
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective reaction of 2-carboxythioesters-1,3-dithiane with nitroalkenes: an organocatalytic strategy for the asymmetric addition of a glyoxylate anion equivalent†
Elisabetta
Massolo
a,
Maurizio
Benaglia
*a,
Andrea
Genoni
a,
Rita
Annunziata
a,
Giuseppe
Celentano
b and
Nicoletta
Gaggero
*b
aDipartimento di Chimica, Universita’ degli Studi di Milano, via Golgi 19, I-20133 Milano, Italy. E-mail: maurizio.benaglia@unimi.it; Fax: +390250314159
bDipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, via Mangiagalli, 25, Milano, Italy
First published on 7th April 2015
Abstract
An efficient organocatalytic methodology has been developed to perform the stereoselective addition of 2-carboxythioesters-1,3-dithiane to nitroalkenes. Under mild reaction conditions γ-nitro-β-aryl-α-keto esters with up to 92% ee were obtained, realizing a formal catalytic stereoselective conjugate addition of the glyoxylate anion synthon. The reaction products are versatile starting materials for further synthetic transformations; for example, the simultaneous reduction of the nitro group and removal of the dithiane ring was accomplished, allowing the preparation of a GABAB receptor agonist baclofen.
Introduction
Chiral α-keto esters are considered products of great importance, as starting materials for the preparation of highly functionalized compounds via synthetic manipulation of their functional groups.1 They also find successful applications in biochemistry and are an integral part of several biologically active natural compounds.2 Therefore, it is not surprising that the stereoselective synthesis of chiral α-keto esters is still the object of intense studies. Recently a novel approach was proposed by Johnson, who described a copper(II)-catalyzed aerobic oxidation reaction as a key step for the preparation of β-substituted α-keto esters, in a procedure where acetoacetate esters were used as glyoxylate anion synthons.3However, most of the reported synthesis of chiral α-keto esters rely on the umpolung strategy that allows the introduction of a nucleophilic acyl anion synthon,4N-heterocyclic carbenes are known for their ability to reverse the polarity of aldehydes, to generate acyl anion equivalents that have been widely employed in benzoin and Stetter-type reactions.5 More traditional methods involve the use of metalated aminonitriles6 and 1,3-dithiane-protected glyoxylates,7 prepared by deprotonation with stoichiometric amounts of a strong base.8 It must be noted that all of these methods suffer from intrinsic limitations and problems (limited general applicability, low yields, modest stereoselectivity, and strongly depending on reactant substrates); in addition, examples of direct synthesis of enantiomerically enriched keto esters are still very rare, especially those related to the preparation of enantiopure β-aryl-α-keto esters, where the strong acidity of the proton in the β position represents a real issue for any successful stereoselective methodology.
With the goal to develop a metal-free catalytic strategy for the synthesis of chiral β-substituted-α-keto esters, we decided to study the addition of 1,3-dithiane-2-carboxy derivatives to nitroalkenes. Surprisingly, a catalytic stereoselective version of this powerful transformation seems to be unknown.9 Here we wish to report the first enantioselective organocatalytic conjugate addition of 2-carboxythioesters-1,3-dithiane to nitroalkenes.
At the beginning of our investigation the chemical reactivity of different 2-carboxyester 1,3-dithianes was investigated in the presence of a cinchona-derived bifunctional catalyst A (Scheme 1)10.
 | ||
| Scheme 1 Preliminary studies of organocatalytic addition of dithianes to nitrostyrene. | ||
Commercially available 1,3-dithiane-2-ethylcarboxylate 2 in the presence of 20 mol% of quinine-thiourea derivative A did not react at all with nitrostyrene. The corresponding S-phenyl thioester 4 showed poor reactivity, affording the corresponding addition product 5 in low yields, even after prolonged reaction times, although in a remarkable 89% enantioselectivity (entries 2 and 3, Table 1). As expected 2-pyridylthioester 6 afforded adduct 7 in higher yields; unfortunately the reaction product turned out to be not very stable, thus preventing the determination of its enantiomeric excess. Finally the use of 2-S-trifluoroethyl carboxy-thioester-1,3-dithiane 8 gave the best results; after 24 hours at RT the stable conjugate addition product 9 was obtained in 71% yield and 87% ee.11
| Entrya | X | Product | t (h) | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|
| a Typical reaction conditions: 0.1 mmol of nitroalkene, 0.2 mmol of dithiane, 0.02 mmol of catalyst A, 1 mL of toluene. b Yields were determined after chromatographic purification. c Enantiomeric excess determined by HPLC on a chiral stationary phase. | |||||
| 1 | OEt | 3 | 24 | — | — |
| 2 | SPh | 5 | 24 | 16 | 89 |
| 3 | SPh | 5 | 72 | 25 | 87 |
| 4 | SPy | 7 | 24 | 41 | n.d. |
| 5 | SCH2CF3 | 9 | 24 | 71 | 87 |
The use of 1,1,1-trifluoroethyl thioester was documented for the first time by Barbas in an organocatalytic Michael reaction.12 Later, our group took advantage of low pKa of the protons in the α position of the carboxy group in the trifluoroethyl thioesters to realize the first organocatalytic direct aldol reaction between a thioester and an aromatic aldehyde.13 Due to its favorable reactivity profile (entry 5, Table 1), thioester 8 was selected as a reagent of choice and its addition to nitrostyrene was studied in the presence of different chiral bifunctional organocatalysts (Scheme 2).
 | ||
| Scheme 2 Chiral organocatalysts investigated in the 1,3-dithiane addition to nitrostyrene. | ||
In the model reaction performed in toluene for 24 hours at room temperature 2 mol eq. of thioester 8 were reacted with 1 mol eq. of nitrostyrene in the presence of 0.2 mol eq. of a catalyst. Based on the good results obtained with catalyst A, another successful thiourea-based bifunctional catalyst, Takemoto catalyst B,14 was tested; the product was isolated in higher yield but lower enantioselectivity than the cinchona-derived catalyst A (entries 1 and 2 of Table 2, 67% ee for catalyst Bvs. 87% ee for A).
|
|
|||
|---|---|---|---|
| Entrya | Catalyst | Yieldb (%) | eec (%) |
| a Typical reaction conditions: 0.1 mmol of nitroalkene, 0.2 mmol of dithiane, 0.02 mmol of catalyst A, 1 mL of toluene. b Yields were determined after chromatographic purification. c Enantiomeric excess determined by HPLC on a chiral stationary phase. | |||
| 1 | A | 71 | 87 |
| 2 | B | 81 | 67 |
| 3 | C | 55 | 75 |
| 4 | D | 53 | 51 |
| 5 | E | 41 | 37 |
| 6 | F | 53 | <5 |
| 7 | G | 55 | 31 |
| 8 | H | 73 | 37 |
| 9 | I | 15 | <5 |
| 10 | L | 51 | 21 |
| 11 | M | 47 | 7 |
| 12 | N | 57 | <5 |

Therefore we turned our attention to the variation of different structural elements of quinine-derived catalyst A; for example with the cinchonidine derivative C, lacking the methoxy group on the quinolone ring, a little decrease in enantioselection was observed (75% vs. 87%). A modification of the same alkoxy group was deleterious for enantioselectivity (see entries 4 and 5, catalysts D and E). Compounds F–I clearly demonstrated the crucial role exerted by the thiourea moiety which cannot be replaced by alternative hydrogen bond donor groups. In addition, when squaramides L–N,15 derived from quinine or trans-diamine-cyclohexane, were tested, very low enantioselectivities were obtained.
Once compound A was selected as a catalyst of choice, a few reaction parameters were optimized (Table 3).
| Entrya | Solvent | Temp. (°C) | Yieldb (%) | eec (%) |
|---|---|---|---|---|
| a Typical reaction conditions: 0.1 mmol of nitroalkene, 0.2 mmol of dithiane, 0.02 mmol of catalyst A, 1 mL of toluene. b Yields were determined after chromatographic purification c Enantiomeric excess determined by HPLC on a chiral stationary phase. d Reaction run with 10 mol% of a catalyst. e Reaction run with 5 mol% of a catalyst. | ||||
| 1 | Toluene | 25 | 71 | 87 |
| 2 | DCM | 25 | 55 | 80 |
| 3 | CH3CN | 25 | 80 | 41 |
| 4 | Hexane | 25 | 75 | 57 |
| 5 | Et2O | 25 | 73 | 67 |
| 6 | Toluene | 0 | 60 | 92 |
| 7 | Toluene | −20 | 61 | 66 |
| 8d | Toluene | 25 | 55 | 81 |
| 9e | Toluene | 25 | 47 | 80 |
The reaction tolerates different solvents, although only dichloromethane guaranteed the same level of ee as toluene (80% ee at RT). Lowering the reaction temperature to 0 °C had a positive effect, allowing isolating the product in 92% ee; lower temperatures did not give satisfactory results. It is noteworthy that the loading of the catalyst could be decreased to 10 mol% and even to 5 mol% with only a marginal loss of enantioselectivity, albeit with diminished chemical yield. The general applicability of the methodology was then investigated (Scheme 3, eqn (a)).
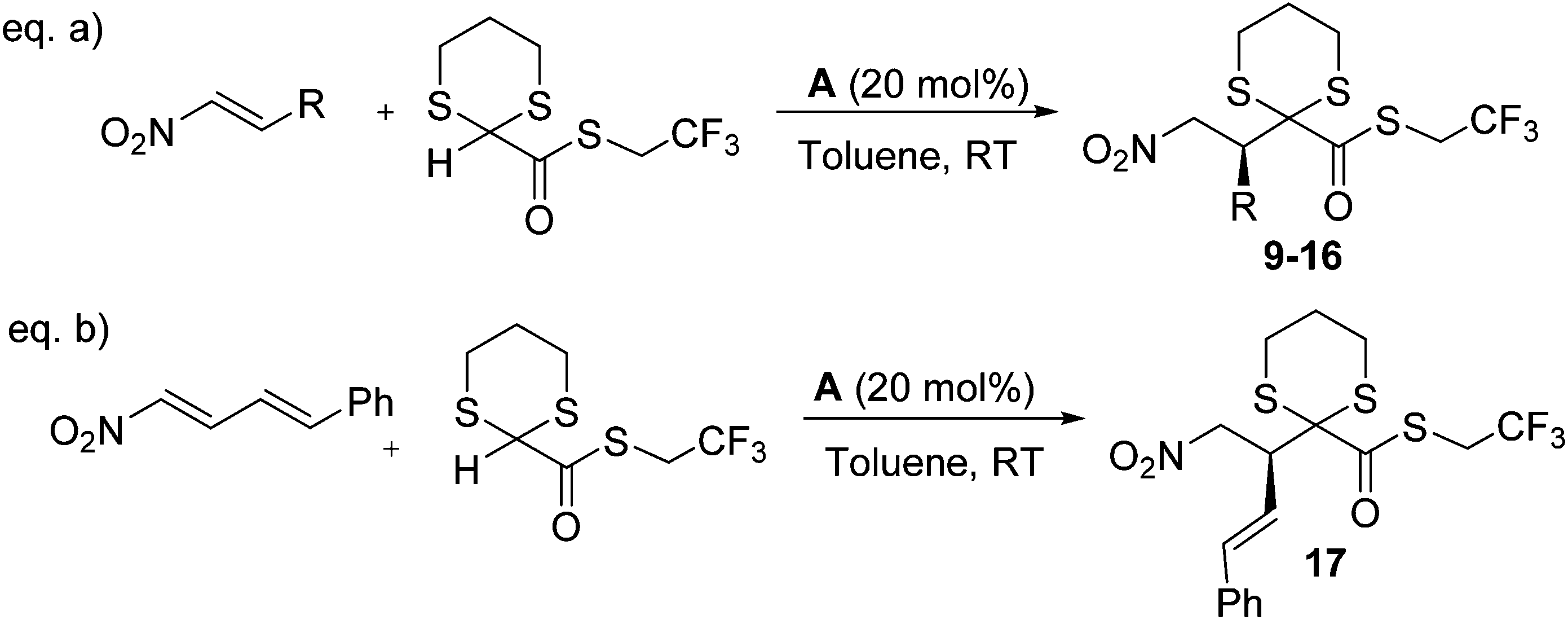 | ||
| Scheme 3 Organocatalytic 1,3-dithiane addition to different nitroalkenes. | ||
Differently substituted nitro alkenes were easily prepared11 and reacted with dithiane 8 in the presence of catalyst A (Table 4).
| Entrya | R | Product | Yieldb (%) | eec (%) |
|---|---|---|---|---|
| a Typical reaction conditions: 0.1 mmol of nitroalkene, 0.2 mmol of dithiane, 0.02 mmol of catalyst A, 1 mL of toluene at 25 °C. b Yields were determined after chromatographic purification. c Enantiomeric excess determined by HPLC on a chiral stationary phase. d Reaction run at 0 °C. | ||||
| 1 | Ph | 9 | 71 | 87 |
| 2d | Ph | 9 | 60 | 92 |
| 3 | 4-CF3Ph | 10 | 41 | 53 |
| 4 | 4-ClPh | 11 | 81 | 86 |
| 5 | 4-CH3Ph | 12 | 71 | 73 |
| 6 | 2-CH3Ph | 13 | 43 | 67 |
| 7 | 4-OCH3Ph | 14 | 75 | 73 |
| 8 | 2-OCH3Ph | 15 | 80 | 71 |
| 9 | 2-OAcPh | 16 | 55 | 85 |
| 10 | i-Pr | n.r. | — | — |
| 11 | CH2CH2Ph | n.r. | — | — |
The reaction works with nitrostyrenes substituted both in ortho and in para positions, with electron rich or poor substituents, leading to products with enantioselectivities ranging typically between 70% and 92%. Typically less reactive alkyl nitro alkenes16 did not react under the present conditions (entries 10 and 11); however, 4-nitro-1-phenyl butadiene reacted smoothly to afford product 17, bearing a styryl moiety in 61% yield and 71% ee (eqn (b), Scheme 3).
In order to establish the absolute configuration of the addition products, compound 11 was treated with powdered zinc and 6 M HCl17 to afford enantiomerically enriched lactam 18, which was determined to be in the (S) configuration by comparison of optical rotation power values. Indeed final hydrolysis with hydrochloridric acid afforded (S)-baclofen 19 (Scheme 4).18
 | ||
| Scheme 4 Synthesis of baclofen. | ||
Therefore, the method offers a straightforward access to a direct precursor of baclofen-type derivatives, GABAB receptor agonists used in the treatment of spasticity and alcohol dependence.19
Finally, conversion of the addition products into the corresponding α-ketothioesters was achieved by reaction with N-bromosuccinimide in acetone (Scheme 5).20
 | ||
| Scheme 5 Synthesis of β-aryl-α-keto thioesters. | ||
For example, compounds 9–11 were quantitatively converted to α-ketothioesters 20–22. Interestingly, upon treatment of 20–22 with silver trifluoroacetate, β-nitro esters 23–25 were obtained (Scheme 5).21
The procedure afforded compounds 20–22 without affecting the stereochemical integrity of the starting materials. For instance, a sample of (R)-9 (53% ee) was quantitatively transformed into α-ketothioester 20 through a 4 hour reaction with NBS in acetone at 0 °C, and then reacted with CF3CO2Ag in methanol to give the known (R)-β-nitro methylester 2322 in 70% yield, its absolute configuration reflecting that of the starting compound 9 without appreciable variation of the enantiomeric excess.23
In proposing a stereoselection model, the coordination of both reaction partners to the bifunctional catalyst may be envisaged, the substrate nitro group being engaged in hydrogen bonds by the thiourea moiety, and the charged quinuclidine nitrogen possibly forming an ion pair with the nucleophile (Fig. 1).
 | ||
| Fig. 1 Proposed stereoselection model. | ||
Accordingly, with the quinine-derived catalyst, the thioester attack occurs on the nitroalkene Si face, affording the experimentally observed (S)-configured product.
In conclusion, the enantioselective organocatalyzed conjugate addition of a newly activated thioester acting as an acyl anion mimic has been performed. The use of chiral bifunctional catalysts to control the stereochemical outcome through a hydrogen bond network imposed by the thiourea moiety afforded good yields and enantioselectivities up to 92%. This methodology represents an entry to highly functionalized, enantiomerically enriched products, such as γ-nitro-β-aryl-α-keto thioesters, valuable precursors of a wide variety of chiral organic compounds.
Acknowledgements
M. B. thanks COST action CM9505 “ORCA” Organocatalysis. E. M. and A. G. thank the University of Milan for PhD fellowships.References
- A review: M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853 CrossRef CAS PubMed . See also: Q. Kang, Z. A. Zhao and S. L. You, Adv. Synth. Catal., 2007, 349, 1657 CrossRef PubMed.
- M. J. Kiefel and M. von Itzstein, Chem. Rev., 2002, 102, 471 CrossRef CAS PubMed . See also: S. Hanessian, Total Synthesis of Natural Products: The Chiron Approach, Pergamon Press, New York, 1983, ch. 2 Search PubMed.
- K. M. Steward and J. S. Johnson, Org. Lett., 2011, 13, 2426 CrossRef CAS PubMed . See also: M. W. Leighty, B. Shen and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 15233 CrossRef PubMed . For a similar approach in the synthesis of chiral ketones through catalytic oxidative C–C bond cleavage of aldehydes by oxygen, see: B. Tiwari, J. Zhang and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 1911 CrossRef PubMed.
- (a) D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239 CrossRef PubMed. For a review on organocatalytic umpolung, see: (b) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC. For some recent papers on organocatalytic additions of acyl anion synthons, see: (c) T. Hashimoto, M. Hirose and K. Maruoka, J. Am. Chem. Soc., 2008, 130, 7556 CrossRef CAS PubMed; (d) M. Rueping, E. Sugiono, T. Theissman, A. Kuenkel, A. Kiickritz, A. Pews-Davtyan, N. Nemati and M. Beller, Org. Lett., 2007, 1065 CrossRef CAS PubMed; (e) B. Alonso, E. Reyes, L. Carillo, J. L. Vicario and D. Badia, Chem. – Eur. J., 2011, 17, 6048 CrossRef CAS PubMed; (f) M. Fernández, U. Uria, J. L. Vicario, E. Reyes and L. Carrillo, J. Am. Chem. Soc., 2012, 134, 11872 CrossRef PubMed; (g) D. Monge, A. M. Crespo-Peña, E. Martín-Zamora, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 12912 CrossRef PubMed; (h) A. M. Crespo-Peña, D. Monge, E. Martín-Zamora, E. Álvarez, R. Fernández and J. M. Lassaletta, Chem. – Eur. J., 2013, 19, 8421 CrossRef PubMed.
- Reviews: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) H. U. Vora, P. Wheleer and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617 CrossRef CAS PubMed; (c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC. For more recent contributions, see: (d) O. Bortolini, G. Fantin, V. Ferretti, M. Fogagnolo, P. P. Giovannini, A. Masso, S. Pacifico and D. Ragno, Adv. Synth. Catal., 2013, 355, 3244 CrossRef CAS PubMed and references cited therein; (e) M. M. D. Wilde and M. Gravel, Angew. Chem., Int. Ed., 2013, 52, 12651 CrossRef CAS PubMed See also the review: S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC.
- D. Enders, M. Bonten and G. Raabe, Angew. Chem., Int. Ed., 2007, 46, 2314 CrossRef CAS PubMed . For a recent contribution, see: K. S. Yang, A. E. Nibbs, Y. E. Turkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050 CrossRef PubMed . See also: P. A. Evans, S. Oliver and J. J. Chae, J. Am. Chem. Soc., 2012, 134, 19314 CrossRef PubMed.
- limited examples, see: (a) M. Amat, M. Perez, N. Llor and J. Bosch, Org. Lett., 2002, 4, 2787 CrossRef CAS PubMed; (b) I. Coldham, K. M. Crapnell, J.-C. Fernandez, J. D. Moseley and R. Rabot, J. Org. Chem., 2002, 67, 6181 CrossRef CAS PubMed; (c) T. Ichige, A. Miyake, N. Kanoh and M. Nakata, Synlett, 2004, 1686 CAS and references cited therein.
- For a recent catalytic, non-stereoselective, conjugate addition of acyl anion equivalents derived from 2-silyl-1,3-dithianes to unsaturated ketones, see: S. E. Denmark and L. R. Cullen, Org. Lett., 2014, 16, 70 CrossRef CAS PubMed . For a Pd-catalyzed coupling of acetyltrimethylsilane, see: S. Ramgren and N. K. Garg, Org. Lett., 2014, 16, 824 CrossRef PubMed.
- For a review of organocatalytic asymmetric conjugate addition, see: (a) D. Enders, C. Wang and J. X. Liebich, Chem. – Eur. J., 2009, 15, 11058 CrossRef CAS PubMed; (b) S. Sulzer-Mosse’ and A. Alexakis, Chem. Commun., 2007, 3123 RSC; (c) D. Roca-Lopez, D. Sadaba, I. Delso, R. P. Herrera, T. Tejero and P. Merino, Tetrahedron: Asymmetry, 2010, 21, 2561 CrossRef CAS PubMed. Recent contributions: (d) D. Uraguchi, Y. Ueki and T. J. Ooi, Chem. Sci., 2012, 3, 842 RSC and; (e) G. K. S. Prakash, F. Wang, Z. Zhang, C. Ni, R. Haiges and G. A. Olah, Org. Lett., 2012, 14, 3260 CrossRef CAS PubMed.
- (a) L.-Q. Lu, X.-L. An, J.-R. Chen and W.-J. Xiao, Synlett, 2012, 490 Search PubMed; (b) O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051 RSC; (c) Y. Xi and X. Shi, Chem. Commun., 2013, 8583 RSC.
- As expected, the reaction performed in the presence of the “quasi” enantiomeric catalyst derived from quinine afforded the addition product with the opposite absolute configuration, in comparable yield (67%) and marginally lower enantioselectivity (83% ee).
- D. A. Alonso, S. Kitagaki, N. Utsumi and C. F. Barbas, Angew. Chem., Int. Ed., 2008, 47, 4588 CrossRef CAS PubMed.
- (a) S. Rossi, M. Benaglia, F. Cozzi, A. Genoni and T. Benincori, Adv. Synth. Catal., 2011, 353, 848 CrossRef CAS PubMed; (b) M. Bonsignore, M. Benaglia, F. Cozzi, A. Genoni, S. Rossi and L. Raimondi, Tetrahedron, 2012, 68, 8251 CrossRef CAS PubMed.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 135, 12672 CrossRef CAS PubMed . Review: Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299 Search PubMed.
- Review: J. Aleman, A. Parra, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 689 CrossRef PubMed.
- For a theoretical work on nitroalkenes reactivity, see: V. Rai and I. N. N. Namboothiri, Eur. J. Org. Chem., 2006, 4693 CrossRef CAS PubMed.
- T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed.
- By employing (R,R)-diaminocyclohexane-derived catalyst B addition products of the (R)-configuration are obtained.
- H. Gotoh, H. Ishikawa and Y. Hayashi, Org. Lett., 2007, 9, 5307 CrossRef CAS PubMed and references cited therein.
- E. J. Corey and B. W. Erickson, J. Org. Chem., 1971, 36, 3553 CrossRef CAS.
- This approach accesses enantiomerically enriched β-substituted β-nitro esters starting from readily available β-nitroalkenes. The exact mechanism of the metal-promoted decarboxylation reaction is currently under study.
- H. Liu, J. Xu and D. M. Du, Org. Lett., 2007, 9, 4725 CrossRef CAS PubMed.
- See the ESI† for HPLC traces.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for nitrostyrene derivatives synthesis, catalysts preparation and organocatalytic reactions. Characterization details for Michael addition products and Baclofen synthesis. Determination of absolute configuration of the reaction products. 1H-NMR, 13C-NMR and 19F-NMR spectra and HPLC chromatograms on the chiral stationary phase of addition products. See DOI: 10.1039/c5ob00492f |
| This journal is © The Royal Society of Chemistry 2015 |