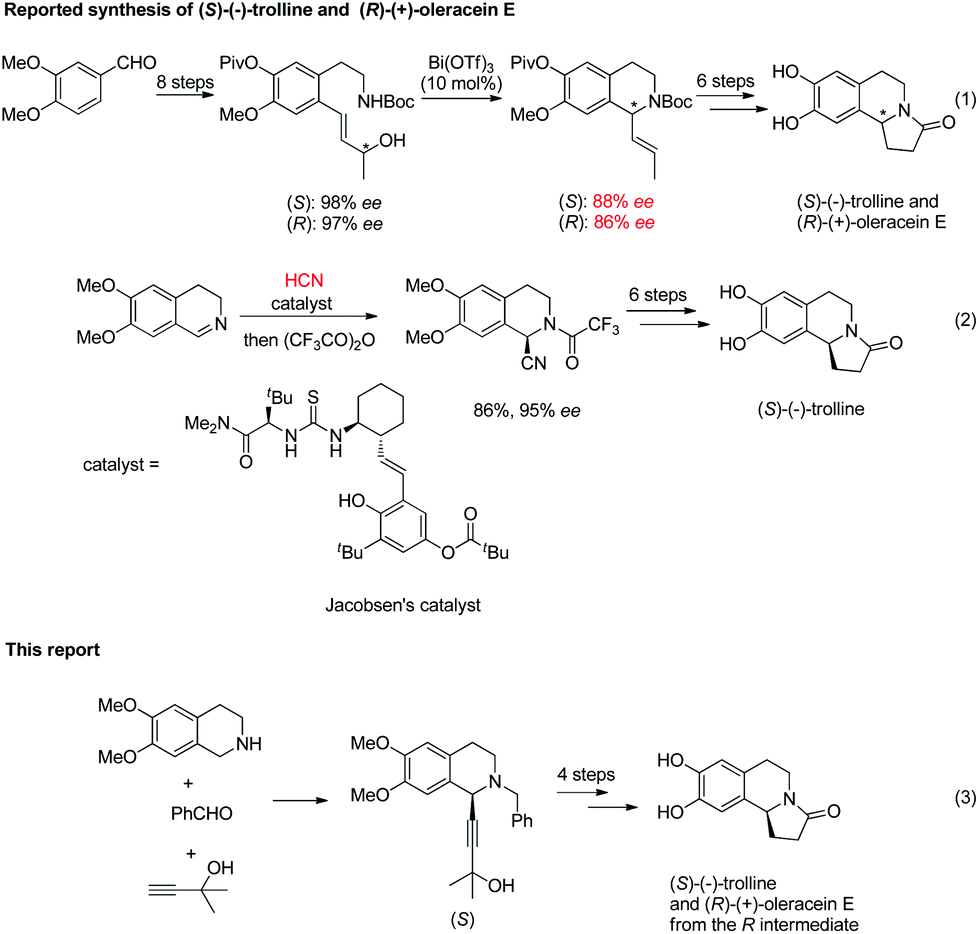 | ||
| Scheme 1 Known routes to (S)-(−)-trolline and (R)-(+)-oleracein E and our synthesis from readily available materials. | ||
Results and discussion
Synthesis of racemic trolline
We started this study by developing the synthetic route to racemic (±)-trolline. In this case, 6,7-dimethoxyl-1,2,3,4-tetrahydroisoquinoline was treated with methyl propiolate 2a, which is an electron-deficient terminal alkyne, initially. However, only the Michael addition product 4 was obtained, the desired product 3 was not formed (Scheme 2).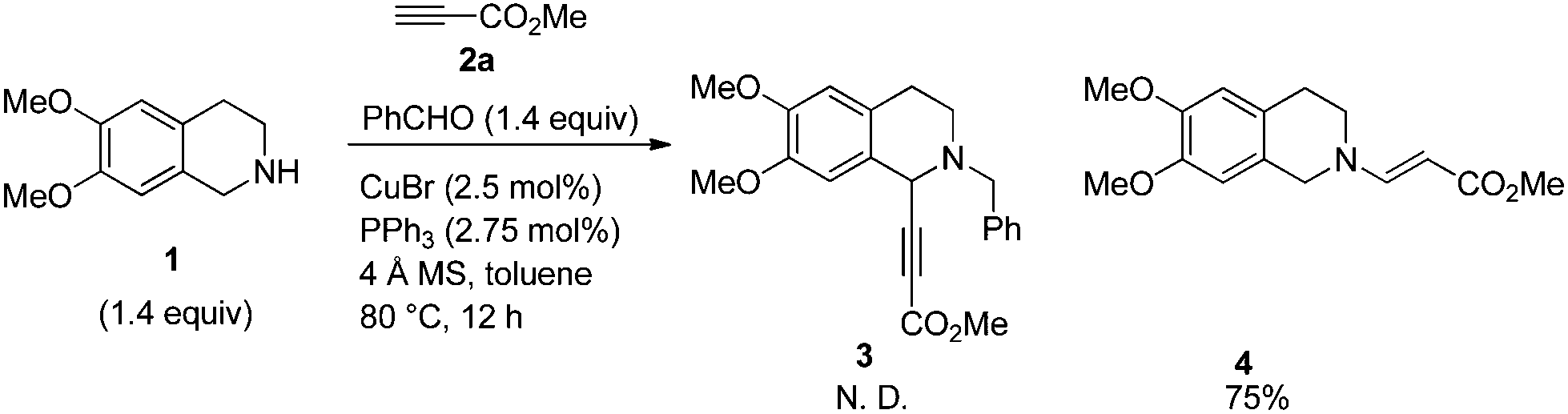 | ||
| Scheme 2 The reaction of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline 1 with 2a. | ||
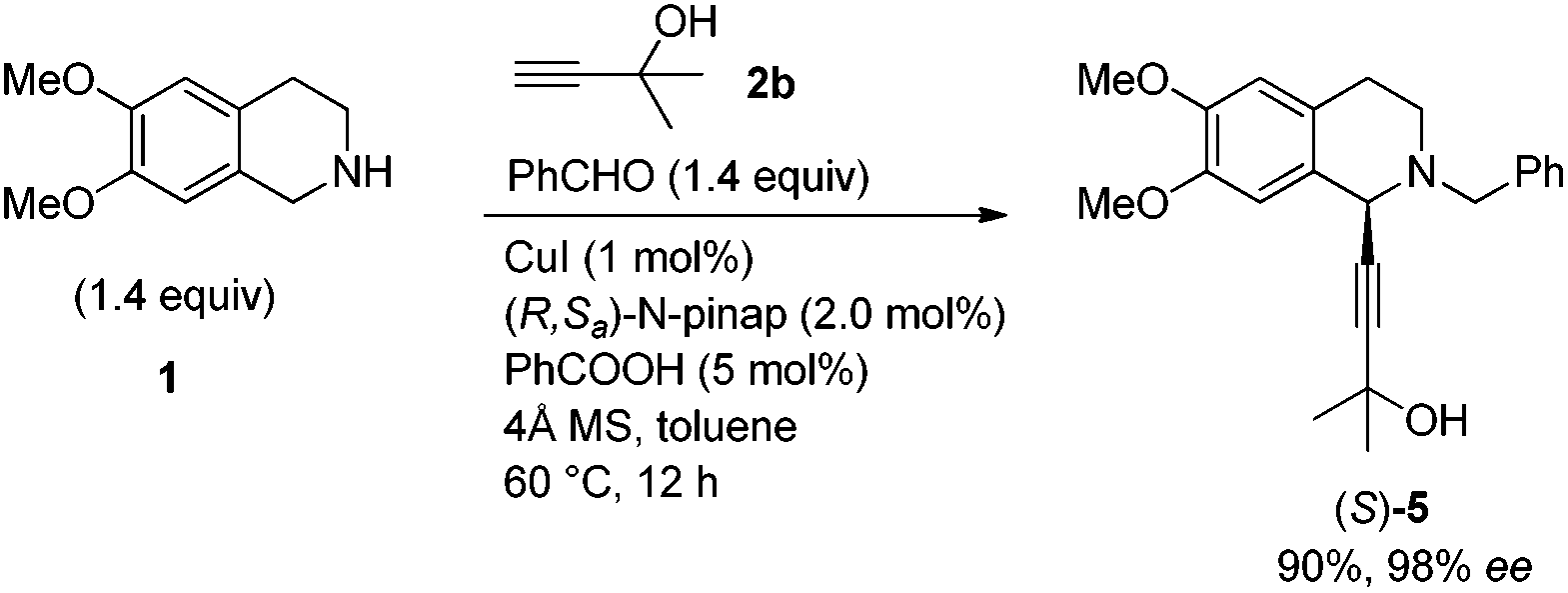
To our delight, the use of the commercially available 2-methyl-3-butyn-2-ol 2b could afford 5 in 89% yield smoothly. The key intermediate 3 was easily obtained via removing the 2-hydroxy-2-propyl group under the treatment with NaH followed by deprotonation with n-BuLi and subsequent treatment with ClCO2Me (Scheme 3).
 | ||
| Scheme 3 An alternative synthetic route to 3. | ||
When compound 3 was then exposed to 1 atm of H2 under the catalysis of Pd/C for the purpose of hydrogenation of C–C triple bonds and debenzylation, the desired product 7 could be obtained in 58% yield along with 32% yield of lactamization product 8 (Scheme 4, entry 1). With the addition of Et3N and NaOAc, the conversion from 7 to 8 was not better (Scheme 4, entries 2 and 3).
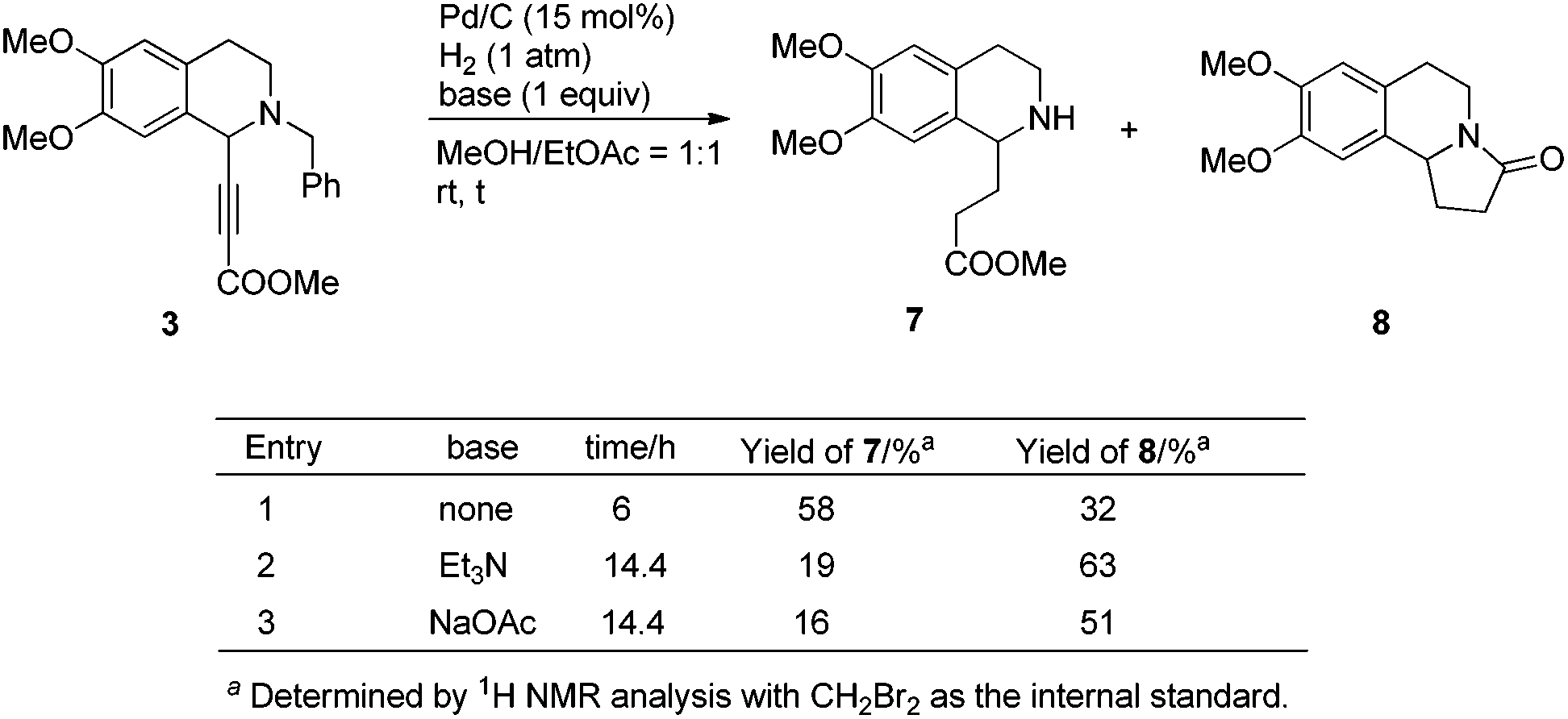 | ||
| Scheme 4 Initial hydrogenation along with cyclization. | ||
Delightedly, a complete conversion from 7 to lactam 8 was achieved by using toluene as the solvent in 84% yield. The racemic (±)-trolline could be easily obtained from lactam 8 in 80% yield via double demethylation (Scheme 5).
 | ||
| Scheme 5 Synthesis of racemic trolline. | ||
Asymmetric synthesis of (S)-(−)-trolline
We next turned to the asymmetric synthesis of (S)-(−)-trolline. It is exciting to observe that the expected propargylic amine (S)-5 was obtained in 90% yield and 98% ee with CuI and (R,Sa)-N-pinap as the chiral catalyst (Scheme 6). And gladly, (S)-3 could also be easily obtained in 97% ee after removing the 2-hydroxy-2-propyl group followed by the reaction with ClCO2Me with the help of n-BuLi. However, upon exposing (S)-3 to 1 atm of H2 under the catalysis of Pd/C, the desired product (S)-8 was obtained in only 88% ee – serious racemization was observed.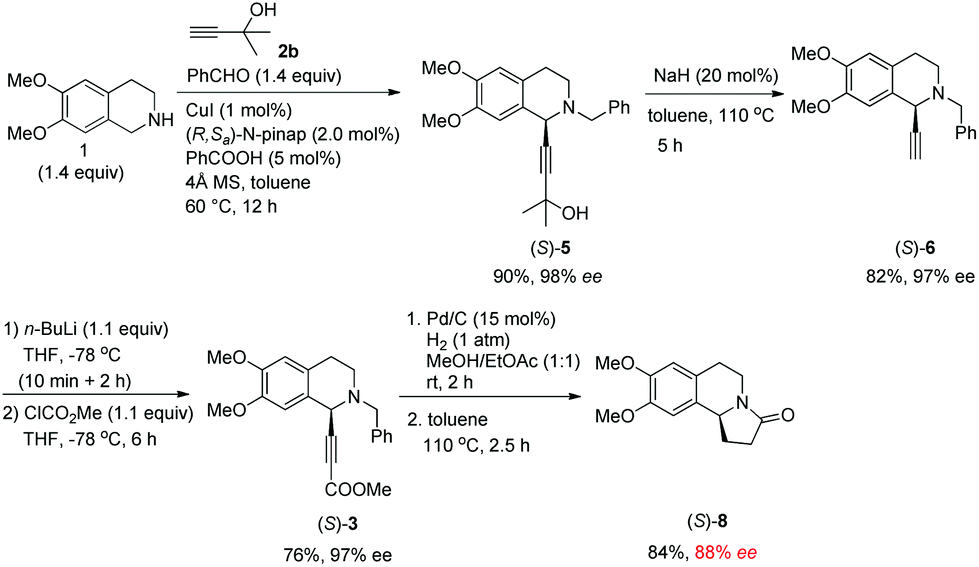 | ||
| Scheme 6 Initial enantioselective synthesis of (S)-8. | ||
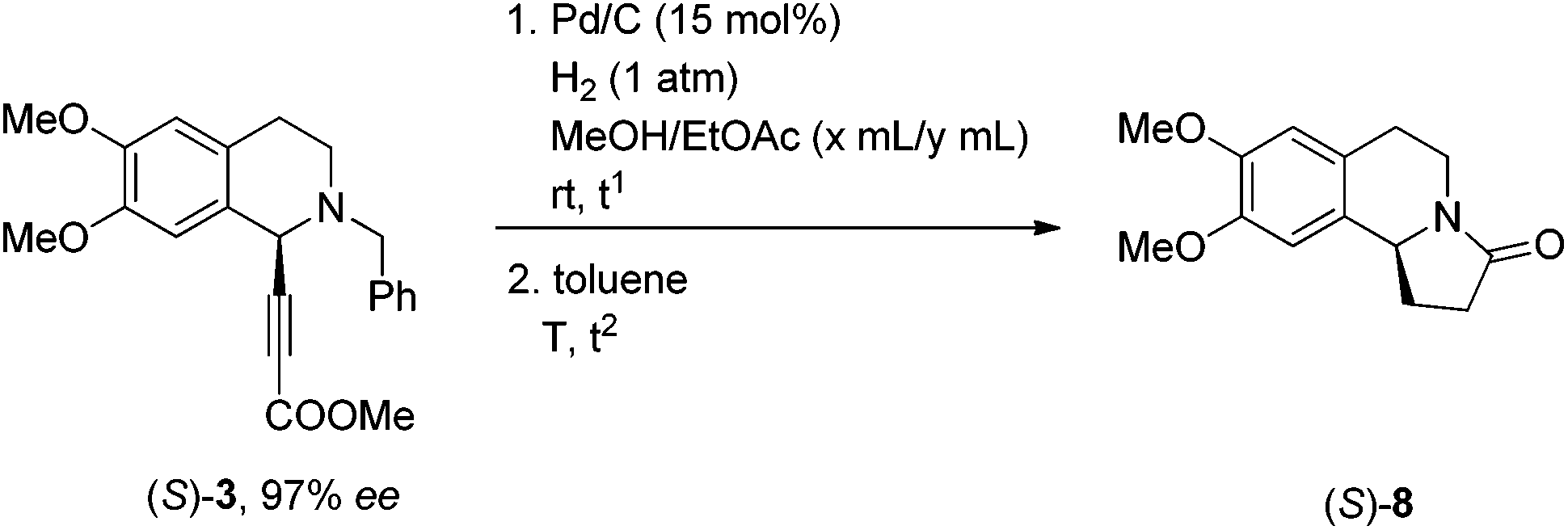
To achieve a highly enantioselective synthesis of (S)-8, the optimization of reaction conditions for hydrogenation under the catalysis of Pd/C was firstly conducted. Excitingly, screening of the solvent effect led to the observation that the hydrogenation in ethyl acetate could improve the ee to 95% (Table 1, entries 1–4). Furthermore, it was found that the cyclization could smoothly afford the desired product (S)-8 with 95% ee when conducting the second step at 40 °C, suggesting a slight racemization in the process of first step-hydrogenation (Table 1, entries 5 and 6). Then, the hydrogenation of (S)-3 was conducted under a hydrogen pressure of 10 atm or at 0 °C finally affording (S)-8 (Table 1, entries 7 and 8). Cyclization may also be conducted at rt (Table 1, entry 9).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | x | y | t 1/h | T/°C | t 2/h | Yield/% | ee (%) |
| a The reaction was conducted on the 0.1 mmol scale. b The hydrogenation was conducted at 0 °C. c The reaction was conducted under a hydrogen pressure of 10 atm. d The reaction was conducted on the 1 mmol scale. | |||||||
| 1 | 1 | 1 | 2 | 110 | 2 | 84 | 89 |
| 2 | 0.6 | 1.4 | 2 | 110 | 2 | 84 | 87 |
| 3 | 0.2 | 1.8 | 2 | 110 | 2 | 84 | 91 |
| 4 | 0 | 2 | 4 | 110 | 2 | 86 | 95 |
| 5 | 0 | 2 | 4 | 80 | 2 | 85 | 95 |
| 6 | 0 | 2 | 3 | 40 | 3 | 81 | 95 |
| 7b | 0 | 2 | 27 | 40 | 3 | 82 | 96 |
| 8c | 0 | 2 | 2 | 40 | 3 | 85 | 96 |
| 9 | 0 | 15 | 3 | rt | 5 | 87 | 95 |
However, (S)-(−)-trolline was obtained in 94% ee with a slight racemization at −20 °C in the process of demethylation (Scheme 7, entry 1). Gladly, demethylation could be achieved with 95% ee when executing this step at −40 °C (Scheme 7, entry 2).
 | ||
| Scheme 7 Optimization of the demethylation. | ||
After addressing these issues, (S)-(−)-trolline was afforded in 41% overall yield in five steps from commercially available 6,7-dimethoxy-3,4-dihydroisoquinoline (Scheme 8).
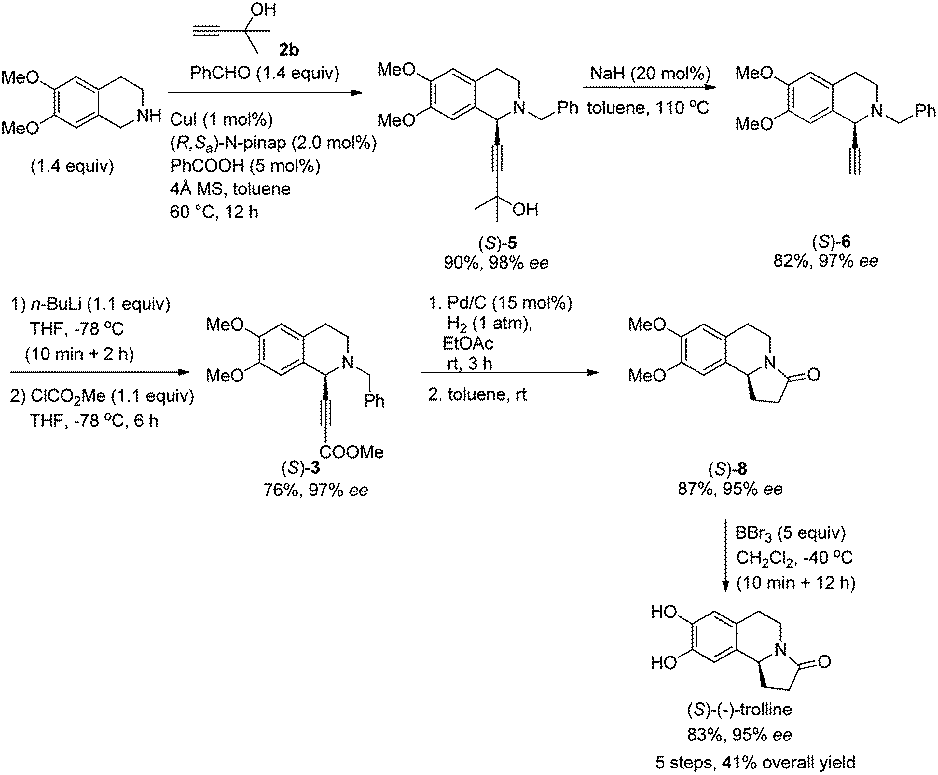 | ||
| Scheme 8 Asymmetric synthesis of (S)-(−)-trolline. | ||
Asymmetric synthesis of (R)-(+)-oleracein E
The same strategy was also easily applied to the highly enantioselective synthesis of (R)-(+)-oleracein E, the enantiomer of (S)-(−)-trolline. As expected, the expected propargylic amine (R)-5 was obtained in 94% yield together with 98% ee smoothly by using the corresponding axially chiral (R,Ra)-N-pinap. Accordingly, after a series of transformation, (R)-(+)-oleracein E was synthesized in 39% overall yield with high enantiopurity (96% ee) in five steps (Scheme 9).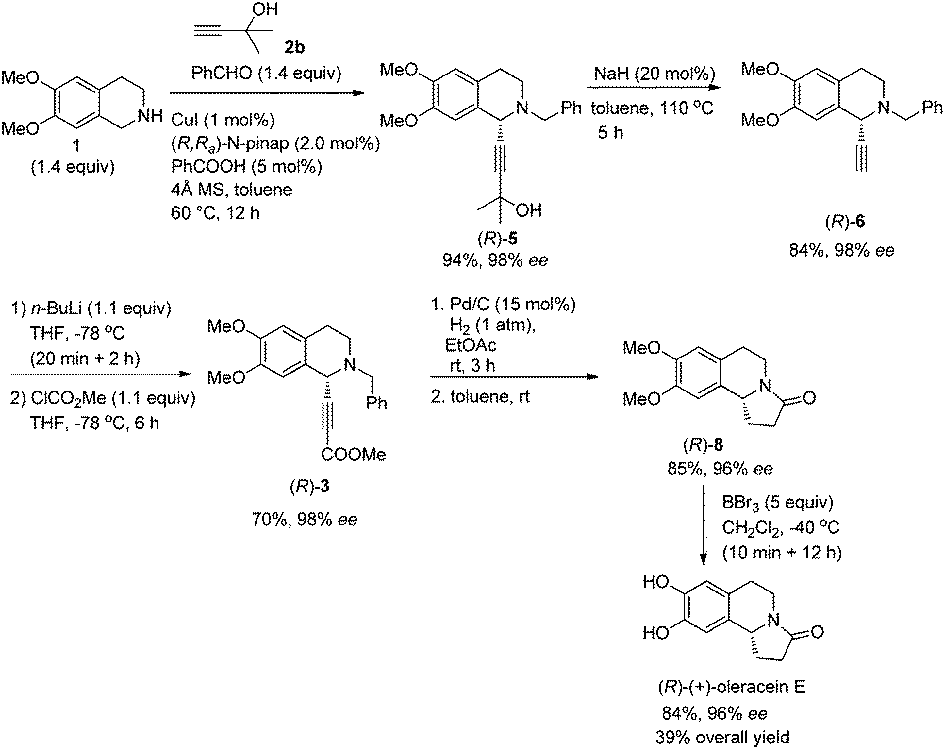 | ||
| Scheme 9 Asymmetric synthesis of (R)-(+)-oleracein E. | ||
Conclusions
In conclusion, we have achieved the highly efficient asymmetric synthesis of a pair of enantiomers, (S)-(−)-trolline (95% ee) and (R)-(+)-oleracein E (96% ee), from commercially available and very cheap 6,7-dimethoxy-3,4-dihydroisoquinoline, benzaldehyde, and 2-methyl-3-butyn-2-ol in five steps with high enantiopurity by applying the catalytic enantioselective C1-alkynylation of 6,7-dimethoxy-3,4-dihydroisoquinoline. Further studies on achieving the synthesis of such biological natural isoquinoline alkaloids are being actively pursued in this laboratory.Experimental
General information
CuI (99.5%) and CuBr (99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd; (R,Ra)-N-pinap (97%) and (R,Sa)-N-pinap (97%) were purchased from Strem Chemicals and kept in a glove box; 4 Å molecular sieves were purchased from Alfa Aesar and kept in a glove box after activation; Pd/C (10% in mass) was purchased from Sigma-Aldrich. Aldehydes were distilled right before use. Toluene and THF were dried over sodium wire and distilled freshly before use with benzophenone as an indicator. Other reagents were used without further treatment. All the temperatures are referred to the oil baths used.Experimental details and analytical data
Synthesis of (E)-methyl 3-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)-2-propenoate (4).
To a flame-dried Schlenk tube were added CuBr (3.6 mg, 0.025 mmol), PPh3 (7.3 mg, 0.0275 mmol), 4 Å molecular sieves (305.1 mg), and toluene (2 mL) sequentially under an Ar atmosphere. The resulting mixture was then stirred at rt for 30 min. PhCHO (149.1 mg, 1.4 mmol)/toluene (1 mL), 1 (271.1 mg, 1.4 mmol)/toluene (1 mL), and 2a (84.2 mg, 1.0 mmol)/toluene (2 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 80 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtered through a short column of silica gel eluted with ethyl acetate (50 mL). After evaporation, the residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 4/1) to afford 4 (207.1 mg, 75%) as an oil: 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 12.8 Hz, 1 H,
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.63 (s, 1 H, ArH), 6.60 (s, 1 H, ArH), 4.70 (d, J = 13.2 Hz, 1 H, CH), 4.25 (s, 2 H, NCH2Ar), 3.86 (s, 6 H, OCH3 × 2), 3.67 (s, 3 H, OCH3), 3.48 (s, 2 H, NCH2), 2.82 (t, J = 5.4 Hz, 2 H, CH2); 13C NMR (100 MHz, CDCl3) δ 169.8, 151.4, 147.63, 147.60, 125.8, 111.1, 108.7, 84.1, 55.8, 55.7, 50.3; MS (EI, 70 eV) m/z 277 (M+, 27.34), 262 (100); IR (neat): ν = 2943, 2910, 2836, 1682, 1606, 1516, 1362, 1254, 1140, 1109, 1011 cm−1; HRMS calcd for C15H19NO4 (M+): 277.1314. Found: 277.1306.
CH), 6.63 (s, 1 H, ArH), 6.60 (s, 1 H, ArH), 4.70 (d, J = 13.2 Hz, 1 H, CH), 4.25 (s, 2 H, NCH2Ar), 3.86 (s, 6 H, OCH3 × 2), 3.67 (s, 3 H, OCH3), 3.48 (s, 2 H, NCH2), 2.82 (t, J = 5.4 Hz, 2 H, CH2); 13C NMR (100 MHz, CDCl3) δ 169.8, 151.4, 147.63, 147.60, 125.8, 111.1, 108.7, 84.1, 55.8, 55.7, 50.3; MS (EI, 70 eV) m/z 277 (M+, 27.34), 262 (100); IR (neat): ν = 2943, 2910, 2836, 1682, 1606, 1516, 1362, 1254, 1140, 1109, 1011 cm−1; HRMS calcd for C15H19NO4 (M+): 277.1314. Found: 277.1306.
Synthesis of 2-benzyl-1-(3-hydroxy-3-methylbutynyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (5) (lwl-8-93).
To a flame-dried Schlenk tube were added CuBr (0.0073 g, 0.05 mmol), PPh3 (0.0145 g, 0.055 mmol), 4 Å molecular sieves (3.0021 g), and toluene (20 mL) sequentially under an Ar atmosphere. The resulting mixture was then stirred at rt for 30 min. PhCHO (1.4861 g, 14 mmol)/toluene (10 mL), 1 (2.7047 g, 14 mmol)/toluene (10 mL), and 2b (0.8412 g, 10 mmol)/toluene (20 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 80 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtered through a short column of silica gel eluted with ethyl acetate (50 mL). After evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3/1) to afford 5 (3.2511 g, 89%) as an oil: 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 6.8 Hz, 2 H, ArH), 7.33 (t, J = 7.2 Hz, 2 H, ArH), 7.27 (t, J = 6.4 Hz, 1 H, ArH), 6.66 (s, 1 H, ArH), 6.57 (s, 1 H, ArH), 4.47 (s, 1 H, NCH), 3.88 (d, J = 13.2 Hz, 1 H, one proton of NCH2Ar), 3.83 (s, 3 H, OCH3), 3.81 (s, 3 H, OCH3), 3.76 (d, J = 12.8 Hz, 1 H, one proton of NCH2Ar), 2.98–2.84 (m, 2 H, CH2), 2.77–2.63 (m, 2 H, CH2), 2.06 (brs, 1 H, OH), 1.54 (s, 6 H, CH3 × 2); 13C NMR (100 MHz, CDCl3) δ 148.0, 147.2, 138.2, 129.2, 128.2, 127.2, 127.1, 125.9, 111.2, 110.3, 91.3, 80.0, 65.2, 59.4, 55.9, 55.8, 53.2, 45.9, 31.73, 31.70, 28.5; MS (EI, 70 eV) m/z 366 (M+ + 1, 7.29), 365 (M+, 30.02), 91 (100); IR (neat): ν = 3425, 3028, 2977, 2931, 2831, 1612, 1517, 1463, 1454, 1360, 1334, 1255, 1223, 1168, 1127, 1017 cm−1; HRMS calcd for C23H27NO3 (M+): 365.1991. Found: 365.1988.
2-Benzyl-1-ethynyl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquino-line (6)12.
To a flame-dried Schlenk tube were added 5 (2.9241 g, 8.0 mmol), toluene (30 mL), NaH (0.0641 g, 60% in mineral oil, 1.6 mmol), and toluene (10 mL) sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 110 °C with stirring for 5 h as monitored by TLC. After cooling to room temperature, water (20 mL) and ethyl acetate (20 mL) were added. After the organic layer was separated, the aqueous layer was extracted with ethyl acetate (20 mL × 3). The combined organic layer was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 10/1) to afford 6 (2.0400 g, 83%) as an oil: 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.2 Hz, 2 H, ArH), 7.33 (t, J = 7.4 Hz, 2 H, ArH), 7.30–7.24 (m, 1 H, ArH), 6.66 (s, 1 H, ArH), 6.58 (s, 1 H, ArH), 4.48 (s, 1 H, NCH), 3.90–3.76 (m, 8 H, NCH2Ar and OCH3 × 2), 3.01–2.87 (m, 2 H, CH2), 2.84–2.74 (m, 1 H, one proton of CH2), 2.71–2.61 (m, 1 H, one proton of CH2), 2.47 (d, J = 1.6 Hz, 1 H,
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH); 13C NMR (100 MHz, CDCl3) δ 148.2, 147.3, 138.2, 129.2, 128.3, 127.1, 126.8, 125.9, 111.3, 110.1, 81.7, 74.5, 59.3, 55.9, 55.8, 53.1, 45.5, 28.5; MS (EI, 70 eV) m/z 308 (M+ + 1, 11.74), 307 (M+, 64.16), 91 (100); IR (neat): ν = 3280, 2914, 2831, 1612, 1519, 1464, 1454, 1360, 1256, 1226, 1130, 1018 cm−1; HRMS calcd for C20H21NO2 (M+): 307.1572. Found: 307.1566.
CH); 13C NMR (100 MHz, CDCl3) δ 148.2, 147.3, 138.2, 129.2, 128.3, 127.1, 126.8, 125.9, 111.3, 110.1, 81.7, 74.5, 59.3, 55.9, 55.8, 53.1, 45.5, 28.5; MS (EI, 70 eV) m/z 308 (M+ + 1, 11.74), 307 (M+, 64.16), 91 (100); IR (neat): ν = 3280, 2914, 2831, 1612, 1519, 1464, 1454, 1360, 1256, 1226, 1130, 1018 cm−1; HRMS calcd for C20H21NO2 (M+): 307.1572. Found: 307.1566.
Methyl 3-(2-benzyl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl)-2-propiolate (3)13.
To a flame-dried Schlenk tube were added 6 (1.4751 g, 4.8 mmol) and THF (20 mL). Then the solution was cooled to −78 °C and n-BuLi (2.1 mL, 2.5 M in hexane, 5.3 mmol) was added dropwise within 8 minutes. The resulting mixture was stirred for 2 h at −78 °C. Then ClCO2Me (0.5001 g, 5.3 mmol)/THF (5 mL) was added and the mixture was stirred at −78 °C for another 6 h. Sat. aq. NH4Cl (20 mL) was added. After the organic layer was separated, the aqueous layer was extracted with diethyl ether (20 mL × 3). The extract was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 10/1) to afford 3 (1.1801 g, 67%) as an oil: 1H NMR (400 MHz, CDCl3) δ 7.56–7.15 (m, 5 H, ArH), 6.60 (s, 2 H, ArH), 4.59 (s, 1 H, NCH), 4.01–3.61 (m, 11 H, OCH3 × 3 and CH2), 3.05–2.69 (m, 3 H, CH2 and one proton of CH2), 2.72–2.55 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 153.8, 148.4, 147.3, 137.5, 129.0, 128.2, 127.2, 126.2, 124.5, 111.3, 109.9, 85.9, 78.5, 59.3, 55.8, 55.7, 52.9, 52.5, 45.6, 28.2; MS (EI, 70 eV) m/z 366 (M+ + 1, 8.90), 365 (M+, 36.51), 91 (100); IR (neat): ν = 3000, 2831, 2218, 1711, 1611, 1516, 1453, 1434, 1360, 1223, 1125, 1098 cm−1. HRMS calcd for C22H23NO4 (M+): 365.1627. Found: 365.1636.
1,5,6,10b-Tetrahydro-8,9-dimethoxy-pyrrolo[2,1-a]isoquinolin-3(2H)-one (8)7.
To a Schlenk tube were added 3 (365.4 mg, 1 mmol), ethyl acetate (10 mL), methanol (10 mL), and Pd/C (160.0 mg, 0.15 mmol, 10% in mass) sequentially, which was purged three times with hydrogen. The suspension was stirred for 2 h at room temperature under a hydrogen pressure of 1 atm, and then was filtered through a short column of Celite eluted with methanol (20 mL). After evaporation, the crude product was then subjected to the next step without further purification and characterization.

To another Schlenk tube were added the crude product (prepared in the previous step) and toluene (10 mL) sequentially. The mixture was stirred for 2.5 h at 110 °C. After evaporation, the residue was purified by silica gel column chromatography (eluent: dichloromethane/methanol/ammonia solution = 40/1/0.1) to provide 8 (207.5 mg, 84%) as an oil: 1H NMR (400 MHz, CDCl3) δ 6.62 (s, 1 H, ArH), 6.57 (s, 1 H, ArH), 4.73 (t, J = 8.0 Hz, 1 H, NCH), 4.31 (ddd, J = 12.7, 5.9, 1.9 Hz, 1 H, one proton of NCH2), 3.88 (s, 3 H, OCH3), 3.87 (s, 3 H, OCH3), 3.02 (td, J = 12.1, 4.3 Hz, 1 H, one proton of NCH2), 2.94–2.82 (m, 1 H, one proton of CH2), 2.74–2.41 (m, 4 H, CH2 × 2), 1.91–1.76 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 173.0, 147.9, 147.7, 129.2, 125.4, 111.5, 107.5, 56.4, 55.9, 55.8, 36.9, 31.7, 27.9, 27.6; MS (EI, 70 eV) m/z 248 (M+ + 1, 10.45), 247 (M+, 66.62), 246 (100); IR (neat): ν = 3455, 2932, 2836, 1672, 1612, 1514, 1439, 1422, 1362, 1255, 1228, 1117, 1009 cm−1.
Synthesis of (±)-trolline7.
To a Schlenk tube were added 8 (49.6 mg, 0.2 mmol), CH2Cl2 (0.5 mL), and the solution was cooled to −20 °C. Then BBr3 (1.0 mL, 1.0 mmol, 1 M in CH2Cl2) was added dropwise, and the resulting mixture was stirred for 13 h at −20 °C. Methanol (5 mL) was added to quench the reaction, and the resulting mixture was evaporated. The residue was purified by silica gel column chromatography (eluent: dichloromethane/methanol = 20/1) to provide (±)-trolline (35.0 mg, 80%) as a solid: m.p. 240–242 °C (MeOH) (lit.7a m.p. 245–247 °C (MeOH)); 1H NMR (400 MHz, (CD3)2SO) δ 8.82 (s, 2 H, OH × 2), 6.51 (s, 1 H, ArH), 6.49 (s, 1 H, ArH), 4.58 (t, J = 7.8 Hz, 1 H, NCH), 4.03–3.88 (m, 1 H, one proton of NCH2), 2.97–2.84 (m, 1 H, one proton of NCH2), 2.65–2.52 (m, 3 H, CH2 and one proton of CH2), 2.47–2.34 (m, 1 H, one proton of CH2), 2.22 (dd, J = 16.4, 9.2 Hz, 1 H, CH), 1.67–1.52 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, (CD3)2SO) δ 172.1, 144.2, 144.0, 128.4, 123.7, 115.4, 111.7, 55.6, 36.6, 31.3, 27.4, 27.3; MS (EI, 70 eV) m/z 219 (M+, 61.4), 218 (100); IR (neat): ν = 3265, 1648, 1522, 1452, 1359, 1277, 1234, 1111 cm−1.
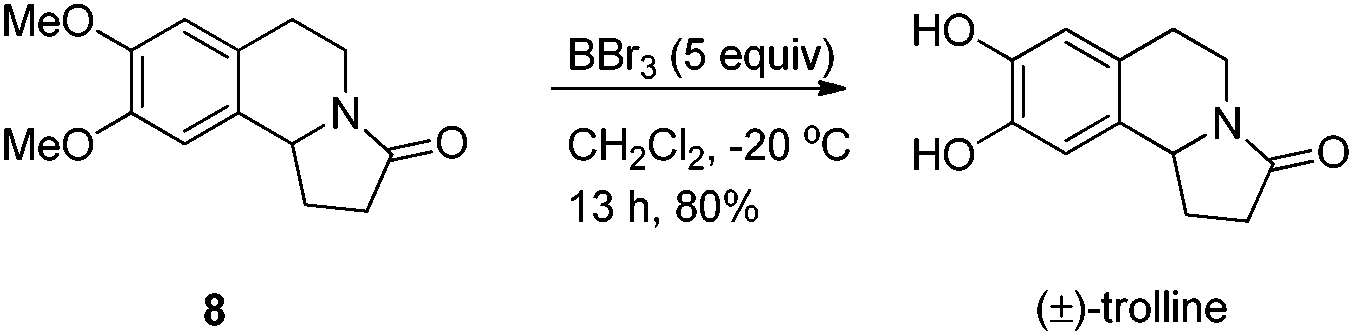
(S)-2-Benzyl-1-(3-hydroxy-3-methylbutynyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline ((S)-5).
To a flame-dried Schlenk tube were added CuI (0.0190 g, 0.1 mmol), (R,Sa)-N-pinap (0.1112 g, 0.2 mmol), 4 Å molecular sieves (3.0021 g), and toluene (20 mL) sequentially under an Ar atmosphere. The Schlenk tube was then stirred at rt for 30 min. PhCOOH (0.0615 g, 0.5 mmol), PhCHO (1.4858 g, 14 mmol)/toluene (10 mL), 1 (2.7055 g, 14 mmol)/toluene (10 mL), and 2b (0.8412 g, 10 mmol)/toluene (20 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 60 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtered through a short column of silica gel eluted with ethyl acetate (150 mL). After evaporation, the residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 3/1) to afford (S)-5 (3.2719 g, 90%) as an oil: 98% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 80/20, 0.3 mL min−1, λ = 214 nm, tR(major) = 14.5 min, tR(minor) = 11.8 min); [α]27D = +53.8 (c = 1.09, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.6 Hz, 2 H, ArH), 7.37–7.23 (m, 3 H, ArH), 6.66 (s, 1 H, ArH), 6.56 (s, 1 H, ArH), 4.47 (s, 1 H, NCH), 3.87 (d, J = 13.2 Hz, 1 H, one proton of NCH2Ar), 3.83 (s, 3 H, OCH3), 3.81 (s, 3 H, OCH3), 3.76 (d, J = 13.2 Hz, 1 H, one proton of NCH2Ar), 2.98–2.82 (m, 2 H, CH2), 2.77–2.60 (m, 2 H, CH2), 2.16 (s, 1 H, OH), 1.54 (s, 6 H, CH3 × 2); 13C NMR (100 MHz, CDCl3) δ 147.9, 147.1, 138.1, 129.2, 128.2, 127.13, 127.08, 125.9, 111.1, 110.2, 91.3, 79.9, 65.2, 59.4, 55.8, 55.7, 53.2, 45.8, 31.69, 31.66, 28.5; MS (EI, 70 eV) m/z 366 (M+ + 1, 14.45), 365 (M+, 62.99), 364 (100); IR (neat): ν = 3511, 2978, 2932, 2831, 1612, 1517, 1454, 1361, 1335, 1256, 1223, 1168, 1127, 1097 cm−1; HRMS calcd for C23H27NO3 (M+): 365.1991. Found: 365.1989.
(S)-2-Benzyl-1-ethynyl-6,7-dimethoxy-1,2,3,4-tetrahydroiso-quinoline ((S)-6).
To a flame-dried Schlenk tube were added (S)-5 (2.9251 g, 8.0 mmol), toluene (30 mL), NaH (0.0651 g, 60% in mineral oil, 1.6 mmol), and toluene (10 mL) sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 110 °C with stirring for 5 h as monitored by TLC. After cooling to room temperature, water (20 mL) and ethyl acetate (20 mL) were added. After the organic layer was separated, the aqueous layer was extracted with ethyl acetate (20 mL × 3). The extract was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 10/1) to afford (S)-6 (2.0191 g, 82%) as an oil: 97% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 95/5, 0.5 mL min−1, λ = 214 nm, tR(major) = 11.4 min, tR(minor) = 12.6 min); [α]27D = +81.5 (c = 1.06, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.2 Hz, 2 H, ArH), 7.37–7.20 (m, 3 H, ArH), 6.64 (s, 1 H, ArH), 6.57 (s, 1 H, ArH), 4.48 (s, 1 H, NCH), 3.93–3.71 (m, 8 H, NCH2Ar and OCH3 × 2), 3.01–2.85 (m, 2 H, CH2), 2.83–2.73 (m, 1 H, one proton of CH2), 2.71–2.59 (m, 1 H, one proton of CH2), 2.47 (s, 1 H,
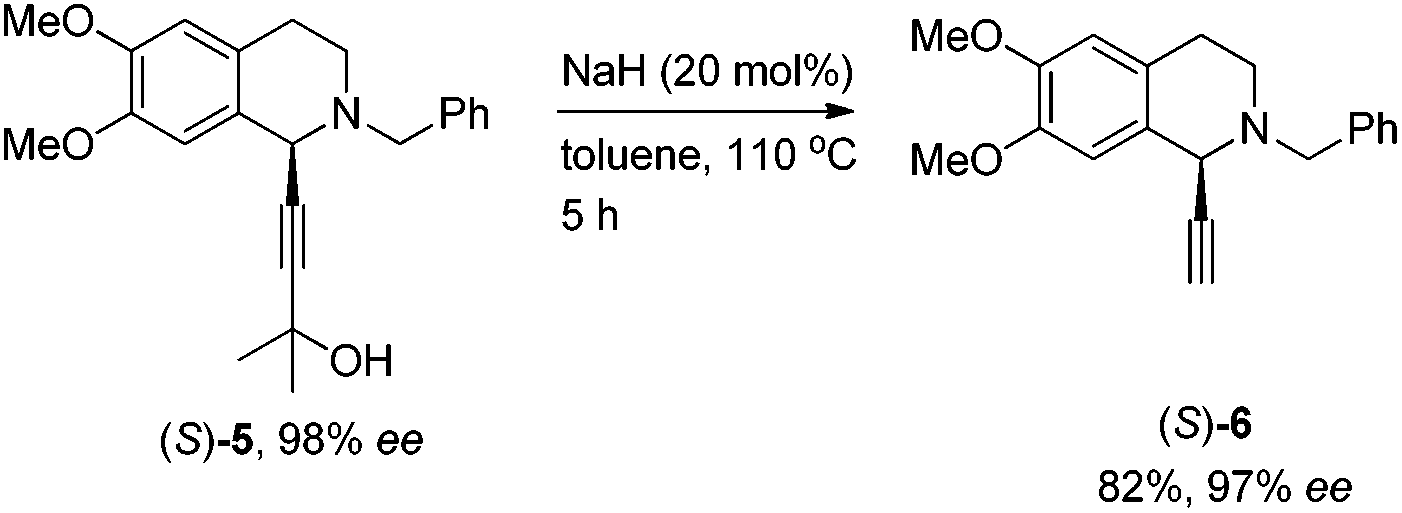 CH); 13C NMR (100 MHz, CDCl3) δ 148.0, 147.2, 138.0, 129.1, 128.2, 127.0, 126.7, 125.8, 111.2, 109.9, 81.5, 74.4, 59.2, 55.72, 55.66, 52.9, 45.4, 28.4; MS (EI, 70 eV) m/z 307 (M+, 19.30), 91 (100); IR (neat): ν = 3283, 3030, 2831, 1612, 1518, 1463, 1359, 1334, 1256, 1225, 1127, 1097, 1018 cm−1; HRMS calcd for C20H21NO2 (M+): 307.1572. Found: 307.1575.
CH); 13C NMR (100 MHz, CDCl3) δ 148.0, 147.2, 138.0, 129.1, 128.2, 127.0, 126.7, 125.8, 111.2, 109.9, 81.5, 74.4, 59.2, 55.72, 55.66, 52.9, 45.4, 28.4; MS (EI, 70 eV) m/z 307 (M+, 19.30), 91 (100); IR (neat): ν = 3283, 3030, 2831, 1612, 1518, 1463, 1359, 1334, 1256, 1225, 1127, 1097, 1018 cm−1; HRMS calcd for C20H21NO2 (M+): 307.1572. Found: 307.1575.
(S)-Methyl 3-(2-benzyl-6,7-dimethoxy-1,2,3,4-tetrahydroiso-quinolin-1-yl)-2-propiolate ((S)-3).
To a flame-dried Schlenk tube were added (S)-6 (1.2301 g, 4.0 mmol) and THF (15 mL). Then the solution was cooled to −78 °C and n-BuLi (1.8 mL, 2.5 M in hexane, 4.4 mmol) was added dropwise within 10 minutes. The resulting mixture was stirred for 2 h at −78 °C. Then ClCO2Me (417.2 mg, 4.4 mmol)/THF (5 mL) was added and the mixture was stirred at −78 °C for another 6 h. Sat. aq. NH4Cl (20 mL) was added. After the organic layer was separated, the aqueous layer was extracted with diethyl ether (20 mL × 3). The extract was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 10/1) to afford (S)-3 (1.1091 g, 76%) as an oil: 97% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 95/5, 0.7 mL min−1, λ = 214 nm, tR(major) = 19.7 min, tR(minor) = 22.1 min); [α]27D = +57.5 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.2 Hz, 2 H, ArH), 7.37–7.21 (m, 3 H, ArH), 6.60 (s, 1 H, ArH), 6.59 (s, 1 H, ArH), 4.59 (s, 1 H, NCH), 3.96–3.62 (m, 11 H, OCH3 × 3 and CH2), 3.04–2.78 (m, 3 H, CH2 and one proton of CH2), 2.72–2.56 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 153.8, 148.4, 147.3, 137.5, 129.0, 128.2, 127.2, 126.2, 124.5, 111.3, 109.9, 85.9, 78.5, 59.3, 55.8, 55.7, 52.9, 52.5, 45.6, 28.2; MS (EI, 70 eV) m/z 365 (M+, 34.07), 91 (100); IR (neat): ν = 2950, 2918, 2828, 2220, 1711, 1608, 1513, 1446, 1359, 1231, 1126, 1029 cm−1. HRMS calcd for C22H23NO4 ([M]+): 365.1627. Found: 365.1621.
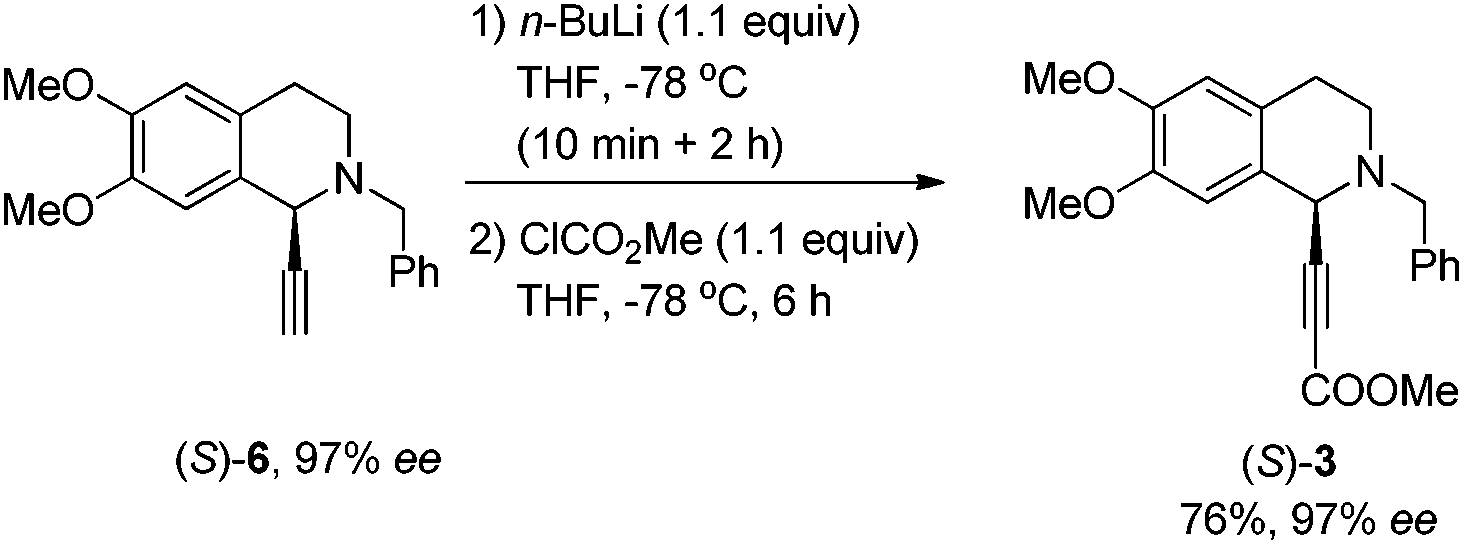
(S)-1,5,6,10b-Tetrahydro-8,9-dimethoxy-pyrrolo[2,1-a]isoquino-lin-3(2H)-one ((S)-8)8b.
To a Schlenk tube were added (S)-3 (367.5 mg, 1 mmol), ethyl acetate (15 mL), and Pd/C (159.9 mg, 0.15 mmol, 10% in mass) sequentially, which was purged three times with hydrogen. The suspension was stirred for 3 h at room temperature under a hydrogen pressure of 1 atm, and then filtered through a short column of Celite eluted with a mixed solvent (30 mL, methanol/ethyl acetate = 1/1 by volume). After evaporation, the crude product was then subjected to the next step without further purification and characterization.
To another Schlenk tube were added the crude product (prepared in the previous step) and toluene (10 mL) sequentially. The mixture was stirred for 5 h at rt. After evaporation, the residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 1/4 to dichloromethane/methanol = 100/1) to provide (S)-8 (216.0 mg, 87%) as an oil: 95% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 60/40, 0.9 mL min−1, λ = 214 nm, tR(major) = 21.4 min, tR(minor) = 14.7 min); [α]27D = −237.5 (c = 0.61, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.63 (s, 1 H, ArH), 6.58 (s, 1 H, ArH), 4.73 (t, J = 7.6 Hz, 1 H, NCH), 4.31 (dd, J = 12.6, 5.4 Hz, 1 H, one proton of NCH2), 3.88 (s, 3 H, OCH3), 3.87 (s, 3 H, OCH3), 3.02 (td, J = 12.2, 3.5 Hz, 1 H, one proton of NCH2), 2.95–2.81 (m, 1 H, one proton of CH2), 2.75–2.41 (m, 4 H, CH2 × 2), 1.91–1.76 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 173.0, 147.9, 147.7, 129.2, 125.3, 111.5, 107.4, 56.4, 55.9, 55.8, 36.9, 31.7, 27.9, 27.6; MS (EI, 70 eV) m/z 247 (M+, 66.04), 246 (100); IR (neat): ν = 3455, 2932, 2836, 1670, 1612, 1513, 1438, 1362, 1254, 1228, 1116, 1009 cm−1.
Synthesis of (S)-(−)-trolline8.
To a Schlenk tube were added (S)-8 (173.9 mg, 0.7 mmol) and CH2Cl2 (1.0 mL), and the solution was cooled to −40 °C. Then BBr3 (3.5 mL, 3.5 mmol, 1 M in CH2Cl2) was added dropwise within 10 minutes, and the resulting mixture was stirred for 12 h at −40 °C. Methanol (10 mL) was added to quench the reaction, and the resulting mixture was evaporated. The residue was purified by silica gel column chromatography (eluent: dichloromethane/methanol = 20/1) to provide (S)-(−)-trolline (127.9 mg, 83%) as a solid: 95% ee (HPLC conditions: Chiralcel AD-H column, hexane/i-PrOH = 80/20, 0.7 mL min−1, λ = 214 nm, tR(major) = 26.7 min, tR(minor) = 31.3 min); [α]28D = −195.3 (c = 0.81, MeOH) (lit.4 [α]20D = −197 (c = 0.8, MeOH)); m.p. 240–242 °C (MeOH) (lit.14 m.p. 257–259 °C (acetone)); 1H NMR (400 MHz, (CD3)2SO) δ 8.83 (s, 2 H, OH × 2), 6.51 (s, 1 H, ArH), 6.50 (s, 1 H, ArH), 4.58 (t, J = 7.6 Hz, 1 H, NCH), 3.97 (d, J = 11.2 Hz, 1 H, one proton of CH2), 2.98–2.85 (m, 1 H, one proton of CH2), 2.65–2.52 (m, 3 H, CH2 and one proton of CH2), 2.46–2.33 (m, 1 H, one proton of CH2), 2.22 (dd, J = 16.0, 9.2 Hz, 1 H, one proton of CH2), 1.66–1.50 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, (CD3)2SO) δ 172.0, 144.2, 144.0, 128.4, 123.7, 115.4, 111.7, 55.6, 36.6, 31.3, 27.4, 27.3; MS (EI, 70 eV) m/z 219 (M+, 59.68), 218 (100); IR (neat): ν = 3256, 1646, 1522, 1452, 1359, 1277, 1235, 1111 cm−1.
(R)-2-Benzyl-1-(3-hydroxy-3-methylbutynyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline ((R)-5).
To a flame-dried Schlenk tube were added CuI (0.0190 g, 0.1 mmol), (R,Ra)-N-pinap (0.1118 g, 0.2 mmol), 4 Å molecular sieves (3.0020 g), and toluene (20 mL) sequentially under an Ar atmosphere. The Schlenk tube was then stirred at rt for 30 min. PhCOOH (0.0602 g, 0.5 mmol), PhCHO (1.4862 g, 14 mmol)/toluene (10 mL), 1 (2.7055 g, 14 mmol)/toluene (10 mL), and 2b (0.8412 g, 10 mmol)/toluene (20 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 60 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtered through a short column of silica gel eluted with ethyl acetate (150 mL). After evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3/1) to afford (R)-5 (3.4315 g, 94%) as an oil: 98% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 80/20, 0.3 mL min−1, λ = 214 nm, tR(major) = 7.2 min, tR(minor) = 8.1 min); [α]30D = −54.5 (c = 0.98, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.6 Hz, 2 H, ArH), 7.32 (d, J = 7.2 Hz, 2 H, ArH), 7.28–7.22 (m, 1 H, ArH), 6.65 (s, 1 H, ArH), 6.55 (s, 1 H, ArH), 4.46 (s, 1 H, NCH), 3.86 (d, J = 13.2 Hz, 1 H, one proton of NCH2Ar), 3.81 (s, 3 H, OCH3), 3.79 (s, 3 H, OCH3), 3.75 (d, J = 13.2 Hz, 1 H, one proton of NCH2Ar), 2.97–2.82 (m, 2 H, CH2), 2.76–2.60 (m, 2 H, CH2), 2.35 (s, 1 H, OH), 1.53 (s, 6 H, CH3 × 2); 13C NMR (100 MHz, CDCl3) δ 147.9, 147.1, 138.0, 129.1, 128.1, 127.1, 127.0, 125.8, 111.1, 110.2, 91.3, 79.8, 65.0, 59.3, 55.8, 55.7, 53.1, 45.7, 31.64, 31.60, 28.4; MS (EI, 70 eV) m/z 365 (M+, 72.86), 364 (100); IR (neat): ν = 3509, 2977, 2932, 2831, 1612, 1517, 1453, 1360, 1334, 1256, 1223, 1168, 1127, 1017 cm−1; HRMS calcd for C23H27NO3 (M+): 365.1991. Found: 365.1989.
(R)-2-Benzyl-1-ethynyl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline ((R)-6).
To a flame-dried Schlenk tube were added (R)-5 (2.9201 g, 8.0 mmol), toluene (30 mL), and NaH (0.0645 g, 60% in mineral oil, 1.6 mmol), and toluene (10 mL) sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 110 °C with stirring for 5 h as monitored by TLC. After cooling to room temperature, water (20 mL) and ethyl acetate (20 mL) were added. After the organic layer was separated, the aqueous layer was extracted with ethyl acetate (20 mL × 3). The extract was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 10/1) to afford (R)-6 (2.0711 g, 84%) as an oil: 98% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 95/5, 0.5 mL min−1, λ = 214 nm, tR(major) = 12.2 min, tR(minor) = 11.2 min); [α]30D = −80.5 (c = 0.99, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.6 Hz, 2 H, ArH), 7.32 (t, J = 7.6 Hz, 2 H, ArH), 7.29–7.20 (m, 1 H, ArH), 6.65 (s, 1 H, ArH), 6.57 (s, 1 H, ArH), 4.48 (s, 1 H, NCH), 3.89–3.75 (m, 8 H, NCH2Ar and OCH3 × 2), 3.00–2.86 (m, 2 H, CH2), 2.83–2.73 (m, 1 H, one proton of CH2), 2.70–2.59 (m, 1 H, one proton of CH2), 2.46 (d, J = 1.2 Hz, 1 H,
 CH); 13C NMR (100 MHz, CDCl3) δ 148.1, 147.2, 138.0, 129.1, 128.2, 127.0, 126.7, 125.8, 111.2, 110.0, 81.5, 74.4, 59.2, 55.74, 55.68, 52.9, 45.4, 28.4; MS (EI, 70 eV) m/z 308 (M+ + 1, 16.29), 307 (M+, 79.46), 306 (100); IR (neat): ν = 3279, 3028, 2915, 2831, 1612, 1518, 1463, 1256, 1224, 1125, 1095, 1016 cm−1; HRMS calcd for C20H21NO2 ([M]+): 307.1572. Found: 307.1561.
CH); 13C NMR (100 MHz, CDCl3) δ 148.1, 147.2, 138.0, 129.1, 128.2, 127.0, 126.7, 125.8, 111.2, 110.0, 81.5, 74.4, 59.2, 55.74, 55.68, 52.9, 45.4, 28.4; MS (EI, 70 eV) m/z 308 (M+ + 1, 16.29), 307 (M+, 79.46), 306 (100); IR (neat): ν = 3279, 3028, 2915, 2831, 1612, 1518, 1463, 1256, 1224, 1125, 1095, 1016 cm−1; HRMS calcd for C20H21NO2 ([M]+): 307.1572. Found: 307.1561.
(R)-Methyl 3-(2-benzyl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl)-2-propiolate ((R)-3).
To a flame-dried Schlenk tube were added (R)-6 (1.2301 g, 4.0 mmol) and THF (15 mL). Then the solution was cooled to −78 °C and n-BuLi (1.8 mL, 2.5 M in hexane, 4.4 mmol) was added dropwise within 20 minutes. The resulting mixture was stirred for 2 h at −78 °C. Then ClCO2Me (0.4183 g, 4.4 mmol) was added and the mixture was stirred at −78 °C for another 6 h. An aqueous NH4Cl solution (20 mL) was added. After the organic layer was separated, the aqueous layer was extracted with diethyl ether (20 mL × 3). The extract was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 10/1) to afford (R)-3 (1.0221 g, 70%) as an oil: 98% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 95/5, 0.7 mL min−1, λ = 214 nm, tR(major) = 19.7 min, tR(minor) = 17.8 min); [α]30D = −56.4 (c = 1.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.6 Hz, 2 H, ArH), 7.37–7.23 (m, 3 H, ArH), 6.60 (s, 1 H, ArH), 6.59 (s, 1 H, ArH), 4.59 (s, 1 H, NCH), 3.89–3.71 (m, 11 H, OCH3 × 3 and CH2), 3.02–2.81 (m, 3 H, CH2 and one proton of CH2), 2.72–2.57 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 153.8, 148.4, 147.3, 137.5, 129.0, 128.3, 127.2, 126.2, 124.5, 111.3, 109.9, 85.9, 78.5, 59.3, 55.8, 55.7, 52.9, 52.5, 45.6, 28.2; MS (EI, 70 eV) m/z 366 (M+ + 1, 22.63), 365 (M+, 89.71), 364 (100); IR (neat): ν = 2953, 2935, 2831, 2218, 1711, 1611, 1516, 1453, 1360, 1223, 1125, 1098 cm−1. HRMS calcd for C22H23NO4 (M+): 365.1627. Found: 365.1632.
(R)-1,5,6,10b-Tetrahydro-8,9-dimethoxy-pyrrolo[2,1-a]isoquinolin-3(2H)-one ((R)-8)8b.
To a Schlenk tube were added (R)-3 (367.9 mg, 1 mmol), ethyl acetate (15 mL), and Pd/C (160.0 mg, 0.15 mmol, 10% in mass) sequentially. The reaction vessel was purged three times with hydrogen. The suspension was then stirred for 3 h at room temperature under a hydrogen pressure of 1 atm, and then filtered through a short column of Celite eluted with a mixed solvent (30 mL, methanol/ethyl acetate = 1/1 by volume). After evaporation, the crude product was then subjected to the next step without further purification and characterization.
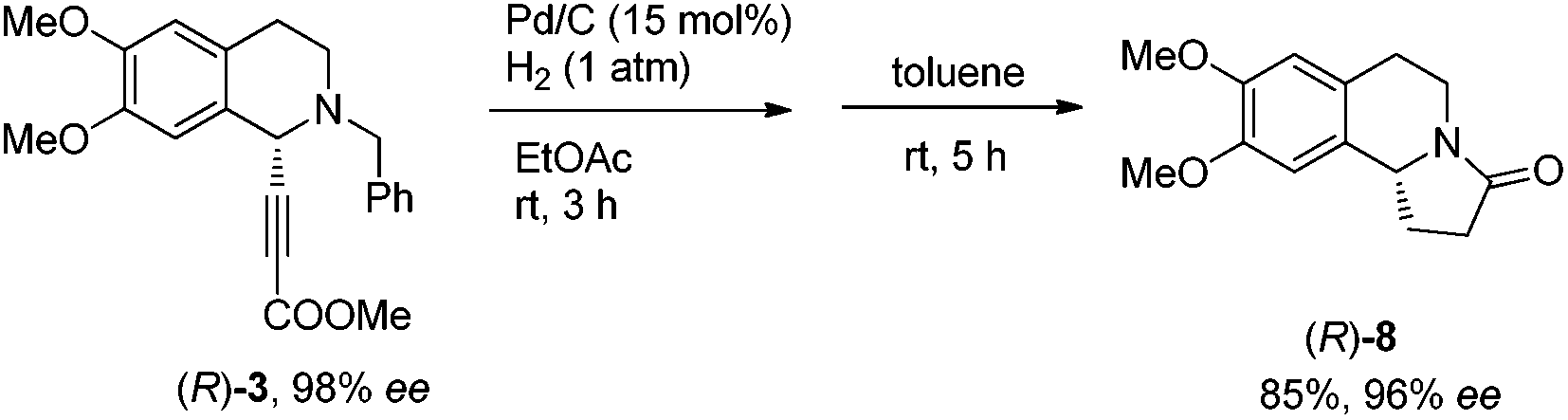
To another Schlenk tube were added the crude product (prepared in the previous step) and toluene (10 mL) sequentially. The mixture was stirred for 5 h at rt. After evaporation, the residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 1/4 to dichloromethane/methanol = 100/1) to provide (R)-8 (211.5 mg, 85%) as an oil: 96% ee (HPLC conditions: Chiralcel AS-H column, hexane/i-PrOH = 60/40, 0.9 mL min−1, λ = 214 nm, tR(major) = 14.7 min, tR(minor) = 22.4 min); [α]28D = +240.7 (c = 0.64, CHCl3) (lit.15 [α]20D = +216 (c = 0.6, CHCl3)); 1H NMR (400 MHz, CDCl3) δ 6.63 (s, 1 H, ArH), 6.58 (s, 1 H, ArH), 4.73 (t, J = 7.8 Hz, 1 H, NCH), 4.31 (dd, J = 13.2, 6.0 Hz, 1 H, one proton of CH2), 3.88 (s, 3 H, OCH3), 3.87 (s, 3 H, OCH3), 3.02 (td, J = 12.0, 4.0 Hz, 1 H, one proton of CH2), 2.95–2.83 (m, 1 H, one proton of CH2), 2.74–2.40 (m, 4 H, CH2 × 2), 1.90–1.76 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 173.0, 147.9, 147.7, 129.2, 125.4, 111.5, 107.4, 56.4, 55.9, 55.8, 36.9, 31.7, 27.9, 27.6; MS (EI, 70 eV) m/z 248 (M+ + 1, 11.35), 247 (M+, 62.56), 246 (100); IR (neat): ν = 3437, 2937, 2837, 1666, 1608, 1513, 1455, 1362, 1255, 1228, 1116, 1008 cm−1.
Synthesis of (R)-(+)-oleracein E8b.
To a Schlenk tube were added (R)-8 (171.1 mg, 0.7 mmol) and CH2Cl2 (1.0 mL), and the solution was cooled to −40 °C. Then BBr3 (3.5 mL, 3.5 mmol, 1 M in CH2Cl2) was added dropwise within 10 minutes, and the resulting mixture was stirred for 12 h at −40 °C. Methanol (10 mL) was added to quench the reaction, and the resulting mixture was evaporated. The residue was purified by silica gel column chromatography (eluent: dichloromethane/methanol = 20/1) to provide (R)-(+)-oleracein E (127.9 mg, 84%) as a solid: 96% ee (HPLC conditions: Chiralcel AD-H column, hexane/i-PrOH = 80/20, 1.0 mL min−1, λ = 214 nm, tR(major) = 18.8 min, tR(minor) = 16.0 min); [α]27D = +194.0 (c = 0.74, MeOH) (lit.8a [α]20D = +187 (c = 0.44, MeOH)); m.p. 241–243 °C (MeOH) (lit.5 m.p. 238–240 °C (MeOH)); 1H NMR (400 MHz, (CD3)2SO) δ 8.83 (s, 2 H, OH × 2), 6.51 (s, 1 H, ArH), 6.50 (s, 1 H, ArH), 4.58 (t, J = 7.8 Hz, 1 H, NCH), 4.02–3.91 (m, 1 H, one proton of CH2), 2.97–2.85 (m, 1 H, one proton of CH2), 2.65–2.52 (m, 3 H, CH2 and one proton of CH2), 2.47–2.35 (m, 1 H, one proton of CH2), 2.22 (dd, J = 16.0, 8.8 Hz, 1 H, one proton of CH2), 1.66–1.52 (m, 1 H, one proton of CH2); 13C NMR (100 MHz, (CD3)2SO) δ 172.0, 144.2, 144.0, 128.4, 123.7, 115.4, 111.7, 55.6, 36.6, 31.3, 27.4, 27.3; MS (EI, 70 eV) m/z 219 (M+, 61.23), 218 (100); IR (neat): ν = 3243, 1645, 1522, 1452, 1359, 1303, 1277, 1234, 1110 cm−1.
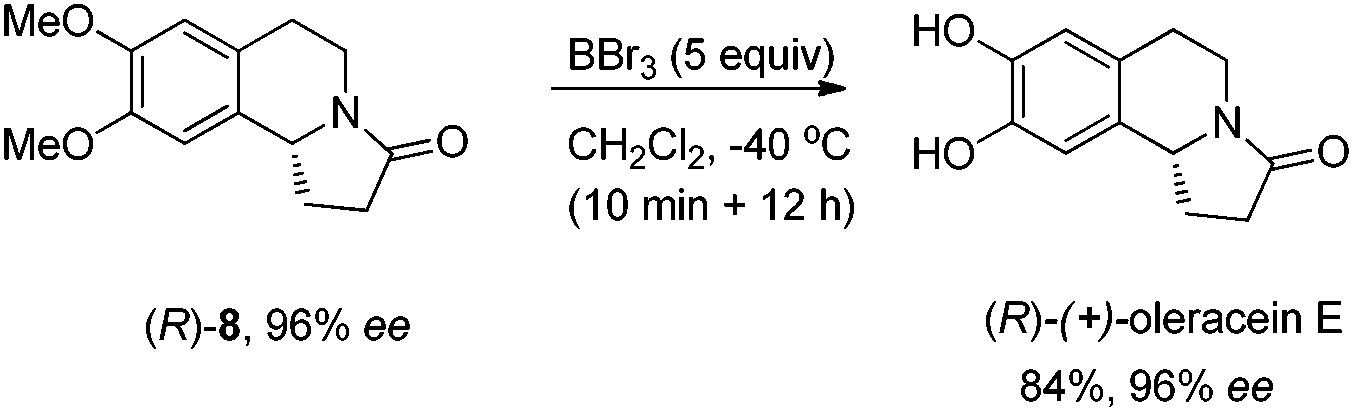
Acknowledgements
Financial support from the National Natural Science Foundation of China (21232006) and the National Basic Research Program (2015CB856600) is greatly appreciated. We thank Mr Yulin Han in this group for reproducing the transformation of (R)-3 to (R)-8 presented in Scheme 9.Notes and references
- (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (b) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444 RSC.
- (a) A. J. Aladesanmi, C. J. Kelly and J. D. Leary, J. Nat. Prod., 1983, 46, 127 CrossRef CAS; (b) E. Tojo, M. A. Onur, A. J. Freyer and M. Shamma, J. Nat. Prod., 1990, 53, 634 CrossRef CAS; (c) A. J. Freyer, M. H. Abu Zarga and S. Firdous, J. Nat. Prod., 1987, 50, 684 CrossRef CAS.
- E. Tojo, J. Nat. Prod., 1989, 52, 909 CrossRef CAS.
- R. Wang, X. Yang, C. Ma, S. Cai, J. Li and Y. Shoyama, Heterocycles, 2004, 63, 1443 CrossRef CAS.
- L. Xiang, D. Xing, W. Wang, R. Wang, Y. Ding and L. Du, Phytochemistry, 2005, 66, 2595 CrossRef CAS PubMed.
- Z. Yang, C. Liu, L. Xiang and Y. Zheng, Phytother. Res., 2009, 23, 1032 CrossRef CAS PubMed.
- For total synthesis of (±)-trolline, see: (a) Q. Q. He, C. M. Liu and K. Li, Chin. Chem. Lett., 2007, 18, 651 CrossRef CAS; (b) E. Shirakawa, N. Uchiyama and T. Hayashi, J. Org. Chem., 2011, 76, 25 CrossRef CAS PubMed; (c) J. Selvakumar, R. S. Rao, V. Srinivasapriyan, S. Marutheeswaran and C. R. Ramanathan, Eur. J. Org. Chem., 2015, 2175 CrossRef CAS.
- For asymmetric synthesis of (S)-(−)-trolline and (R)-(+)-oleracein E, see: (a) N. Kawai, M. Matsuda and J. Uenishi, Tetrahedron, 2011, 67, 8648 CrossRef CAS; (b) T. Kanemitsu, Y. Yamashita, K. Nagata and T. Itoh, Heterocycles, 2007, 74, 199 CrossRef CAS.
- (a) W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu, W. Yuan, J. Zhang, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 277 CrossRef CAS PubMed; (b) W. Lin and S. Ma, Org. Chem. Front., 2014, 1, 338 RSC.
- For some of the related studies, see: D. Das, A. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 3765 CrossRef CAS PubMed.
- For representative review or account on the redox-A3 reaction, see: (a) D. Seidel, Org. Chem. Front., 2014, 1, 426 RSC; (b) D. Seidel, Acc. Chem. Res., 2015, 48, 317 CrossRef CAS PubMed.
- S. J. Havens and P. M. Hergenrother, J. Org. Chem., 1985, 50, 1763 CrossRef CAS.
- D. A. Rooke and E. M. Ferreira, Angew. Chem., Int. Ed., 2012, 51, 3225 CrossRef CAS PubMed.
- Y. Xiang, Y. Li, J. Zhang, P. Li and Y. Yao, Yaoxue Xuebao, 2007, 42, 618 CAS.
- E. Mons, M. J. Wanner, S. Ingemann, J. H. van Maarseveen and H. Hiemstra, J. Org. Chem., 2014, 79, 7380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and HPLC spectra of all the products. See DOI: 10.1039/c7qo00062f |
| This journal is © the Partner Organisations 2017 |