
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic blood plasma separation for medical diagnostics: is it worth it?
W. S.
Mielczarek
ab,
E. A.
Obaje
b,
T. T.
Bachmann
b and
M.
Kersaudy-Kerhoas
*ab
aInstitute of Biological Chemistry, Biophysics and Bioengineering, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: m.kersaudy-kerhoas@hw.ac.uk
bDivision of Infection and Pathway Medicine, College of Medicine and Veterinary Medicine, The University of Edinburgh, Edinburgh EH16 4SB, UK
First published on 1st August 2016
Abstract
Circulating biomarkers are on the verge of becoming powerful diagnostic tools for various human diseases. However, the complex sample composition makes it difficult to detect biomarkers directly from blood at the bench or at the point-of-care. Blood cells are often a source of variability of the biomarker signal. While the interference of hemoglobin is a long known source of variability, the release of nucleic acids and other cellular components from hemocytes is a new concern for measurement and detection of circulating extracellular markers. Research into miniaturised blood plasma separation has been thriving in the last 10 years (2006–2016). Most point-of-care systems need microscale blood plasma separation, but developed solutions differ in complexity and sample volume range. But could blood plasma separation be avoided completely? This focused review weights the advantages and limits of miniaturised blood plasma separation and highlights the most interesting advances in direct capture as well as smart blood plasma separation.
1 Introduction
Human plasma is the most important and one of the most convenient sources of circulating biomarkers. Studies on plasma proteome, transcriptome and metabolome have rapidly increased the spectrum of diagnostic targets for a wide range of diseases from cancer1 to Alzheimer's2 to sepsis.3 Likewise, antibodies as well as foreign nucleic acids and antigens present in plasma, allow for the diagnosis of serious infectious diseases such as those caused by Ebola4 or Zika5 viruses during recent outbreaks.The quality of biomarkers often depends not only on biological factors such as physical condition and age of the patient, but also on technical factors such as the lack of standardisation of sample collection and preparation.6–8 Amongst other technical factors, in vitro hemolysis is a well-known source of biomarker variability. Blood cells such as leukocytes, or white blood cells, can also be a source of contamination in relation to biomarkers such as cell-free DNA targets. Therefore, plasma separation from hemocytes is very desirable in most cases. In the lab-on-a-chip community, a growing number of groups are tackling the challenge of micro-scale blood plasma separation (BPS), with often good results in yield or purity, but rarely achieving both at the same time. Recent lab-on-chip (LOC) developments, which demonstrate some level of sample preparation and/or detection of circulating blood biomarkers, still omit the blood plasma separation part. In a large number of cases, the operator of the LOC relies on a bench-top centrifuge to produce plasma as the input sample even if the required volume is in the range of a few microlitres. Therefore, continuing improvement of microscale blood plasma separation seems needed to achieve truly integrated and point-of-care (POC) devices. However, given the complexity of microscale plasma separation, could this step be avoided altogether? The improvements in direct capture now allow some circulating biomarkers to be detected in the blood matrix, allowing “sample preparation free” solutions. Is microfluidic blood plasma separation still worth it? Could it really be integrated on-chip or could it be achieved with a centrifuge next to a point-of-care testing (POCT) device?
A review we published in 2013 produced a detailed map of the miniaturised blood plasma separation state-of-the-art, listing and describing analytes of interest, characterisation and the challenges of blood plasma separation at the microscale.9 Tripathi et al. recently reviewed the design rules of passive blood plasma separation techniques.10 Here we provide a balanced overview of miniaturised blood plasma separation by considering the arguments for omitting integrated BPS in POC devices as well as highlighting the most interesting advances in the field between 2013 and 2016.
2 Avoiding blood plasma separation: rationale and solutions
The main rationale for avoiding BPS arises from the shortcomings of currently available BPS technologies.10,11 These shortcomings are (i) inconsistencies with diluting blood samples; as observed in the literature, majority of the POC systems require dilution of whole blood which introduces inaccuracies into the measurements and reduced concentration of target analytes, subsequently leading to reduced assay sensitivity. (ii) Complex designs that are cost-ineffective to integrate into microfluidic platforms; these tend to be a feature of active BPS techniques which generally involve the use of external force fields for separation. (iii) Time consuming workflow systems with low extraction rates; these tend to be associated with passive BPS systems that rely solely on the properties of blood flow at the microscale, leading to potential low throughput and slow flow rates.To alleviate the aforementioned issues, recent technological advances have led to the development of simplified microfluidic systems that facilitate biomarker detection directly from whole blood samples, thereby bypassing the need for on/off-chip BPS and its inherent complexities. Examples of these novel POC systems can be broadly categorised according to the factors that have made direct detection of analytes from whole blood possible.
Improvements in analyte characterisation and analyte-capture technology
Exhaustive research into understanding and characterising analyte binding efficiencies has facilitated the development of novel immunoassay capture strategies directly from whole blood.12,13 For example, extensive research into cardiac markers (analytes) like troponin and N-terminal pro b-type natriuretic peptide (NT-proBNP) have led to the development of commercially available POC assays like the Radiometer AQT90 FLEX analyser, a 20 minute BPS-free sandwich immunoassay test directly from 2 mL of blood for effective cardiovascular diagnosis and treatment.14 Additionally, better characterisation of genomic marker sequences has led to optimised molecular diagnostic assays. As an example, the mitochondrial cytochrome oxidase subunit 1 gene is targeted in an isothermal amplification assay (loop-mediated isothermal amplification (LAMP)) for malarial DNA from a sample of heat-treated host's blood without nucleic acid purification in about 30 minutes (∼1 parasite per μL blood). This assay is capable of distinguishing P. falciparum from P. vivax15 confirming the obvious benefits of improved analyte characterisation by increased knowledge of target sequences.Improvements in signal transduction and amplification mechanisms
Coupling the advances in analyte capture technology with innovative signal transduction and amplification strategies has enabled the development of new POCTs bypassing BPS. For example, an integrated, multiplexed digital-analogue microfluidic immunoassay platform based on the principles of mechanically induced trapping of molecular interactions (MITOMI),16 used for enhanced monitoring of binding affinities of molecular interactions like protein-antibody has recently been validated for rapid, specific and sensitive (100 pM) detection of anti-Ebola IgG in ultralow volume of whole blood samples (5 μL). The dynamic range of the immunoassay was significantly enhanced by combining digital MITOMI (i.e. enzyme conjugated) with analogue MITOMI (fluorophore-labelled) readout systems. Subsequently a low-cost portable prototype system was capable of sensitively distinguishing between three common Ebola strains in a multiplexed assay format.17Novel microfluidic composite material
Advances in the development of composite materials for microfluidic devices have led to improved platforms for immobilising capture probes. An affordable enzyme-linked immunosorbent assay (ELISA) test for the prostate-specific antigen (PSA) tumour marker normally performed on standard microtiter plates (MTP), was ported successfully onto a POC device utilising a melt-extruded fluoropolymer microcapillary film (MCF) for sensitive, direct and affordable whole blood PSA screening in low resource settings. The new PSA MCF ELISA was shown to be 20 times faster than the standard MTP ELISA, whilst maintaining similar assay performance with respect to precision and limit of detection (LoD).18,19Alternative strategies
The developments of novel nanoparticle-based bioassays are also lending themselves to providing suitable BPS-free POC solutions that can overcome the matrix effects of whole blood coupled with inexpensive readout systems based on common consumer electronics. For example, a POCT was recently constructed to sensitively detect thrombin activity at 18 NIH units per mL in 12 μL of blood (by monitoring hydrolase activity), using an integration of immobilised semiconductor quantum dots (QD) and Förster resonance energy transfer (FRET) with a smartphone imaging readout on a paper-in-PDMS chip. The system was further adapted for sensitive streptavidin detection (200 nM–10 μM) from 50% whole blood, via a competitive binding assay highlighting its potential for detecting multiple protein analyte classes while using common consumer electronics as a viable POC diagnostic platform.20Some of the example assays mentioned above still need to be optimised before being adapted into commercially available POCTs. However, it is becoming more evident that rapid technological advances are leading to increasing number of sensitive, smart and simplified POCT examples bypassing BPS, hence creating a rationale for its exclusion.
3 Blood plasma separation: when and why do you need it?
With both established and novel biomarkers, reproducibility and accuracy of the analytical methods are paramount. However, the quality of the analytical methods does not always guarantee the diagnostic usefulness of the test. An important yet sometimes overlooked issue relates to variabilities in sample collection, storage and processing. Indeed, preanalytical variables are recognised as the predominant source of mistakes in laboratory medicine, accounting for up to 70% of all laboratory errors.21 Therefore, the need for improving preanalytical procedures has been highlighted in a number of publications reviewing diagnostic methods.22 As described in the previous section, diagnostic tests are being successfully designed for the direct detection of biomarkers in blood, removing the need for integrating BPS in POCTs. However, the presence of blood cells in the sample disrupts the detection of a range of biomarkers in a number of ways. We review some of these effects.Influence of hemolysis in blood samples
The breakdown of erythrocyte (red blood cells) membranes causing the release of hemoglobin in blood in- or ex vivo is a leading problem during the preanalytical phase. in vitro hemolysis in laboratory samples accounts for up to 40–70% of sample rejections in laboratories, and has a direct financial impact.23,24 The variability of microRNA levels due to hemolysis has been well documented.25 Not all, but a large fraction of microRNAs identified so far are affected by sample handling.26 As illustrated in Fig. 1, Kirschner et al. have shown that the levels of miRNA are affected even by low hemolysis, typically undetectable by eye, and thus difficult to systematically exclude through a simple visual inspection.25 Proteins are also affected by hemolysis. An important example regards the use of cardiac troponins I (cTnI) and T (cTnT) to diagnose acute myocardial infarction and other cardiac diseases. cTnI and cTnT have been used in POC tests directly on blood samples as detailed in the previous sections and have been reported to be unaffected by moderate hemolysis.27 However, a study by Bais using a different cTn assay has shown that hemolysis index of around 150 (hemoglobin concentration of 1.9 g L−1) caused a 20% change in cTn, above the critical level, suggestive of an acute myocardial infarction.28 Although these examples relate to extreme physiological conditions rather than common cases, they highlight the advantage of rapidly obtaining hemolysis-free samples.29,30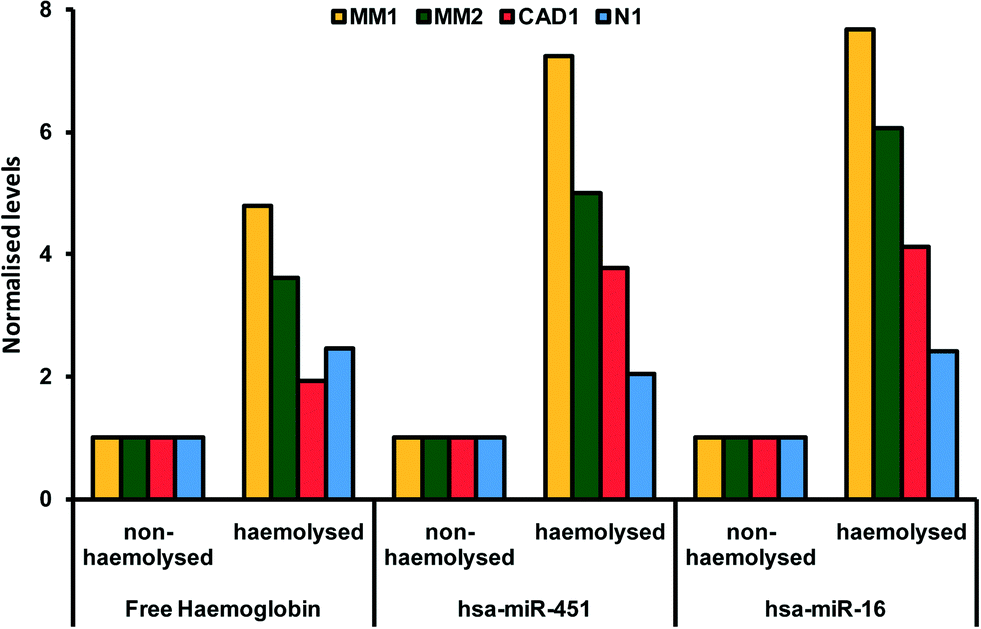 | ||
| Fig. 1 Paired hemolysed and non-hemolysed samples from four patients (coded MM1, MM2, CAD1, N1) showing different levels of two microRNAs (hsa-miR-451 and hsa-miR-16) commonly used as reference genes in microRNA assays. Reproduced from ref. 25 under Creative Commons Attribution (CC BY) license. | ||
The third example of the influence of hemolysis on circulating biomarkers relates to metabolites. Hemolysis can falsely decrease or increase values of metabolites, like potassium, lactate dehydrogenase, and aspartate aminotransferase.31 In particular, potassium concentration in RBCs is over 20 times that of plasma, making it notoriously sensitive to hemolysis.31 It was shown that over a third of hypokalemic cases (potassium deficiency) could be missed due to hemolysis when using whole blood to estimate potassium level.29 The elevation of potassium due to hemolysis has been reported to be more important in capillary blood samples compared to venous blood samples, which is of special interest to POCT applications.32 It was also shown that temperature during transport might lead to misdiagnosis and “pseudo hyperkalemia”, or so-called “seasonal pseudohyperkalemia”.33 To alleviate this issue, a study showed that the introduction of centrifuges in UK GP surgeries led to a reduction of these cases, thereby proving the benefits of rapid, point-of-care plasma extraction.34 While temperature control during sample transport might be possible in some cases, the universal removal of blood cells prior to transport is an attractive idea. We have discussed the impact of hemolysis on various important and common diagnostic tests. However, it is important to note that not all analytes are sensitive to hemolysis and a number of them can withstand delayed plasma separation for several days without changing significantly (less than 4% per day).35
Influence of leukolysis
Leukocytes lysis, or leukolysis, also has significant effects on circulating biomarkers, specifically molecular markers, in situations where the released background genomic DNA acts as noise on the molecular marker signal. This situation is specifically encountered in non-invasive, nucleic-acid-based cancer and prenatal diagnostic. A low concentration of cell-free DNA (cfDNA), around 10–50 ng ml−1, is normally detectable in plasma from healthy subjects prepared by standard centrifugation (1600g), representing hundreds or thousands of genome copies per ml. In the case of prenatal diagnostics, fetal DNA is thought to make up typically 10–15% of the total circulating DNA, depending strongly on gestation age and perinatal health.36,37 Fluctuations in the detectable fetal nucleic acid fraction can be attributed to the release of maternal DNA during maternal cell lysis. Extracting plasma at the site of blood draw has been shown to efficiently eliminate the contamination from maternal cells.38 Similarly, cancer tumours lead to the presence of free circulating tumour DNA (ctDNA) which can be used for early cancer diagnostic and treatment monitoring. However, the abundance of genomic DNA dilutes the tumour derived ctDNA and hinders robust detection of mutations.39 In both of these applications, pre-analytical factors such as the rapid removal or the stabilisation of blood cells have been shown to improve the reliability of the detection.40Microfluidics, an attractive technology to standardise the preanalytical phase
Physical cell preservation solutions do not alleviate sample preparation and introduce an additional cost on top of the total sample preparation cost (individual commercial cell stabilisation tubes generally cost above $6). While hemolysis due to delayed blood preparation constitutes only a fraction of all in vitro hemolysis cases, in general, miniaturised, microfluidic-based solutions for blood plasma sample preparation at the site of blood draw might help to standardise the preparation through robust near-patient solutions, decreasing the variability of biomarker measurements. In several of the cases presented earlier, plasma extraction at the site of blood draw has been shown to effectively remove detrimental blood cell contamination. At present, regardless of the required sample volume, plasma is obtained from whole blood using a centrifuge that has a significant footprint, needs batch-processing and might not always be available. Integrated sample preparation and in particular the removal of blood cells needed for the reliable detection of circulating biomarkers, remain a bottleneck in the development of a number of POC devices. In the next section, we look at the most recent solutions with a focus on performance factors such as plasma recovery. Table 1 provides a summary of the advantages and limitations of the two approaches.| Centrifugation | Microfluidic based techniques | |
|---|---|---|
| Advantages | • High yield | • Can be integrated with continuous flow processes |
| • High purity depending on parameters | • Standardisation possible | |
| • Reproducible | • Portable | |
| • Well established | • Contained | |
| Limits | • Batch-processing | • New method, lack of reproducibility studies |
| • Bulky | • Variable efficiencies | |
| • Relatively expensive | • Low yield | |
| • Potential for low-cost |
4 Separation strategies
The number and type of tests to perform dictates the volume of plasma required, and subsequently the starting volume of blood. For example, in monitoring chronic hepatitis B patients 6 to 8 tests are typically performed with a requirement of at least 200 μL of plasma in total,41 while measurements on circulating nucleic acid can require as much as several millilitres of plasma.42 Here we classify the techniques by processed sample volume to offer clarity to end-users looking for the most suitable approaches (Table 2).| Blood volumes | Finger-prick volumes up to a few microlitres | Finger-prick or venous blood volumes in the microlitre range | Venous blood volumes in the millilitre range |
|---|---|---|---|
| Typical plasma analytes | Blood gas, electrolytes and metabolites (BGEM) | BGEM | Protein and peptide markers |
| Protein markers | Protein and peptide markers | Antigens | |
| Antigens | Antigens | Circulating nucleic acids (CNAs) | |
| Pathogenic DNA/RNA | Pathogenic DNA/RNA | ||
| microRNA | Illegal drugs and their metabolites | ||
| Microfluidic plasma separation methods | Filtration | Filtration-based | High-throughput hydrodynamic |
| Sedimentation | Centrifugal (CD) | Sedimentation–filtration | |
| Centrifugal (CD) | Hydrophoresis | ||
| Capillary hydrodynamic | Dielectrophoresis | ||
| Active methods | |||
| Examples of technology |
|
|
|
4.1 Finger-prick volumes up to a few microlitres
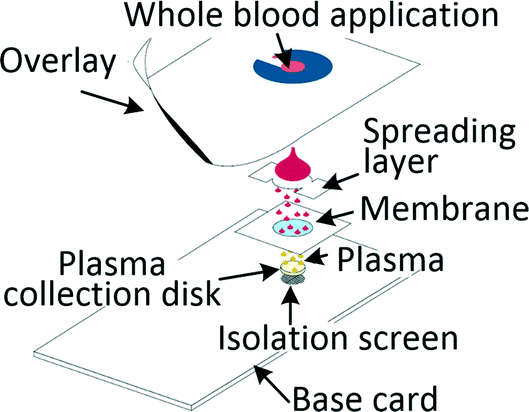
Many diagnostic tests such as blood gas, electrolytes and metabolites (BGEM) tests can be carried out on plasma from a drop of blood,43 enabling traditional venepuncture to be substituted with a finger-prick procedure, an important asset in POC setting. Filtration and paper-based technologies are a common approach when working with blood volumes in the microlitre range. Kim et al. developed a filtration-based tool for separating 2.5 μl of plasma in 3 min and its subsequent deposition on a collection disc, which can be air-dried and sent for analysis. Measurements of vitamin D and spiked-in yeast enolase revealed that the extracted plasma was equivalent to the venepuncture-derived control.44 Other filtration methods required blood dilution to increase filter lifetime.45,46 Kuo et al. tested a capillary-driven chip, in which cells are eliminated through size-exclusion. Plasma can be extracted in 75 s from 10 μl of whole blood at 15% yield through a fishbone microchannel network.47 Another capillary driven chip with filtering microstructures was capable of extracting 0.1 μl plasma from 5 μl of undiluted blood.48 Recent developments in centrifugal disc (CD) microfluidic systems, or so called lab-on-a-disc systems, allowed for separation of plasma from undiluted blood and achieved highly controlled actuation of small sample volumes, which is required for integrated analyte detection. The disc is spun on a rotating platform and separation was achieved within a few seconds,49 while other systems typically removed 99.5% of cells in 10 min.50Separation of plasma from microlitre amounts of undiluted blood was also achieved by methods ranging from capillary chip with asymmetric superhydrophilic coating,51 to cotton thread,52 to phase-guide assisted sedimentation53 and to dielectrophoresis (DEP).54,55 Different levels of blood dilution were needed for systems based on sedimentation (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (ref. 56) and 1:9 (ref. 57)), magnetophoresis (1:10)58 and a combination of hydrodynamic separation with DEP (1:1).59
:3 (ref. 56) and 1:9 (ref. 57)), magnetophoresis (1:10)58 and a combination of hydrodynamic separation with DEP (1:1).59
4.2 Finger-prick or venous blood volumes in the microlitre range
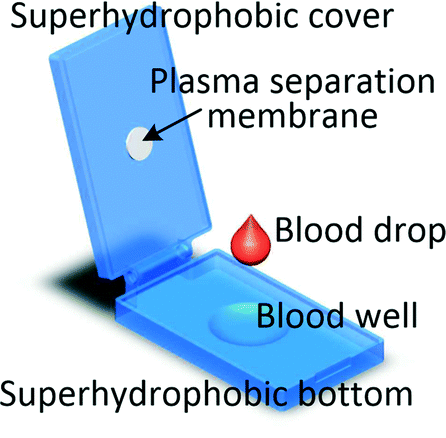
It is worth mentioning that even if the blood volume needed for the test is between 1 μl and several hundred microlitres it does not imply that capillary blood can be used. As levels of analytes can be different in capillary and venous blood, for some tests microlitre blood volume will still be collected through venepuncture.60 Recent advancements in processing blood samples of volumes up to a few hundred microlitres have been achieved with membrane filtration, passive hydrodynamic separation, centrifugal microfluidics and active separation methods. A centrifugal system for liver function screening at resource poor settings separates plasma from 150 μl of capillary blood and performs a 5-parameter liver assay panel in less than 20 min.61 Liu et al. demonstrated a device in which up to 200 μl of undiluted blood is sandwiched between two superhydrophobic surfaces. The device is left for 10 min allowing the cells to settle down and plasma to fill a collection chamber through a membrane placed at the top of the blood reservoir. Yield of 33% is achieved and 85% recovery of spiked-in S. mansoni genomic DNA was demonstrated.62 An electrochemical paper-based analytical device (ePAD) using blood separation paper (VF2, Whatman) was developed by Noiphung et al. Plasma from 250 μl of undiluted blood was separated in 4 min and subsequently the glucose level was measured.63 An unorthodox approach was employed by Kim and Choi who created hand-held “smart-pipette” with a microfluidic plasma separating tip relying on hydrophoresis. A 5 min operation produced 60 μl of plasma with 93% separation efficiency.64 A capillary driven chip with low-voltage (1 V) DEP for capturing RBCs from 150 μl undiluted blood was developed by Chen et al. The device was capable of removing close to 90% of RBCs with around 40% volumetric plasma yield. However, the removal efficiency of WBCs and platelets were not measured.654.3 Venous blood volumes in the millilitre range
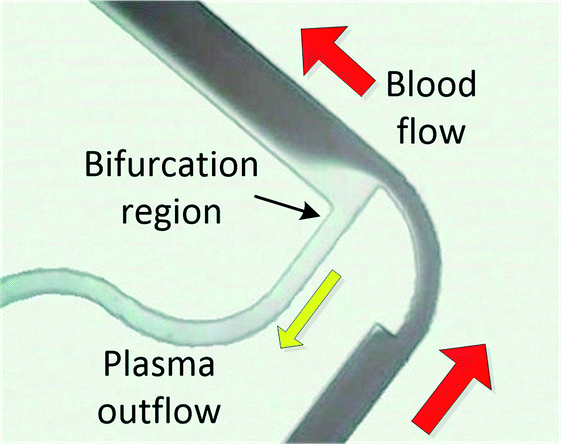
When processing large blood volumes, avoiding the saturation issues encountered in filtration is crucial, and this has been traditionally achieved by hydrodynamic plasma separation, a method employed in the seminal work of Yang et al.66 The most widely used systems rely on plasma skimming effect sometimes referred to as the Zweifach–Fung effect.9 Plasma is extracted in a channel bifurcation through a side channel of higher fluidic resistance while cells flow through a main, low resistance channel. Usually the separation is enhanced by additional geometrical features like constriction–expansion regions improving both the cell focussing and plasma skimming effects. This approach requires external pumping equipment but often achieves high throughputs. However, the usual trade-off is high blood dilution (typically 1:1 to 1:20). Agarwal and co-workers published several papers exploring the impact of different bifurcation geometries, e.g. varying constriction length and bend, on plasma extraction.67–70 In the recent work chips were tested at a wide range of hematocrits (7–62%) achieving separation efficiency close to 100% and plasma quality was validated by measuring levels of glucose and human chorionic gonadotropin (hCG) hormone. Although plasma yield was rather low (1–6%), the devices could potentially produce a few hundreds of microlitres of plasma due to relatively high throughputs (0.3–0.5 ml min−1) with reported reduced clogging.68 Marchalot et al. developed a BPS platform with two constriction–expansion units placed in series. The group studied the effect of system geometry on development of cell free layer and cell-focussing at relatively high dilutions (1:10–1:20). Numerical calculations of their network's hydraulic resistance helped to reach a 25% volumetric extraction yield of highly diluted plasma with a purity of 99% or higher.71 Others have tried to vary the constriction geometry to enhance the formation of a microvortex, with limited success (1:20 blood dilution, separation efficiency 90%).72
Inertial microfluidics, developed for cell sorting73 have been applied to separate plasma from blood. Cell removal through inertial focussing with secondary flow has been achieved in a constriction–expansion array74 and serpentine channel.75,76 The last device, comprised of 16 multiplexed slanted spiral channels, achieved a particularly high separation efficiency of 100% at yield close to 50% and massive throughput of 24 ml min−1. However, the system is only suitable for highly diluted blood (Hct = 0.5–1%).76 The limiting factor is the interaction between cells, that was found to prevent cell focussing at Hct higher than several percent.75
Commercially available plasma separating membranes, although predominantly aimed at microlitre blood volumes, have also been used for large sample volumes. Liu et al. developed a hybrid sedimentation–filtration based system, in which plasma is extracted on 2 sides of a vertical sample loading chamber. Filter clogging was largely reduced through gravitational sedimentation of cells and the device was capable of extracting 275 μl of plasma from 1.8 ml undiluted blood (yield 15%). The group used HIV-spiked blood to evaluate device's utility in diagnostics.77 In another sedimentation–filtration device, blood is drawn through a cell sedimentation chamber with an additional microporous membrane at the end, producing plasma of ∼99% purity. The sedimentation chamber requires priming with isotonic buffer that leads to some plasma dilution (generally lower than 1:1).78 Gong et al. developed a simple cartridge PMMA based device capable of filtering plasma out of millilitre volumes of blood through a membrane. Sample flow was improved by hydrophilic coating of the channels. In ∼5 min the device produced over 100–200 μl of plasma from 0.8–1 ml of blood achieving remarkable separation efficiency of 99.9% and 96.9% for RBCs and WBCs, respectively. The group tested the device in POC setting, performing a number of hepatitis B related tests on clinical samples, achieving results similar to those based on control plasma obtained by centrifugation.41
5 Recommendations and future outlook
In a large number of cases, plasma provides a stable matrix for the detection of analytes and biomarkers susceptible to hemo- and leukolysis, making miniaturised plasma extraction especially attractive for POCT. In parallel, researchers continue to pursue the refinement of strategies aimed at removing the need for blood plasma separation altogether. Here we reviewed some technological considerations and potential challenges for the development of microfluidic BPS technologies.Solving the dilution issue
Methods where high blood dilution is needed have arguable applicability in medical diagnostics. A hypothetical efficient pump-operated device (25% plasma yield, 30 ml h−1 inlet flow rate) separating plasma from blood diluted 1:10 requires 30 min to extract a sample volume containing only 200 μl of undiluted plasma. Based on a 45% Hct the dilution of such plasma sample is approximately 1:18, which can make the detection of rare analytes difficult or impossible. Furthermore, considering major progress in effective self-driven low-impact solutions for extracting the same volumes of undiluted plasma, the development of high throughput systems should demonstrate production of plasma samples suitable for diagnostics purposes.
Technological considerations
Medical diagnostics could potentially benefit from stand-alone microfluidic plasma extraction at the point of blood draw, as it has the potential to circumvent issues around the transport of blood in the context of circulating molecular markers detection. For example, we have demonstrated the use of stand-alone plasma separation in the specific context of prenatal diagnostics,79 while others proposed a BPS device producing plasma-bearing strips ready for transport.44 Integration of BPS modules into full LOC is nevertheless important for the reasons listed in section 3.There is no single best microfluidic BPS technology, with each one having its advantages and limitations. In general, hybrid devices presented here achieved higher yield and sample purity than those employing a single separation mechanism.77,78 Therefore, a hybrid approach, in which advantages of different BPS mechanisms are combined in one device, has a large potential in plasma-based POC diagnostics, especially for processing larger volumes of blood. An inherent issue of all microfluidic technologies relates to clogging. While it is most apparent in filter-based devices (finite lifetime limiting sample volume processed), larger microfluidic channels can also get clogged from cell aggregation, coagulation or presence of debris. In order for a BPS technology to be considered for product development, it needs to produce both high quality plasma sample and demonstrate reliability. For this reason, information on device failure rate would contribute to assessing the efficacy of the method. Moreover, the practicality of a particular solution strongly depends on fabrication. The use of complex fabrication methods and designs as well as the use of external equipment for device operation should be justified by improvements in performance. Additionally, compatibility with mass fabrication methods is crucial for translation to market.
Suitability of the extracted plasma for diagnostic purpose
The majority of papers presenting BPS do not include a comprehensive study of plasma quality such as remaining cell content and hemolysis measurements. For instance, flow cytometry, automated hemocytometry or manual cell counting can be used for measuring residual cell content in plasma but have different precision, and comparison of results obtained with different methods is not always informative. Demonstration of successful biomarker detection enables the evaluation of the device for a specific diagnostic purpose, but the lack of a robust generic biological characterisation hinders conclusive comparison of the available technologies. Therefore, we recommend that more rigorous testing is carried out to assess the performance of new devices in the context of medical diagnostics.Acknowledgements
WSM is funded by a James Watt Scholarship. MKK acknowledges the Royal Academy of Engineering for funding.Notes and references
- G. Cheng, Adv. Drug Delivery Rev., 2015, 81, 75–93 CrossRef CAS PubMed.
- B. Olsson, R. Lautner, U. Andreasson, A. Öhrfelt, E. Portelius, M. Bjerke, M. Hölttä, C. Rosén, C. Olsson, G. Strobel, E. Wu, K. Dakin, M. Petzold, K. Blennow and H. Zetterberg, Lancet Neurol., 2016, 15, 673–684 CrossRef CAS PubMed.
- S. Neugebauer, E. J. Giamarellos-Bourboulis, A. Pelekanou, A. Marioli, F. Baziaka, I. Tsangaris, M. Bauer and M. Kiehntopf, Crit. Care Med., 2016 DOI:10.1097/CCM.0000000000001740.
- WHO Public report for RealStar® Filovirus Screen RT-PCR Kit 1.0 (EA 0002-002-00), 2014.
- R. Charrel, I. Leparc-Goffart and S. Pas, Bull. W. H. O., 2016 DOI:10.2471/BLT.16.171207.
- K. W. Witwer, E. I. Buzás, L. T. Bemis, A. Bora, C. Lässer, J. Lötvall, E. N. N.-‘t Hoen, M. G. Piper, S. Sivaraman, J. Skog, C. Théry, M. H. Wauben and F. Hochberg, J. Extracell. Vesicles, 2013, 2 Search PubMed.
- L. H. Araujo, C. Timmers, K. Shilo, W. Zhao, J. Zhang, L. Yu, T. G. Natarajan, C. J. Miller, A. S. Yilmaz, T. Liu, J. Amann, J. R. Lapa E Silva, C. G. Ferreira and D. P. Carbone, PLoS One, 2015, 10, e0143092 Search PubMed.
- H. Butz and A. Patócs, Exper. Suppl., 2015, 106, 55–71 CrossRef.
- M. Kersaudy-Kerhoas and E. Sollier, Lab Chip, 2013, 13, 3323–3346 RSC.
- S. Tripathi, Y. V. B. Varun Kumar, A. Prabhakar, S. S. Joshi, A. Agrawal, Y. V. B. V. Kumar, A. Prabhakar, S. S. Joshi and A. Agrawal, J. Micromech. Microeng., 2015, 25, 083001 CrossRef.
- D. Gossett, W. Weaver and A. Mach, Anal. Bioanal. Chem., 2010, 397(8), 3249–3267 CrossRef CAS PubMed.
- P. B. Luppa, C. Müller, A. Schlichtiger and H. Schlebusch, TrAC, Trends Anal. Chem., 2011, 30, 887–898 CrossRef CAS.
- P. von Lode, Clin. Biochem., 2005, 38, 591–606 CrossRef CAS PubMed.
- B. T. Ivandic, E. Spanuth and E. Giannitsis, Clin. Lab., 2014, 60, 903–908 Search PubMed.
- S. S. Modak, C. A. Barber, E. Geva, W. R. Abrams, D. Malamud and Y. S. Y. Ongagna, Infect. Dis., 2016, 9, 1–9 Search PubMed.
- J. L. Garcia-Cordero and S. J. Maerkl, J. Lab. Autom., 2016, 21, 356–367 CrossRef PubMed.
- F. Piraino, F. Volpetti, C. Watson and S. J. Maerkl, ACS Nano, 2016, 10, 1699–1710 CrossRef CAS PubMed.
- A. I. Barbosa, A. P. Castanheira, A. D. Edwards and N. M. Reis, Lab Chip, 2014, 14, 2918–2928 RSC.
- B. Hallmark, F. Gadala-Maria and M. R. Mackley, J. Nonnewton. Fluid Mech., 2005, 128, 83–98 CrossRef CAS.
- E. Petryayeva and W. R. Algar, Analyst, 2015, 140, 4037–4045 RSC.
- M. Plebani, Clin. Chim. Acta, 2009, 404, 16–23 CrossRef CAS PubMed.
- P. Carraro, G. Servidio and M. Plebani, Clin. Chem., 2000, 46, 306–307 CAS.
- G. Lippi, N. Blanckaert, P. Bonini, S. Green, S. Kitchen, V. Palicka, A. J. Vassault and M. Plebani, Clin. Chem. Lab. Med., 2008, 46(6), 764–772 CrossRef CAS PubMed.
- S. F. Green, Clin. Biochem., 2013, 46, 1175–1179 CrossRef PubMed.
- M. B. Kirschner, S. C. Kao, J. J. Edelman, N. J. Armstrong, M. P. Vallely, N. van Zandwijk and G. Reid, PLoS One, 2011, 6, e24145 CAS.
- L. Moldovan, K. E. Batte, J. Trgovcich, J. Wisler, C. B. Marsh and M. Piper, J. Cell. Mol. Med., 2014, 18, 371–390 CrossRef CAS PubMed.
- M. Daves, G. L. Salvagno, R. Cemin, M. Gelati, G. Cervellin, G. C. Guidi and G. Lippi, Clin. Lab., 2012, 58, 333–336 Search PubMed.
- R. Bais, Clin. Chem., 2010, 56, 1357–1359 CAS.
- R. C. Hawkins, Clin. Chem., 2003, 49, 1226–1227 CAS.
- C. Puelacher, R. Twerenbold, T. Mosimann, J. Boeddinghaus, M. Rubini Gimenez, K. Wildi, C. Jaeger, T. Reichlin, J. Schneider, U. Honegger, W. Max, C. Schumacher, T. Nestelberger, P. Hillinger, K. Grimm, P. Kreutzinger, Z. Moreno Weidmann, K. Rentsch, C. Arnold, S. Osswald and C. Mueller, Int. J. Cardiol., 2015, 187, 313–315 CrossRef PubMed.
- J. R. Asirvatham, V. Moses and L. Bjornson, N. Am. J. Med. Sci., 2013, 5, 255–259 CrossRef PubMed.
- M. Oostendorp, W. W. van Solinge and H. Kemperman, Arch. Pathol. Lab. Med., 2012, 136, 1262–1265 CrossRef PubMed.
- D. Sinclair, J. Clin. Pathol., 2003, 56, 385–387 CrossRef CAS PubMed.
- H. E. Turner, R. W. A. Peake and J. J. Allison, Ann. Clin. Biochem., 2012, 49, 94–96 CrossRef CAS PubMed.
- S. Clark, Clin. Chem., 2003, 49, 518–520 CAS.
- A. B. Sparks, C. A. Struble, E. T. Wang, K. Song and A. Oliphant, Am. J. Obstet. Gynecol., 2012, 206, 319.e1–319.e9 CrossRef CAS PubMed.
- G. E. Palomaki, E. M. Kloza, G. M. Lambert-Messerlian, J. E. Haddow, L. M. Neveux, M. Ehrich, D. van den Boom, A. T. Bombard, C. Deciu, W. W. Grody, S. F. Nelson and J. A. Canick, Genet. Med., 2011, 13, 913–920 CrossRef CAS PubMed.
- A. N. Barrett, H. A. Thadani, C. Laureano-Asibal, S. Ponnusamy and M. Choolani, Prenatal Diagn., 2014, 34, 1283–1288 CrossRef CAS PubMed.
- X. Xue, M. D. Teare, I. Holen, Y. M. Zhu and P. J. Woll, Clin. Chim. Acta, 2009, 404, 100–104 CrossRef CAS PubMed.
- J. L. Sherwood, C. Corcoran, H. Brown, A. D. Sharpe, M. Musilova and A. Kohlmann, PLoS One, 2016, 11, e0150197 Search PubMed.
- M. M. Gong, B. D. MacDonald, T. Vu Nguyen, K. Van Nguyen and D. Sinton, Biomicrofluidics, 2013, 7, 44111 CrossRef PubMed.
- R. W. K. Chiu, R. Akolekar, Y. W. L. Zheng, T. Y. Leung, H. Sun, K. C. A. Chan, F. M. F. Lun, A. T. J. I. Go, E. T. Lau, W. W. K. To, W. C. Leung, R. Y. K. Tang, S. K. C. Au-Yeung, H. Lam, Y. Y. Kung, X. Zhang, J. M. G. van Vugt, R. Minekawa, M. H. Y. Tang, J. Wang, C. B. M. Oudejans, T. K. Lau, K. H. Nicolaides and Y. M. D. Lo, BMJ, 2011, 342, c7401 CrossRef PubMed.
- Abaxis Inc., http://www.piccoloxpress.com/wp-content/uploads/centrifugation-whitepaper.pdf.
- J.-H. Kim, T. Woenker, J. Adamec and F. E. Regnier, Anal. Chem., 2013, 85, 11501–11508 CrossRef CAS PubMed.
- J. Chen, D. Chen, T. Yuan, X. Chen, Y. Xie, H. Fu, D. Cui, X. Fan and M. K. Khaing Oo, Microelectron. Eng., 2014, 128, 36–41 CrossRef CAS.
- J. H. Son, S. H. Lee, S. Hong, S. Park, J. Lee, A. M. Dickey and L. P. Lee, Lab Chip, 2014, 14, 2287–2292 RSC.
- J.-N. Kuo and Y.-H. Zhan, Microsyst. Technol., 2013, 21, 255–261 CrossRef.
- M. Hojjat, C.-T. Jasmina and M. Mahdi, Biofabrication, 2015, 7, 25007 CrossRef PubMed.
- J.-N. Kuo and X.-F. Chen, Microsyst. Technol., 2015, 22, 861–869 CrossRef.
- Z. Cai, J. Xiang and W. Wang, Sens. Actuators, B, 2015, 221, 257–264 CrossRef CAS.
- K. K. Lee and C. H. Ahn, Lab Chip, 2013, 13, 3261–3267 RSC.
- M. F. Ulum, L. Maylina, D. Noviana and D. H. B. Wicaksono, Lab Chip, 2016, 1492–1504 RSC.
- L. Xu, H. Lee, M. V. Brasil Pinheiro, P. Schneider, D. Jetta and K. W. Oh, Biomicrofluidics, 2015, 9, 014106 CrossRef PubMed.
- C. Szydzik, K. Khoshmanesh, A. Mitchell and C. Karnutsch, Biomicrofluidics, 2015, 9, 64120 Search PubMed.
- S.-H. Liao, C.-Y. Chang and H.-C. Chang, Biomicrofluidics, 2013, 7, 24110 CrossRef PubMed.
- C. Kuroda, Y. Ohki, H. Ashiba, M. Fujimaki, K. Awazu, T. Tanaka and M. Makishima, in Proceedings of IEEE Sensors, December 2014, vol. 2014-Decem, pp. 1854–1857 Search PubMed.
- Y. Xie, D. Chen, S. Lin, Z. Wang and D. Cui, RSC Adv., 2016, 6, 30722–30727 RSC.
- P. Kim, E. H. Ong, K. H. H. Li, Y.-J. Yoon, S. H. G. Ng and K. Puttachat, Biomicrofluidics, 2016, 10, 24110 CrossRef PubMed.
- M. Mohammadi, H. Madadi, J. Casals-Terré and J. Sellarès, Anal. Bioanal. Chem., 2015, 407, 4733–4744 CrossRef CAS PubMed.
- Australian PoCT Practitioner's Network.
- C. E. Nwankire, M. Czugala, R. Burger, K. J. Fraser, T. M. O'Connell, T. Glennon, B. E. Onwuliri, I. E. Nduaguibe, D. Diamond and J. Ducrée, Biosens. Bioelectron., 2014, 56, 352–358 CrossRef CAS PubMed.
- C. Liu, S.-C. Liao, J. Song, M. G. Mauk, X. Li, G. Wu, D. Ge, R. M. Greenberg, S. Yang and H. H. Bau, Lab Chip, 2016, 16, 553–560 RSC.
- J. Noiphung, T. Songjaroen, W. Dungchai, C. S. Henry, O. Chailapakul and W. Laiwattanapaisal, Anal. Chim. Acta, 2013, 788, 39–45 CrossRef CAS PubMed.
- B. Kim and S. Choi, Small, 2016, 12, 190–197 CrossRef CAS PubMed.
- C.-C. Chen, P.-H. Lin and C.-K. Chung, Lab Chip, 2014, 14, 1996–2001 RSC.
- S. Yang, A. Ündar and J. D. Zahn, Lab Chip, 2006, 6, 871–880 RSC.
- A. Prabhakar, Y. V. B. V. Kumar, S. Tripathi and A. Agrawal, Microfluid. Nanofluid., 2015, 18, 995–1006 CrossRef CAS.
- S. Tripathi, Y. V. B. Kumar, A. Agrawal, A. Prabhakar and S. S. Joshi, Sci. Rep., 2016, 6, 26749 CrossRef CAS PubMed.
- S. Tripathi, A. Prabhakar, N. Kumar, S. G. Singh and A. Agrawal, Biomed. Microdevices, 2013, 15, 415–425 CrossRef PubMed.
- S. Tripathi, Y. V. B. Varun Kumar, A. Prabhakar, S. S. Joshi and A. Agrawal, J. Micromech. Microeng., 2015, 25, 084004 CrossRef.
- J. Marchalot, Y. Fouillet and J.-L. Achard, Microfluid. Nanofluid., 2014, 17, 167–180 CrossRef CAS.
- A. Haller, A. Spittler, L. Brandhoff, H. Zirath, D. Puchberger-Enengl, F. Keplinger and M. J. Vellekoop, Micromachines, 2015, 6, 239–251 CrossRef.
- D. Di Carlo, Lab Chip, 2009, 9, 3038–3046 RSC.
- M. G. Lee, J. H. Shin, S. Choi and J.-K. Park, Sens. Actuators, B, 2014, 190, 311–317 CrossRef CAS.
- J. Zhang, S. Yan, W. Li, G. Alici and N.-T. Nguyen, RSC Adv., 2014, 4, 33149–33159 RSC.
- M. Rafeie, J. Zhang, M. Asadnia, W. Li and M. E. Warkiani, Lab Chip, 2016, 16, 2791–2802 RSC.
- C. Liu, M. Mauk, R. Gross, F. D. Bushman, P. H. Edelstein, R. G. Collman and H. H. Bau, Anal. Chem., 2013, 85, 10463–10470 CrossRef CAS PubMed.
- C. Galligan, J. Nichols, E. Kvam, P. Spooner, R. Gettings, L. Zhu and C. M. Puleo, Lab Chip, 2015, 15, 3274–3277 RSC.
- M. Kersaudy-Kerhoas, M. Desmulliez and J. Norman, Arch. Dis. Child. Fetal Neonatal Ed., 2014, 99, A2 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |