
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers with pendant ferrocenes
Rudolf
Pietschnig
Universität Kassel, Institut für Chemie und CINSaT, Heinrich-Plett-Straße 40, 34132 Kassel, Germany. E-mail: pietschnig@uni-kassel.de
First published on 9th May 2016
Abstract
The tailoring of smart material properties is one of the challenges in materials science. The unique features of polymers with pendant ferrocene units, either as ferrocenyl or ferrocenediyl groups, provide electrochemical, electronic, optoelectronic, catalytic, and biological properties with potential for applications as smart materials. The possibility to tune or to switch the properties of such materials relies mostly on the redox activity of the ferrocene/ferricenium couple. By switching the redox state of ferrocenyl units – separately or in a cooperative fashion – charge, polarity, color (UV-vis range) and hydrophilicity of polymers, polymer functionalized surfaces and polymer derived networks (sol–gel) may be controlled. In turn, also the vicinity of such polymers influences the redox behavior of the pendant ferrocenyl units allowing for sensing applications by using polymer bound enzymes as triggering units. In this review the focus is set mainly on the literature of the past five years.
 Rudolf Pietschnig | Rudolf Pietschnig studied chemistry at the University of Bonn and earned his PhD degree in 1996. After postdoctoral studies at the University of Wisconsin Madison, USA (DFG-Fellowship) and Ruhr-University Bochum, Germany (Liebig-Fellowship) he moved to the University of Graz (Austria), where he started his own research group focusing on main-group organometallic chemistry. He completed his habilitation and received his venia in 2005 and was appointed as Professor for Chemical Hybrid Materials at the University of Kassel (Germany) in 2011. Since 2012 he has been the elected member of the Center for Interdisciplinary Nanostructure Science and Technology (CINSaT). His research interests are centered around main-group organometallic chemistry ranging from inorganic π-systems and multiple bonds, with respect to their unusual bonding situation and reactivity, to organosilanols as building blocks for molecular and supramolecular structures and surface modification/sorption processes and their thermal aspects. |
Key learning points• Pendant ferrocenyl units may substantially alter the properties of the polymer and its backbone• The redox properties of the ferrocenyl units may be used to tune charge, polarity, color and hydrophilicity of polymers and polymer functionalized interfaces and networks • The connection between the polymer vicinity and the ferrocene redox couple via the polymer backbone enables sensing applications • Pendant ferrocenyl units increase the thermal stability of polymer backbones • Ferrocene containing polymers may be used as precursors for patterned metal oxides and alloys |
1. Introduction and scope
Ferrocene-containing polymers are an outstanding family of metal-containing polymers raising considerable interest, mainly owing to the unique properties and high chemical stability of ferrocene. In principle there are two ways of incorporating covalently bound ferrocene into a polymer: (a) incorporation into the main chain (backbone) or (b) attachment of ferrocenyl units as (or to) pendant groups adjacent to the polymer backbone. Polymers containing ferrocene in the backbone usually involve the covalent attachment at both Cp rings (Cp = cyclopentadienyl) via divalent 1,1′-ferrocenylene (or ferrocenediyl) units within the main chain. In principle, the attachment via only one of the two Cp rings involving 1,2- or 1,3-ferrocenylene units is also possible, however is less common. There are excellent reviews available on polymers containing metallocenes in the main chain covering synthetic and structural aspects as well as the fascinating properties and applications of these compounds which are mostly devoted but not limited to ferrocene.1–3In this tutorial review the focus is set on polymers with pendant ferrocene units and key aspects of their synthesis and properties together with emerging applications of this fascinating class of compounds. Related to this topic, the synthesis and applications of several ferrocene-functionalized polymer brushes have been summarized recently.4 Further reviews have covered polymers with pendant ferrocene units together with polymers containing ferrocene only in the backbone.5,6 One might be tempted to assume that the main chain controls the properties of the polymer without exhibiting the typical features of organometallic polymers, owing to the distance of the Fe centers. Therefore, this aspect will be explicitly addressed and from the examples presented the effect of the ferrocene moieties on the overall properties of the polymer will be outlined. As a consequence, polymers with pendant ferrocene units combine the unique properties of ferrocene with features of the backbone giving rise to materials featuring redox activity, thermal and oxidative stability, moisture resistance, biological activity and unusual rheological properties. In such materials the ferrocene unit may ultimately serve as the thermo-, mechanical-, acoustic-, and even chemical-stimulus responsive switch depending on the boundary conditions. In combination with the chemical and physiological inertness, high flexibility and high permeability of inorganic polymer chains, such materials may result in smart inorganic polymers in the literal sense. Smart materials with responsive and switchable properties are especially attractive since they can automatically adapt their properties to different settings. The aim of this tutorial review is to provide an entry into the most recent developments of polymers with pendant ferrocenes over roughly the last 5 years in light of smart polymers and applications.
2. Synthetic aspects
2.1 Starting from ferrocene containing monomers
A straightforward access to polymers with pendant ferrocenyl units starts from monomers containing a polymerizable functionality already connected to the ferrocenyl group (Chart 1). This methodology gives access to homopolymers as well as to block copolymers. Monomers which have been used for the purpose involve functional alkenes like ferrocenylmethyl methacrylate (FMMA),7,8 (2-(methacryloyloxy)ethyl ferrocenecarboxylate) (FcMA),9,10 2-(acryloyloxy)ethyl ferrocenecarboxylate) AEFc,11 vinylferrocene (VFc),8,9,12 ferrocenyl glycidyl ether (FcGE)13 and vinyl ferrocenyl glycidyl ether (VFcGE).14 The latter monomer is an especially valuable starting material because it contains two functionalities with orthogonal reactivity (Scheme 1). Thus, the vinyl group in VFcGE is available for radical polymerization leaving the epoxy unit unaffected, while the epoxy function is prone to anionic polymerization leaving the vinyl functionality for postsynthetic functionalisation without the need for a protection group.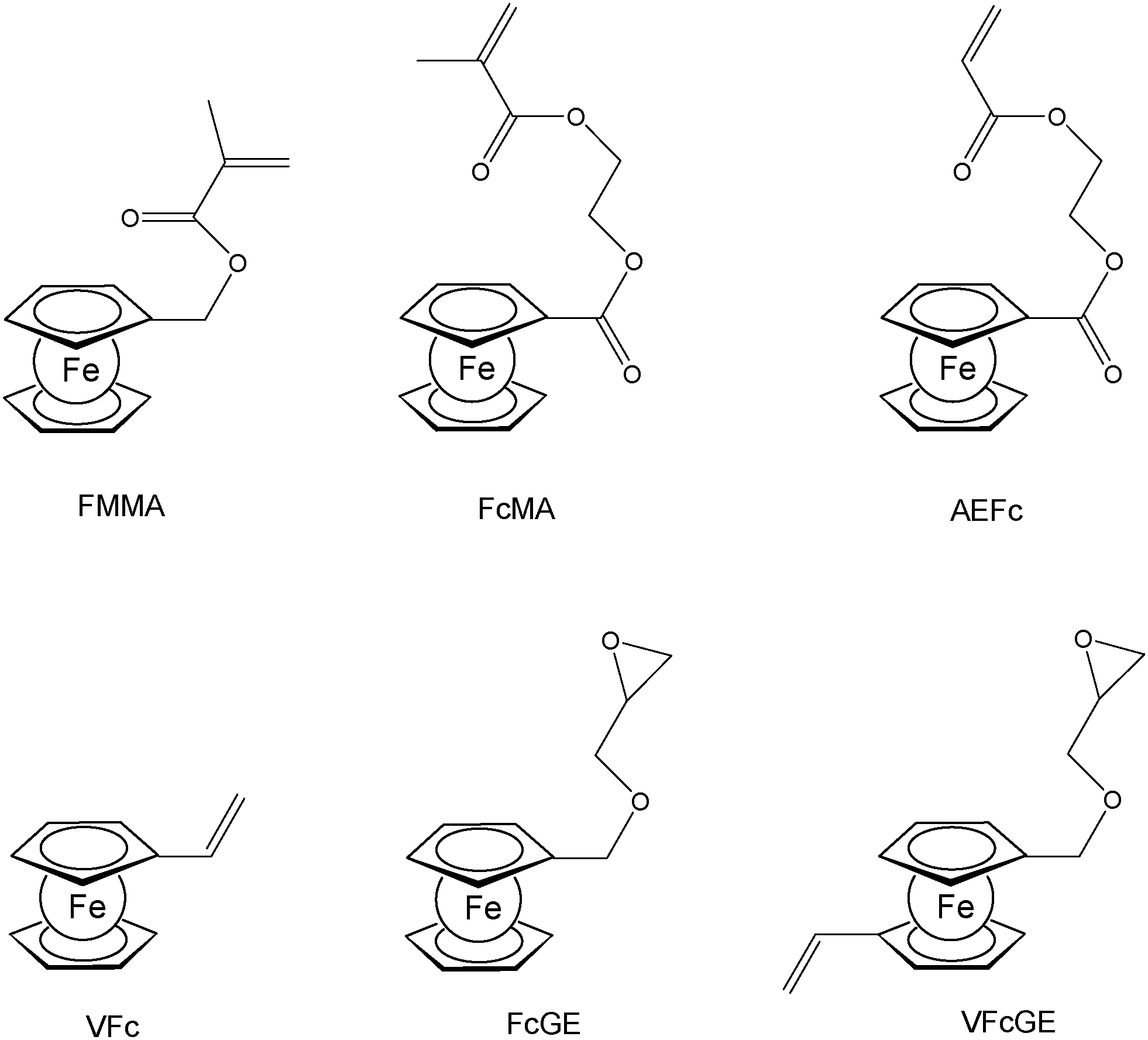 | ||
| Chart 1 Typical ferrocenyl functionalized monomers. | ||
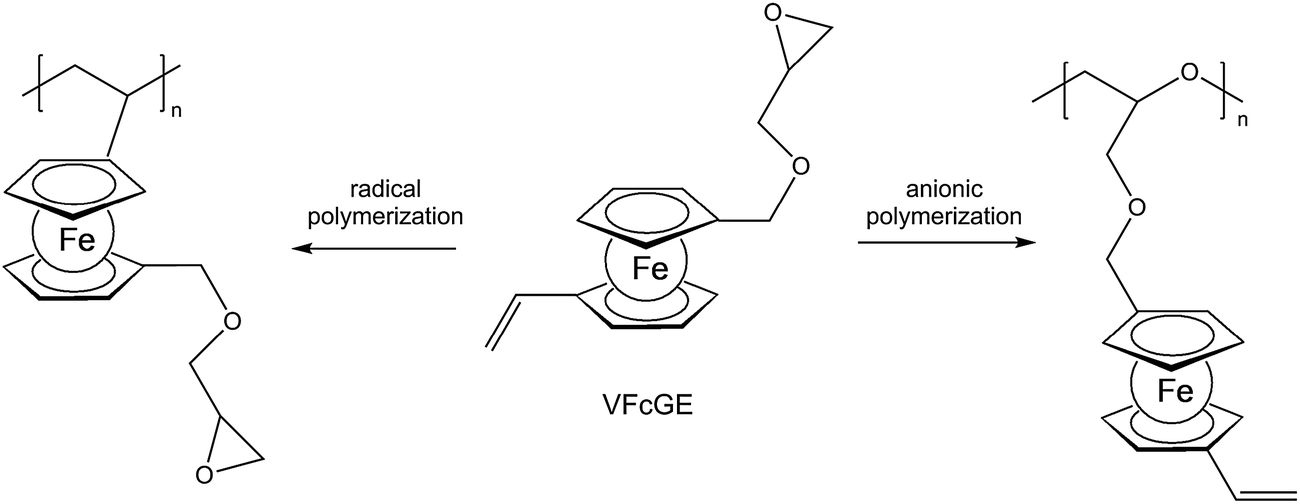 | ||
| Scheme 1 Orthogonal reactivity of VFcGE. | ||
In general, the methods being used to polymerize the above mentioned unsaturated monomers are solution based radical processes,7,12,14 including sequential atom transfer radical polymerization11 or surface-initiated atom transfer radical polymerization (SI-ATRP).9,10 Moreover, anionic polymerizations have been used successfully for VFc, VfcGE and FMMA as well.8,9,14 Living anionic polymerizations are especially sensitive to impurities causing chain termination and require therefore monomers of high purity and polymerization procedures under strict inert conditions.8
The use of unsaturated functionalities is generally well suited for organic backbones where such unsaturated monomers are stable. To achieve the formation of an inorganic polymer backbone, however, methods like ring opening polymerization (ROP) are more suitable. For an unusual example where a pendant metallocene unit (in this case a ruthenocenyl group) is present, besides a ferrocene in the main chain, the corresponding ruthenocenyl substituted [1]ferrocenophane has been employed as a monomer for the photocontrolled ring-opening polymerisation yielding the respective polymer with a poly-ferrocenylsilane backbone and pendant ruthenocenyl units (Scheme 2, top).15 Moreover, diblock copolymers consisting of polycarbosilane and poly-FMMA or poly-VFc sections have been synthesised via living anionic polymerization, following sequential monomer addition protocols.8 In the initial step the carbosilane block is generated via anionic ring opening of 1,1-dimethylsilacyclobutane (Scheme 2, bottom).
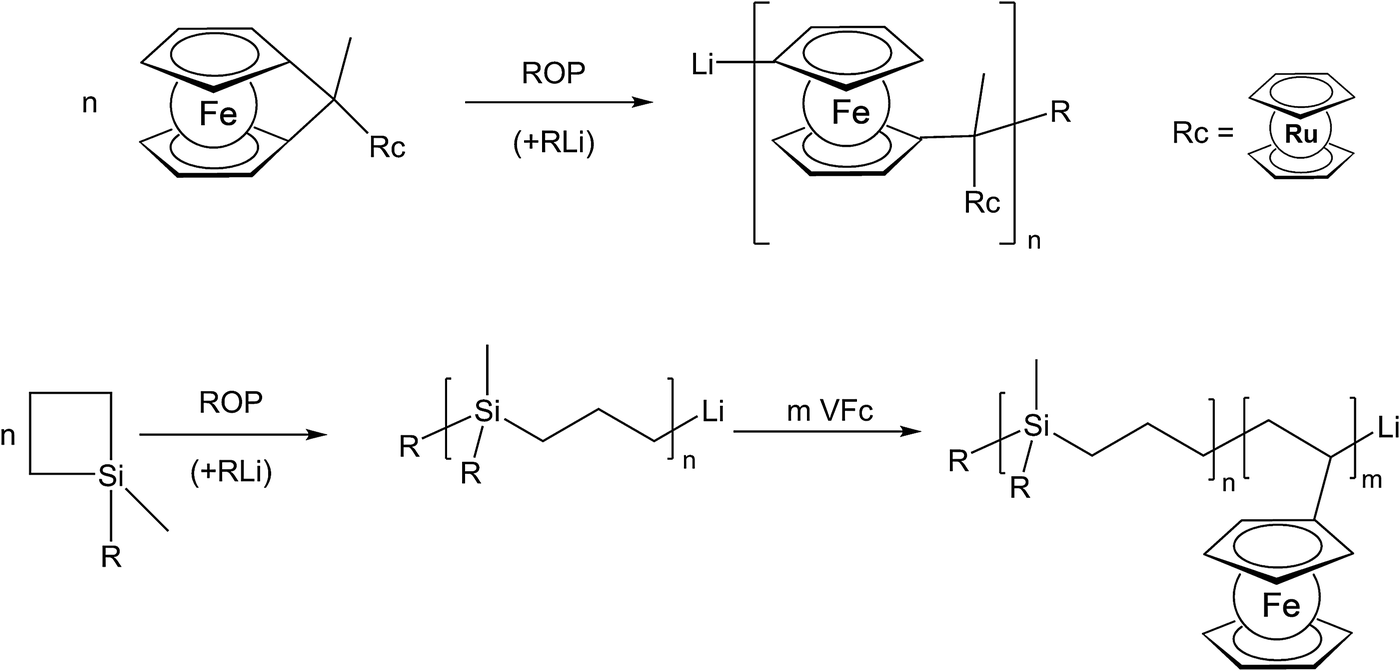 | ||
| Scheme 2 Examples of anionic ring opening polymerization (ROP). | ||
Recently, a very unique and elegant approach to inorganic polymer backbones has been established for phosphinoboranes. Starting from various primary phosphine borane adducts, polymers were obtained using catalytic dehydrocoupling employing a Rh(I) catalyst (Scheme 3).16,17 The special appeal of this reaction arises from the possibility to vary the structure of the monomer easily and thus planar chiral and also ionic monomers have been successfully polymerized using this novel approach.16
 | ||
| Scheme 3 Dehydrocoupling to polymeric phosphinoboranes. | ||
For the formation of surface bonded polymers and polymer films electropolymerization is a suitable method as well. Closely related to the well-established electrochemical deposition of polyaniline (PANI) on anodic surfaces, the deposition of analogous polymers with pendant ferrocene groups has been accomplished using N-(ferrocenylmethyl)-o-phenylenediamine (FMOPD) as an electropolymerizable monomer (Chart 2).18 Moreover, 3,4-ethylenedioxythiophene (EDOT) bearing monomers with pendant ferrocene units were synthesized and electrochemically polymerized.19,20 Finally, α,ω-diiodoalkanes with incorporated 1,1′-ferrocenylene units, so called di(iodoalkyl ferrocenes) (DIAFs), have been successfully used as unusual monomers for grafting onto electrode surfaces under cathodic conditions.21
 | ||
| Chart 2 Typical electropolymerizable ferrocenyl functionalized monomers (Ph = phenyl; Naph = 2-naphthyl; n = 4, 6, 10, 12). | ||
Besides the possibility to polymerize ferrocene containing monomers, it is equally possible to generate the polymer backbone first and to attach the ferrocenyl units via suitable functionalities in a subsequent step. This strategy of post-synthetic modification is especially suitable for inorganic polymer backbones but has been employed for organic ones as well (v.i.). A borderline case between these alternative strategies is the simultaneous conduction of SI-ATRP and click chemistry starting from ethynylferrocene and functional organic azides.24 It was suggested that following an initial click reaction 5-ferrocene-triazolyl methacrylate (PFTMA) may occur as an intermediate, which would then be classified as a ferrocene containing monomer as well (Scheme 4). Likewise in the reaction mixture of this simultaneous protocol, an initial polymerization of the acrylate based azide may occur followed by the subsequent addition of ethynylferrocene via a click-reaction which then would be a post-synthetic modification.
 | ||
| Scheme 4 One pot click reaction with competing pre- or post-synthetic ferrocene attachment to the polymer. | ||
2.2 Post-synthetic modification with ferrocenyl units
As previously mentioned, post-synthetic modification of polymer backbones is especially suitable for inorganic polymers. However, this is not only a consequence of the lack of the easy availability of unsaturated inorganic functionalities. In fact, inorganic polymer backbones provide reactivity patterns which reach beyond those of their organic counterparts. In polyphosphazenes, for instance, methyl groups adjacent to the backbone may be functionalized simply by deprotonation (Scheme 5). This behavior is a consequence of the unusual bonding situation where the methyl groups are attached to formal phosphonium centers, which stabilize negative charges in the α-position via negative hyperconjugation and therefore increase the acidity of these methyl groups. Using this approach, formylferrocene has been added to these carbanions giving rise to pendant ferrocene groups connected via carbinol units.25 Similarly hydrogen atoms at an inorganic backbone have been employed to attach ferrocenyl units. For example Si–H functionalized polysiloxanes have been used to graft dendritic wedges with ferrocenyl units to the backbone via hydrosilylation (Scheme 6).26 In turn, the attachment of ferrocene containing hydrosilanes has been used to attach these units via hydrosilylation to a carbosilane backbone as well.27,28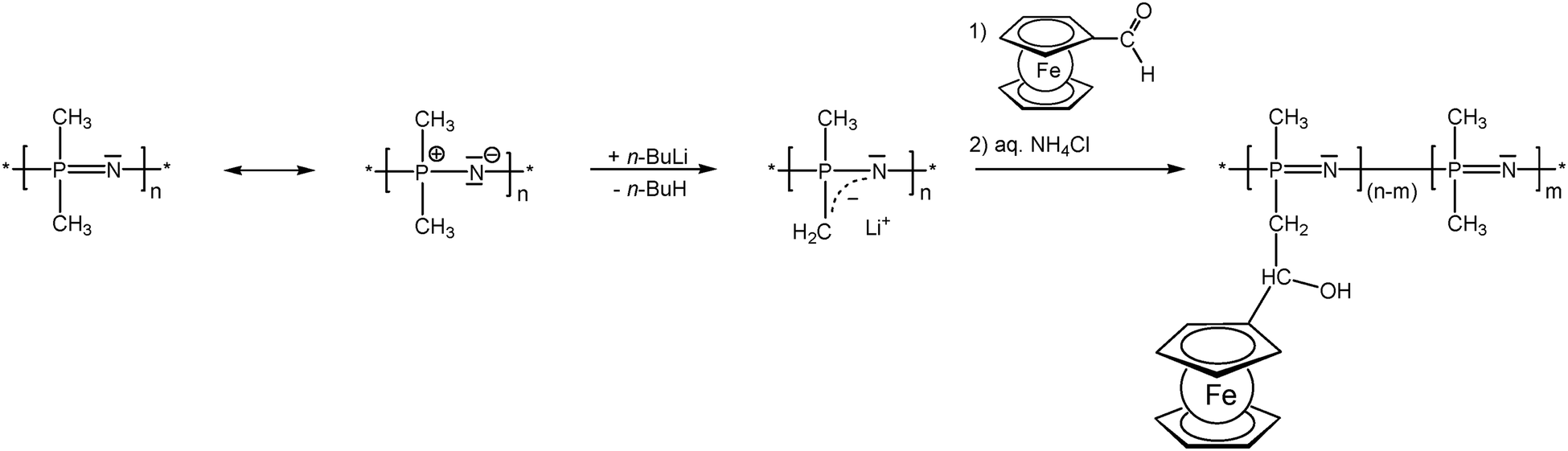 | ||
| Scheme 5 Deprotonation–substitution sequence in the post-synthetic modification of polyphosphazene backbones. | ||
 | ||
| Scheme 6 Postsynthetic polymer functionalization via reactive hydrogen atoms. | ||
Starting from thiol functionalized polysiloxanes the regioselective addition of the –SH units to vinylferrocene has been established as a new synthetic tool for macromolecules with pendant ferrocenyl functionalisation (Scheme 6).29 Both thermally and UV-photochemically initiated thiol–ene reactions with vinylferrocene tolerate a broad range of chemical functionalities, and proceed in good yields. Generally, the UV light initiated thiol–vinylferrocene reactions were considerably faster in the presence of a photoinitiator and more efficient than the thermally initiated ones. In addition, vinyl units with organic polyether backbones can be postmodified with thiol containing amino acids like cysteine.13
The high flexibility of inorganic backbone architectures contrasts the rather rigid backbone of conjugated polymers. Insoluble polythiophene carrying sulfonamide functionalities has been post-synthetically modified via a transamination reaction with a ferrocenealkylamine.30
Pendant ferrocenyl units have been attached to the backbone of linear poly(ethyleneimine) polymers (LPEI) using nucleophilic substitution reactions at haloalkylferrocenes via halide displacement.31 This procedure allows simple variation of the connecting spacer unit (C3 to C6). Alternatively the reductive amination of ferrocenyl carbonyl compounds has been employed to obtain attachment via 1-carbon spacers (Scheme 7). Moreover, the reductive amination approach has been used to attach ferrocene to polyallylamine and chitosane starting from ferrocene carboxaldehyde.32 Biocompatible polyaspartamide polymers have been functionalized at the available amino functions starting from 4-ferrocenylbutanoic acid (Scheme 6, right).33 The above mentioned attachment of electrophilic ferrocene derivatives to polymeric amines is complemented by the similar reactions with polymer bound alcohols. Thus, the functionalization of poly-2-hydroxyethylmethacrylate (HEMA) has been achieved with ferrocenecarbonyl chloride.34,35 In contrast to polymers with nucleophilic reaction sites at the backbone, polymers with strongly electrophilic functionalities require different reaction strategies and reagents. As an example, borylated polystyrene derivatives with pendant electron-rich ferrocene moieties at boron along with aryl groups have been prepared via post-synthetic modification of the corresponding dibromoborane with trimethylstannyl ferrocene (Scheme 8).36,37
 | ||
| Scheme 7 Post-synthetic modification of linear poly(ethyleneimine) (LPEI) with varying spacer length to the pendant ferrocenyl unit. | ||
 | ||
| Scheme 8 Post-synthetic modification of borylated polystyrene with stannylferrocene. | ||
3. Polymer properties
3.1 Switchable polarity
Once the pendant ferrocenyl units are attached to the backbone, the main chain will affect the redox properties of the adjacent ferrocene in various respects, be it by additional attractive/repulsive secondary interactions, direct electronic interactions or kinetic aspects like reduced diffusional access. In turn, the ferrocenyl units will affect the backbone as well. Especially when the neutral ferrocenyl units become cationic upon oxidation, the polarity of the polymer may be inverted and in the case of several pendant ferrocenyl units the electrostatic repulsion among them may change the arrangement and coiling of the polymer as well and its interaction towards further reaction partners. Using these principles hydrophobic polymers may be turned into their polar and hydrophilic counterparts simply by using pendant ferrocene units as redox switches. The application of this idea to surfaces dates back to Whitesides et al. who, two decades ago, reported the change in surface wettability of self-assembled monolayers of a ferrocene compound on a gold surface upon oxidation.38 Meanwhile the underlying principle has been significantly expanded to other surfaces and polymeric systems. Very instructive in this respect is a study in which ferrocene polymers have been attached to a silicon-based surface allowing for switchable surface wettability (Scheme 9). The polymers used for this comparative study are polyvinylferrocene (PVFc) and poly(2-(methacryloyloxy)ethyl ferrocenecarboxylate) (PFcMA) on silica wafers. The attachment to the surface was accomplished by applying a surface-initiated atom transfer radical polymerisation (SI-ATRP) protocol to PFcMA, while end-functionalized PVFc was immobilized by using a grafting onto approach. In the case of PFcMA, a remarkable contact angle (CA) drop for water of approximately 70° could be observed upon oxidation, while the effect for immobilized PVFc was less pronounced (CA drop after oxidation of approximately 30°). In the case of PFcMA, the effect of chain length was additionally studied, showing a more significant CA drop for PFcMA chains with higher molar masses.9 The reversible nature of switching the surface wettability has been demonstrated and different oxidants have been used to trigger the redox stimuli. | ||
| Scheme 9 Sketch of a surface with tunable wettability. | ||
Closely related to this, a methacrylate polymer with pendant ferrocene units has been attached to a gold surface. Again a reversible switching between the hydrophobic surface coating and its oxidized hydrophilic counterpart can be achieved as shown by water contact angle measurements and other methods. In addition to the interaction with solvent, the attachment and detachment of β-cyclodextrin (β-CD) polymers from such surfaces have been studied (Scheme 10, top). While the neutral hydrophobic PFTMA brush is capped by the β-CD polymer, the surface properties can be switched to hydrophilic via a redox stimulus addressing the ferrocene/ferrocenium couple entailing de-capping of the β-CD polymer. Reversible switching between the hydrophilic and the hydrophobic surface situation suggests a well-regulated and completely reversible surface oxidation and reduction process.24 The same host–guest interaction between ferrocenyl units and β-cyclodextrin has also been used the other way round: a gold surface covered with β-CD-modified self-assembled monolayers (SAMs) interacts with a linear ferrocenyl-grafted polymer in its neutral hydrophobic state forming a multivalent host–guest complex (Scheme 10, bottom). Electrochemical switching of this layered structure leads to a change in charge and polarity of the Fc-grafted polymer and detachment from the β-CD units at the surface which in total can be described as a host–guest driven controlled adsorption/desorption of ferrocenyl-functionalized linear polymers.32
 | ||
| Scheme 10 Sketch of switchable host–guest interaction on surfaces. | ||
While the above mentioned examples are immobilized surface layers on gold which likewise serve as an electrode and interface to apply the redox stimulus, the fruitful host–guest interaction between ferrocenyl units and β-cyclodextrin can be used directly in solution with striking rheological consequences. Thus, the mixture of two hydrophilic copolymers with β-CD or ferrocene pendant groups in solution forms a hydrogel for which the host–guest interaction between β-CD and ferrocene is the dominant driving force. The hydrogel undergoes a gel–sol transition after the addition of an oxidizing agent like FeCl3 (oxidation to Fc+ and detachment) and returned to its gel state when a reducing agent like ascorbic acid was added (reduction to Fc and attachment). The strategy has been used for the preparation of stimuli-responsive polymer networks with reversible linkers (Scheme 11).34
 | ||
| Scheme 11 Principle of redox responsive gel–sol transition. | ||
The underlying concept has been developed further to prepare an electrochemically driven redox responsive supramolecular self-healing hydrogel by the same group. The self healing properties are endowed by the dynamic nature of the host–guest interaction between the polymer chains.35 Using a cholesterol based gelator, ferrocene has been introduced as a pendant unit into dicholesteryl derivatives and the gelation behavior of these compounds has been explored.23 Interestingly, the gel of one of these gelators with n-decane shows thermo-, mechanical-, acoustic-, and even chemical-stimulus responsive sol–gel phase transition properties but not the expected redox responsiveness to the gel systems. The latter finding is particularly surprising and may be attributed to the choice of the special oxidant used in this investigation.
With phospholipids, a closely related natural building block for layered self-assemblies, liposomes with covalently bound ferrocenyl groups as pendant redox active units have been designed for studying membrane-bound electrochemical reactions, which are important in the design of redox-sensitive liposome delivery systems.22 This organometallic functionalization of the liposome surface shows improved stability compared with quinone-functionalized liposomes which are known to be unstable over time. The redox activity of the ferrocenyl units is suggested to control the permeability and/or integrity of the liposome for drug delivery purposes depending on the redox state.
Owing to their topological difference in solubility, mechanical flexibility, redox activity, polarity etc., block copolymers are useful polymer architectures especially with respect to ordered polymer assemblies. Triblock polymers of the type A–B–A in which a rather soft polyisobutylene block (B) connects two rather hard terminal polyvinylferrocene blocks (A) assemble even in dilute (10−2–10−3 mg ml−1) toluene or THF solutions.39 Nevertheless, thin films of the copolymers show reversible single electron redox behavior similar to that of ferrocene. Moreover, polyvinylferrocene has been combined with polylactide as a degradable polyester component to form diblock and miktoarm star polymers (Scheme 12).40 In miktoarm star polymers the arms attached to the core unit are not identical (greek: miktos = mixed). The synthesis starts from polyvinylferrocene (PVFc) obtained via carbanionic polymerization followed by termination affording mono- and dihydroxyl-functionalized PVFcs for the subsequent polymerization of L-lactide via catalytic ring-opening polymerization. Both the linear and the star-shaped block copolymers self assemble into spherical structures with well-defined diameters when exposed to a selective solvent for polylactide.
 | ||
| Scheme 12 Degradable diblock and miktoarm star polymers. | ||
VfcGE based copolymers obtained via anionic ring-opening copolymerization of VfcGE and ethylene oxide (EO) generate stimuli-responsive, multifunctional poly[(vinyl ferrocenyl glycidyl ether)-co-(ethylene oxide)] (P[VfcGE-co-EO]) copolymers with an intact vinyl function at ferrocene. An interesting feature of these organometallic copolymers is the tunable cloud point by partial or complete oxidation of the ferrocene moieties to ferrocenium ions, based on the water insolubility of the neutral polymer and an increased polarity and solubility of the oxidized form.14 The free radical polymerization of the same monomer (VfcGE) leads to polymers with epoxide side chains at each pendant ferrocene unit. Such a polymer was used to generate redox-responsive protein nanoparticles with bovine serum albumin (BSA) by nucleophilic ring-opening of the pendant epoxides.14 To show the redox responsivity of these nanoparticles, the ferrocene units have been oxidized with hydrogen peroxide under slightly acidic conditions. Upon oxidation, the formation of large aggregates has been observed probably due to electrostatic interactions. Transmission electron microscopy (TEM) imaging showed no distinct nanoparticles after oxidation, but the formation of supramolecular aggregates opening the way to redox-controlled precipitation.
3.2 Modified charge transport and electric potential
One of the salient features of polymers with pendant ferrocenyl units is their ability to undergo multiple redox processes at the peripheric ferrocene cores affecting the properties of the backbone via conjugative or electrostatic effects. Depending on the dynamic situation such redox processes may occur individually or in a cooperative fashion. Therefore, the charge transport and the communication between redox centers are of interest in such redox polymers. In general, redox polymers are conducting macromolecules that contain spatially and electronically localized redox sites which means that they are electroactive; however, they lack the delocalized electronic structure of conjugated polymers with the charge transport therefore occurring via a site-to-site hopping mechanism. Casting of such redox polymers on electrode surfaces allows us to trigger a redox stimulus via electrochemical means which is a way of electronically switching the molecular properties, or in turn, changing the molecular properties accounting for an electronic signal. This connection provides an interface between molecular systems and electronic devices, which offers huge potential for various applications (see Section 4).Electrodeposited films of carbosilane-based hyperbranched polymers bearing alkyl chain branches of different lengths (C3 and C11) with pendant ferrocenyl units have shown to be efficient redox mediators for the electrocatalytic reduction of oxygen as well as the reduction and oxidation of hydrogen peroxide. The polymer-coated electrodes have been used successfully as electrochemical sensors showing the characteristic behaviour of interacting ferrocene units. Films of the more flexible polymethyldiundecenylsilane with C11 branches exhibit higher electrocatalytic activity than those with C3 branches leading to improved sensitivity and detection limit values.27 Moreover, the same polymers have been employed as amperometric biosensors for NADH in combination with Pt nanoparticles (PtNPs). The catalytic synergy of PtNPs with the interacting ferrocenes is related to the polymer structure as well as protecting the surface against deterioration. In addition, the polymer/PtNPs/Pt electrodes give the attractive possibility of lowering the potential of the NADH oxidation, thus opening the way to serve as an electrochemical support for the incorporation of dehydrogenase enzymes such as alcohol dehydrogenase (ADH). Based on these findings, novel amperometric alcohol biosensors were successfully prepared with ADH showing a higher affinity for methanol than for ethanol, with a wide linear range to 30 mM and sensitivities of 0.957 and 0.756 μA mM−1 cm−2.28 Similarly, a biosensing system for xanthine detection has been designed by immobilizing xanthine oxidase (XO) via covalent attachment on a redox copolymer with pendant epoxy and ferrocene moieties (poly(glycidyl methacrylate-co-vinylferrocene = P(GMA-co-VFc)) on an electrode surface (Scheme 13).
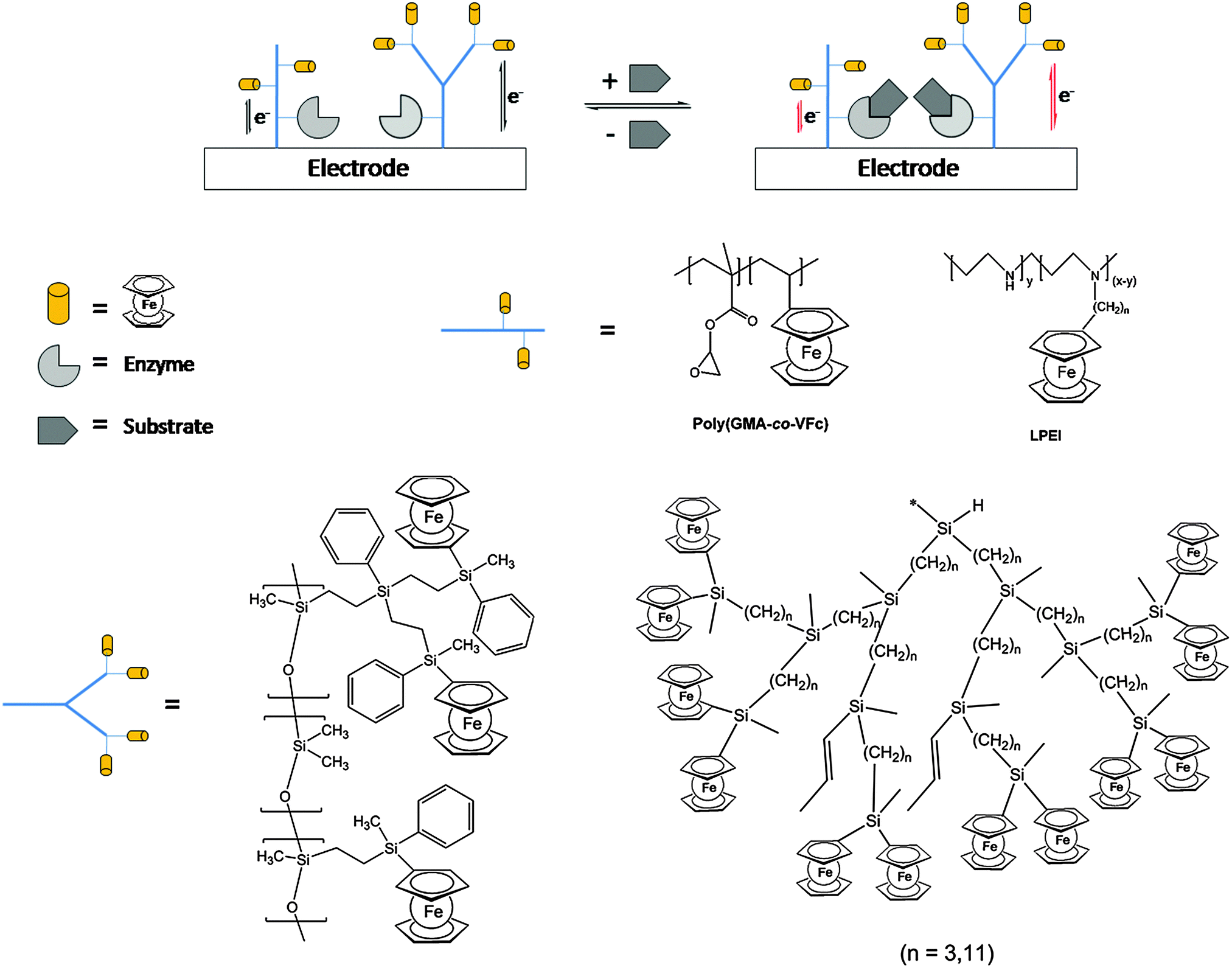 | ||
| Scheme 13 Sketch of the enzyme activity influencing the redox interaction of the redox polymer with the electrode surface for selected polymer architectures. | ||
The enzyme activity affects the ferrocene redox couple and therefore the electrode potential which can be used to track the xanthine concentration.12 In redox polymers the distance between the redox active pendant group and the polymer backbone, and the flexibility of the spacer tethering the pendant group can have a significant impact on the overall electrochemical properties which has been systematically explored for the linear poly(ethyleneimine) polymers (LPEI) (Schemes 7 and 13) in which the ferrocenyl units are connected to the backbone via a single-, three or six-carbon spacer. It has been suggested that electron transfer in redox films is primarily due to collisions between reduced and oxidized redox centers; therefore, a longer tether increases electron transfer by allowing the redox center to sweep out a larger volume element thereby increasing the number of successful electron-transferring collisions. The findings could nicely demonstrate that the extension of the ferrocene groups away from the LPEI backbone eliminates the multiwave redox behavior and oxidative degradation observed in cross-linked films of Fc-C1-LPEI. An additional benefit of increasing the lengths of tethers linking redox centers to the polymer backbone is the possibility to assess the rate at which electrons are transferred between an enzyme's redox center and the polymer's redox sites. However, no correlation between mediator spacing and electron transport or electronic communication with the enzyme glucose oxidase (GOX) was observed, in contrast to previous reports of electron transport increasing with spacer length. Nevertheless, it was nicely demonstrated that both Fc-C3-LPEI and Fc-C6-LPEI are able to exchange electrons with the FAD centers in GOX producing current densities at saturation of ∼600–1000 μA cm−2. Surprisingly, the redox polymer (Fc-C3-LPEI) with the lowest electron transport produced the highest enzymatic response (>1 mA cm−2), suggesting that other factors (e.g., polymer–enzyme complexation) are relevant as well. Increasing the spacer resulted in a six-fold increase in the electrochemical stability of cross-linked redox polymer films. The stability observed in the electrochemical response of the cross-linked Fc-C3-LPEI and Fc-C6-LPEI films translates to an enhanced stability in the sensor response under continuous operation in comparison with Fc-C1-LPEI.31 As mentioned above, the motional flexibility of the redox active ferrocenyl groups increases with the distance (spacer length) of pendant ferrocenyl units from the polymer backbone. Another way of achieving these effects is to take advantage of an inorganic polymer backbone and its inherent flexibility and improved diffusion properties. Functionalized polysiloxanes bearing small appended redox-active dendritic wedges, with electron-donor ferrocenyl units are air- and moisture-stable and soluble in common organic solvents. Solution electrochemical studies showed that all the ferrocenyl redox units present in the ferrocenyl dendronized polysiloxanes are electrochemically independent. Using these properties, the feasibility of modifying the electrode surfaces with stable electroactive films of these siloxane-based polyferrocenyl dendronized molecules has been demonstrated (Scheme 13).26 The polyphosphazene backbone is isoelectronic and isosteric to that of polysiloxanes. Polyphosphazene polymers with varying degrees of functionalization with pendant ferrocenyl units are able to show high torsional mobility as reflected by low Tg values. Such polymers have been derived from backbones comprised of nearly equal portions of randomly distributed [(Ph)(Me)PN] and [Me2PN] units in which the methyl groups serve as attachment sites for the tethered ferrocenyl units (Scheme 14). Compared with analogous polymers in which all phosphorus atoms carry one phenyl group rather than smaller Me groups, mobility and diffusion coefficients of the terpolymers are larger in solution and appear to be mostly independent of the degree of substitution with ferrocene groups. Reversible electrochemistry was observed for the ferrocene moiety in polymers with the phosphazene backbone acting as an electron-withdrawing group in the range of 433–492 mV (vs. Ag/AgCl). In evaporatively cast films, the degree of coverage with electroactive groups decreases by over 2 orders of magnitude when the degree of substitution with Fc groups decreases from 50 to 15%. This suggests that a lower number of ferrocenyl groups are effectively redox active due to the morphology of the cast film and the increased separation distance between them, lowering electron diffusion via through space electron hopping. Indeed, scanning electron microscopy revealed that the evaporatively cast films and the films deposited during redox cycling in solution have different morphologies.25
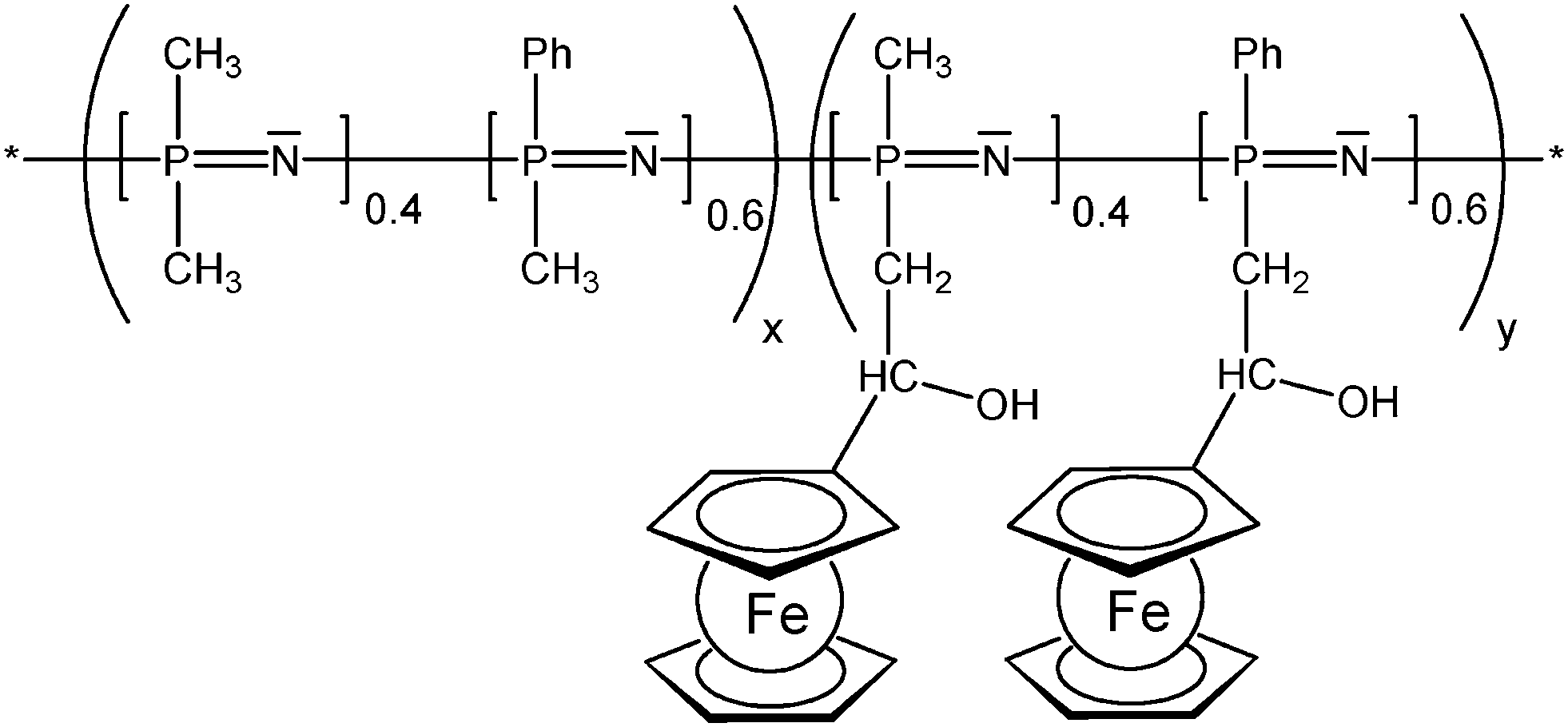 | ||
| Scheme 14 Randomly distributed pendant ferrocenyl units at a polyphosphazene backbone (x = 0.5-0.85, y = 0.5-0.15). | ||
Similar to the electron withdrawing properties of the phosphazene backbone, borylated pendant units containing ferrocene are available with polystyrene backbones. Conjugation of the electron rich ferrocene unit to boron gives rise to polymers, the Lewis acidity of which may be tuned by the oxidation of the metallocene. In support of this the oxidation potential of the pendant ferrocenyl groups was found to be elevated due to the electron withdrawing nature of the adjacent boron center. Anion complexation with fluoride has been studied and showed a distinct polymer effect compared with analogous low molecular model compounds.36,37
For redox polymers immobilized on electrode surfaces both the spacer between the redox center and the polymer backbone and the distance between the redox center and the electrode surface become relevant. Therefore, the preparation of block copolymer brushes with varying sequences and chain lengths of FcMA segments was conducted to interrogate the effects of spacing from the indium tin oxide (ITO) electrode surface on the electrochemical properties of a tethered electroactive film (Scheme 15). Cyclovoltammetry (CV) measurements of these tethered thin films confirmed that the block sequence directly altered the electrochemistry of the films, as observed by differences in cathodic and anodic peak shape and potential. Moreover, significant solvent effects were observed in the CV of polymer brushes which was anticipated to arise from both impeded counterion diffusion into tethered thin films and differences in electron transfer rates between electroactive groups.10
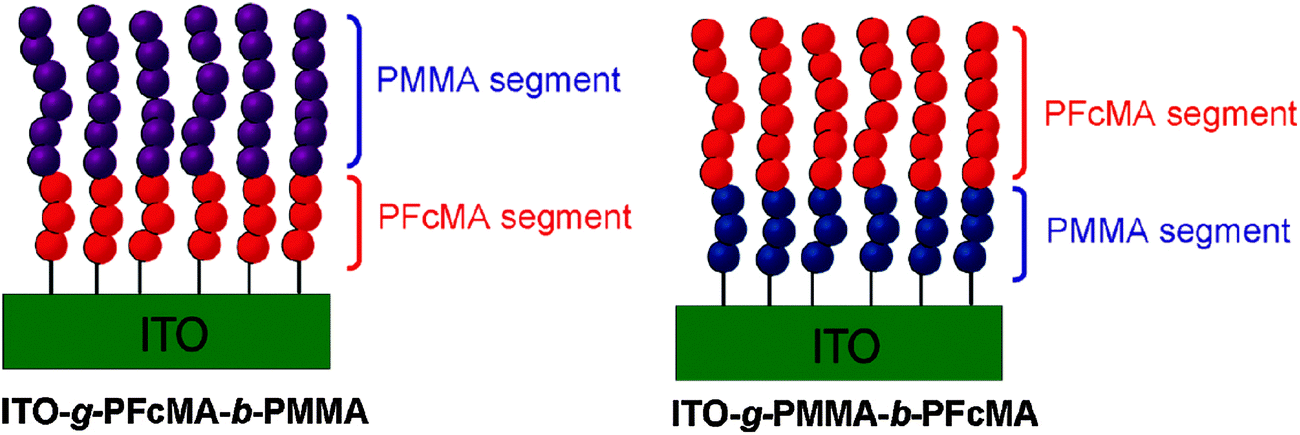 | ||
| Scheme 15 Variation of the distance between PFcMA and the electrode (ITO) in surface bonded block-copolymers. Reprinted with permission from ref. 10. Copyright (2010) American Chemical Society. | ||
Closely related to this, microdomains of different sizes and shapes in thin films of ferrocene-containing diblock copolymers have been prepared on a gold electrode with varying contents of the ferrocenyl substituted component. Based on ellipsometry, AFM and electrochemical measurements it was suggested that electron propagation across ferrocene moieties in the PAEFc microdomains took place via electron hopping accompanied by counterion migration through the solvent-swollen films, as with ferrocene-containing homopolymer films. The electrochemical behavior suggests that films with a larger PAEFc fraction like PS154-b-PAEFc51 and PS154-b-PAEFc26 contain continuous PAEFc microdomains extending from the electrode to the surface, in contrast to lower PAEFc fraction films (PS154-b-PAEFc12) which contain isolated PAEFc microdomains buried within the polystyrene (PS) matrix (Scheme 16). Electron propagation took place only through PAEFc microdomains that could electrically communicate with the underlying electrode. These results provide guidance for designing redox-active metalloblock copolymers for various applications, which include electrocatalysis, electrochemical mediation in enzyme sensors, and redox-controlled molecular deposition.11
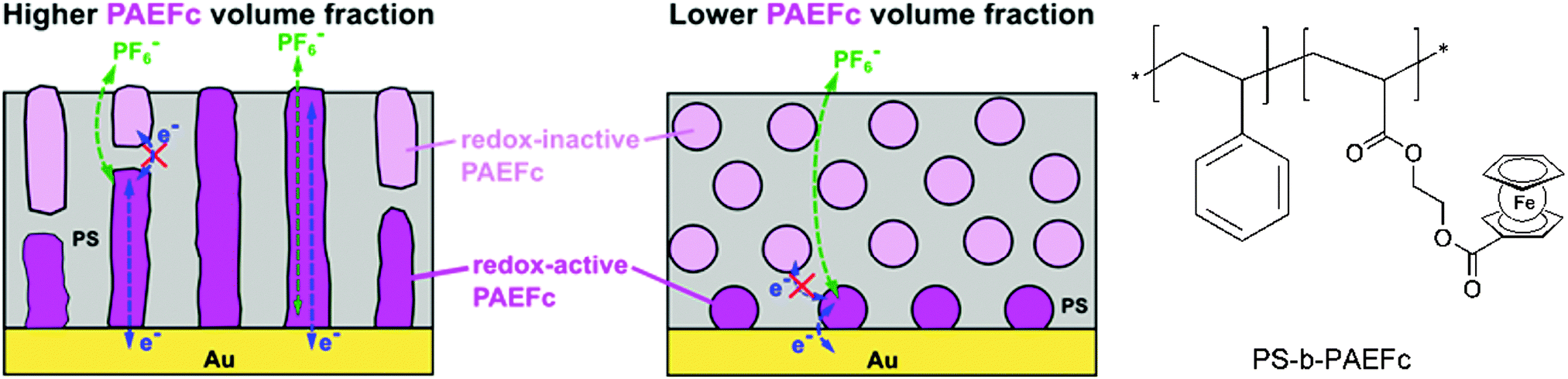 | ||
| Scheme 16 Microdomains of different sizes and shapes in thin films of ferrocene-containing diblock copolymers (left, reprinted with permission from ref. 11. Copyright (2015) American Chemical Society) and sketch of the polymer structure (right). | ||
With respect to rigid backbones as in conjugated polymers, a polymer-modified electrode has been prepared based on a polythiophene network with pendant ferrocenyl units for which the electronic interaction between the backbone and the pendant units has been demonstrated. The ferrocenyl oxidation was assisted by the conducting polymer chain while exposure to ferrocene in solution resulted in a prompt increase as the conductivity of the film is switched “ON”.30 Ferrocenylene units have been attached to glassy carbon electrode surfaces via alkylidene chains for which the redox activity of the pendant ferrocene units can be employed to assess the coverage of the surface with these grafted strands. Coverage levels were found to be high, and surface concentrations of ferrocene units >10−8 mol cm−2 were reached producing chemically and electrochemically stable layers suitable for efficient storage of electric charge.21
3.3 Electrochromic properties
Attachment of pendant ferrocenyl groups to a conjugated polymer backbone endows these materials with electrochromic behavior. By adjusting an appropriate electric potential difference the redox active ferrocene unit can be switched from the neutral ferrocene to the cationic ferricenium form and vice versa. These changes are accompanied by characteristic color changes between the reduced and oxidized forms. However, more than two colored states may be accessible because the interaction of the whole ensemble of ferrocene units at the same or neighboring polymer chains may be relevant and, in addition, the redox properties of the conjugated polymer may interfere or simply overlay the ferrocene properties (Scheme 17). | ||
| Scheme 17 Sketch of combinations of redox steps in the main chain and/or at the pendant units enabling multicolored electrochromism. | ||
For example EDOT-Fc based polymers with pendant ferrocenyl groups (cf.Chart 2) can be switched between purple and blue. The visible absorption of Fc+ groups translated into a blue colored oxidized state for the polymer film. Moreover, the partial oxidation of the polymer allowed the detection of a gray colored transition state as a consequence of mixing different colored states of the main chain and ferrocenyl groups. The effect of the ferrocenyl unit is clearly visible by substitution with other units which leads to the variation of the optical band gaps in the range 1.69–1.77 eV and different numbers of achievable colored states.19 Furthermore, the incorporation of the ferrocenyl substituent enhanced the optical contrast in the visible region which in turn resulted in better transparency for the oxidized state. The multicolored electrochromism observed can be attributed to the independent electrochromic switching of the pendant ferrocenyl units (cf. A and B in Scheme 13).20
A ferrocene functionalized, p- or n-dopable conjugated polymer was synthesized by the electrochemical polymerization of FMOPD on platinum and transparent ITO electrodes using potential cycling and constant potential electrolysis.18 The potentiodynamic technique was found to be more suitable for the electropolymerization of FMOPD, because films produced potentiodynamically are very stable and adherent to the electrode surface compared to the ones produced potentiostatically. This is in agreement with the decrease in optical contrast after 300 cycles for p-doping and n-doping processes, which is 13% at maximum. The electroactivity of the polymer film was retained even after hundreds of cycles between their redox states. Spectroelectrochemical analysis demonstrated that the polymer film reveals a reversible cycling with distinctive color changes between neutral and reduced/oxidized forms involving metal-to-ligand charge transfer (MLCT) and/or intraligand charge transfer (ILCT) from Fc moieties to the polymer skeleton (Scheme 18).
 | ||
| Scheme 18 Sketch of metal-to-ligand charge transfer (MLCT) and/or intraligand charge transfer (ILCT) from Fc moieties to the polymer skeleton in poly-FMOPD. | ||
3.4 Thermal stability
The thermal stability of polymers can benefit from the presence of ferrocenyl units as pendant substituents which extends the possible temperature range for applications. On the other hand, metal containing polymers may serve as useful precursors for ceramic materials or metal based catalysts as well. For such purposes the possibility of processing the preceramic polymer into films or fibers is an additional important aspect. Inorganic polymers like polysiloxanes are thermally more robust than polymers with organic backbones. Usual degradation pathways of polysiloxanes involve hydrolytic cleavage of the Si–O backbone at high temperatures and in the presence of basic impurities or oxidative attack on the pendant side chains. In turn, ferrocene as a pendant unit is able to improve the thermal stability of siloxanes.29For certain siloxane based polymers structural aspects can be correlated with the thermal stability of the polymer. In this case the ferrocenyl units have been attached to the backbone as part of dendritic wedges (Scheme 19, left). The thermal stability of these ferrocenyl polysiloxanes was found to be strongly dependant on the size of the ferrocenyl dendritic fragment appended to the siloxane backbones: the more extended (Fc-rich) the wedges are, the higher the thermal stability is. The latter polymers are thermally stable to ca. 200–250 °C under N2 and yield ceramic residues in relatively high amounts at more elevated temperatures as followed by TGA.26 For a related polymer, phosphorus atoms have been used as nodes to which several ferrocenyl units are attached via ethylidene spacers with the pendant nodes to the polymethacryl main chain.41 This polymer is a remarkable polyelectrolyte with the positive charge localized at the phosphonium center and not at the ferrocenyl units as in many other examples (Scheme 19, right). The thermal decomposition of such polymers with varying chain lengths occurs in the range 310–500 °C. Pyrolysis of thin films at higher temperatures resulted in ceramization furnishing magnetite besides carbon-, phosphorus- and oxygen-rich phases. The extension of this approach to bimetallic systems has been accomplished starting from heterobimetallic diblock copolymers in which blocks with pendant ferrocenyl units have been combined with blocks containing pendant cobaltocenium units.42 Depending on the ratio of the lengths of the cobaltocenium and the ferrocene containing blocks the products of pyrolytic treatment vary between Fe/Co-phosphides and Fe/Co-alloys with different magnetic properties (Scheme 20). The metal rich phases are obtained embedded in carbon matrices derived from the organic part of the pre-ceramic polymers which were shown to consist of amorphous carbon films and multiwalled carbon nanotubes. By contrast, the formation of carbon matrices can be avoided by pyrolysis upon exposure to UV light and ozone. This has been demonstrated for the relatively carbon-rich triblock copolymer poly(ethylene oxide)-b-poly(2-(methacryloyloxy)-ethyl ferrocenecarboxylate)-b-polystyrene where ordered Fe2O3 nanoparticles have been obtained at 1200 °C.43
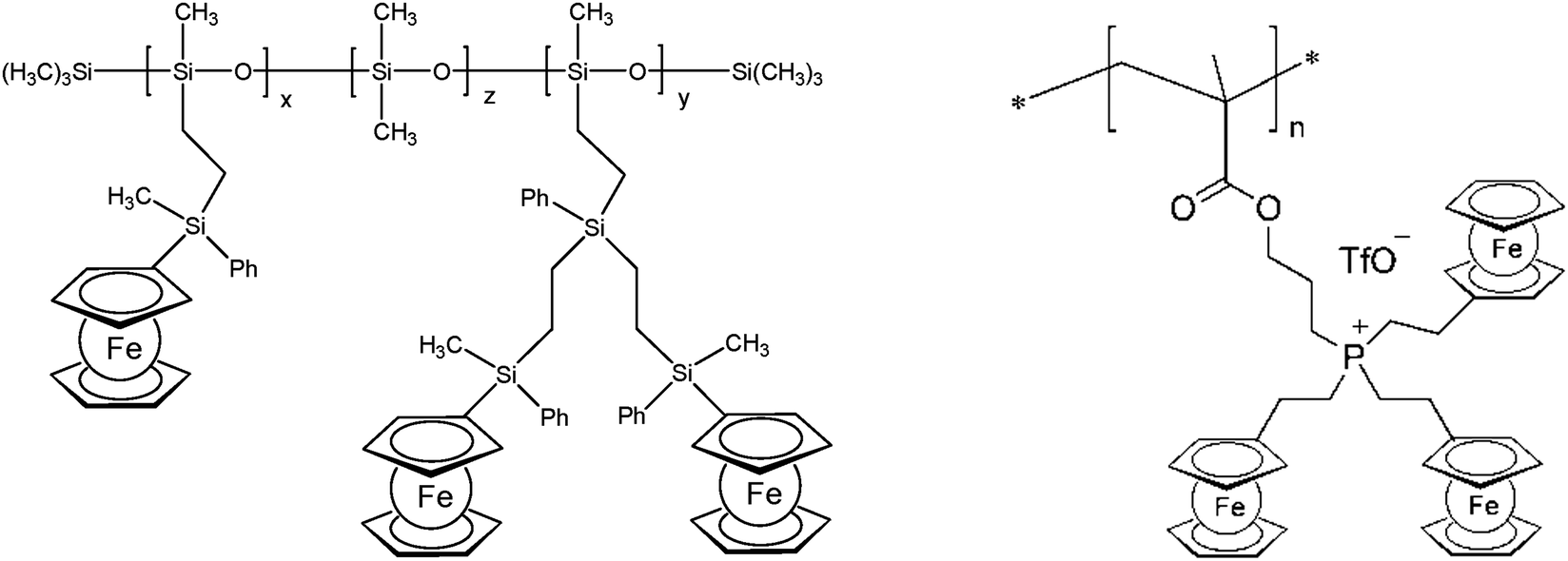 | ||
| Scheme 19 Left: Polysiloxanes with ferrocene containing dendritic wedges (x + y = 30–35%, z = 65–70%). Right: Phosphonium center as the attachment point for ferrocene. | ||
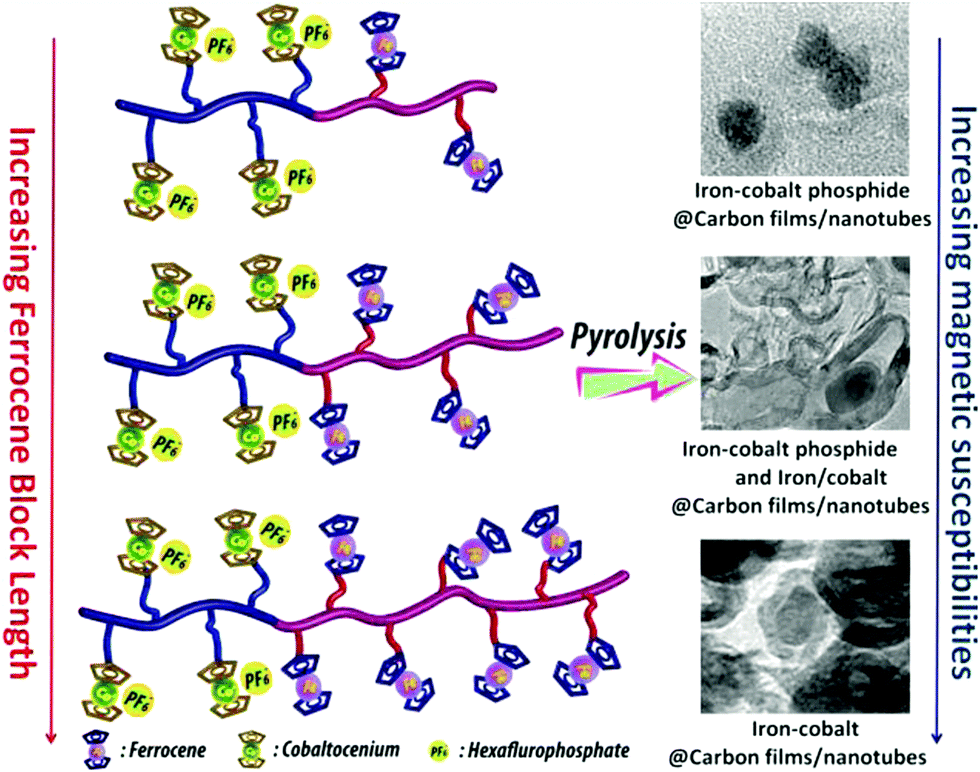 | ||
| Scheme 20 Variation of the metal ratio in heterobimetallic diblock copolymers and influence of the magnetic behavior. Reprinted with permission from ref. 42. Copyright (2014) American Chemical Society. | ||
As a further example for heterobimetallic polymers the accumulation of metallocene units in the backbone in addition to the pendant units has been achieved in polyferrocenylsilane with pendant ruthenocenyl groups available from the corresponding [1]ferrocenophane which is a promising preceramic material in light of the known catalytic activity of FeRu nanoparticles.15 Likewise, nanostructured ceramic particles have been obtained by thermal treatment (1000 °C) of ferrocenyl substituted phosphinoborane polymers.16 Interestingly, the thermal stability of such polymers is with 250 °C already high for the unsubstituted ferrocenyl case and increases further to 320 °C by attaching aminoborane and especially quaternary ammonium units to the ferrocenyl system (cf.Scheme 3). With respect to the stability of polymers under high energetic conditions hybrid organic–organometallic polymers with pendant ferrocene units have been used to prepare photoresist materials for applications in nanopatterning using extreme ultraviolet lithography (EUVL) where the pendant ferrocene units improved the thermal stability of the resultant hybrid system.7
4. Polymer based devices and prospective applications
Given the wealth of desirable properties emerging from the interplay of polymer backbones with pendant ferrocenyl units, various applications in the fields of sensors, molecular electronics, self-healing composites, drug delivery, switchable surfaces (color/wettability) can be envisioned. While some of them have already been realized others are currently being developed with more to come in the future and prototypes of devices based on such material starting to emerge as well. In the following section the current status on devices and application perspectives are briefly outlined. Based on their operational mode and properties, they have been subdivided into sections where polymers with pendant ferrocenyl units are attached to either charged or uncharged interfaces or being employed as bulk material.4.1 Charged interfaces
Interfaces to which a voltage is, or may be, applied are especially prone to interact with redox polymers in vicinity. In principle, polymers may be connected by covalent or ionic bonds, chemisorbed or physisorbed. Electrodes are typically charged interfaces and offer the possibility of controlling and switching molecular properties when a voltage is applied. For polymers with pendant ferrocenyl units changes in the optical properties in the visible region can be observed especially with conjugated polymer backbones (cf. Section 3.3). These systems give rise to multicolored electrochromic devices where ferrocenyl substitution results in enhanced optical contrast in the visible region and in turn better transparency for the oxidized state.20 Using a translucent electrode system potentiodynamically produced redox polymer films have been shown to be chemically and mechanically very stable and adherent to the ITO electrode surface. The longevity of the polymer film has been demonstrated by cycling between their redox states after which the electroactivity was retained even after several hundred cycles.18 Therefore, films of conducting polymers with pendant ferrocene units have high potential for application in electrochromic devices which may include displays or screen applications while electrochromic windows can be used for protection against (sun) light aiming at improved temperature and energy management in buildings.Sensing applications have been demonstrated by monitoring the activity of enzymes attached to a polymer with pendant ferrocene moieties on an electrode surface. Thus, a ferrocene based electrochemical biosensing system for xanthine detection in the μM range has been designed (Scheme 21). Since the xanthine level is a marker for the metabolic degradation of adenine, it is related to freshness of food or certain clinical disorders. Eventually, ferrocene based redox polymers may help to maintain food safety and to provide a device for medical monitoring of this parameter.12
 | ||
| Scheme 21 Sketch of a xanthine biosensor. Reproduced from ref. 12 with permission from Elsevier. | ||
Owing to their flexibility and diffusion properties, polysiloxanes are attractive backbones for redox polymers with respect to stable electroactive films. With pendant ferrocenyl groups of various kinds, such polymers are air- and moisture-stable and soluble in common organic solvents which is relevant for processing these materials into devices. Moreover, the thermal stability of such polymers increases with the number of pendant ferrocenyl groups. Electrode surfaces coated with films of such polymers behave as chemically modified electrodes and display nearly ideal behavior.26,29 Incorporation of sulfur atoms into the side chains provides outstanding adsorption properties of precious metal surfaces and electrodes. Taking into account the exceptional reactivity of sulfur and its importance in polymer chemistry, biology, and medicine, sulfur-rich polysiloxanes are highly desirable materials.44–46
4.2 Dielectric interfaces
On dielectric surfaces the charge transport via the surface is usually negligible. A redox stimulated responsive behavior may be triggered, however, by redox active components in solution in contact with the respective redox polymer. The resulting polarity change can be used for devices with tunable properties. A prominent example is the switchable surface wettability which owing to its reversible nature opens the way to smart surfaces which are hydrophilic or water repellant depending on their redox states.9 Besides tuning the rather unspecific interaction of smart surfaces with solvents of different polarity, quite specific host–guest chemistry and molecular recognition can be switched using such principles (cf. Section 3.1). Multifunctional polymers can be attached or detached depending on the redox state. The well-regulated and completely reversible nature of such processes can be achieved and used for the assembly and disassembly of layered polymer based devices.24Aiming at catalytic applications periodic lamellar structures in the nanometer range have been obtained by the self-assembly of block copolymers derived from vinylferrocene and isoprene. The crosslinked isoprene blocks serve as supports for the polyvinylferrocene chains which upon oxidation show good catalytic activity in Michael addition reactions.47 Unlike the situation of catalysts on siliceous supports, the rigidity and diffusion properties of the supporting isoprene blocks can be modified via the degree of crosslinking in such self-assembled heterogeneous catalysts.
Looking at electronic devices, the redox activity of pendant ferrocenyl units may be used to prepare components applicable as logic gates. The combination of a conjugated polythiophene polymer network with grafted ferrocenyl units can be used to switch the conductivity of the film to “ON” depending on other stimuli.30 Redox polymers with pendant ferrocene units have been used to prepare photoresist materials for applications in the nanopatterning of semiconductors using extreme ultraviolet lithography (EUVL) where the reduced wavelengths allow the fabrication of smaller features and patterns (Fig. 1).7 A poly(styrene–vinylferrocene) copolymer has been employed as a micropatterned catalyst for the formation of carbon nanotubes which allowed the fabrication of flexible carbon nanotube composites for which the location and density of conducting channels within the composite can be easily controlled by soft lithography patterning.48 Such composites can then be used for applications in devices that require conductive channels within a flexible matrix.
 | ||
| Fig. 1 Nanopatterns obtained from ferrocene based photoresist. Reproduced from ref. 7 with permission from The Royal Society of Chemistry. | ||
4.3 Unsupported polymer networks
Even without the boundaries and special features of interfaces redox polymers with pendant ferrocenyl units show fascinating ways of interaction which may be accompanied by significant changes in their rheological properties. These switchable properties do not only affect rheological parameters but in addition open the way to self healing composites and drug delivery systems.Stimuli-responsive polymer networks have been prepared using combinations of two hydrophilic copolymers with β-CD (Fig. 2(b)) and ferrocene (Fig. 2(a)) pendant groups for which the host–guest interaction between β-CD and ferrocene is the dominant driving force. Reversible transitions from gel (c) to sol (d, e) can be triggered via oxidation and from sol to gel via reduction giving rise to an electrochemically driven redox responsive supramolecular self-healing hydrogel.
 | ||
| Fig. 2 Redox switchable sol–gel system. Reproduced from ref. 35 with permission from The Royal Society of Chemistry. | ||
In principle one might envision sequestration or release mechanisms triggered by reducing or oxidizing metabolites of living cells. Therefore the biocompatibility of the material has been established by cytotoxicity measurements using mice cells.34,35 In addition, vesicular structures like liposomes with covalently bound ferrocenyl groups are available as well, which are important in the design of redox-sensitive liposome or carrier polymer delivery systems from which the bioactive compound may be released in the intracellular space.22,33
5. Conclusions and perspectives
The field of stimuli-responsive redox polymers is a growing and diverse area of study. While the redox sites can in principle be incorporated directly into the polymer backbone, covalently attached as pendant groups, or electrostatically bound, this review has focused on polymers with pendant ferrocene units in the condensed phase. Since the electrochemical properties (e.g., redox potential, electron transport) of a redox polymer depend on a number of variables like the type of polymer backbone, the type of redox mediator, the concentration of the redox mediator, and the polymer charge, the interplay between these parameters is critical in tailoring the properties of a redox polymer for a specific purpose. Recent reports of polymers with pendant ferrocene units indicate progress in the design of stimuli-responsive systems. Redox switchable systems triggered by oxidants or electrochemically via electrodes are especially popular owing to the stability and reversible nature of the ferrocene/ferricenium couple. Using this approach, charge, polarity, color (UV-vis range) and hydrophilicity of polymers, polymer functionalized surfaces and polymer derived networks may be tuned or switched in a controlled fashion, based on which applications like smart surfaces, windows, blinds, drug delivery and energy storage systems started to emerge. The redox activity can be used the other way round as well, by influencing the redox couple by polymer entangled or immobilized receptors, the status of which can be monitored electrochemically, giving rise to sensing devices especially with respect to biosensing applications and consumer protection.However, the potential of ferrocene grafted polymers clearly goes beyond their redox properties, and dynamic host–guest systems allow the construction of self healing composites and gels that are responsive to thermal, mechanical, acoustic, and even chemical stimuli. The design of such polymers and polymer networks remains a central challenge for synthetic chemists. Based on these efforts, the design of new adaptive materials is in progress with implications for the fascinating intersection of chemistry, physics and biology.
Acknowledgements
The author would like to thank the Deutsche Forschungsgemeinschaft (DFG) and the Austrian Science Fund (FWF) for continued financial support and the EU-COST network CM1302 “Smart Inorganic Polymers” (SIPs).References
- P. Nguyen, P. Gomez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515–1548 CrossRef CAS PubMed.
- I. Manners, Macromol. Symp., 2003, 196, 57–62 CrossRef CAS.
- I. Manners, Science, 2001, 294, 1664–1666 CrossRef CAS PubMed.
- Ferrocene-functionalized polymer brushes: synthesis and applications, ed. X. L. Qun, K. En-Tang and F. G. Dong, CRC Press, 2012 Search PubMed.
- Y. Gao and J. M. Shreeve, J. Inorg. Organomet. Polym. Mater., 2007, 17, 19–36 CrossRef CAS.
- A. Abd-El-Aziz and I. Manners, J. Inorg. Organomet. Polym. Mater., 2005, 15, 157–195 CrossRef CAS.
- V. S. V. Satyanarayana, V. Singh, V. Kalyani, C. P. Pradeep, S. Sharma, S. Ghosh and K. E. Gonsalves, RSC Adv., 2014, 4, 59817–59820 RSC.
- M. Gallei, S. Tockner, R. Klein and M. Rehahn, Macromol. Rapid Commun., 2010, 31, 889–896 CrossRef CAS PubMed.
- J. Elbert, M. Gallei, C. Ruettiger, A. Brunsen, H. Didzoleit, B. Stuehn and M. Rehahn, Organometallics, 2013, 32, 5873–5878 CrossRef CAS.
- B. Y. Kim, E. L. Ratcliff, N. R. Armstrong, T. Kowalewski and J. Pyun, Langmuir, 2010, 26, 2083–2092 CrossRef CAS PubMed.
- G. Ghimire, Y. Yi, M. A. Derylo, L. A. Baker and T. Ito, Langmuir, 2015, 31, 12307–12314 CrossRef CAS PubMed.
- S. Z. Bas, E. Maltas, B. Sennik and F. Yilmaz, Colloids Surf., A, 2014, 444, 40–47 CrossRef CAS.
- A. Alkan, A. Natalello, M. Wagner, H. Frey and F. R. Wurm, Macromolecules, 2014, 47, 2242–2249 CrossRef CAS.
- A. Alkan, L. Thomi, T. Gleede and F. R. Wurm, Polym. Chem., 2015, 6, 3617–3624 RSC.
- M. Erhard, K. Lam, M. Haddow, G. R. Whittell, W. E. Geiger and I. Manners, Polym. Chem., 2014, 5, 1264–1274 RSC.
- S. Pandey, P. Lönnecke and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2014, 2456–2465 CrossRef CAS.
- S. Pandey, P. Lönnecke and E. Hey-Hawkins, Inorg. Chem., 2014, 53, 8242–8249 CrossRef CAS PubMed.
- H. Gulce, A. Yetkin, E. Akgul and A. Gulce, Thin Solid Films, 2013, 545, 81–88 CrossRef CAS.
- H. Akpinar, A. Balan, D. Baran, E. K. Unver and L. Toppare, Polymer, 2010, 51, 6123–6131 CrossRef CAS.
- S. Özdemir, A. Balan, D. Baran, Ö. Dogan and L. Toppare, React. Funct. Polym., 2011, 71, 168–174 CrossRef.
- V. Jouikov and J. Simonet, J. Appl. Electrochem., 2012, 42, 527–537 CrossRef CAS.
- D. Correia-Ledo, A. A. Arnold and J. Mauzeroll, J. Am. Chem. Soc., 2010, 132, 15120–15123 CrossRef CAS PubMed.
- P. He, J. Liu, K. Liu, L. Ding, J. Yan, D. Gao and Y. Fang, Colloids Surf., A, 2010, 362, 127–134 CrossRef CAS.
- L. Q. Xu, D. Wan, H. F. Gong, K.-G. Neoh, E.-T. Kang and G. D. Fu, Langmuir, 2010, 26, 15376–15382 CrossRef CAS PubMed.
- D. C. Kraiter, P. Wisian-Neilson, C. Zhang and A. L. Crumbliss, Macromolecules, 2012, 45, 3658–3668 CrossRef CAS.
- M. Zamora, S. Bruna, B. Alonso and I. Cuadrado, Macromolecules, 2011, 44, 7994–8007 CrossRef CAS.
- G. de la Cruz, H. Schüle, J. Losada, M. P. García-Armada, H. Frey, B. Alonso and C. M. Casado, Eur. J. Inorg. Chem., 2013, 44–53 CrossRef CAS.
- A. Jimenez, M. P. G. Armada, J. Losada, C. Villena, B. Alonso and C. M. Casado, Sens. Actuators, B, 2014, 190, 111–119 CrossRef CAS.
- I. Martinez-Montero, S. Bruna, A. M. Gonzalez-Vadillo and I. Cuadrado, Macromolecules, 2014, 47, 1301–1315 CrossRef CAS.
- T. V. Thang and C. Cougnon, J. Electroanal. Chem., 2011, 657, 79–83 CrossRef CAS.
- S. A. Merchant, M. T. Meredith, T. O. Tran, D. B. Brunski, M. B. Johnson, D. T. Glatzhofer and D. W. Schmidtke, J. Phys. Chem. C, 2010, 114, 11627–11634 CAS.
- G. V. Dubacheva, A. Van Der Heyden, P. Dumy, O. Kaftan, R. Auzely-Velty, L. Coche-Guerente and P. Labbe, Langmuir, 2010, 26, 13976–13986 CrossRef CAS PubMed.
- A. I. Mufula and E. W. Neuse, J. Inorg. Organomet. Polym. Mater., 2012, 22, 134–148 CrossRef CAS.
- H. Zhang, L. Peng, Y. Xin, Q. Yan and J. Yuan, Macromol. Symp., 2013, 329, 66–69 CrossRef CAS.
- L. Peng, H. Zhang, A. Feng, M. Huo, Z. Wang, J. Hu, W. Gao and J. Yuan, Polym. Chem., 2015, 6, 3652–3659 RSC.
- K. Parab and F. Jäkle, Macromolecules, 2009, 42, 4002–4007 CrossRef CAS.
- H. Li, A. Sundararaman, T. Pakkirisamy, K. Venkatasubbaiah, F. Schödel and F. Jäkle, Macromolecules, 2011, 44, 95–103 CrossRef CAS.
- N. L. Abbott and G. M. Whitesides, Langmuir, 1994, 10, 1493–1497 CrossRef CAS.
- F. Yan, T. Higashihara, R. Mosurkal, L. Li, K. Yang, R. Faust and J. Kumar, J. Macromol. Sci., Part A: Pure Appl.Chem., 2008, 45, 910–913 CrossRef.
- J. Morsbach, A. Natalello, J. Elbert, S. Winzen, A. Kroeger, H. Frey and M. Gallei, Organometallics, 2013, 32, 6033–6039 CrossRef CAS.
- A. Rabiee Kenaree, B. M. Berven, P. J. Ragogna and J. B. Gilroy, Chem. Commun., 2014, 50, 10714–10717 RSC.
- J. Zhang, Y. Yan, J. Chen, W. M. Chance, J. Hayat, Z. Gai and C. Tang, Chem. Mater., 2015, 26, 3185–3190 CrossRef.
- C. G. Hardy, L. Ren, S. Ma and C. Tang, Chem. Commun., 2013, 49, 4373–4375 RSC.
- A. Ciborska, E. Conterosito, M. Milanesio, K. Kazimierczuk, K. Rzymowska, K. Brzozowski and A. Dołęga, Eur. J. Inorg. Chem., 2015, 3059–3065 CrossRef CAS.
- Y. Li, H. Zhu, D. M. Andrada, G. Frenking and H. W. Roesky, Chem. Commun., 2014, 50, 4628–4630 RSC.
- S. Spirk, F. Belaj, N. Hurkes and R. Pietschnig, Chem. Commun., 2012, 48, 8398–8400 RSC.
- D. A. Durkee, H. B. Eitouni, E. D. Gomez, M. W. Ellsworth, A. T. Bell and N. P. Balsara, Adv. Mater., 2005, 17, 2003–2006 CrossRef CAS.
- E. Lahiff, C. Y. Ryu, S. Curran, A. I. Minett, W. J. Blau and P. M. Ajayan, Nano Lett., 2003, 3, 1333–1337 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |