DOI:
10.1039/C3MH00023K
(Review Article)
Mater. Horiz., 2014,
1, 46-57
Molecular crystalline materials with tunable luminescent properties: from polymorphs to multi-component solids
Received
6th May 2013
, Accepted 12th July 2013
First published on 10th September 2013
Abstract
Tuning and controlling the luminescent properties of molecular materials by changing the orientation and arrangement of the fluorophores within a solid has played an important role in realizing multi-color emission. The formation of polymorphs and multi-component molecular solids have attracted considerable interest as new ways of achieving controllable luminescence and other photophysical properties for application in the next generation of photofunctional materials. In this article, recent advances in the synthesis of fluorescent polymorphs and multi-component materials and potential photo-related applications of the resulting materials are described. We first review the methods of preparation of polymorphs with tunable static luminescence, and the switching of the dynamic luminescence between polymorphs for potential sensor applications is also introduced. Attention is then focused on the supramolecular design (making use of hydrogen bonding and halogen bonding interactions) and methods of fabrication of multi-component molecular solids, and their color-tunable fluorescence and phosphorescence together with their stimuli-responsive properties for use as sensors. The use of density functional theory to study intramolecular and intermolecular energy transfer as well as the electronic structures of multi-component molecular solids is also outlined. Finally, we briefly discuss perspectives for the further development of these luminescent molecular solid-state materials.
 Dongpeng Yan | Dongpeng Yan obtained his PhD degree under the supervision of Prof. Xue Duan at Beijing University of Chemical Technology (BUCT) in 2012, In 2011, as a visiting student, he studied at the Department of Chemistry, University of Cambridge under Prof. William Jones. Now he is an associate professor at BUCT. His main research topics are experimental and computer simulation studies of organic photofunctional crystals and new layered materials. He was received an Award Nomination of Annual Figure among Chinese University Students (2011), a “Talent Model” Award in the Universities in Beijing (2010), the Tang Aoqing Chemical Scholarship (2011), and many other awards. |
 David G. Evans | David G. Evans studied as both undergraduate and research student at Jesus College, Oxford and obtained a DPhil under the supervision of Prof. D. M. P. Mingos FRS. After postdoctoral work at Bristol University with Prof. F. G. A. Stone FRS, he was appointed to a lectureship at Exeter University in 1985. Several visits to Chinese university chemistry departments in the early 1990s convinced him of China's great potential for development in this area and he moved to Beijing University of Chemical Technology in 1996. His research interests focus on intercalation in layered solids. He was awarded an International Scientific and Technological Cooperation Award of the People's Republic of China in 2005. |
1 Introduction
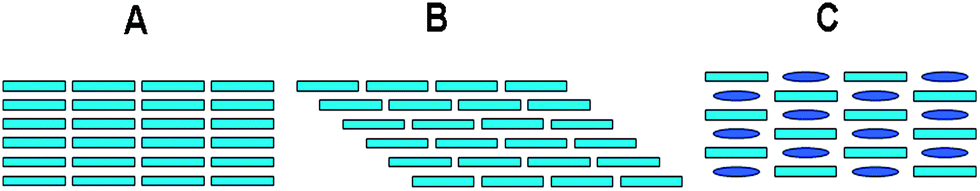
Organic solid-state luminescent materials have been widely studied during the past 20 years because of their excellent optoelectronic characteristics leading to applications in light-emitting diodes,1 lasers,2 sensors,3 and biological imaging.4 Their low cost, facile processing, and flexibility also lead to their potential uses in large-scale flat panel displays and soft optical device applications. In order to develop new types of multi-color displays and tunable dye lasers,5 it is necessary to find ways of tailoring the luminescent properties of a particular organic chromophore. For low-molecular-weight organic solid materials, it has been recognized that the bulk luminescent/optical properties are not just governed by the photo-activity of single molecules but by their collective arrangement and interaction modes.6 During the last decade, a number of pure organic solid-state chromophore systems with controllable emission properties have been developed. The luminescent properties of these materials are generally tailored by altering the molecular packing mode within the crystal structure, i.e. by forming polymorphs7—different solid crystalline phases of a specific compound resulting from different arrangements of the molecules (Scheme 1A and B) within the solid state. Such tunable polymorph-based emissive behavior not only paves an effective way to tailor the luminescence of molecular materials, but also provides an insight into the relationship between the molecular arrangement/packing fashion and bulk optical properties. As an alternative to forming polymorphs of a single material, the synthesis of multi-component molecular solids8 is also of great current interest in the fields of molecule-based materials chemistry and crystal engineering and has already been extensively used, for example, to improve bioavailability, stability and pharmacological activity of pharmaceutical formulations.9 As generally used in the literature, the term “multi-component solid” can cover co-crystals, mixed crystals, salts, adducts, and complexes. Incorporating the target chromophore in a multi-component solid along with a variety of co-assembled units (co-formers) clearly offers more flexibility than the formation of polymorphs of the pure material. Moreover, from a material design perspective, the development of supramolecular synthesis strategies10 means that the stacking structures of the resulting multi-component solids can be more predictable than for polymorphs, which makes it easier to design solid-state materials with the desired properties. Thus the construction of well-organized multi-component materials (Scheme 1C) by the assembly of two or more units has recently emerged as an alternative method to obtain tunable fluorescence, although the examples are still more limited than those of polymorph-dependent emission.
 |
| | Scheme 1 Polymorphs (A and B) of a single chromophore (cyan color) and a multi-component solid (C) of the same chromophore assembled with a co-former (blue color). The different packing modes can give different fluorescent emissions from the same chromophore. | |
Pioneering reviews from the groups of Kato and Sagara11a and Das and Varghese11b have mainly focused on the dynamic luminescent properties of π-conjugated molecular systems, and provide a good understanding of the way in which solid-state optical properties depend on molecular aggregation. Due to the rapid development in the design and fabrication of both polymorphs and, particularly, multi-component solids with different static luminescent properties, we believe it is timely and necessary to summarize recent progress in this area and thus hopefully stimulate the design of new types of photofunctional molecular solids. In addition, we also discuss some recent theoretical calculations on supramolecular multi-component systems which have provided a better understanding of structure–luminescence relationships. In this article, we focus—from both materials chemistry and crystal engineering perspectives—on strategies for tuning the fluorescence of molecular materials by forming polymorphs and multi-component solids. Firstly, we review methods for the synthesis of polymorphs and the static polymorph-dependent luminescence of different materials. We then review the design and construction of multi-component solid materials with color-tunable luminescence and the way in which the dynamic switching of their luminescence is achieved for potential applications in sensors. The new physical and chemical insights into these materials arising from theoretical calculations are also discussed. Finally, perspectives for the further development of luminescent solid-state materials are addressed.
2 Polymorphs with tunable emission
2.1 Structure–luminescence relationships
(a) Packing polymorphs.
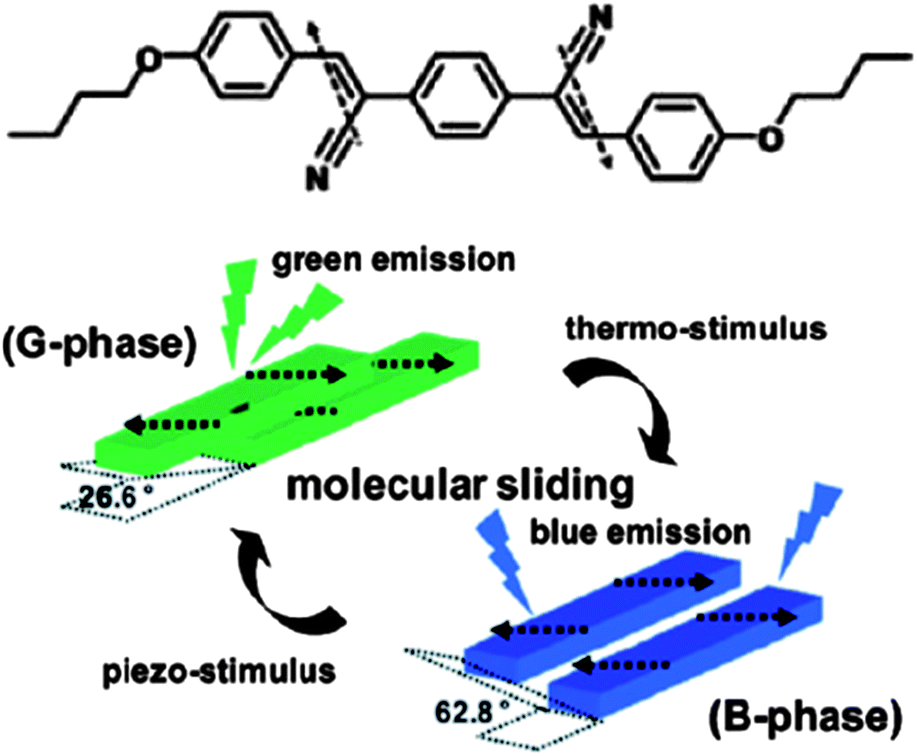
In theory, packing arrangements, molecular conformations, intermolecular interactions, and lattice energies of the same molecules in different crystalline polymorphs can all influence their fluorescence properties. Up to now, different packing arrangements that result in different intermolecular interactions have played the most significant role in tailoring the luminescence.12–15 In the case of planar π-conjugated molecular systems, the emission behavior is largely dependent on the relative orientations and degree of overlap between the chromophores: H- and J-type aggregates correspond to θ > 54.7° and θ < 54.7° respectively, where θ is the angle between the transition dipole vector and the line between the centers of the two photoactive molecules.16 Normally, the presence of H- or J-type dimers results in blue or red-shifts, respectively, compared with the monomer luminescence.17 For example, Park et al.18 have reported that the metastable G-phase (θ = 26.6°) and thermodynamically stable B-phase (θ = 62.8°) polymorphs of a cyano distyrylbenzene derivative, (2Z,2′Z)-2,2′-(1,4-phenylene)bis(3-(4-butoxyphenyl))acrylonitrile (DBDCS, Fig. 1) exhibit green and blue emission respectively. The two phases can also be interconverted by changing the temperature or pressure (Fig. 1).
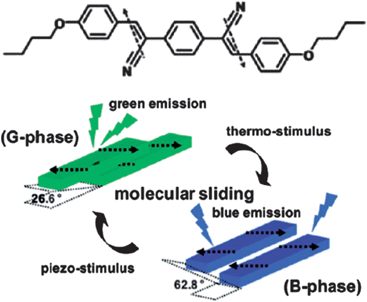 |
| | Fig. 1 Chemical structure of DBDCS; illustration of the two different modes of slip-stacking in molecular sheets, dictated by different ways of antiparallel/head-to-tail coupling of local dipoles. Reprinted with permission from ref. 18. © American Chemical Society, 2010. | |
(b) Conformational polymorphs.
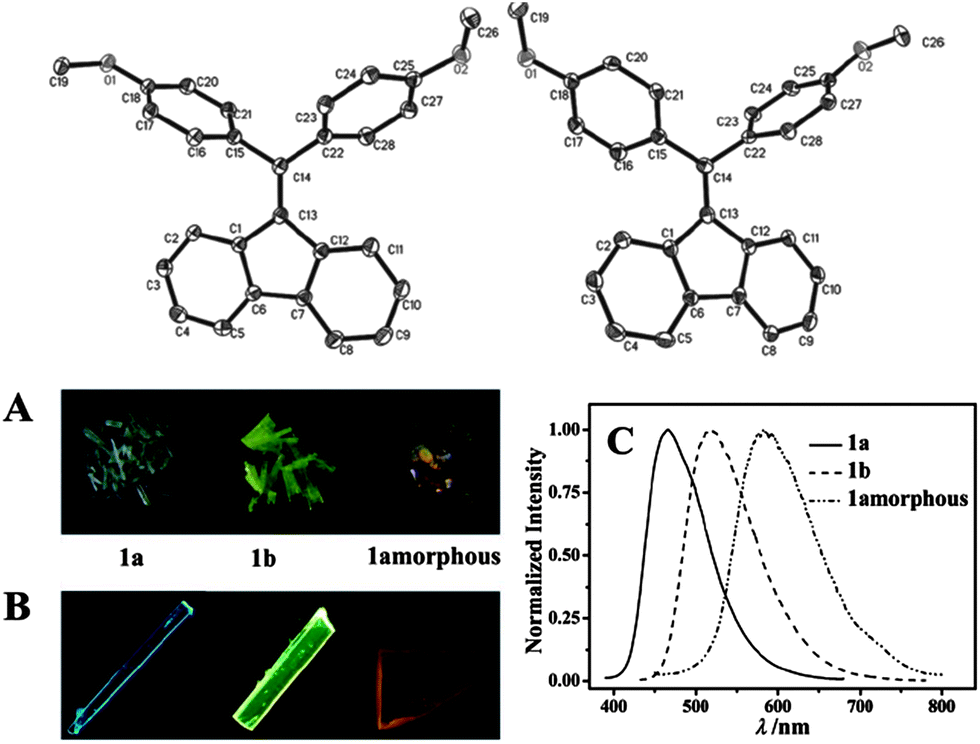
The occurrence of conformational polymorphs can also change the absorption and emission behaviors of molecular solids, since differences in the extent of conjugation of the molecules induced by different conformations can influence the energy levels and band gap of the solids. For example, Gu et al.19 have systematically studied the fluorescence of conformational polymorphs of di(p-methoxylphenyl)dibenzofulvene (a and b, shown in Fig. 2). The polymorphs a and b exhibit strong fluorescence with the maximum emission wavelengths (λmaxem) of 466 and 518 nm, and fluorescence lifetimes of 0.95 and 0.62 ns, respectively. The twisted conformations of a and b exclude any strong intermolecular interactions, suggesting the absence of intermolecular H- or J-aggregates within the crystals. In polymorph a, the torsion angles C13–C14–C15–C16 and C13–C14–C22–C28 are 74.40° and 57.95°, respectively, while the two torsion angles in b are 65.43° and 44.83°, respectively (Fig. 2, top). Thus, the fluorene ring in polymorph b exhibits more coplanarity with the two benzene rings than in polymorph a. Therefore, the different absorption (Fig. 2A) and emission (Fig. 2B and C) behaviors of polymorphs a and b, and the amorphous solid can be mainly attributed to the changes in the molecular conformations. This conclusion is confirmed by theoretical calculations, which show that the two stable optimized conformations in the gas phase result in emission maxima at 454 and 539 nm, reasonably close to the experimental results.
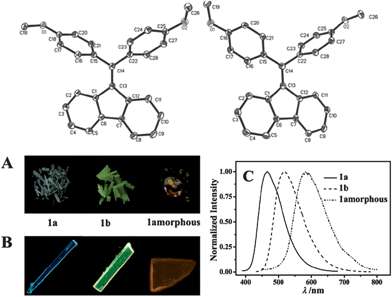 |
| | Fig. 2 (Top) Molecular structures of a (left) and b (right); (bottom) daylight photos (A) and fluorescence microscope images (B) of the blue emission from crystals of polymorph a and the yellow-green emission from crystals of polymorph b and the amorphous solid. (C) The fluorescence spectra of polymorphs a and b, and the amorphous solid. Reprinted with permission from ref. 19. © Wiley-VCH, 2012. | |
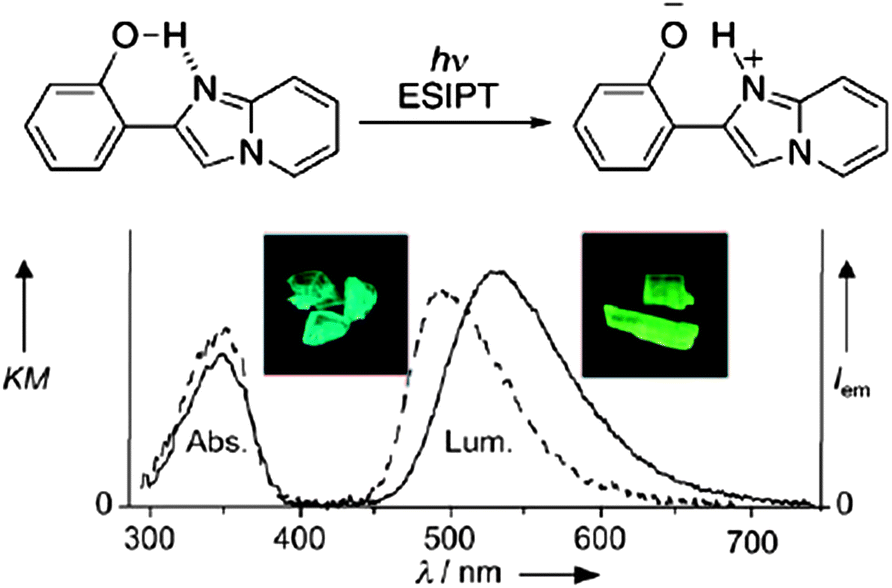
For chromophores containing an intramolecular hydrogen bond, the excited-state intramolecular proton-transfer (ESIPT)20 process can be another effective mechanism to obtain polymorphism-dependent luminescence. ESIPT luminescence is usually sensitive to the external environment and the mode of molecular packing in the solid state. Araki and co-workers14a have developed two crystal polymorphs (BG and Y) of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine (Fig. 3 (top)), which exhibit bright blue-green and yellow fluorescence respectively (inset in Fig. 3 (bottom)). Single crystal diffraction shows the presence of O–H⋯N intramolecular hydrogen bonds in the molecular solids, and that BG and Y are conformational polymorphs, in which the major conformational difference involves the dihedral angle between the phenyl and imidazopyridine rings: the two aromatic rings are nearly coplanar (with a torsion angle of 1.38°) in polymorph Y, while the conformation is twisted in polymorph BG (with a torsion angle of 5.88°). This work shows that differences in the molecular conformation can affect the ESIPT process and result in tunable ESIPT luminescence within solids.
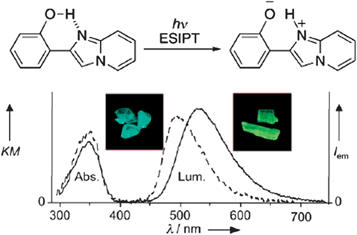 |
| | Fig. 3 (Top) Schematic illustration of the ESIPT of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine; (bottom) absorption and luminescence spectra of polymorph BG (dotted line) and Y (solid line). Inset: pictures of both polymorphs under UV irradiation (365 nm). Reprinted with permission from ref. 14a. © Wiley-VCH, 2008. | |
2.2 Synthesis methods
(a) Solution processes.
The most common method to obtain luminescent polymorphs is to dissolve the fluorophore compounds in various solvents and grow single crystals by slow evaporation of the solvents until there is evidence of a new crystalline state. Empirical solution crystallization is a trial-and-error process,21 and the number of polymorphs obtained is usually roughly proportional to the number of attempts,22 since it is still difficult to predict a priori whether or not any product will be a new polymorph. In an early example, Ibanez et al.23 obtained two polymorphs of 1-cyano-1-(4-nitrophenyl)-2-(4-methoxyphenyl)ethene (CMONS) from toluene and dichloromethane solutions. Although the detailed crystal structures were not obtained, differential scanning calorimetry (DSC) and powder X-ray diffraction (XRD) confirmed the formation of two different polymorphs, with λmaxem located at 539 and 557 nm. In addition, the two polymorphs of CMONS also exhibit different second harmonic generation (SHG) behavior.
(b) Vacuum sublimation.
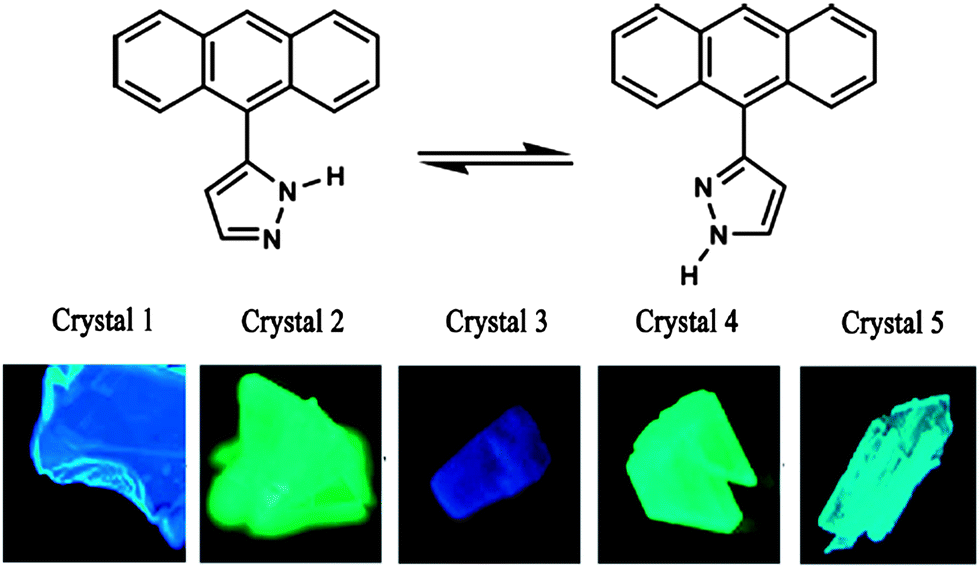
The development of solvent-free crystallization methods is a prerequisite to large-scale applications of such materials, since it is relatively difficult and environmentally unfriendly to evaporate organic solvents in large quantities. In addition to not requiring any solvent, vacuum sublimation also offers other advantages: in the sublimation process, the type and amount of carrier gas, and the temperature of both source zone and growth zone can be finely tuned, which makes it possible to get several new polymorphs. For example, by careful control of the temperature of the condensation area (24–40 °C) during sublimation, Wang et al.12a obtained five high-quality crystalline polymorphs of 3(5)-(9-anthryl)pyrazole (ANP, top in Fig. 4), which exhibited emission ranging from blue to green in color (bottom in Fig. 4). In another example, Ma et al.24 obtained a new polymorph of trans-1,4-distyrylbenzene by sublimation, which exhibits thin and plate-like morphology with regular edges. Such high-quality crystals produced by vacuum sublimation can be used in optically pumped lasers.
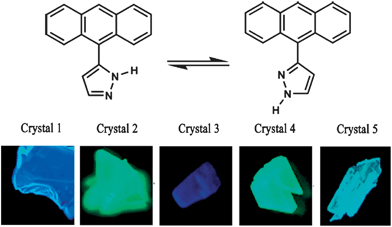 |
| | Fig. 4 (Top) Structure of 3(5)-(9-anthryl)pyrazole (ANP); (bottom) photographs of crystals 1–5 excited under UV light (365 nm). Reprinted with permission from ref. 12a. © Wiley-VCH, 2006. | |
(c) External stimuli.
The free energy differences between different crystalline polymorphs are usually very small (<10 kJ mol−1),25 which suggests that new stacking modes in crystalline solids may be obtained by external perturbation (such as heat or mechanical pressure). Thermal interconversion between luminescent polymorphs was first developed by Araki and co-workers,6a who obtained two polymorphs of 2′,2′′:6′,2′′-terpyridine (tpy), where the needle- and plate-like crystalline states exhibit essentially no luminescence (photoluminescence quantum yield (PLQY) < 0.01) and blue luminescence (PLQY = 0.2) respectively. When the needle-like crystals were heated at 89.5 °C for 10 min and then cooled to room temperature, the solid showed a strong blue emission. XRD and DSC confirmed that the needle-to-plate crystal phase transition occurs on heating. Thermally driven formation of new fluorescent polymorphs has also been reported for several other molecular solids, such as cyano-substituted diphenylbutadienes,26 (4-biphenylyl)phenyldibenzofulvene,27 and trifluoromethyl-substituted aromatic amines.12c Mechanical pressure is another effective way to obtain new polymorphs. For example, Zhang et al.28 have reported that two polymorphs of difluoroboron avobenzone (a boron complex of a commercial sunscreen material) can be interconverted by alternate crushing and physical smearing, which results in a reversible change in luminescence between green and yellow. The above dynamic responsive behaviors are known as thermochromic and mechanochromic emission, respectively. Such fluorescent polymorphs are potential candidates for rewritable optical materials and use in information storage devices.29
3 Tuning fluorescence by forming multi-component solids
3.1 The design of multi-component structures
The formation of multi-component molecular solids as a means of tuning fluorescence is based on the expectation that by appropriate selection and design of the chromophore and its co-assembled components (co-formers), and of the interaction type (such as hydrogen bonding, halogen bonding and π–π interactions), the stacking of the fluorescent molecules in the solid state can be modified. Generally, luminescent multi-component molecular solids can be designed and constructed based on the supramolecular synthon method.10,30
(a) Hydrogen-bond design.
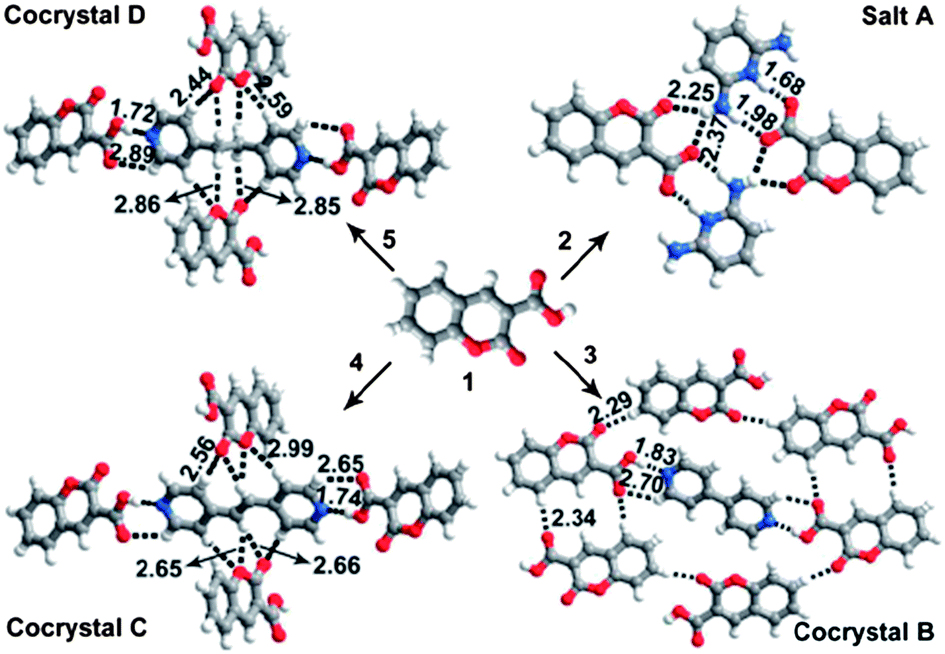
The hydrogen bond interaction has been widely used to design luminescent multi-component crystalline products.31–41 Carboxylic acids are a large family of hydrogen bond donors, which can form intermolecular interactions with a range of hydrogen bond acceptors. For example, coumarin-3-carboxylic acid (C3C)42—a well-documented chromophore for photofunctional applications—can form hydrogen bonds with pyridine-based molecules, and the luminescence of the resulting multi-component crystals with different crystal arrangements (Fig. 5) can vary over a wide range (400–460 nm) in the blue region.31 Assembly of carboxylic acid-containing compounds with amine derivatives, often results in proton transfer from the carboxylic acid group to the amine nitrogen atom, resulting in the formation of a salt. For example, Imai et al.32 have reported that 2-naphthalenecarboxylic acid forms salts with three phenylethylamine derivatives, namely, 1-phenylethylamine, 1-(p-tolyl)ethylamine, and 1-(4-methoxyphenyl)ethylamine. These multi-component fluorophores have one-dimensional (1D) columnar network structures formed by the carboxylate oxygen of a 2-naphthalenecarboxylate anion and the ammonium hydrogen of a protonated amine. The fluorescence emission and PLQY value vary in the range 349–368 nm and 0.02–0.10, respectively. Dong et al.39 exploited the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H hydrogen bond interaction to develop two co-crystals of 1-(5-(anthracen-9-yl)-3-(4-methoxyphenyl)-4,5-dihydropyrazol-1-yl)ethanone (AMPE) with acetic acid and malonic acid. The resulting co-crystals exhibit fluorescence peaks centered at 478 and 497 nm, which are red-shifted and broader than the emission band of pure AMPE crystals. Typical examples of luminescent multi-component solids based on hydrogen bond assembly are listed in Table 1.
O⋯H hydrogen bond interaction to develop two co-crystals of 1-(5-(anthracen-9-yl)-3-(4-methoxyphenyl)-4,5-dihydropyrazol-1-yl)ethanone (AMPE) with acetic acid and malonic acid. The resulting co-crystals exhibit fluorescence peaks centered at 478 and 497 nm, which are red-shifted and broader than the emission band of pure AMPE crystals. Typical examples of luminescent multi-component solids based on hydrogen bond assembly are listed in Table 1.
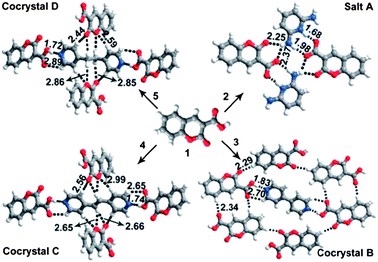 |
| | Fig. 5 Crystal structures of multi-component solids A–D assembled from C3C and co-formers (pyridine-2,6-diamine, 4,4′-bipyridine, (E)-1,2-di(pyridin-4-yl)ethane, and 1,2-di(pyridin-4-yl)ethane). Intermolecular hydrogen bonds and C–H⋯O contacts are indicated by dashed lines: H⋯X distances are given in Ångstrom units. Reprinted from ref. 31. © Royal Society of Chemistry, 2012. | |
Table 1 Typical examples of luminescent multi-component molecular solids based on assembly via hydrogen bonding
| Chromophore |
Co-formers |
References |
| Coumarin-3-carboxylic acid |
Pyridine-2,6-diamine |
31
|
| 4,4′-Bipyridine |
| (E)-1,2-Di(pyridin-4-yl)ethene |
| 1,2-Di(pyridin-4-yl)ethane |
| 2-Naphthalenecarboxylic acid |
1-Phenylethylamine |
32 and 38
|
| 1-(p-Tolyl)ethylamine 1-(4-methoxyphenyl)-ethylamine |
| 2-Fluorobenzylamine 3-fluorobenzylamine |
| Benzylamine |
| 5-Pyridin-4-yl-pyrimidine |
Resorcinol |
33
|
| Hydroquinone |
| 5-(4-Imidazol-1-yl-phenyl)-pyrimidine |
Resorcinol |
33
|
| Hydroquinone |
| 2,4,5-Triphenylimidazole |
4-Bromotetrafluorobenzoic acid |
34
|
| Tetrafluorosuccinic acid |
| Anthracene-2,6-disulfonic acid |
CH3(CH2)(NH2)n (n = 0–5) |
35
|
| 2-Cyano-3(4-(diphenylamino)phenyl) acrylic acid |
4-Picoline; 3-picoline |
36 and 37
|
| 2-Picoline; pyridine |
| Pyrrolidine; piperidine |
| Morpholine |
| 1-(5-(Anthracen-9-yl)-3-(4-methoxyphenyl)-4,5-dihydropyrazol-1-yl)ethanone |
Acetic acid |
39
|
| Malonic acid |
| 1-Acetyl-3-(4-methoxyphenyl)-5-(1-pyrenyl)-pyrazoline |
Phenol |
40
|
| 2-Naphthol |
| 7-Amino-2,4-dimethylquinoline |
Hydrogen chloride |
41
|
| Formic acid |
| Acetic acid |
| Benzoic acid |
|
L-Tartaric acid |
| 1,4-Bis-p-cyanostyrylbenzene |
Resorcinol |
49
|
| 1,4-Dibromotetrafluorobenzene carboxylic acid |
| Pentafluorophenol |
(b) Halogen-bond design.
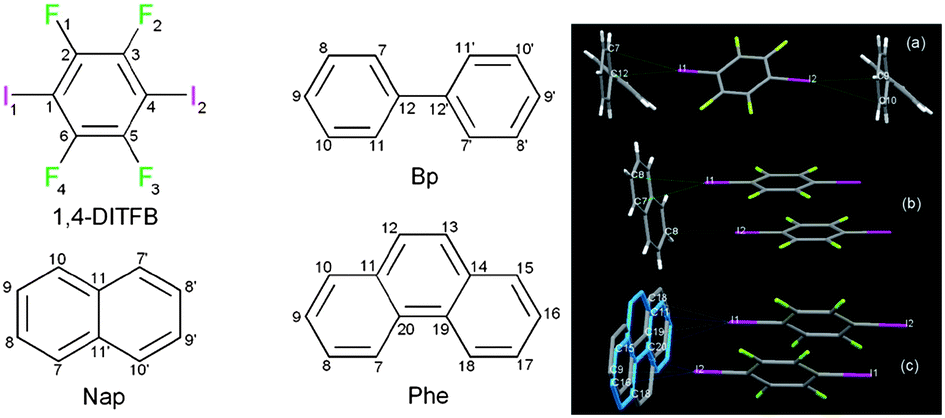
Halogen bonding is an intermolecular non-covalent interaction which is formed between an electron-deficient covalent halogen atom and an electron-rich atom or group.43 Halogen bonding has become an increasingly important and effective way to assemble functional co-crystal materials.44 1,4-Diiodotetrafluorobenzene (1,4-DITFB) is an excellent halogen bond building block due to the electron-withdrawing effect of the fluorine atoms, which enhance the electron-accepting ability of the neighboring iodine atom.45 Jin et al.46 have developed several organic co-crystal systems composed of 1,4-DITFB and aromatic chromophores (such as fluorene, dibenzofuran, dibenzothiophene, phenanthrene, biphenyl, naphthalene, phenanthrene, and pyrene). For example, carbazole (dibenzopyrrole) and 1,4-DITFB form crystals with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry containing infinite chains constructed by C–I⋯π halogen bonding between carbazole and 1,4-DITFB and edge to edge π–π stacking between two 1,4-DITFB molecules. The calculated halogen bonding energy is ca. 23.0 kJ mol−1 based on the single crystal structure, indicative of relatively strong halogen bonding. Similar C–I⋯π interactions also appear in the co-crystal systems assembled from 1,4-DITFB and biphenyl/naphthalene/phenanthrene molecules46c as shown in Fig. 6.
:1 stoichiometry containing infinite chains constructed by C–I⋯π halogen bonding between carbazole and 1,4-DITFB and edge to edge π–π stacking between two 1,4-DITFB molecules. The calculated halogen bonding energy is ca. 23.0 kJ mol−1 based on the single crystal structure, indicative of relatively strong halogen bonding. Similar C–I⋯π interactions also appear in the co-crystal systems assembled from 1,4-DITFB and biphenyl/naphthalene/phenanthrene molecules46c as shown in Fig. 6.
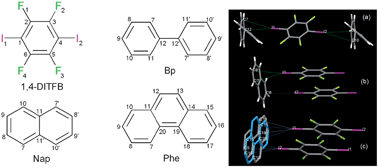 |
| | Fig. 6 Chemical structures of 1,4-DITFB and biphenyl (Bp), naphthalene (Nap), and phenanthrene (Phe); C–I⋯π interactions in co-crystals assembled by halogen bonding donors and acceptors. Reprinted from ref. 46c. © Royal Society of Chemistry, 2012. | |
3.2 Methods of forming luminescent multi-component molecular solids
(a) Solution processes.
Based on intermolecular interactions, molecular recognition between a fluorescent molecule and co-formers can sometimes be achieved in solution, with new co-crystals gradually being formed by slow evaporation of solvent. For example, Draper and co-workers36 have developed supramolecular luminescence systems based on 2-cyano-3(4-(diphenylamino)phenyl)acrylic acid (CDPA) and four amine-based co-formers (pyridine, pyrrolidine, piperidine and morpholine) with CH3CN as the solvent. Compared with pure solid CDPA, the as-obtained multi-component materials exhibit blue-shifted emission and enhanced solid-state luminescence intensity, which might be due to the deaggregation of CDPA in the solid co-crystal matrix.
(b) Physical vapor transport methods.
The vapor transport method can be a suitable strategy to build mixed multi-component crystal solids in which the two or more components have similar shapes and structures.47 In these mixed multi-component crystal systems, the luminescence and energy transfer efficiency can be optimized by adjusting the doping ratio of the guest chromophores within the host matrix. By using this method, Li et al.47a have synthesized single crystals of 1,3,6,8-tetramethylpyrene (TMPY) doped within a perylene host with different ratios in the range from 1% to 15%. Fluorescence spectra indicated that effective energy transfer was achieved in the mixed crystal systems and the luminescence efficiency reached as high as 78%. Sun et al.47b have also obtained large-sized mixed crystals of tetracene- and pentacene-doped 1,4-bis(4-methylstyryl)-benzene (BSB-Me); the PLQY values of the as-prepared tetracene-doped BSB-Me and pentacene-doped BSB-Me crystals were as high as 79% and 47%, and the energy transfer efficiencies from the host BSB-Me to the guests tetracene and pentacene were 49.7% and 73.1%, respectively.
(c) Grinding and sonocrystallization.
Compared with the growth of single-component crystalline polymorphs in solution, co-crystallization is a relatively difficult process particularly when the components have very different solubilities and weak interactions. This has restricted the widespread use of co-crystallization as a means to optimize and tune the luminescence of chromophores. From the perspective of molecular recognition, the necessary force to bring about weak interactions between two components may be supplied by a continuous source of external energy, such as grinding.48 For example, fluorescent co-crystals of 1,4-bis-p-cyanostyrylbenzene (bpcb) and 1,4-DITFB cannot be co-crystallized by conventional solution-based crystallization methods. However, a new crystalline phase with strong blue-green emission can be obtained by either grinding of the two solids, or liquid-assisted grinding of the components, in a ball mill.49 The as-prepared powdered product can in turn be used to seed the growth of 1:1 co-crystals from a solution. Such a two-step process provides an indirect yet effective way to obtain multi-component materials in the form of single crystals.50 A similar grinding and subsequent seeding process was also used to prepare co-crystallized powders and single crystals by assembly of 2,4,5-triphenylimidazole (TPI) as the chromophore and 4-bromotetrafluorobenzene carboxylic acid or tetrafluorosuccinic acid as co-formers.34
Although ultrasonic radiation has been widely used to synthesize hydrogen-bond-organized co-crystals,51 examples of its use in the assembly of luminescent materials are rather rare. In a recent report, it was demonstrated that co-crystals of bpcb and 1,4-DITFB assembled by halogen bonding can be prepared by simultaneously injecting solutions of the two precursors into a sonicated mixture of ethanol and water (v/v = 1:5).52 The as-obtained products have a more uniform particle size on the nanometer scale than the materials produced by the earlier grinding–seeding strategy, which can be attributed to the rapid nucleation of the co-crystals.
3.3 Luminescent properties of multi-component solids
(a) Structure–fluorescence relationships.
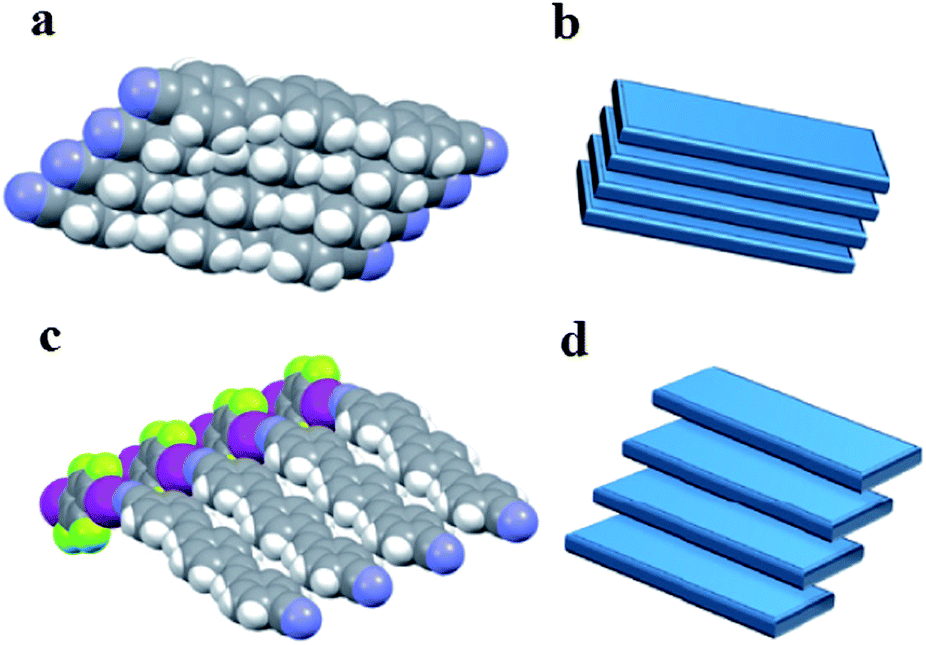
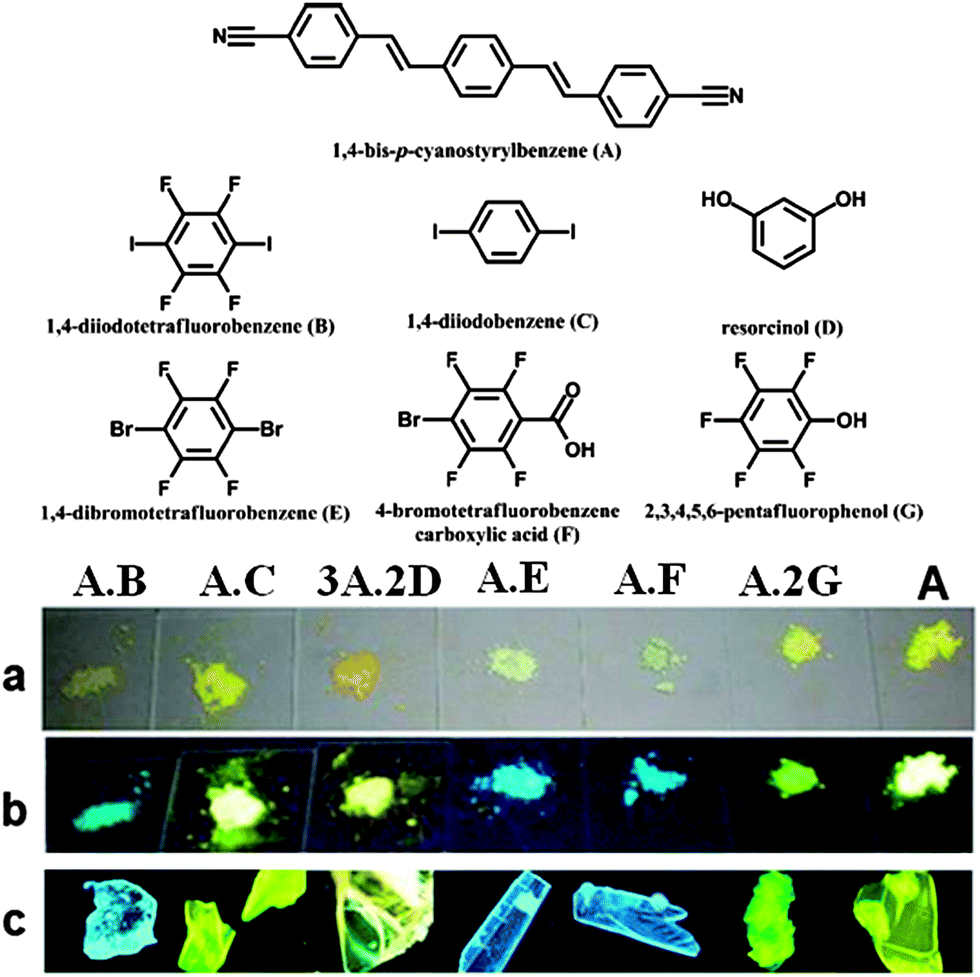
The presence of the co-former within multi-component crystals often alters the packing arrangement of the chromophores, which results in different emission characteristics compared with the pristine solid chromophore. Taking bpcb-based multi-component crystals as an example,50 in the pure bpcb crystal, the bpcb molecules interact with each other through face-to-face stacking (average centre-to-centre distance: 3.79 Å, Fig. 7a). This stacking structure involves strong π–π aromatic interactions (Fig. 7b), which facilitate the formation of molecular excimers with relatively long fluorescence lifetimes and long-wavelength emission. By assembling bpcb with the co-former 1,4-DITFB, the two components stack alternately with one another, which disrupts the strong face-to-face stacking of bpcb molecules (Fig. 7c) and thus the chromophores become more widely separated (Fig. 7d). The change in the packing structure greatly alters the luminescent properties, including emissive color, fluorescence lifetime and PLQY value. Similar behavior is also observed for other co-crystals (as both powders (Fig. 8a and b) and single crystals (Fig. 8c)) of bpcb with other co-formers (such as 1,4-diiodobenzene, resorcinol, 1,4-dibromotetrafluorobenzene, 4-bromotetrafluorobenzene carboxylic acid, and 2,3,4,5,6-pentafluorophenol), which show tunable color and photoemission properties. This example further confirms that organic solids with multi-color emission (blue, green and yellow) can be obtained based on a single chromophore.
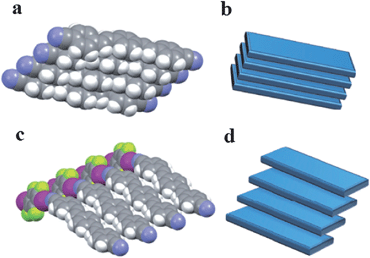 |
| | Fig. 7 Comparison of the crystallization of the chromophore bpcb on its own and its co-crystallization with 1,4-DITFB: (a) partial view of the structure of crystals of pure bpcb; (b) representation of the face-to-face aromatic stacking of molecules of bpcb in the pure crystals; (c) partial view of the structure of 1:1 co-crystals of bpcb and co-former 1,4-DITFB; (d) representation of the offset packing of molecules of bpcb in the co-crystals. Reprinted from ref. 50. © Nature Publishing group, 2012. | |
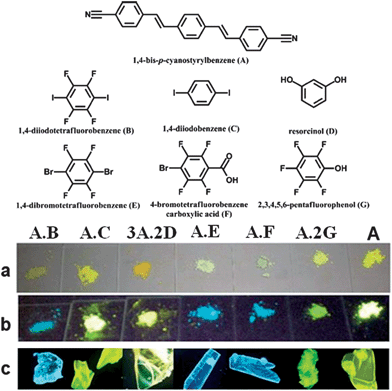 |
| | Fig. 8 (Top) Chemical structures of the fluorescent molecule (A, bpcb) and its co-formers ((B) 1,4-diiodotetrafluorobenzene; (C) 1,4-diiodobenzene; (D) resorcinol; (E) 1,4-dibromotetrafluorobenzene; (F) 4-bromotetrafluorobenzene carboxylic acid; (G) 2,3,4,5,6-pentafluorophenol). (Bottom) Photographs of solid-state co-crystal samples (from left to right: A·B; A·C; 3A·2D; A·E; A·F; A·2G) and A: (a and b) powder samples under daylight and UV (365 nm); (c) the single crystal samples under UV (365 nm). Reprinted with permission from ref. 49. © Wiley-VCH, 2011. | |
(b) Color-tunable phosphorescence.
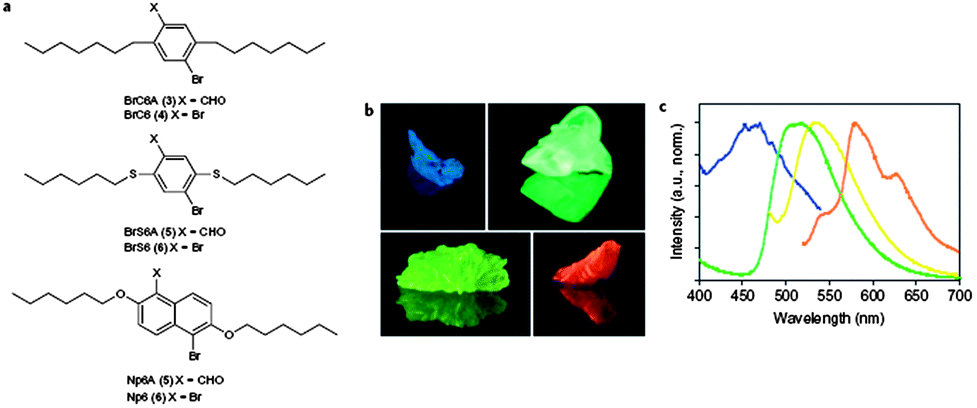
Phosphorescent materials have been widely used in electronic devices53 as well as in chemical and biological detection.54 However, examples of pure organic solids with phosphorescent emission are still very limited compared with their organometallic and inorganic counterparts,55 due to the lack of triplet states. Recently, Kim et al.56 have designed aromatic carbonyl-containing organic materials, which exhibit highly efficient ambient phosphorescence with the aid of heavy atom effects and halogen bonding. Promotion of spin–orbit coupling at the carbonyl oxygen atom in these aromatic carbonyl compounds effectively generates intrinsic triplet states through intersystem crossing. Moreover, by introduction of the carbonyl compounds as the guest in an isostructural bi-halogenated chromophore host matrix (Fig. 9a), the resulting mixed multi-component crystals can exhibit color-tunable phosphorescence emission (Fig. 9b and c) with high phosphorescence quantum yields due to the inhibition of emissive self-quenching. The highest reported room-temperature phosphorescence quantum yield in such multi-component crystals is 55%. The formation of halogen bonding within organic solids can be regarded as a direct heavy atom effect allowing efficient solid-state phosphorescence to occur. In the example of a C–I⋯π halogen-bonding-facilitated co-crystal system composed of 1,4-DITFB and carbazole with a 2:1 ratio discussed above,46a the phosphorescence intensity is significantly enhanced (58-fold) compared with the pure carbazole. The phosphorescence lifetime of the co-crystal is ca. 2.75 ms.
 |
| | Fig. 9 (a) Structures of host/aldehyde co-crystals designed to produce shifts in emission wavelength; (b) photographs of BrC6A/BrC6, Br6A/Br6, Np6A/Np6 and BrS6A/BrS6 (clockwise from top left) mixed crystals under UV light (365 nm), each containing 1 wt% aldehyde chromophore and 99 wt% host compound; (c) normalized photoluminescence emission spectra of BrC6A/BrC6 (blue trace), Br6A/Br6 (green trace, repeated for reference), BrS6A/BrS6 (yellow trace) and Np6A/Np6 (orange trace) crystals. Reprinted from ref. 56. © Nature Publishing Group, 2011. | |
3.3 Theoretical insights
In recent years, due to the rapid development in computer hardware and software capabilities, much attention has been focused on theoretical calculations as a way of investigating molecular solids at the electronic structure level, since these provide a complementary tool to experimental procedures for the study and prediction of the structures and properties of new types of functional materials. For multi-component crystals, the introduction of co-formers may not only tune the aggregation and packing of the chromophore molecules by non-covalent interactions, but can also adjust the frontier orbital distribution, the energy gap, electronic structures, and interactions between the building blocks. Many examples of intramolecular and intermolecular energy transfer in multi-component systems have been confirmed by density functional theoretical (DFT) calculations.
(a) Intramolecular energy transfer.
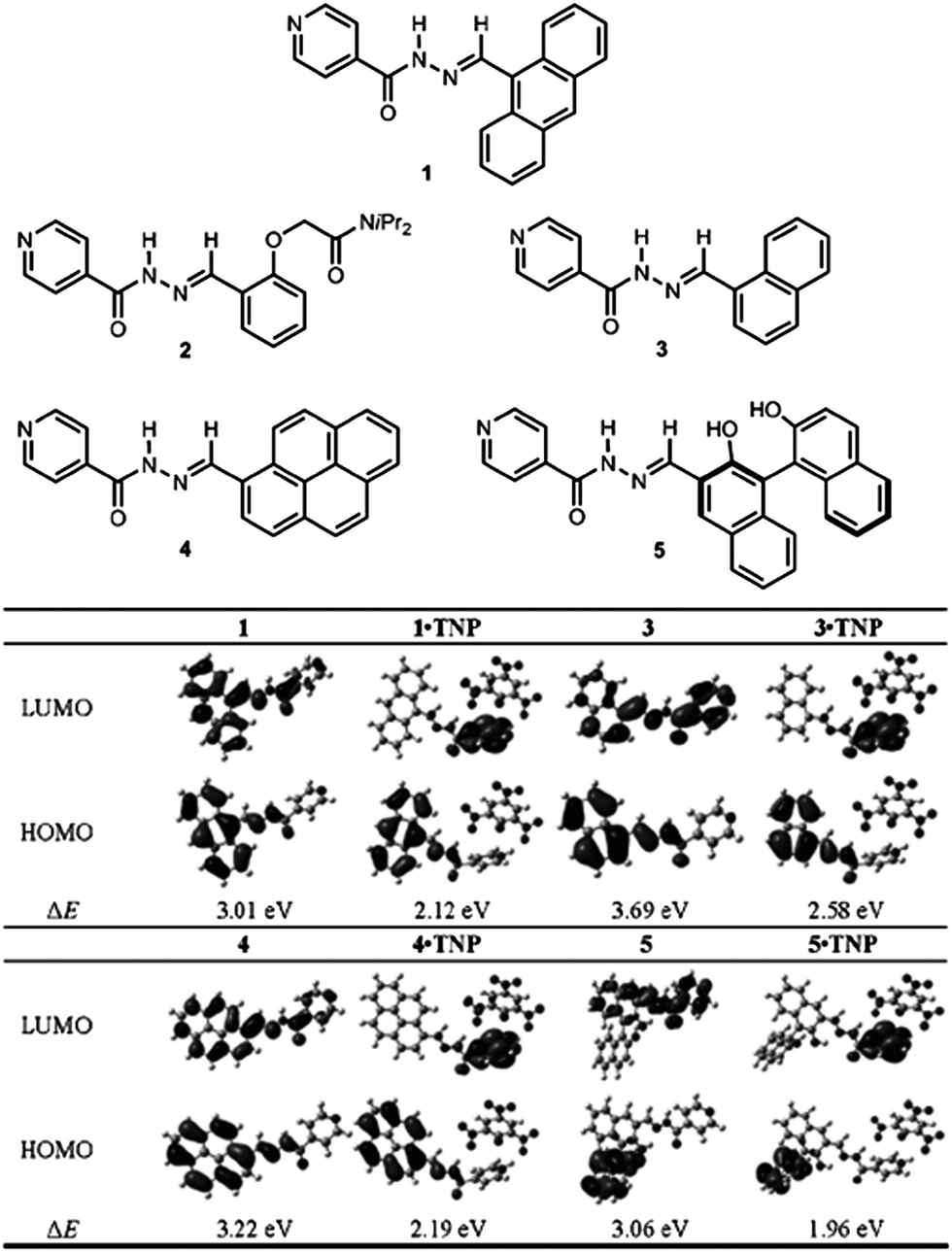
Recently, Peng et al.57 have developed a series of fluorescent co-crystals of N-acylhydrazone derivatives (shown in 1–5 in Fig. 10) and 2,4,6-trinitrophenol (TNP). Single crystal analysis shows that supramolecular multi-component structures were formed by self-assembly between the N-acylhydrazone derivatives and TNP by hydrogen bonding and π–π interactions, and that the pyridine group of these derivatives is the key site for interaction with TNP. Fluorescence spectra show that the co-crystals of 1·TNP, 3·TNP, 4·TNP, and 5·TNP undergo fluorescence quenching and red-shift relative to the pure samples of 1, 3, 4, and 5. DFT calculations show that for pristine 1, 3, 4, and 5, the electron densities of the highest occupied molecular orbitals (HOMOs) are mainly localized at the chromophores, whilst the electron densities of the lowest unoccupied molecular orbitals (LUMOs) are delocalized over the whole molecule (Fig. 10). For the co-crystals of 1·TNP, 3·TNP, 4·TNP, and 5·TNP, whilst the HOMOs are still mainly located at the chromophores, the LUMOs are now localized on the pyridine group. This can be attributed to the protonation of the pyridine group upon interaction with TNP. The calculations show that intramolecular energy transfer from the fluorophore (donor) to the pyridine group (acceptor) occurs, which results in the change of fluorescence in the emission spectra as observed experimentally. Based on their specific intramolecular energy transfer features, compounds 1, 3, 4, 5 can serve as chemosensors for recognition of TNP.
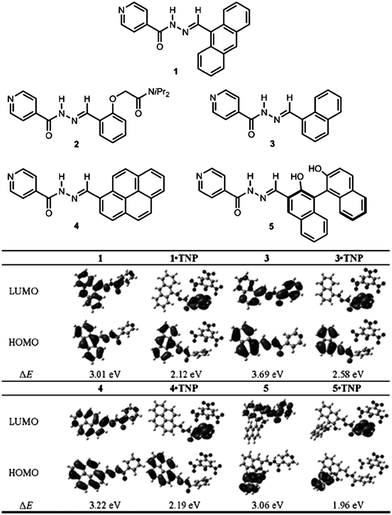 |
| | Fig. 10 (Top) Molecular structures of N-acylhydrazone derivatives (1–5). (Bottom) Molecular orbital plots of the HOMOs and LUMOs of 1, 3, 4, 5, 1·TNP, 3·TNP, 4·TNP, and 5·TNP. Reprinted with permission from ref. 57. © Wiley-VCH, 2013. | |
(b) Intermolecular energy transfer.
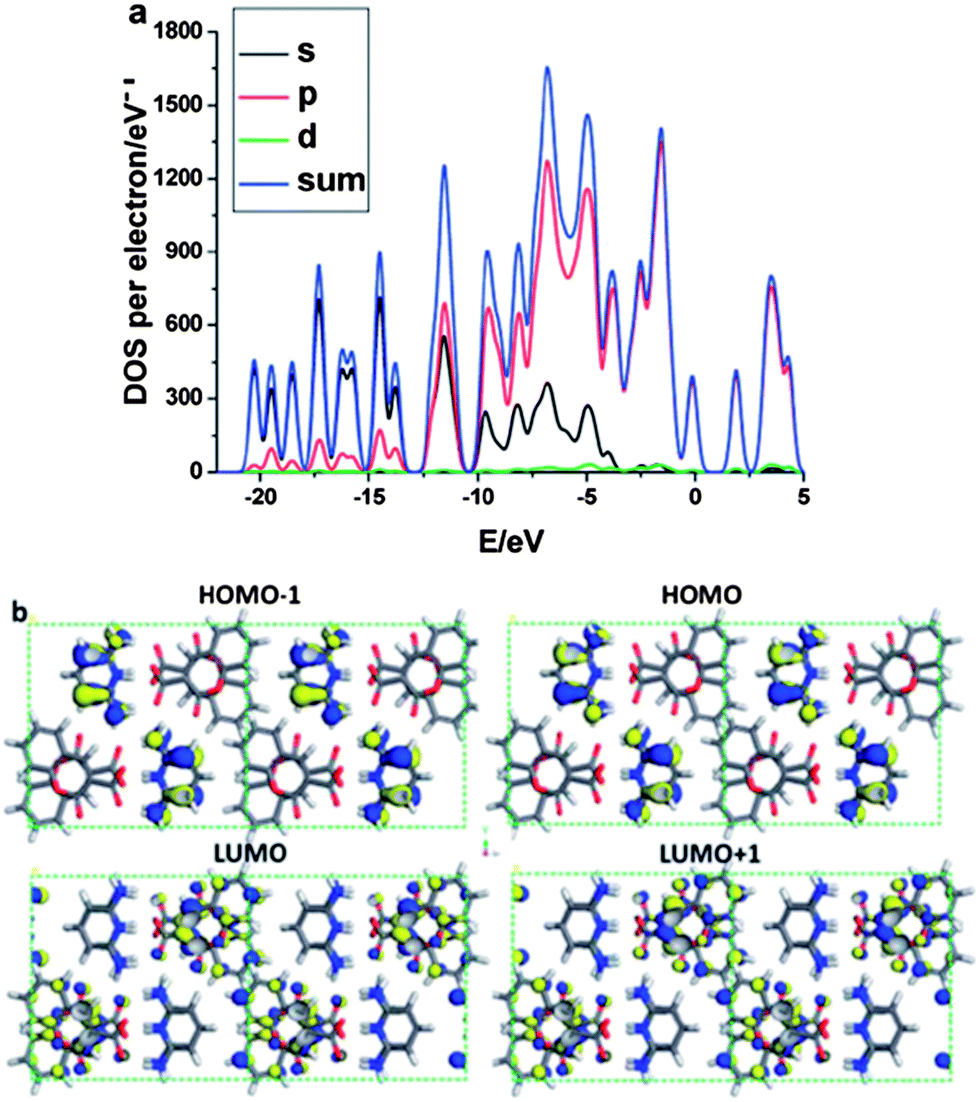
Intermolecular energy transfer between the chromophore and co-formers has been observed in C3C-based multi-component crystals.31 For the pure C3C crystal, periodic DFT calculations show that the HOMOs and LUMOs are distributed over the whole π-conjugated system in the C3C molecule, suggesting that no intramolecular energy transfer occurs during the photoexcitation process. Upon formation of a salt of C3C with pyridine-2,6-diamine, total electronic densities of states (TDOS) and partial electronic densities of states (PDOS) analyses (Fig. 11a) reveal that HOMO−1 and HOMO are mainly populated on the C and N atoms in the co-former pyridine-2,6-diamine, whereas the LUMO and LUMO+1 are mainly distributed on the C and O atoms in the C3C molecule (Fig. 11b), showing that intermolecular energy transfer occurs from the co-former pyridine-2,6-diamine to C3C. A similar energy transfer behavior can also be observed in co-crystals assembled from C3C and the co-former 4,4′-bipyridine, where the HOMO−1 and HOMO are populated on C3C and the LUMO and LUMO+1 are mainly distributed on the co-former 4,4′-bipyridine. This energy transfer between the supramolecular building blocks is likely to account for the observed red-shift of the luminescence of co-crystals composed of C3C and 4,4′-bipyridine. The calculations indicate that the fluorescence excitation/emission and energy transfer processes of the chromophore can be engineered by choosing suitable co-formers with appropriate energy levels relative to those of the pristine chromophore. A similar energy transfer process has also been observed in crystals containing the TPI chromophore co-assembled with 4-bromotetrafluorobenzene carboxylic acid.34
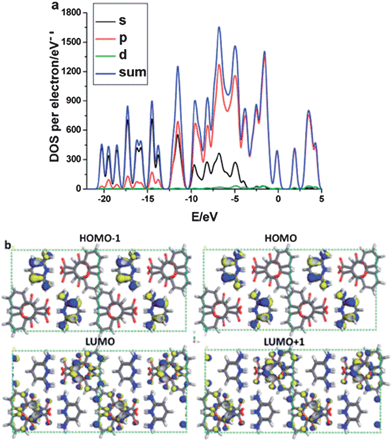 |
| | Fig. 11 (a) Total and partial electronic densities of state (TDOS and PDOS) and (b) frontier orbital profiles (HOMO−1, HOMO, LUMO, LUMO+1) of the salt composed of C3C and pyridine-2,6-diamine (blue/yellow colors denote electronic wavefunctions). The Fermi energy level EF was set to zero. Reprinted from ref. 31. © Royal Society of Chemistry, 2012. | |
(c) The origin of the changes in fluorescence.
Calculations of the energy levels and band gap can also provide further understanding of the mechanism responsible for the tunable fluorescence. For the pure bpcb system,52 the calculated energy gap decreases from 2.05 eV (for a single molecule) to 1.70 eV upon formation of crystalline solid bpcb. In addition, intermolecular energy transfer can also be observed between the HOMO and LUMO of the crystal unit cell, suggesting that strong molecular aggregation occurs. Upon formation of co-crystals of bpcb·1,4-DITFB, energy band analysis showed that the sample has a band gap of 1.94 eV. The difference between the calculated band gaps of the co-crystal and pure bpcb (0.24 eV) is close to the experimentally observed blue-shift of the luminescence (0.32 eV) of the co-crystal bpcb·1,4-DITFB relative to the pure bpcb crystals. The frontier orbital distribution further shows that electronic densities in the co-former 1,4-DITFB are mostly populated in HOMO−1 and part of HOMO. This can be attributed to the strong electron-withdrawing effect of the fluorine atoms in the co-former 1,4-DITFB, which reduces the electron density in bpcb molecules within the crystal.
3.4 Sensor and optoelectronic applications
(a) Stimuli-responsive sensors.
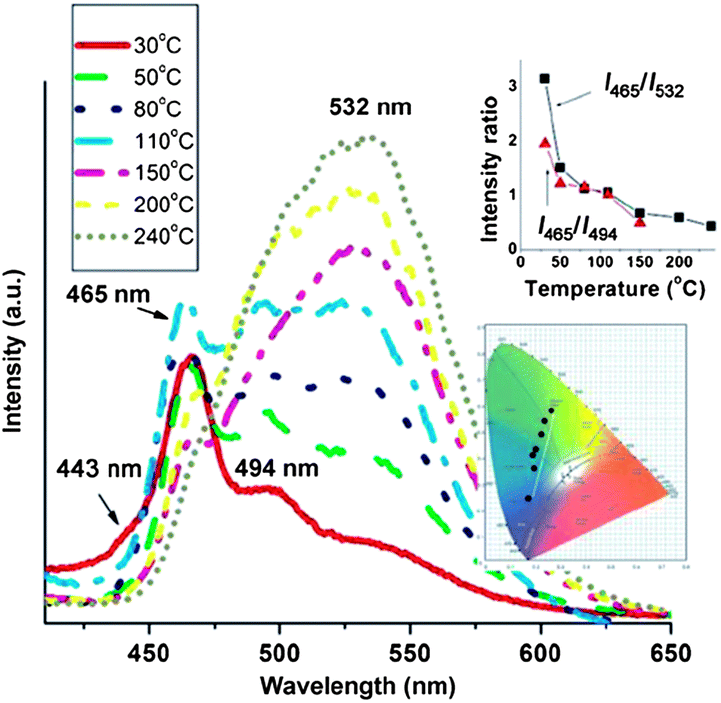
The weak non-covalent interactions between the chromophore and its co-formers within multi-component crystals can be modified under external stimuli, which further induces changes in the fluorescence properties. As mentioned above, temperature is one of the most common environmental factors, and the temperature-induced fluorescent chromic materials (known as thermochromic luminescent (TCL) materials) can potentially be used as memory chips and security inks.29 The main principle governing the construction of multi-component crystals with TCL properties is the decomposition of the multi-component system and its transformation into the single-component pure chromophore solid. Therefore, the multi-component crystals should meet two prerequisites: both the melting point and the emission of the chromophore must differ from those of the co-former. In the co-crystals composed of bpcb and 1,4-DITFB,52 the luminescence intensities of the photoemission peaks at 494 and 537 nm gradually increase upon heating from 30 to 110 °C. Once the temperature increases to 150 °C, the fluorescence emission at around 532 nm becomes dominant. The emissive intensity also increases upon further increasing the temperature in the range 150–240 °C. The intensity ratio I465/I532 decreases systematically from 3.15 to 0.43 across the whole temperature range studied (inset of Fig. 12). Therefore, the co-crystals exhibit an obvious temperature-dependent luminescence with a color change from blue-green (color coordinates (0.171, 0.250)) to yellow (color coordinates (0.259, 0.560)) on going from 30 to 240 °C, as shown in the inset of Fig. 12. The change in luminescence is due to the structural changes associated with the removal of the co-former 1,4-DITFB from the co-crystals. Based on the response of the fluorescence intensity ratios for different wavelengths to changes in temperature, the multi-component sample may potentially serve as a new type of temperature sensor.
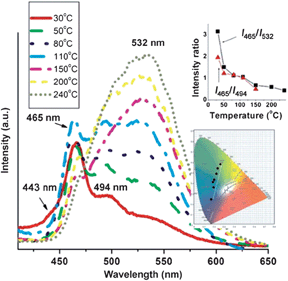 |
| | Fig. 12 Fluorescence spectra of co-crystals of bpcb and 1,4-DITFB in the temperature range 30–240 °C (insets: variation of the fluorescence intensity ratios I465/I494 and I465/I532 and the color coordinates with temperature). Reprinted with permission from ref. 52. © Wiley-VCH, 2013. | |
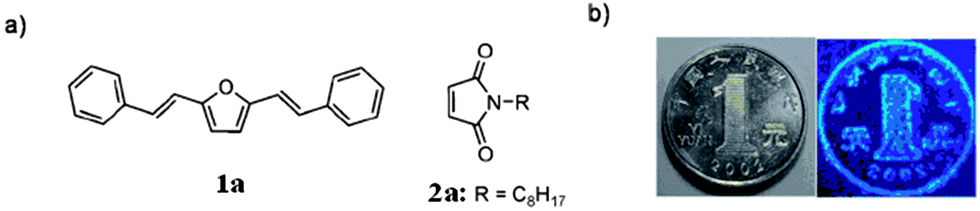
Like TCL materials, pressure- or mechanically-induced chromic luminescent (PCL and MCL) materials also offer significant applications in luminescence switches and pressure sensors.11a Recently, Luo and co-workers58 have designed a binary co-crystal system, in which two components (1a and 2a, shown in Fig. 13a) act as a fluorescent donor (D) and a non-emissive acceptor (A), respectively. The luminescence changes from the quenched (black) to the emitting (colored) state when a mechanical force is applied. Although detailed structural information about the co-crystal was not obtained, powder XRD analysis shows that the PCL behavior of the D–A system can be explained by a phase separation mechanism under pressure, i.e., in the co-crystal of 1a·2a, the N-alkyl substituted maleimide acceptor can induce the fluorescence quenching of the donor via a photo-induced electron transfer process. Under external pressure, the stable dense packing of the chromophore 1a leads to its self-crystallization and phase separation from the D–A co-crystals, which results in stronger luminescence. Based on this strategy, the range of available PCL materials can be extended from single-component materials to co-crystal solid systems by the rational choice of donor and acceptor molecules.
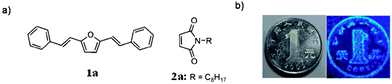 |
| | Fig. 13 Piezochromic luminescence from a binary complex. (a) Chemical structures of the selected donor and acceptor. (b) Transfer of the protruding pattern of a coin by stamping. The image mirrors the pattern of the coin. The substrate is paper which has been saturated with a 1:1 complex of 1a and 2a. Reprinted with permission from ref. 58. © Wiley-VCH, 2011. | |
Several multi-component systems exhibit solvent-dependent solid-state luminescent properties. For example, Draper et al.37 have observed that the luminescence of the supramolecular assembly of CDPA and piperidine is sensitive to different organic solvents. The pristine CDPA·piperidine co-crystal system exhibits luminescence at 510 nm. Exposure of powdered CDPA·piperidine to different solvent vapors (EtOAc, CH2Cl2, CHCl3, MeOH, EtOH, toluene and H2O) for 3–5 min leads to red-shifts to 518–522 nm, while the emissive band of the material is located at 536 nm in CH3CN vapor, suggesting that there is selective fluorescent recognition of CH3CN in the crystal lattice of CDPA·piperidine. It was notable that solvent-dependence of the solid-state luminescence was not found in related multi-component systems such as CDPA·pyrrolidine and CDPA·morpholine, suggesting that the choice of a suitable co-former plays an important role in achieving solvent-dependent luminescence. From this example, it can be seen that the formation of multi-component systems has become an important strategy to understand molecular recognition and in fluorescent detection.
(b) Optoelectronic applications.
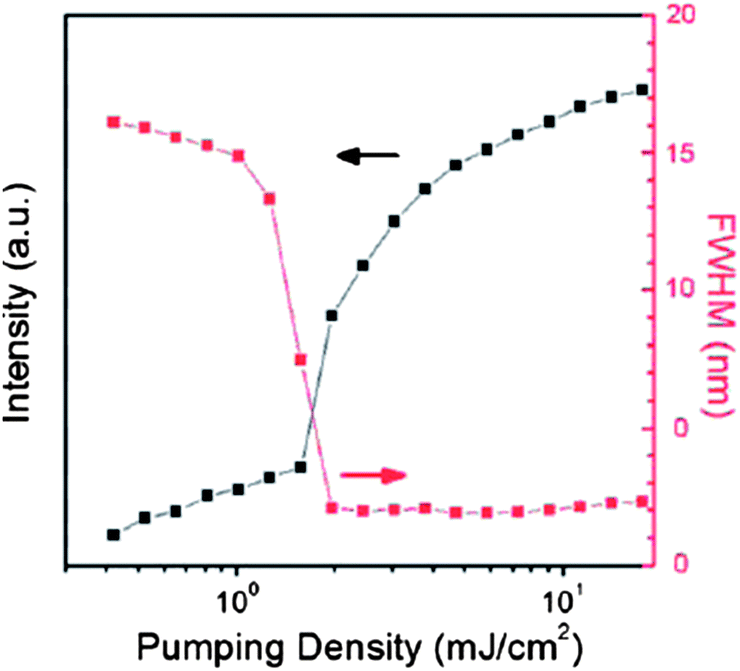
By virtue of their tunable optical/luminescent properties, multi-component solids have been employed in optoelectronic devices, although actual examples reported to date are still rather few. In co-crystals of 1,3,6,8-tetramethylpyrene (TMPY) doped in perylene, laser emission with the highest luminescence efficiency was obtained for a 5% doped sample.47a With a low pumping density (0.65 mJ cm−2) the luminescence spectra exhibited a broad feature above 475 nm. As the pumping density was increased, the intensity of the peak near 490 nm significantly increased and the full width at half maximum (FWHM) became much narrower. These results indicate that amplified spontaneous emission (ASE) can be obtained in TMPY/perylene co-crystal systems. The gain-narrowing threshold was 1.54 mJ cm−2 and the FWHM remained about 2 nm above the threshold energy (Fig. 14). An optically pumped laser with highly efficient ASE has also been observed with 8% tetracene and pentacene doped trans-1,4-distyrylbenzene (trans-DSB) multi-component crystals,47c which exhibit green and red luminescence, respectively, whereas the pure trans-DSB has blue emission; the shift in emission wavelengths result from the energy transfer and suppression of intermolecular interactions in the doped systems.
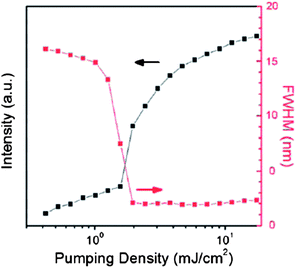 |
| | Fig. 14 Luminescence intensities and FWHM as a function of pumping density in the co-crystals of 5% TMPY doped in perylene. Reprinted from ref. 47a. © Royal Society of Chemistry, 2011. | |
Based on the intermolecular interactions within the multi-component solid, a co-crystal system can also serve as a high-performance organic field-effect transistor. Recently, Park et al.59 have developed a 1:1 co-crystal system comprising two isometric distyrylbenzene- and dicyanodistyrylbenzene-based chromophores, which exhibit high and ambipolar field-effect mobilities of holes and electrons of up to 6.7 × 10−3 and 6.7 × 10−2 cm2 V−1 s−1, respectively. The high mobility can be attributed to the donor–accepter (D–A) charge transfer interaction between the two components. Such a D–A approach affords an effective and versatile way to fabricate highly efficient opto-electronic materials.
4 Conclusions and outlook
Crystal engineering involving the formation of different polymorphs of fluorophores, and of multi-component solids containing these materials, has been shown to be a powerful strategy for tuning and tailoring the luminescent properties of molecular solids leading to new functional materials. The development of new synthesis methods (such as physical vapor transport, grinding and ultrasonication) has also promoted developments in this field. Recent theoretical studies on these luminescent systems have supplied new physicochemical insights into the interactions between components within the crystals (such as intramolecular and intermolecular energy transfer processes). The systems reported to date have helped to give us a preliminary understanding of molecular structure/crystal structure–luminescence relationships. However, it is still difficult to design polymorph materials with a specific desired luminescence due to the lack of predictability. Supramolecular synthon strategies for the fabrication of multi-component solid systems (co-crystals and salts) may be a more effective way to design and synthesize new luminescent materials. However a detailed picture of the interactions between chromophores still requires single crystal structure analysis. In this regard, the development of computational technology for crystal structure prediction60 is highly desirable in order to obtain the optimized stable/metastable structures of both polymorphs and co-crystallization. This will allow the luminescent properties of molecular solids to be predicted to some extent before the materials have been synthesized, and thus provide a roadmap for the synthesis of new materials. From the application perspective, these molecular crystalline materials have already emerged as good candidates for applications in luminescent sensors; however, observations of a dynamic luminescent response are still rather rare for multi-component systems when compared with polymorph-based materials. The fabrication of such multi-component crystals needs new and effective mechanisms to be developed for switching organic solid-state luminescence and research in this area is continuing in our laboratory and many others worldwide.
Acknowledgements
We would like to thank all the co-workers cited in the references below for their invaluable contributions to the work described here. This work was supported by the National Natural Science Foundation of China, the 973 Program (Grant no. 2011CBA00504), the 111 Project (Grant no. B07004), Central University Research Funds, and Program for Changjiang Scholars and the Innovative Research Team in University (PCSIRT: IRT1205).
Notes and references
-
(a) C. W. Tang and S. A. Van Slyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS;
(b) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes and R. N. Marks, Nature, 1999, 397, 121 CrossRef CAS PubMed;
(c) J. H. Lee, Y. Y. Yuan, Y. J. Kang, W. L. Jia, Z. H. Lu and S. N. Wang, Adv. Funct. Mater., 2006, 16, 681 CrossRef CAS;
(d) C. A. Strassert, C. Chien, M. D. G. Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946 CrossRef CAS PubMed.
-
(a) C. Zhang, C. Zou, Y. Yan, R. Hao, F. Sun, Z. Han, Y. S. Zhao and J. N. Yao, J. Am. Chem. Soc., 2011, 133, 7276 CrossRef CAS PubMed;
(b) F. Gao, Q. Liao, Z. Xu, Y. Yue, Q. Wang, H. Zhang and H. Fu, Angew. Chem., Int. Ed., 2010, 49, 732 CrossRef CAS PubMed.
-
(a) L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 CrossRef CAS PubMed;
(b) Y. Che and L. Zang, Chem. Commun., 2009, 5106 RSC;
(c) Z. Ning, Z. Chen, Q. Zhang, Y. Yan, S. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799 CrossRef CAS.
-
(a) N. Tessler, V. Medvedev, M. Kazes, S. Kan and U. Banin, Science, 2002, 295, 1506 CrossRef PubMed;
(b) T. Cheng, Y. Xu, S. Zhang, W. Zhu, X. Qian and L. Duan, J. Am. Chem. Soc., 2008, 130, 161 Search PubMed;
(c) K. Kundu, S. F. Knight, N. Willett, S. Lee, W. R. Taylor and N. Murthy, Angew. Chem., Int. Ed., 2009, 48, 299 CrossRef CAS PubMed.
-
(a) C.-L. Chiang, S.-M. Tseng, C.-T. Chen, C.-P. Hsu and C.-F. Shu, Adv. Funct. Mater., 2008, 18, 248 CrossRef CAS;
(b) D. P. Yan, J. Lu, M. Wei, S. H. Qin, L. Chen, S. T. Zhang, D. G. Evans and X. Duan, Adv. Funct. Mater., 2011, 21, 2497 CrossRef CAS.
-
(a) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685 CrossRef CAS PubMed;
(b) Y. Sonoda, M. Goto, S. Tsuzuki and N. Tamaoki, J. Phys. Chem. A, 2007, 111, 13441 CrossRef CAS PubMed.
-
(a)
J. Bernstein, Polymorphism in Molecular Crystals, Clarendon, Oxford, 2002 Search PubMed;
(b) A. J. Matzger, Cryst. Growth Des., 2008, 8, 2 CrossRef CAS.
-
(a)
P. Vishweshwar, J. A. McMahon and M. J. Zaworotko, in Frontiers in Crystal Engineering, ed. E. R. T. Tiekink and J. J. Vittal, Wiley, Chichester, 2006, pp. 25–49 Search PubMed;
(b) A. Nangia, Cryst. Growth Des., 2008, 8, 1079 CrossRef CAS.
-
(a) S. Takahashi, H. Miura, H. Kasai, S. Okada, H. Oikawa and H. Nakanishi, J. Am. Chem. Soc., 2002, 124, 10944 CrossRef CAS PubMed;
(b) C. Karunatilaka, D.-K. Bučar, L. R. Ditzler, T. Friščić, D. C. Swenson, L. R. MacGillivray and A. V. Tivanski, Angew. Chem., Int. Ed., 2011, 50, 8642 CrossRef CAS PubMed;
(c) A. Delori and W. Jones, CrystEngComm, 2011, 13, 6315 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
-
(a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed;
(b) S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863 CrossRef CAS.
-
(a) H. Zhang, Z. Zhang, K. Ye, J. Zhang and Y. Wang, Adv. Mater., 2006, 18, 2369 CrossRef CAS;
(b) J. Wang, Y. Zhao, J. Zhang, J. Zhang, B. Yang, Y. Wang, D. Zhang, H. You and D. Ma, J. Phys. Chem. C, 2007, 111, 9177 CrossRef CAS;
(c) Y. Zhao, H. Gao, Y. Fan, T. Zhou, Z. Su, Y. Liu and Y. Wang, Adv. Mater., 2009, 21, 3165 CrossRef CAS;
(d) C. Dou, L. Han, S. Zhao, H. Zhang and Y. Wang, J. Phys. Chem. Lett., 2011, 2, 666 CrossRef CAS.
- A. D. G. D. Mauro, M. Carotenuto, V. Venditto, V. Petraccone, M. Scoponi and G. Guerra, Chem. Mater., 2007, 19, 6041 CrossRef.
-
(a) T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe and K. Araki, Angew. Chem., Int. Ed., 2008, 47, 9522 CrossRef CAS PubMed;
(b) M. Sase, S. Yamaguchi, Y. Sagara, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2011, 21, 8347 RSC.
- J. W. Chung, Y. You, H. S. Huh, B. K. An, S. J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163 CrossRef CAS PubMed.
- D. P. Yan, J. Lu, M. Wei, J. Ma, D. G. Evans and X. Duan, Phys. Chem. Chem. Phys., 2009, 11, 9200 RSC.
-
B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag GmbH, 2001 Search PubMed.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed.
- X. Gu, J. Yao, G. Zhang, Y. Yan, C. Zhang, Q. Peng, Q. Liao, Y. Wu, Z. Xu, Y. S. Zhao, H. Fu and D. Zhang, Adv. Funct. Mater., 2012, 22, 4862 CrossRef CAS.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803 RSC.
- S. Roy, R. Banerjee, A. Nangia and G. J. Kruger, Chem.–Eur. J., 2006, 12, 3777 CrossRef CAS PubMed.
-
W. C. McCrone, in Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M. Labes and A. Weissberger, Interscience, New York, 1965, vol. II, pp. 726–767 Search PubMed.
- N. Sanz, P. L. Baldeck, J. F. Nicoud, Y. L. Fur and A. Ibanez, Solid State Sci., 2001, 3, 867 CrossRef CAS.
- Z. Xie, H. Wang, F. Li, W. Xie, L. Liu, B. Yang, L. Ye and Y. Ma, Cryst. Growth Des., 2007, 7, 2512 CAS.
- V. S. Senthil Kumar, A. Addlagatta, A. Nangia, W. T. Robinson, C. K. Broder, R. Mondal, I. R. Evans, J. A. K. Howard and F. H. Allen, Angew. Chem., 2002, 114, 4004 CrossRef.
-
(a) R. Davis, N. P. Rath and S. Das, Chem. Commun., 2004, 74 RSC;
(b) R. Davis, N. S. S. Kumar, S. Abraham, C. H. Suresh, N. P. Rath, N. Tamaoki and S. Das, J. Phys. Chem. C, 2008, 112, 2137 CrossRef CAS.
- Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40 RSC.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS PubMed.
-
(a) M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759 CrossRef CAS PubMed;
(b) D. Yan, J. Lu, J. Ma, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 720 CrossRef CAS PubMed.
-
(a)
G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, 1989 Search PubMed;
(b) J. D. Wuest, Chem. Commun., 2005, 5830 RSC;
(c) G. Cavallo, C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22 RSC.
- D. P. Yan, A. Delori, G. O. Lloyd, B. Patel, T. Friščić, G. M. Day, D. K. Bučar, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, CrystEngComm, 2012, 14, 5121 RSC.
-
(a) Y. Kobayashi, T. Sato, Y. Tanaka, T. Harada, R. Kuroda and Y. Imai, CrystEngComm, 2012, 14, 1468 RSC;
(b) Y. Kobayashi, T. Kinuta, T. Sato, T. Harada, R. Kuroda, Y. Matsubara and Y. Imai, CrystEngComm, 2013, 15, 4624 RSC.
- G. G. Hou, H. J. Zhao, J. F. Sun, D. Lin, X. P. Dai, J. T. Han and H. Zhao, CrystEngComm, 2013, 15, 577 RSC.
- D. P. Yan, B. Patel, A. Delori, W. Jones and X. Duan, Cryst. Growth Des., 2013, 13, 333 CAS.
-
(a) Y. Mizobe, N. Tohnai, M. Miyata and Y. Hasegawa, Chem. Commun., 2005, 1839 RSC;
(b) Y. Mizobe, T. Hinoue, A. Yamamoto, I. Hisaki, M. Miyata, Y. Hasegawa and N. Tohnai, Chem.–Eur. J., 2009, 15, 8175 CrossRef CAS PubMed.
- S. P. Anthony, S. Varughese and S. M. Draper, J. Phys. Org. Chem., 2010, 23, 1074 CrossRef CAS.
- S. P. Anthony, S. Varughese and S. M. Draper, Chem. Commun., 2009, 7500 RSC.
- N. Nishiguchi, T. Sato, T. Kinuta, R. Kuroda, Y. Matsubara and Y. Imai, Cryst. Growth Des., 2011, 11, 827 CAS.
- B. Dong, M. Wang, C. Xu, Q. Feng and Y. Wang, Cryst. Growth Des., 2012, 12, 5986 CAS.
- Q. Feng, M. Wang, B. Dong, C. Xu, J. Zhao and H. Zhang, CrystEngComm, 2013, 15, 3623 RSC.
- Q. Su, M. He, Q. Wu, W. Gao, H. Xu, L. Ye and Y. Mu, CrystEngComm, 2012, 14, 7275 RSC.
-
(a) G. Jones and M. A. Rahman, J. Phys. Chem., 1994, 98, 13028 CrossRef CAS;
(b) C. T. Chen, C. L. Chiang, Y. C. Lin, L. H. Chan, C. H. Huang, Z. W. Tsai and C. T. Chen, Org. Lett., 2003, 5, 1261 CrossRef CAS PubMed.
-
(a) A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686 CrossRef;
(b) P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12, 5835 CrossRef CAS;
(c) U. Mayerhöffer and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 5615 CrossRef PubMed;
(d) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. A. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, DOI:10.1351/PAC-REC-12-05-10.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2012, 5, 42 CrossRef PubMed.
-
(a) J. L. Syssa-Magale, K. Boubekeur, P. Palvadeau, A. Meerschaut and B. Schollhorn, CrystEngComm, 2005, 7, 302 RSC;
(b) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013 DOI:10.1021/ar400103r.
-
(a) H. Y. Gao, Q. J. Shen, X. R. Zhao, X. Q. Yan, X. Pang and W. J. Jin, J. Mater. Chem., 2012, 22, 5336 RSC;
(b) Q. J. Shen, H. Q. Wei, W. S. Zou, H. L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 1010 RSC;
(c) Q. J. Shen, X. Pang, X. R. Zhao, H. Y. Gao, H. L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 5027 RSC;
(d) H. Y. Gao, X. R. Zhao, H. Wang, X. Pang and W. J. Jin, Cryst. Growth Des., 2012, 12, 4377 CrossRef CAS;
(e) X. Pang, H. Wang, X. R. Zhao and W. J. Jin, CrystEngComm, 2013, 15, 2722 RSC.
-
(a) J. Li, S. Takaishi, N. Fujinuma, K. Endo, M. Yamashita, H. Matsuzaki, H. Okamoto, K. Sawabe, T. Takenobu and Y. Iwasa, J. Mater. Chem., 2011, 21, 17662 RSC;
(b) H. Wang, F. Li, B. R. Gao, Z. Q. Xie, S. J. Liu, C. L. Wang, D. H. Hu, F. Z. Shen, Y. X. Xu, H. Shang, Q. D. Chen, Y. G. Ma and H. B. Sun, Cryst. Growth Des., 2009, 9, 4945 CrossRef CAS;
(c) H. Wang, B. Yue, Z. Xie, B. Gao, Y. Xu, L. Liu, H. Sun and Y. Ma, Phys. Chem. Chem. Phys., 2013, 15, 3527 RSC.
-
(a) T. Friščić, J. Mater. Chem., 2010, 20, 7599 RSC;
(b) T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621 CrossRef;
(c) M. Frenette, G. Cosa and T. Friščić, CrystEngComm, 2013, 15, 5100 RSC.
- D. P. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483 CrossRef CAS PubMed.
- J. D. Wuest, Nat. Chem., 2012, 4, 74 CrossRef CAS PubMed.
-
(a) J. R. G. Sander, D. K. Bučar, R. F. Henry, G. G. Z. Zhang and L. R. MacGillivray, Angew. Chem., Int. Ed., 2010, 49, 7284 CrossRef CAS PubMed;
(b) D. Yan, W. Jones, G. Fan, M. Wei and D. G. Evans, J. Mater. Chem. C, 2013, 1, 4138 RSC.
- D. P. Yan, D. K. Bučar, A. Delori, B. Patel, G. O. Lloyd, W. Jones and X. Duan, Chem.–Eur. J., 2013, 19, 8213 CrossRef CAS PubMed.
- Y. Shao and Y. Yang, Adv. Mater., 2005, 17, 2841 CrossRef CAS.
- W. L. Rumsey, J. M. Vanderkooi and D. F. Wilson, Science, 1988, 241, 1649 CAS.
-
(a) W. Y. Wong and C. L. Ho, J. Mater. Chem., 2009, 19, 4437 RSC;
(b) S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386 CrossRef CAS.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CAS.
- M. Dong, Y. W. Wang, A. J. Zhang and Y. Peng, Chem.–Asian J., 2013, 8, 1321 CrossRef CAS PubMed.
- J. Luo, L. Y. Li, Y. Song and J. Pei, Chem.–Eur. J., 2011, 17, 10515 CrossRef CAS PubMed.
- S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757 CrossRef CAS PubMed.
- T. Beyer, G. M. Day and S. L. Price, J. Am. Chem. Soc., 2001, 123, 5086 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.