
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matching the organic and inorganic counterparts during nucleation and growth of copper-based nanoparticles – in situ spectroscopic studies†
Malwina
Staniuk
a,
Daniel
Zindel
b,
Wouter
van Beek
c,
Ofer
Hirsch
a,
Niklaus
Kränzlin
a,
Markus
Niederberger
a and
Dorota
Koziej
*a
aLaboratory for Multifunctional Materials, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland. E-mail: dorota.koziej@mat.ethz.ch
bLaboratory of Physical Chemistry, ETH Zurich, Vladimir-Prelog-Weg 2, 8093 Zurich, Switzerland
cSwiss-Norwegian Beamlines at European Synchrotron Research Facility, 71 Avenue des Martyrs, 38043 Grenoble, France
First published on 9th June 2015
Abstract
Although syntheses in organic solvents provide access to a wide range of copper-based nanoparticles, the correlation between organic reactions in solution and nucleation and growth of nanoparticles with defined properties is not well understood. Here, we utilize the Multivariate Curve Resolution-Alternative Least Squares (MCR-ALS) methodology to examine spectroscopic data recorded in situ during the synthesis of copper-based nanoparticles. While earlier studies showed that depending on the temperature copper(II) acetylacetonate reacts with benzyl alcohol and forms either copper oxides or copper nanoparticles, we link the inorganic reaction with their organic counterparts. From X-ray Absorption Near Edge Spectroscopy (XANES) and Ultraviolet-visible spectroscopy (UV-vis) data we learn that copper(I) oxide forms directly from the solution and is the final product at low temperature of 140 °C. We observe in Fourier Transformed Infrared (FTIR) spectra an increasing concentration of benzyl acetate that co-occurs with the formation of a copper enolate and evolution of benzaldehyde, which accompanies the reduction of copper ions. We also record the interaction of organic species at the Cu2O surface, which inhibits a further reduction to metallic copper. When we raise the synthesis temperature to 170 °C it turns out that the Cu2O is just an intermediate species. It subsequently transforms by solid-state reduction to metallic copper accompanied by oxidation of benzyl alcohol to benzaldehyde.
1. Introduction
Twenty years after the initial findings1 the nonaqueous sol–gel process is now an established method used to synthesize crystalline nanoparticles of customizable size, composition and functionalities.2–6 Specially for metal oxides, the advancement has been made possible due to the understanding that solvents are not only reaction medium but may also be an oxygen source and act as stabilizing agent. That knowledge has been gained by studying the organic compounds forming during synthesis.7–13 When benzyl alcohol is used as a solvent, the most common routes are: alkyl halide elimination, ester elimination, ether elimination, aldol condensation and C–C bond formation between benzylic alcohols and alkoxides.5,12,14–16 Remarkably, for some metal oxides with the occurrence of nanoparticles, the organic reactions do not terminate, and in fact, new reactions are initiated.17–21 By far the most intriguing example is the formation of monolithic tungsten oxide-polybenzylene hybrids induced by the nucleation of tungsten oxide nanoparticles and their catalytic activity.20 Moreover, in the last decade, the non-aqueous method has been further adopted to synthesize metal phosphates, -sulfides, -diimides and even metallic nanoparticles.18,22–25 With the increased complexity of the inorganic product also the linkage of the inorganic species to their respective organic counterparts became more challenging.Here, our main focus is the determination of organic and inorganic species formed during the nucleation and growth of copper-based nanoparticles and their interdependence by utilizing different spectroscopic techniques. The latest development of synchrotron- and laboratory-based equipment and analysis tools allow for in situ monitoring the complex reactions in solution.7,26–36 However, the specificity of the individual methods often makes a quantitative analysis or a direct comparison of results difficult. We overcome this problem by selecting two spectroscopic methods, X-ray Absorption Near Edge Spectroscopy (XANES) and Ultraviolet–visible spectroscopy (UV-vis), which even though they probe fundamentally different phenomena, provide information on the chemical composition. In order to determine the changes of the composition of the reaction solution during nucleation and growth of Cu2O and Cu nanoparticles we apply Multivariate Curve Resolution-Alternative Least Squares (MCR-ALS) method. It has been previously successfully used to determine the concentration profile and pure XANES spectra of individual species during in situ studies of Cu-catalysts, Cu-doped V2O3 batteries and to follow the synthesis of Co, TiO2 and Ni-doped MoO2 nanoparticles.18,37–45
Additionally, we track the formation of organic compounds by Fourier Transform Infrared spectroscopy (FTIR) studies that are recorded simultaneously with UV-vis measurements. In the control experiments without copper precursor, we: (a) evaluate the decomposition of benzyl alcohol upon heating, (b) determine the lower detection limit and (c) calibrate the curve for benzaldehyde, benzyl acetate and dibenzyl ether, compounds that are considered the main organic counterparts of copper-based nanoparticle nucleation. This lays the foundation for a quantitative analysis of in situ FTIR studies. We find that only the formation of benzyl acetate correlates directly with the occurrence of copper-based nanoparticles in benzyl alcohol. At low reaction temperature, the formation of benzaldehyde as a product of Cu2O synthesis makes up approximately thirty percent of the total amount of benzaldehyde. The remaining seventy percent are a product of catalytic activity of Cu2O nanoparticles. At high reaction temperature, Cu2O nanoparticles are further reduced to Cu. Interestingly, the formation of copper nanoparticles terminates the catalytic formation of benzaldehyde, and instead triggers the catalytic formation of dibenzyl ether.
2. Experimental
Chemicals
Benzyl alcohol (99–100.5%,), benzaldehyde (≥99%), dibenzyl ether (99%), and copper hydroxide (Cu(OH)2) (technical grade) were supplied by Sigma Aldrich, copper(II) acetylacetonate (98%) by Acros Organics, benzyl acetate (99%), ethanol (99.8%) by Fluka and tetrahydrofuran by Riedel-de-Haen (99.9%, stabilized by butylhydroxytoluol (BHT)). The chemicals were used without any further purification. CuO was obtained by thermal oxidation of Cu2O.46Synthesis procedure
Ex situ characterization
X-ray Absorption Spectroscopy (XAS) data of the corresponding powder pellets were measured at the Swiss Norwegian Beamline (SNBL) BM01B at the European Synchrotron Research Facility (ESRF) from 8.850 to 9.800 keV in continuous scanning mode with two ion chambers, one before the sample and one after the sample. The powders were diluted in cellulose to optimize the edge-step and pressed into pellets. Powder X-ray diffraction (PXRD) patterns of synthesized powders were collected on an Empyrean powder diffractometer (PANalytical B.V., The Netherlands), operating in reflection mode under constant irradiated area conditions with Cu Kα radiation (45 kV, 40 mA). Scanning electron microscopy (SEM) images were taken on a Magellan 400 FEG (FEI, USA) to show the shape and morphology of the nanoparticles. Gas chromatography-mass spectrometry (GC-MS) analysis of the filtered reaction liquids was performed with a Thermo Scientific Trace 1300 (GC) coupled with an ISQ single quad (MS). The samples were diluted in tetrahydrofuran (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). Attenuated total reflection (ATR)-FTIR spectra of organic samples in preheated benzyl alcohol were measured as references on a ReactIR15 (Mettler Toledo AutoChem) with a 6.5 mm AgX DiComp Fiber Conduit probe. 60 mL of benzyl alcohol were heated to 140 or 170 °C and then the corresponding organic species were added. The analyzed range was 800 to 1800 cm−1 and the resolution was 4 cm−1.
:20). Attenuated total reflection (ATR)-FTIR spectra of organic samples in preheated benzyl alcohol were measured as references on a ReactIR15 (Mettler Toledo AutoChem) with a 6.5 mm AgX DiComp Fiber Conduit probe. 60 mL of benzyl alcohol were heated to 140 or 170 °C and then the corresponding organic species were added. The analyzed range was 800 to 1800 cm−1 and the resolution was 4 cm−1.
In situ characterization
Data handling
| D = CST + E | (1) |
Rows of matrix D are spectra acquired during measurement, columns of matrix C and rows in matrix ST are concentration profiles and spectra of resolved components, respectively. Matrix E contains residuals not explained by the model. Eqn (1), generally valid for spectroscopic data governed by the Lambert–Beer law like XANES or FTIR, is solved by ALS algorithm, which iteratively calculates the C and ST matrices that fits best the experimental data. The optimization of C and ST is carried out for a proposed number of components. Initial estimates of C and ST are obtained by using SIMPLe-to-use Interactive Self-Modeling Algorithm (SIMPLISMA), which finds the most different spectra within the dataset that are further used as an input for ALS optimization.
The number of components was selected on the basis of Singular Value Decomposition (SVD). The initial spectra of the components were estimated with the PURE algorithm with the noise level set to 3% (XANES) and 4% (ATR-FTIR). Constraints applied for ALS calculation were as follows: non-negativity of spectra and concentration (XANES), non-negativity of concentration (XANES and ATR-FTIR), unimodality of concentration (XANES and ATR-FTIR), convergence criterion: 0.1 (XAS and ATR-FTIR). For more information please refer to ESI† Fig. S2–S4, Tables S2–S5.
3. Results and discussion
3.1. Synthesis
In a previous study we found that the reaction between copper(II) acetylacetonate and benzyl alcohol resulted in the formation of a metallic copper foil with Cu2O nanoparticles as intermediates.25 Now we adopt this synthesis to a reactor equipped with in situ ATR-UV-vis and -FTIR probes to collect information about the formation mechanism. In order to be able to probe the organic species in solution we prevent formation of the Cu foil at the surface of ATR crystals by vigorous stirring. Similar to the previous report at low reaction temperature cuprous oxide and at high temperature metallic copper are formed, respectively.25 Obviously, the time required for the reduction of cuprous oxide to copper varies because of the differences in reactor volume, stirring velocity and heating rate. Therefore, for the in situ studies we choose temperature conditions at which the product in the Mettler Toledo reactor is phase pure. At 140 °C and 170 °C we obtain Cu2O or Cu nanoparticles, respectively. The corresponding PXRD patterns and SEM images are shown in Fig. 1. | ||
| Fig. 1 (a) PXRD patterns of the nanoparticles synthesized at 140 (black pattern) and 170 °C (red pattern). Reference PXRD patterns of Cu2O (ICDD no. 00-005-0667) and Cu (ICDD no. 01-071-4610). SEM images of (b) Cu2O synthesized at 140 °C and (c) Cu synthesized at 170 °C. | ||
3.2. In situ studies of inorganic species in solution
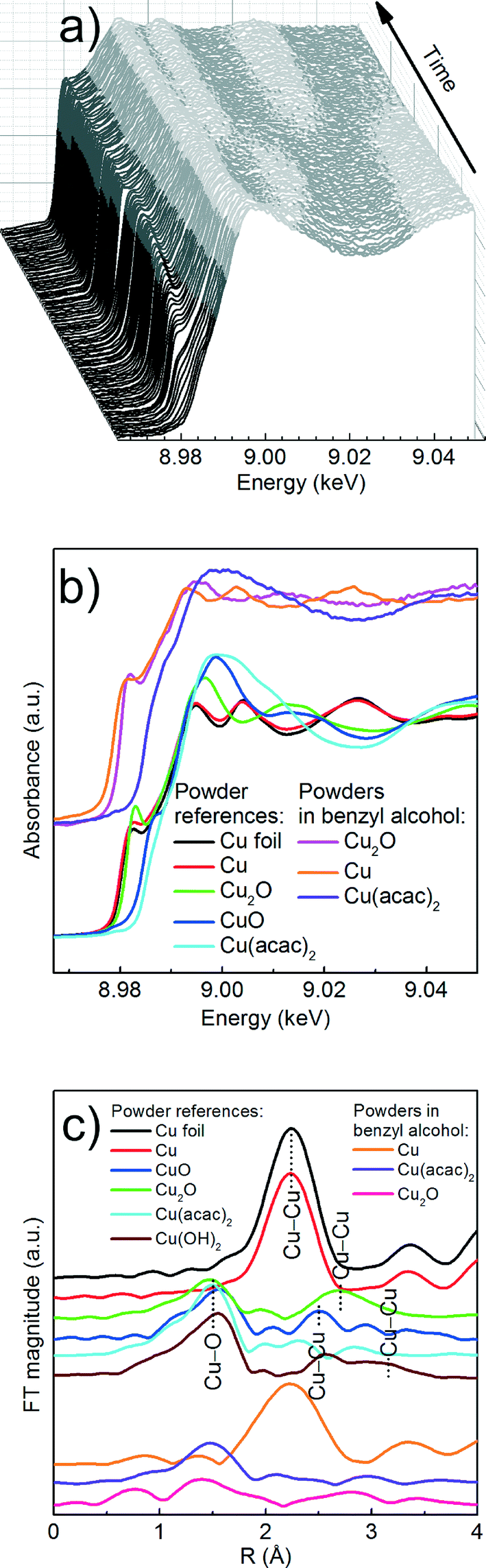 | ||
| Fig. 2 Cu K-edge XAS spectra; (a) XANES spectra taken during the synthesis of copper nanoparticles from Cu(acac)2 in benzyl alcohol at 180 °C; (b) comparison of XANES spectra of powder references (pellets) and powders in benzyl alcohol; Note: the spectrum of Cu2O in benzyl alcohol was recorded at 160 °C; (c) FT EXAFS spectra of powder pellets. The spectra were shifted on the ordinate for better representation. | ||
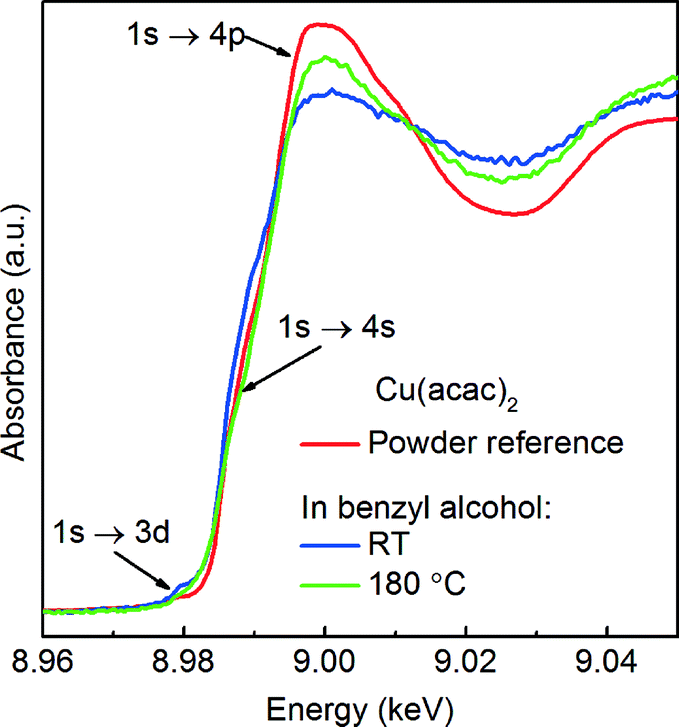 | ||
| Fig. 3 Comparison of Cu K-edge XANES spectra of Cu(acac)2 powder measured in transmission mode (red curve) and Cu(acac)2 dissolved in benzyl alcohol at RT (blue curve) and 180 °C (green curve) measured in fluorescence mode. | ||
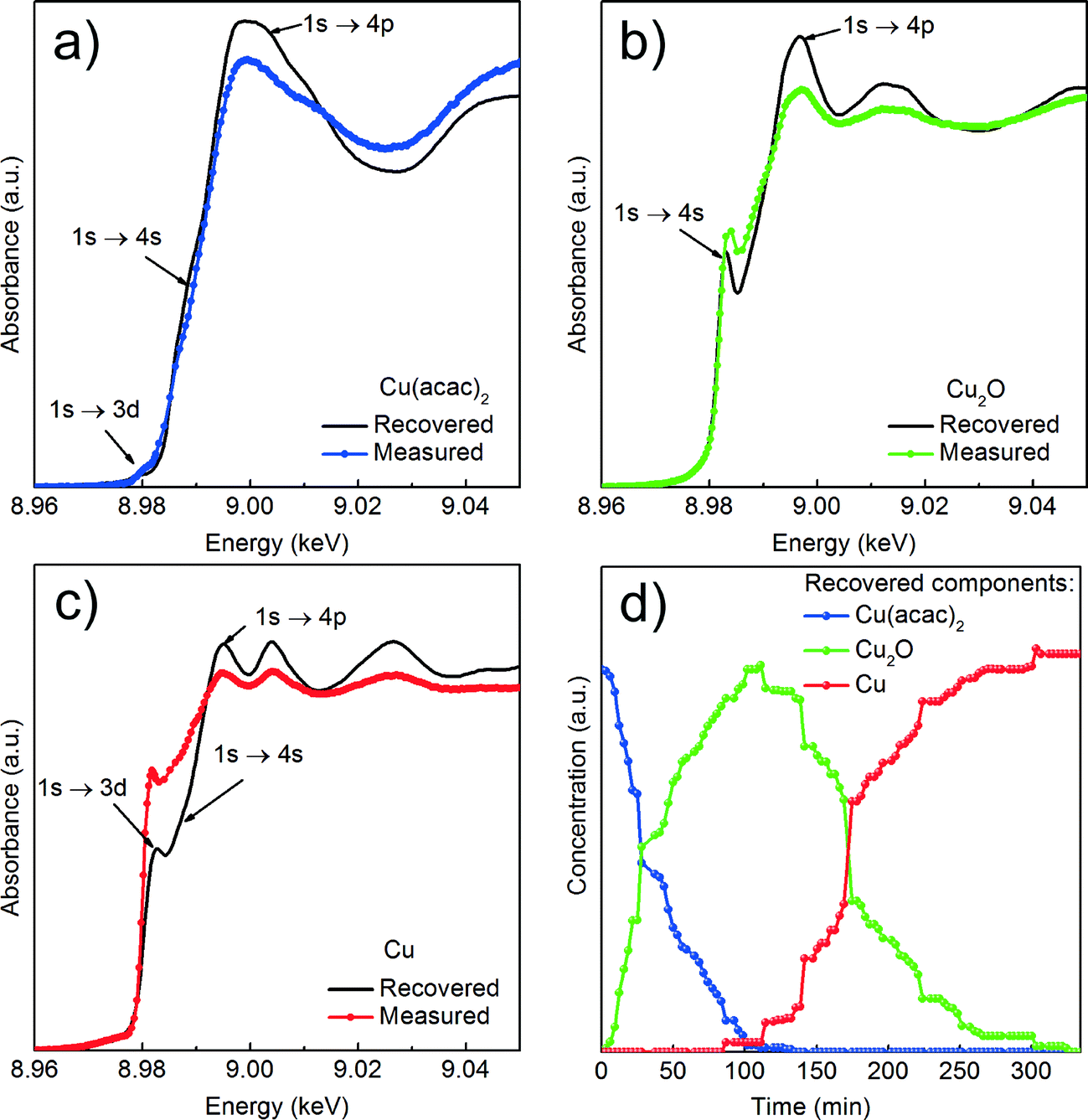 | ||
| Fig. 4 Results of the MCR-ALS analysis of in situ Cu K-edge XANES spectra recorded in fluorescence mode; (a), (b) and (c) recovered spectra of individual components and corresponding reference spectra; The reference compounds were measured as pellets in transmission mode and are only given here as guidance; (d) concentration profiles of the three recovered components during the synthesis at 180 °C. | ||
We first determine the initial oxidation state of copper species upon dissolution in benzyl alcohol at 180 °C as shown in Fig. 3. Then, we utilize the MCR-ALS method to analyze the in situ XANES spectra during nucleation of copper nanoparticles. This solely variance based method allows to determine, without a priori knowledge, the number of components, their spectra and relative concentration in a solution as shown in Fig. 4.
Precursor. Here, a careful comparison of the XANES spectra of the precursor as pellet (red curve), dissolved in benzyl alcohol at RT (blue curve) and at 180 °C (green curve), shown in Fig. 3, reveals the subtle differences in the pre- and post-edge regions. In general, such differences are related to the changes of geometry of the first coordination sphere of the absorbing element.52,53 Above the edge, the spectrum of solid Cu(acac)2 shows more pronounced features in comparison to the Cu(acac)2 in benzyl alcohol. The decrease of their intensity indicates a decrease of multiple scattering that involves second nearest neighbor or higher shells. We assign it to the loss of long-range order due to the dissolution of the precursor in benzyl alcohol. In addition, the deviation in the 1s → 4s transition indicates changes in the coordination of the copper ion, suggesting complexation with benzyl alcohol. The spectra of Cu(acac)2 in benzyl alcohol at 180 °C shows, with respect to RT, an increase of the intensity of the 1s → 4p transition. We do not observe characteristic features either of the CuO or of the Cu(OH)2. Thus, we can conclude that no solid CuO or the CuOH2 species are formed before formation of Cu2O.
Cu reduction at 180 °C in benzyl alcohol. To track the changes from Cu(acac)2 to Cu during the reaction at 180 °C we analyze the in situ XANES data by MCR-ALS as shown in Fig. 4. According to SVD results, three components suffice to fit the data. We use SIMPLISMA based algorithm to find the spectra that are used as initial estimates for MCR-ALS. We show the spectra of powder references recorded at room temperature in transmission mode only as guidance, since the XANES spectra of nanoparticles strongly depend on their size, species adsorbed at their surface and temperature at which the spectrum was measured. Thus, the recovered spectra are the spectra of a given compound at the particular reaction conditions. The first recovered spectrum is the precursor dissolved in benzyl alcohol at 180 °C. The second and third spectrum show characteristic features observed in the reference spectra of Cu2O and Cu, respectively. Therefore, the appearance of the second and third component indicates the nucleation of Cu2O and its consecutive reduction to Cu. The concentration dependence of the three recovered components proves that Cu2O and Cu are not formed concurrently. First, exclusively Cu2O forms and only after 83 minutes it gets reduced to metallic copper, in line with previously reported PXRD studies.25 The direct formation of Cu2O confirms the reduction of Cu2+ to Cu+ in the solution and solid intermediate species like CuO and Cu(OH)2 are excluded.
3.3. Organic species forming during synthesis – complementary in situ ATR-FTIR and GC-MS studies
Simultaneously with UV-vis, we measure in situ FTIR to track the organic species forming in solution during the synthesis of Cu-based nanoparticles at 140 and 170 °C as shown in ESI† Fig. S2a–b. In the following, we first qualitatively describe the in situ FTIR results. Then, the GC-MS studies give complementary information about species forming in the solution below the detection limit of FTIR. Finally, we determine the lower detection limit and calibration curve for the main organic species, which allows us to estimate their quantities and correlate the organic and the inorganic compounds.At both reaction temperatures, as long as some of the precursor is present in the solution, the same organic compounds, benzyl acetate and benzaldehyde, form. At 140 °C the reaction terminates with the reduction of Cu(acac)2 to Cu2O. Only at that point the bands assigned to the species adsorbed at the surface of Cu2O become clearly visible as shown in ESI† Fig. S4a–b. At 170 °C the nucleation of Cu2O is only an intermediate step towards the formation of Cu nanoparticles and we do not observe adsorption of organic species at the Cu2O surface. Instead, we observe the formation of dibenzyl ether as the product of benzyl alcohol condensation, which is a result of the catalytic activity of metallic copper.
In summary, comparing the organic species from these three experiments, benzyl acetate is the only compound that exclusively forms when copper-based nanoparticles crystallize. All other organic compounds may also form as products of parallel, side reactions that are not directly related to the nucleation of the copper-based nanoparticles.
Additionally, we heat benzyl alcohol without Cu(acac)2 at 140 and 170 °C, respectively, and in both cases with FTIR we exclusively detect benzaldehyde as shown in Fig. 5 (gray curves). It is the product of the oxidation of benzyl alcohol according to the reaction shown in Scheme 1.54 The amounts of dibenzyl ether and benzyl benzoate detected by GC-MS are obviously below the detection limit of FTIR.
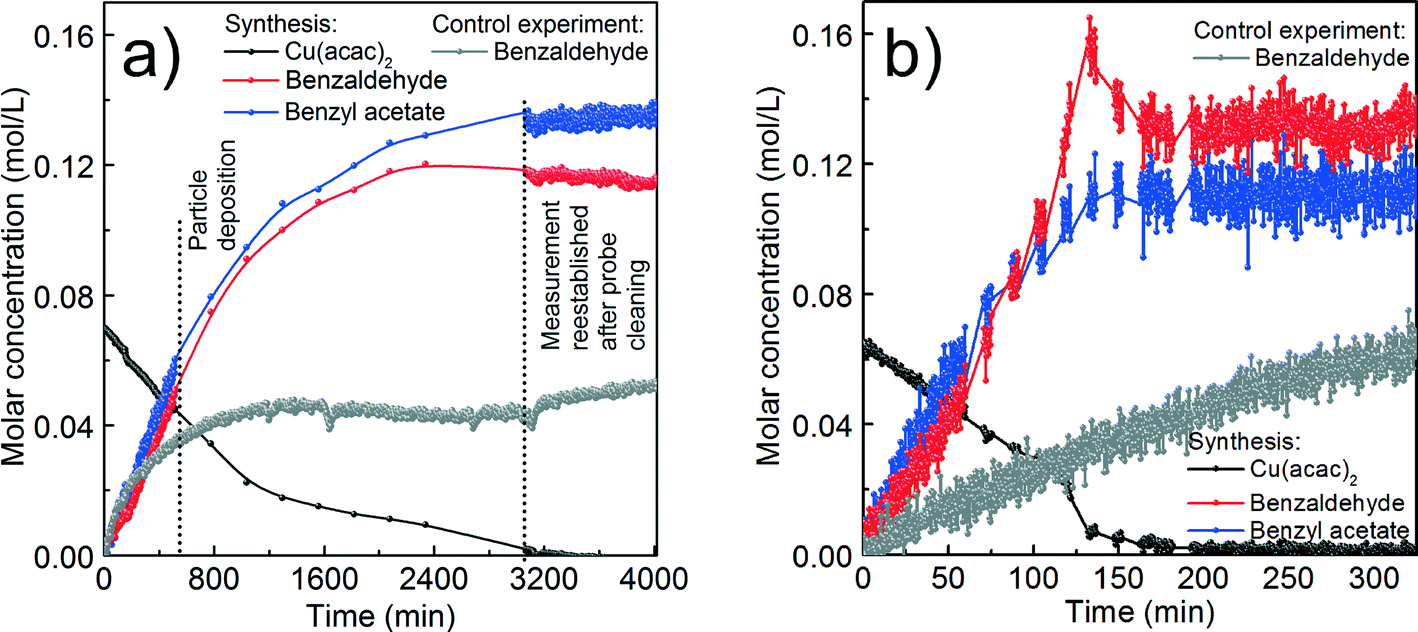 | ||
| Fig. 5 Molar concentration of copper(II) acetylacetonate, benzyl acetate and benzaldehyde during the synthesis of copper-based particles as well as molar concentration of benzaldehyde from the control experiments calculated from in situ FTIR data; (a) reaction at 140 °C and (b) at 170 °C. We cannot fully avoid the deposition of copper oxide and copper on the surface of the ATR crystals. In a) the vertical, dashed lines indicate the start of “particle deposition” and “measurements reestablished after probe cleaning”, respectively. The measurements in this time span were background-corrected. | ||
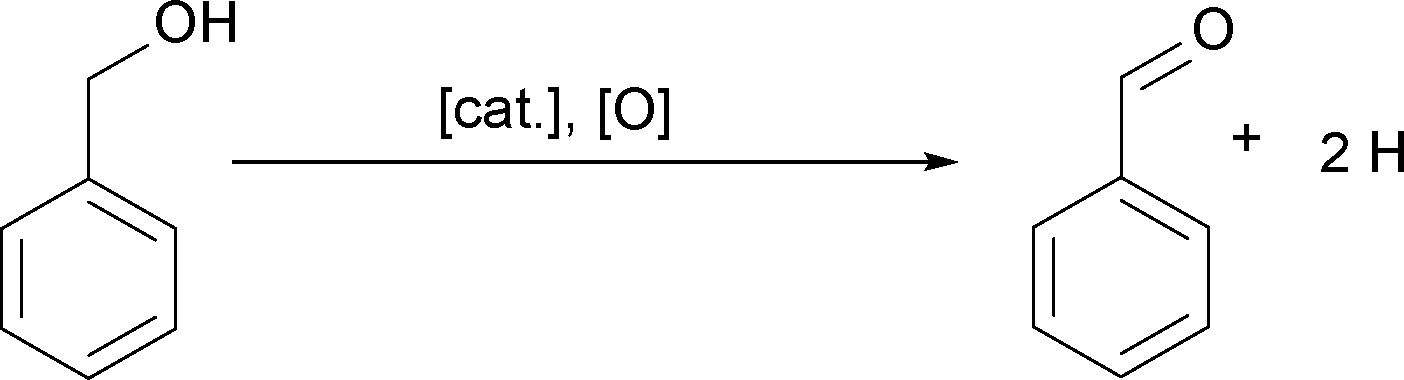 | ||
| Scheme 1 Oxidation of benzyl alcohol to benzaldehyde.54–57 | ||
Now we discuss the quantities of the two compounds benzyl acetate and benzaldehyde we measured in comparison to what would be expected from the proposed chemical reactions. The chemical reactions themselves will be further discussed in section (d).
We assume that benzyl acetate is formed by the reaction of the acetylacetonate ligand with benzyl alcohol, as shown in Scheme 2.
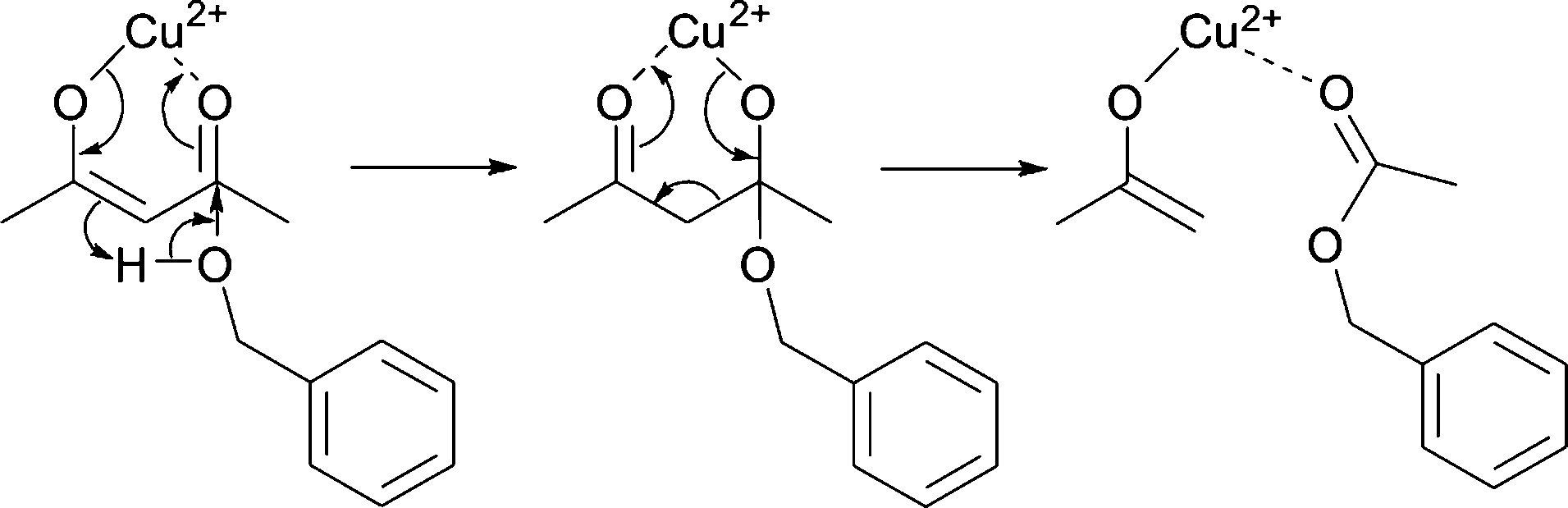 | ||
| Scheme 2 Formation of benzyl acetate by nucleophilic attack of benzyl alcohol on one of the carbonyl groups of the acetylacetonate ligand. | ||
Benzaldehyde is the oxidation product of benzyl alcohol as a result of the reduction of Cu2+ to Cu+ and finally Cu0 according to Scheme 3.
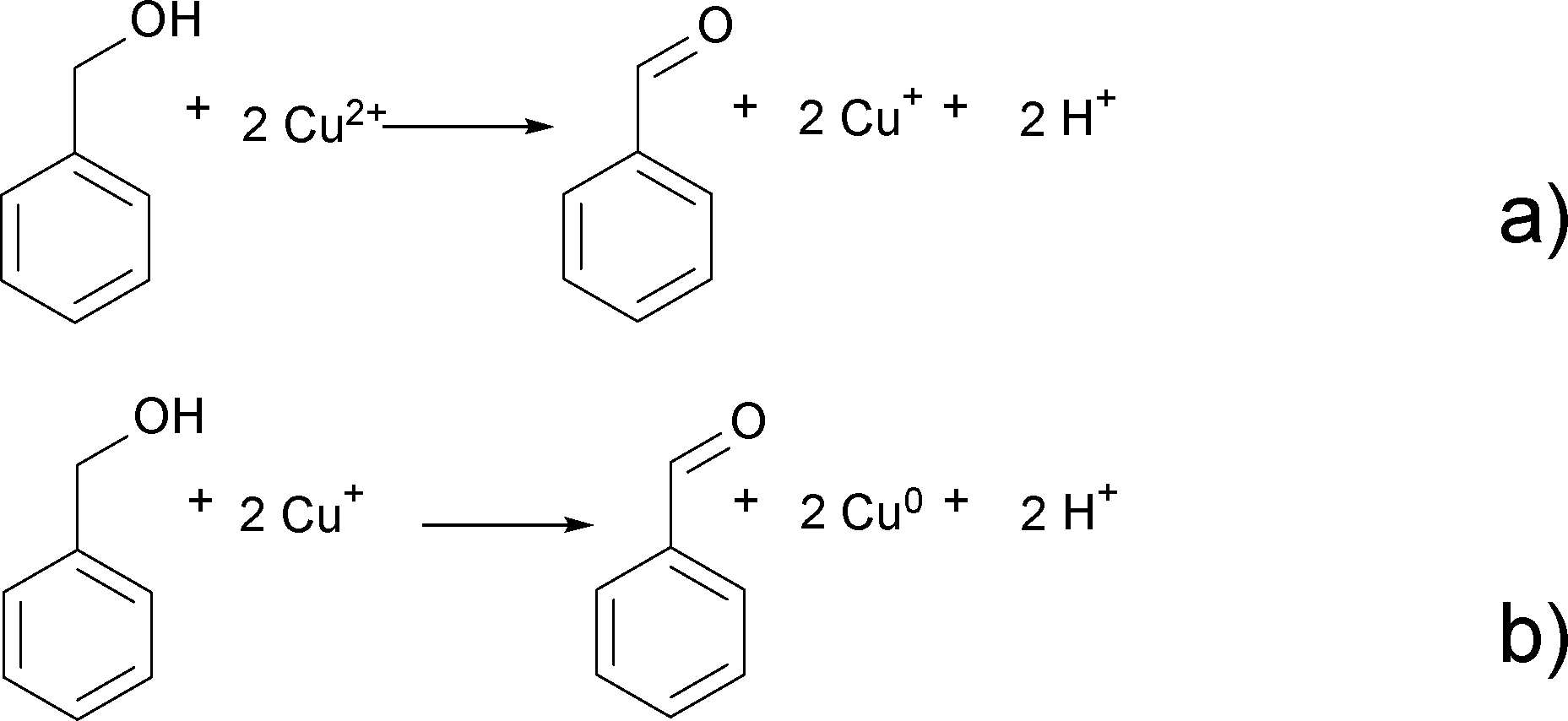 | ||
| Scheme 3 Oxidation of benzyl alcohol to benzaldehyde and reduction of Cu2+ to Cu+ (a) and Cu+ to Cu0 (b). | ||
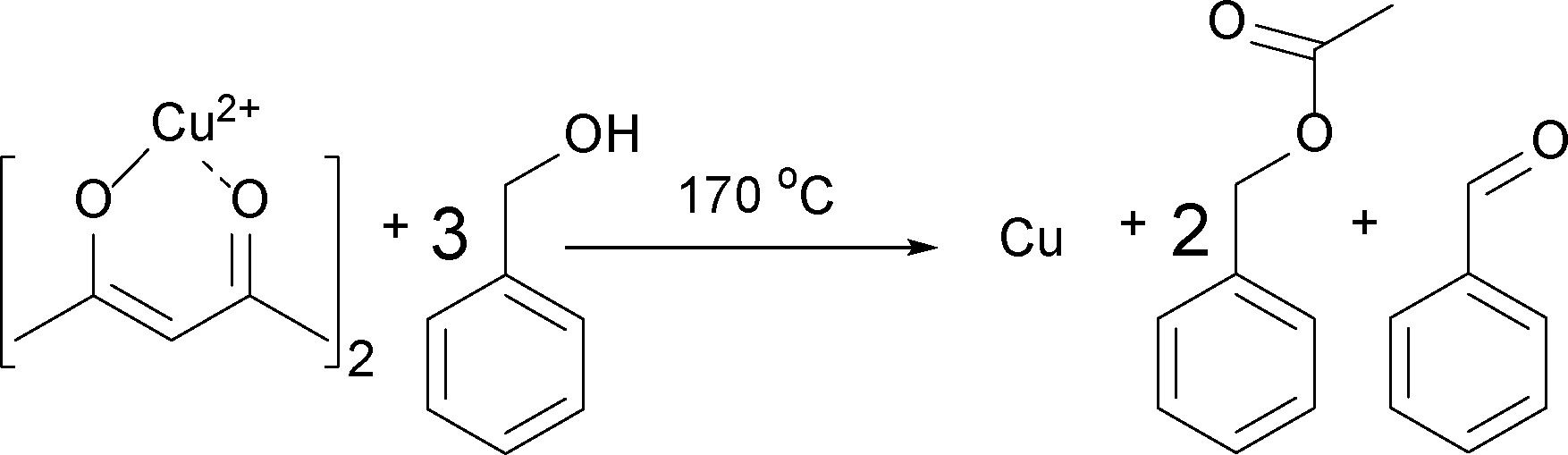
Having these reactions in mind and just considering the organic species detected, we can formulate an overall chemical reaction for the formation of Cu2O, as shown in Scheme 4, and for Cu0 as displayed in Scheme 5.
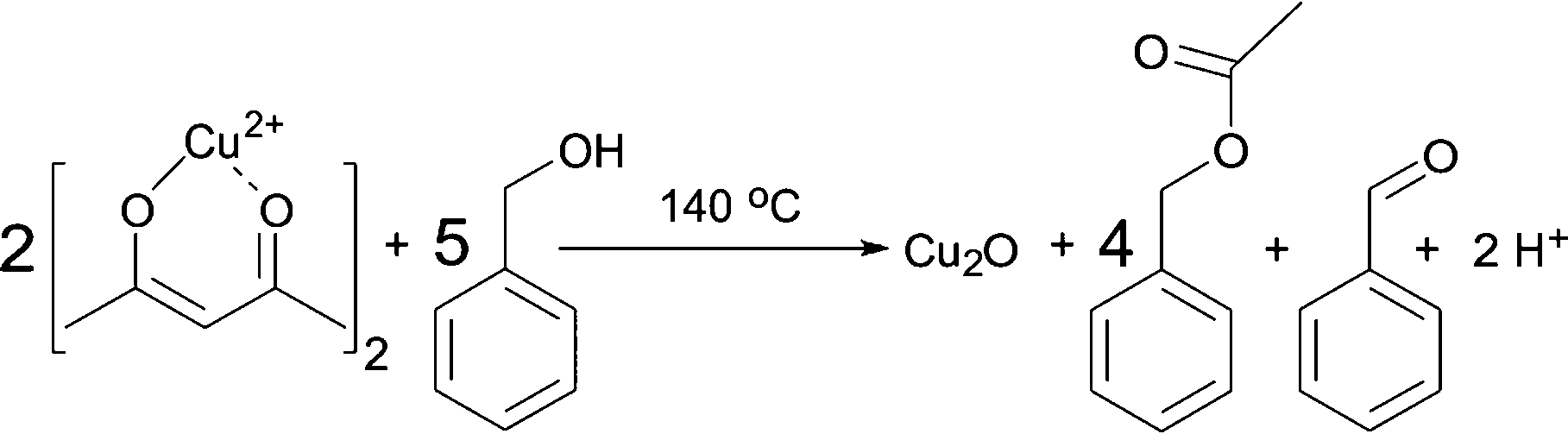 | ||
| Scheme 4 Formation of Cu2O, benzyl acetate and benzaldehyde from Cu(acac)2 and benzyl alcohol at 140 °C. | ||
 | ||
| Scheme 5 Formation of Cu, benzyl acetate and benzaldehyde from Cu(acac)2 and benzyl alcohol at 170 °C. | ||
From Scheme 4 it is evident that the formation of 1 mmol Cu2O produces 4 mmol of benzyl acetate and 1 mmol of benzaldehyde. The initial concentration of Cu(acac)2 in benzyl alcohol is 0.070 M, which means that 0.175 M benzyl alcohol (BnOH) are required, resulting in 0.140 M benzyl acetate, 0.035 M benzaldehyde and 0.070 M Cu+ (or 0.035 M Cu2O) as summarized in eqn (2):
| 0.070 M Cu2+ + 0.175 M BnOH → 0.140 M BnAc + 0.035 M BnAd + 0.070 M Cu+ | (2) |
According to Scheme 5, the formation of Cu at 170 °C follows eqn (3):
| 0.070 M Cu2+ + 0.210 M BnOH → 0.140 M BnAc + 0.070 BnAd + 0.070 M Cu0 | (3) |
For the reaction at 140 °C, the measured concentration of benzyl acetate matches with the nominal value of 0.137 M within the resolution limit of our method, as shown in Fig. 5a and Table 1. At the same time, the sum of the concentrations of benzaldehyde in the control experiment (0.052 M) and the nominal value based on eqn (2) equals to 0.087 M, which is much lower than the actually measured concentration of just 0.115 M. Evidently, the Cu2O nanoparticles are able to catalyze the oxidation of benzyl alcohol58,59 to increase the benzaldehyde concentration beyond the expected value. The high coverage of the surface of the Cu2O nanoparticles as mentioned before terminates the reaction at this point without further reduction and we observe a saturation of the benzaldehyde concentration as evidenced in Fig. 5a.
| Compound | Lower detection limit (M) | Synthesis at 140 °C | Synthesis at 170 °C | ||
|---|---|---|---|---|---|
| Expected value (M) | Measured value (M) | Expected value (M) | Measured value (M) | ||
| Benzyl acetate | 0.007 | 0.140 | 0.137 | 0.140 | 0.112 |
| Benzaldehyde | 0.003 | 0.035 | 0.115 | 0.070 | 0.133 |
| Dibenzyl ether | 0.008 | 0 | 0 | 0 | 0.047 |
| Control experiment | 140 °C | 170 °C | |||
| Benzaldehyde | 0.008 | — | 0.052 | — | 0.062 |
For the reaction at 170 °C, based on the shape of the concentration profile of the organic compounds, the mechanism seems to be even more complex as shown in Fig. 5b. The final concentration of benzyl acetate equals to 0.112 M, which is lower than the nominal value of 0.140 M expected from eqn (3). This can be explained by the observation of acetone and acetylacetonate in the GC-MS chromatogram. Therefore, we assume that ligand exchange reactions, as shown in Scheme 6, are competing for Cu(acac)2 with the reaction shown in Scheme 2.
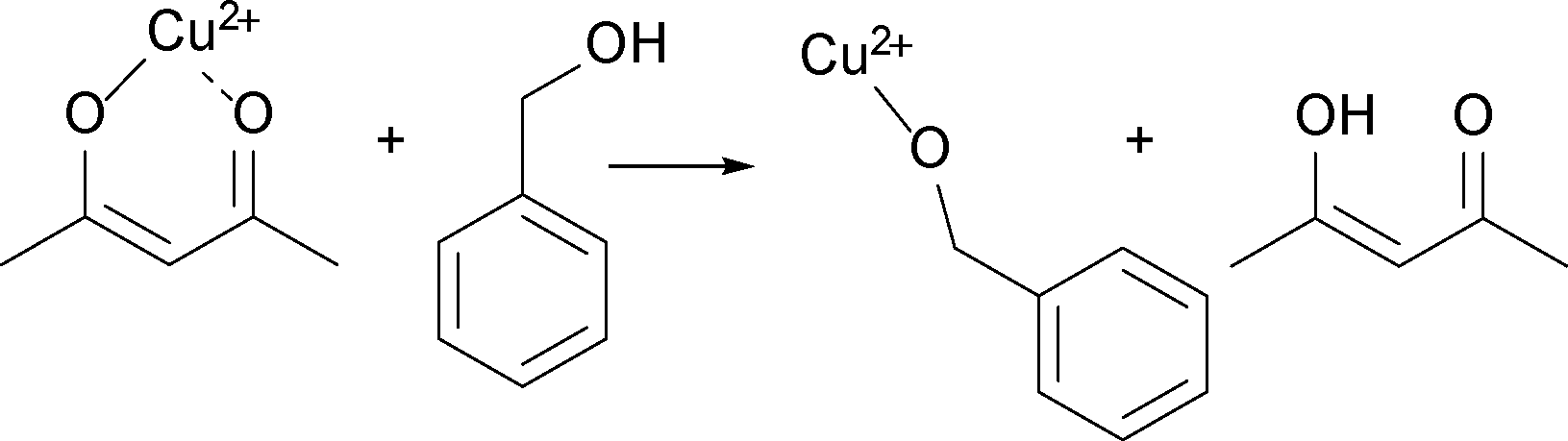 | ||
| Scheme 6 Ligand exchange between acetylacetonate and benzyl alcohol as competition reaction. | ||
In contrast to the synthesis at 140 °C, at 170 °C the measured concentration of benzaldehyde of 0.133 M agrees well with the expected value of 0.132 M, which is the sum of benzaldehyde from the control experiment (0.062 M) and from the reaction shown in eqn (3). This result is not surprising, because with the transformation of Cu2O to Cu also the catalytically active nanoparticles disappear. However, in the very short time frame between 120–150 min we observed a rapid increase of the concentration of benzaldehyde and a decrease of the precursor concentration. It is interesting to note that only at this point of the reaction, the concentration of benzaldehyde does not correlate with the concentration of benzyl acetate, which indicates that the reactions shown in Scheme 6 might become more pronounced.
60 or Zn(acac)2.9 This step starts with the nucleophilic attack of the hydroxyl group of benzyl alcohol on one of the carbonyl groups of the acetylacetonate ligand. As a result, benzyl acetate and an enolate ligand form (Scheme 2). In the next step, benzyl alcohol coordinates to the copper ion. While the benzyl alcohol gets oxidized to benzaldehyde, Cu2+ is reduced to Cu+ as shown in Scheme 3a.
Up to here, we did not discuss, where the oxygen for the formation of the copper oxide comes from. Based on the results described above, we did not find any organic compounds that are typically ascribed to condensation reactions responsible for the formation of a metal–oxygen–metal bond.15 Since the synthesis is performed under ambient conditions, we assume that water impurities in solvent can influence the formation of nanoparticles. In addition hydrogen ion formed during benzyl alcohol oxidation might react with oxygen to form water as proposed in Scheme 7a.57,61,62 Once water is present, two possible scenarios for the formation of a Cu–O bond are possible. A water molecule directly coordinates to the enolate ligand, which leads to the formation of acetone and Cu(I)–OH species as shown in Scheme 7b. Alternatively, a benzyl alcohol molecules reacts with the enolate ligand, which leads to the formation of acetone and a copper benzyl alcoholate. These scenarios are both plausible, because traces of acetone were detected by GC-MS. A consecutive reaction of the copper alkoxide with water leads to a Cu(I)–OH species, as displayed in Scheme 7c. Finally, the Cu(I)–OH species condense with each other under release of water molecules and formation of Cu–O–Cu bonds.
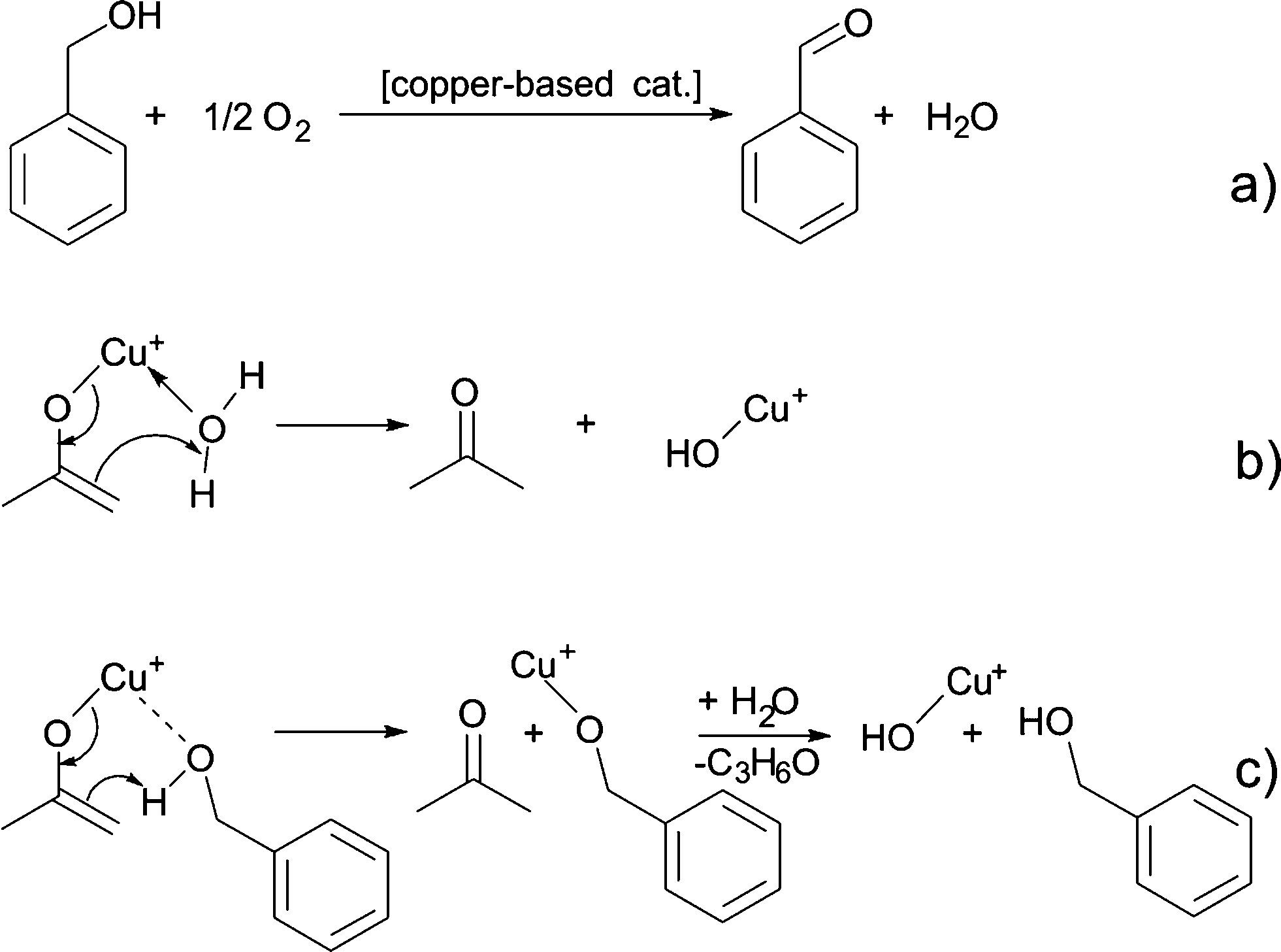 | ||
| Scheme 7 (a) Formation of water, (b) reaction of the copper enolate with water to acetone and copper hydroxide, and (c) reaction of the copper enolate with benzyl alcohol to acetone and copper benzyl alcoholate, which further reacts to copper(I) hydroxide and benzyl alcohol. | ||
4. Conclusions
Our in situ XAS/UV-vis/FTIR experiments reveal the dependence between the reactions leading to nucleation and growth of Cu-based nanoparticles and catalytic activity of newly forming nanoparticles as shown in Fig. 6. The organic species formed directly due to reduction of copper(II) acetylacetonate and nucleation of Cu2O and Cu nanoparticles are shown in red. The nucleation of copper(I) oxide nanocubes co-occurs with the formation of benzyl acetate and benzaldehyde. Cu2O subsequently transforms by solid-state reduction to metallic copper, which is accompanied by oxidation of benzyl alcohol to benzaldehyde. The parallel side reactions are shown in blue. We observe aerobic oxidation of benzyl alcohol to benzaldehyde. Overall, we can quantitatively differentiate between benzaldehyde forming in three independent reactions. Moreover, newly formed Cu nanoparticles show significant catalytic activity towards condensation of benzyl alcohol to dibenzylether. In this work, we demonstrate how to disentangle contributions of main and side reactions forming organic species during nanoparticle synthesis. That is particularly important when the nucleation of nanoparticles is coupled with the changes of the oxidation state of a metal.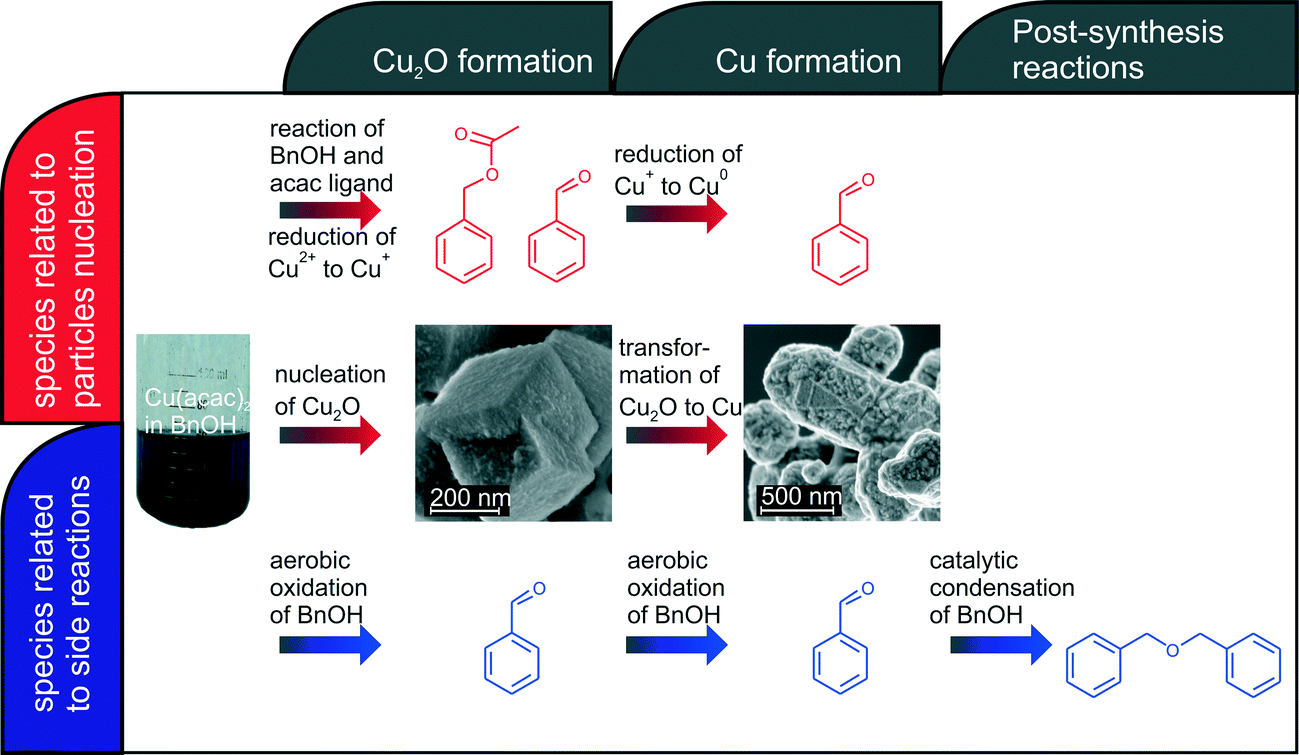 | ||
| Fig. 6 Overview on organic and inorganic reactions taking place during synthesis of copper-based nanoparticles. | ||
Acknowledgements
We acknowledge the Swiss National Science Foundation (200021_137637) for the financial support and SNX Council of SNBL, ESRF for the beam time allocation. We thank the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich for the use of the SEM. We thank G. Grassi and F. Arc (Laboratory of Physical Chemistry, ETH Zurich) for the assistance with GC-MS measurements. MS thanks Dr. I. Bilecka and Dr. F. Heiligtag for their help in the initial phase of the project.References
- M. Niederberger, M. H. Bartl and G. D. Stucky, J. Am. Chem. Soc., 2002, 124, 13642–13643 CrossRef CAS PubMed
.
- C. J. Dalmaschio, E. G. D. Firmiano, A. N. Pinheiro, D. G. Sobrinho, A. F. de Moura and E. R. Leite, Nanoscale, 2013, 5, 5602–5610 RSC
- P. H. Mutin and A. Vioux, J. Mater. Chem. A, 2013, 1, 11504–11512 CAS
- A. Vioux, Chem. Mater., 1997, 9, 2292–2299 CrossRef
- N. Pinna and M. Niederberger, Angew. Chem., Int. Ed, 2008, 47, 5292–5304 CrossRef CAS PubMed
- N. Shi, W. Cheng, H. Zhou, T. X. Fan and M. Niederberger, Chem. Commun., 2015, 51, 1338–1340 RSC
- G. Garnweitner and C. Grote, Phys. Chem. Chem. Phys., 2009, 11, 3767–3774 RSC
- I. M. Grabs, C. Bradtmoller, D. Menzel and G. Garnweitner, Cryst. Growth Des., 2012, 12, 1469–1475 CAS
- B. Ludi, M. J. Suess, I. A. Werner and M. Niederberger, Nanoscale, 2012, 4, 1982–1995 RSC
- B. Ludi and M. Niederberger, Dalton Trans., 2013, 42, 12554–12568 RSC
- I. Bilecka, P. Elser and M. Niederberger, ACS Nano, 2009, 3, 467–477 CrossRef CAS PubMed
- G. Garnweitner and M. Niederberger, J. Mater. Chem., 2008, 18, 1171–1182 RSC
- K. B. Ramos, G. Clavel, C. Marichy, W. Cabrera, N. Pinna and Y. J. Chabal, Chem. Mater., 2013, 25, 1706–1712 CrossRef
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582–596 CrossRef CAS
- M. Niederberger and G. Garnweitner, Chem. – Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed
- N. Pinna, M. Karmaoui and M. G. Willinger, J. Sol-Gel Sci. Technol., 2011, 57, 323–329 CrossRef CAS PubMed
-
M. Staniuk, M. Niederberger and D. Koziej, Nanoengineering: Fabrication, Properties, Optics, and Devices Xi, 2014, 9170
- M. Staniuk, O. Hirsch, N. Kranzlin, R. Bohlen, W. van Beek, P. M. Abdala and D. Koziej, Chem. Mater., 2014, 26, 2086–2094 CrossRef CAS
- D. Koziej, M. D. Rossell, B. Ludi, A. Hintennach, P. Novak, J. D. Grunwaldt and M. Niederberger, Small, 2011, 7, 377–387 CrossRef CAS PubMed
- I. Olliges-Stadler, M. D. Rossell and M. Niederberger, Small, 2010, 6, 960–966 CrossRef CAS PubMed
- F. B. F. Silva, E. C. Paris, G. M. da Costa and C. Ribeiro, RSC Adv., 2014, 4, 53265–53272 RSC
- I. Bilecka, A. Hintennach, I. Djerdj, P. Novak and M. Niederberger, J. Mater. Chem., 2009, 19, 5125–5128 RSC
- B. Ludi, I. Olliges-Stadler, M. D. Rossell and M. Niederberger, Chem. Commun., 2011, 47, 5280–5282 RSC
- D. Koziej, F. Krumeich, R. Nesper and M. Niederberger, J. Mater. Chem., 2009, 19, 5122–5124 RSC
- N. Kranzlin, S. Ellenbroek, D. Duran-Martin and M. Niederberger, Angew. Chem., Int. Ed., 2012, 51, 4743–4746 CrossRef PubMed
- K. M. O. Jensen, C. Tyrsted, M. Bremholm and B. B. Iversen, ChemSusChem, 2014, 7, 1594–1611 CrossRef CAS PubMed
- G. Philippot, K. M. O. Jensen, M. Christensen, C. Elissalde, M. Maglione, B. B. Iversen and C. Aymonier, J. Supercrit. Fluids, 2014, 87, 111–117 CrossRef CAS PubMed
- H. Jensen, M. Bremholm, R. P. Nielsen, K. D. Joensen, J. S. Pedersen, H. Birkedal, Y. S. Chen, J. Almer, E. G. Sogaard, S. B. Iversen and B. B. Iversen, Angew. Chem., Int. Ed., 2007, 46, 1113–1116 CrossRef CAS PubMed
- A. Michailovski, J. D. Grunwaldt, A. Baiker, R. Kiebach, W. Bensch and G. R. Patzke, Angew. Chem., Int. Ed., 2005, 44, 5643–5647 CrossRef CAS PubMed
- Y. Zhou, Y. H. Lin and G. R. Patzke, Prog. Chem., 2012, 24, 1583–1591 CAS
- Y. Zhou, N. Pienack, W. Bensch and G. R. Patzke, Small, 2009, 5, 1978–1983 CrossRef CAS PubMed
- I. Olliges-Stadler, J. Stotzel, D. Koziej, M. D. Rossell, J. D. Grunwaldt, M. Nachtegaal, R. Frahm and M. Niederberger, Chem. – Eur. J., 2012, 18, 2305–2312 CrossRef CAS PubMed
- I. Olliges-Stadler, M. D. Rossell, M. J. Suess, B. Ludi, O. Bunk, J. S. Pedersen, H. Birkedal and M. Niederberger, Nanoscale, 2013, 5, 8517–8525 RSC
- Y. G. Sun, Mater. Today, 2012, 15, 140–147 CrossRef CAS
- J. Szlachetko, J. Sa, M. Nachtegaal, U. Hartfelder, J. C. Dousse, J. Hoszowska, D. L. A. Fernandes, H. Q. Shi and C. Stampfl, J. Phys. Chem. Lett., 2014, 5, 80–84 CrossRef CAS
- M. Wuithschick, B. Paul, R. Bienert, A. Sarfraz, U. Vainio, M. Sztucki, R. Kraehnert, P. Strasser, K. Rademann, F. Emmerling and J. Polte, Chem. Mater., 2013, 25, 4679–4689 CrossRef CAS
- J. Jaumot, R. Gargallo, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS PubMed
- W. H. Cassinelli, L. Martins, A. R. Passos, S. H. Pulcinelli, C. V. Santilli, A. Rochet and V. Briois, Catal. Today, 2014, 229, 114–122 CrossRef CAS PubMed
- Q. Tang, P. E. Carrington, Y. C. Horng, M. J. Maroney, S. W. Ragsdale and D. F. Bocian, J. Am. Chem. Soc., 2002, 124, 13242–13256 CrossRef CAS PubMed
- C. A. Nunes, E. C. Resende, I. R. Guimaraes, A. S. Anastacio and M. C. Guerreiro, Appl. Spectrosc., 2011, 65, 692–697 CrossRef CAS PubMed
- H. W. P. Carvalho, S. H. Pulcinelli, C. V. Santilli, F. Leroux, F. Meneau and V. Briois, Chem. Mater., 2013, 25, 2855–2867 CrossRef CAS
- C. Marquez-Alvarez, I. Rodriguez-Ramos, A. Guerrero-Ruiz, G. L. Haller and M. Fernandez-Garcia, J. Am. Chem. Soc., 1997, 119, 2905–2914 CrossRef CAS
- P. Conti, S. Zamponi, M. Giorgetti, M. Berrettoni and W. H. Smyrl, Anal. Chem., 2010, 82, 3629–3635 CrossRef CAS PubMed
- O. Hirsch, G. B. Zeng, L. Luo, M. Staniuk, P. M. Abdala, W. van Beek, F. Rechberger, M. J. Suess, M. Niederberger and D. Koziej, Chem. Mater., 2014, 26, 4505–4513 CrossRef CAS
- N. Kranzlin, M. Staniuk, F. J. Heiligtag, L. Luo, H. Emerich, W. van Beek, M. Niederberger and D. Koziej, Nanoscale, 2014, 6, 14716–14723 RSC
- N. Kranzlin, W. van Beek, M. Niederberger and D. Koziej, Adv. Mater. Interfaces, 2015 DOI:10.1002/admi.201500094
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- A. de Juan and R. Tauler, Crit. Rev. Anal. Chem., 2006, 36, 163–176 CrossRef CAS PubMed
- A. Gaur, B. D. Shrivastava, K. Srivastava, J. Prasad and V. S. Raghuwanshi, J. Chem. Phys., 2013, 139, 034303 CrossRef PubMed
- S. Vukojevic, O. Trapp, J. D. Grunwaldt, C. Kiener and F. Schuth, Angew. Chem., Int. Ed., 2005, 44, 7978–7981 CrossRef CAS PubMed
- D. E. Doronkin, M. Casapu, T. Gunter, O. Muller, R. Frahm and J. D. Grunwaldt, J. Phys. Chem. C, 2014, 118, 10204–10212 CAS
- S. Gross and M. Bauer, Adv. Funct. Mater., 2010, 20, 4026–4047 CrossRef CAS PubMed
- V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer and G. Ferey, Chem. Commun., 2010, 46, 767–769 RSC
- Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2011, 47, 9275–9292 RSC
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed
- Z. C. Li, C. H. Chen, E. S. Zhan, N. Ta, Y. Li and W. J. Shen, Chem. Commun., 2014, 50, 4469–4471 RSC
- C. Keresszegi, D. Ferri, T. Mallat and A. Baiker, J. Phys. Chem. B, 2005, 109, 958–967 CrossRef CAS PubMed
- J. Fan, Y. Dai, Y. Li, N. Zheng, J. Guo, X. Yan and G. D. Stucky, J. Am. Chem. Soc., 2009, 131, 15568–15569 CrossRef CAS PubMed
- G. Zhao, H. Hu, M. Deng, M. Ling and Y. Lu, Green Chem., 2011, 13, 55–58 RSC
- L. Zhang, G. Garnweitner, I. Djerdj, M. Antonietti and M. Niederberger, Chem. – Asian J., 2008, 3, 746–752 CrossRef PubMed
- Y. Xie, Z. F. Zhang, S. Q. Hu, J. L. Song, W. J. Li and B. X. Han, Green Chem., 2008, 10, 278–282 RSC
- M. Caravati, J. D. Grunwaldt and A. Baiker, Phys. Chem. Chem. Phys., 2005, 7, 278–285 RSC
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5ce00454c |
| This journal is © The Royal Society of Chemistry 2015 |