
Metal porphyrins and metal phthalocyanines as designable molecular model electrocatalysts
Ya-Chen
Feng
ab,
Xiang
Wang
*a and
Dong
Wang
 *ab
*ab
aCAS Key Laboratory of Molecular Nanostructure and Nanotechnology, CAS Research/Education Center for Excellence in Molecular Sciences, Beijing National Laboratory for Molecular Science (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wangxiang@iccas.ac.cn; wangd@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 11th October 2023
Abstract
Metal porphyrins (MP) and metal phthalocyanines (MPc) have precisely tailored structural and electronic properties, and their coordination environment and electronic structure can be optimized at the atomic scale to improve their catalytic performance. Furthermore, MP and MPc are structurally similar to M–N4 single-atom catalysts (SACs), which have shown great potential in the field of electrochemistry due to their high atom utilization and excellent electrocatalytic performance. MP and MPc have the advantages of well-defined M–N4 active sites and can be used as ideal model systems for investigating the catalytic mechanism of M–N4 SACs. In this review, the applications of MP and MPc as model systems for electrocatalysis are summarized systematically. First, the structural characteristics of MP and MPc are introduced. Then, the tailoring, fabrication and modification of MP and MPc as both soluble and insoluble catalysts are presented. The influence of the substituents, axial coordination and complex effects is emphasized. Next, we discuss the construction of practical catalysts (such as metal/covalent organic frameworks) with MP and MPc motifs, and present the electrocatalytic behaviour of different systems. In the following section, the applications of advanced in situ characterization techniques for revealing the interfacial reaction processes and catalytic mechanisms of MP and MPc are discussed. In the end, the challenges and future directions in this field are outlined.
1 Introduction
In recent years, an increasing number of metal porphyrin (MP) and metal phthalocyanine (MPc) compounds have been applied in various electrocatalytic systems, such as the oxygen reduction reaction (ORR),1–4 CO2 reduction reaction (CO2RR),5,6 oxygen evolution reaction (OER)7,8 and hydrogen evolution reaction (HER).4,9,10 As shown in Fig. 1, MP has a π-conjugated macrocyclic structure. By changing the metal centres and the meso-, β- and axial substituents on the macrocycle, the coordination environments and electronic structures of MPs and MPcs can be optimized at the atomic scale to improve the catalytic performance.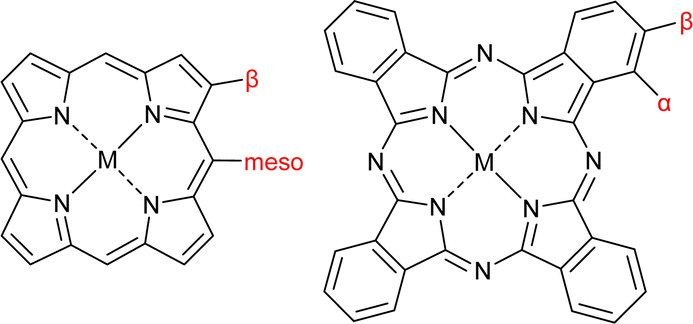 | ||
| Fig. 1 Structures and substituent sites of MP and MPc. | ||
Single-atom catalysts (SACs), generally referring to heterogeneous catalysts with single metal atoms, as emerging catalytic materials in electrocatalysis, have attracted extensive attention in recent years due to their high atom utilization and excellent electrocatalytic performance.11–14 Compared to metallic catalysts, SACs have unique characteristics in terms of electronic structures, surface states and adsorption behaviours, which may contribute to a better catalytic performance. For instance, the electronic structures of the ionic metal centre in SACs are significantly different from those of metals, leading to changes in the adsorption behaviour of intermediates in the reaction, thus affecting the reaction pathways.15–17 In particular, SACs with M–N4 sites have been proven to be one of the promising electrocatalysts. For instance, Tang and co-workers synthesized atomically dispersed Fe on N-doped carbon nanospheres with Fe–N4 active sites, which exhibited superior ORR performance (Eonset = 1.046 V vs. RHE, E1/2 = 0.87 V vs. RHE) and good durability in alkaline solutions.18 Fontecave et al. studied the CO2RR performance of atomically dispersed Fe–N–C catalysts. In 0.5 M NaHCO3, the CO Faradaic efficiency of the best Fe–N–C catalyst can reach 90%, in which the Fe–N4 site plays a crucial role in the high selectivity of CO.19
Investigating the structure–activity relationship and catalytic mechanism of SACs with M–N4 sites is the basis for constructing highly efficient catalysts. Practical M–N4 SACs are usually prepared by pyrolysis, so the structures of the catalytic active sites are nonuniform and complex, which makes it difficult to study the catalytic mechanism. Therefore, model catalytic systems, which possess uniform M–N4 active sites and well-defined structures, are a prerequisite for establishing structure–activity relationships.1–3,20,21 The representative model catalysts include molecular catalysts,1,2 highly crystallized framework materials,3,20 and two-dimensional (2D) materials.21 Among them, MP and MPc molecular catalysts with a well-defined M–N4 coordination structure, precisely tailored structures and electronic properties, have been widely used for investigating the catalytic mechanism of M–N4 sites in SACs.22,23
The catalytic activity and reaction pathway vary with different metal centres in MP or MPc molecules. For instance, the ORR involves a four-electron reduction to produce H2O with early transition metal sites, while for late transition metal sites, H2O2 is the main product.4,24 The changes can be attributed to the differences in the d-occupation and electronic structure of different metal centres.
By introducing substituents at the meso-sites of MP, the activity, stability and selectivity of these molecular catalysts can be changed. Electron-donating substituents can increase the electron density on metal centres, thereby enhancing the ability to combine with O2, facilitating electron transfer and improving electrocatalytic ORR activity. For instance, Ardakani and co-workers optimized the catalytic activity of Co porphyrin by introducing electron-donating methyl and methoxy substituents at the meso-site of the porphyrin ring.25 In addition, the β-substituent on the porphyrin macrocycle significantly affects the electronic structure of the central metal.26 Dey et al. investigated the effect of introducing electron-withdrawing groups at the β-site of Fe porphyrins. An electron-withdrawing ester group at the β-position can reduce the high overpotential for the electrocatalytic ORR. Compared with meso-position substituents, β-position substituents on the porphyrin ring are spatially closer to the metal centre, and the influence on the electronic structure and the catalytic performance is greater.13
The planar structure of MPs and MPcs provides a good platform for the modification of axial ligands due to the lack of steric hindrance. At the same time, the unoccupied d-orbital of the metal active centre can receive the electrons provided by the axial ligand coordination and increase its own electron density, thereby affecting its electrocatalytic activity. Cao et al. studied the ORR catalysed by imidazole-coordinated Fe porphyrin. Compared with catalysts without axial imidazole ligands, the imidazole-coordinated catalyst exhibits a higher binding ability with O2 and better ORR performance, which is due to the increased electron density of Fe.27
In addition, intermolecular or intramolecular coupling can be designed using MP and MPc motifs with different substituents to construct emerging catalytic materials with a larger conjugated structure. For instance, the Suzuki coupling reaction was used to synthesize framework materials with a large conjugated structure with cobalt porphyrin, and the catalytic activity of the framework materials towards the ORR and OER was enhanced due to the presence of high-density CoII sites and efficient electron transport in the π-conjugated framework.28,29
Due to their well-defined M–N4 sites and atomically tailored structure, MP and MPc are ideal model catalysts for investigating the catalytic mechanism of M–N4 sites in SACs. Herein, we review the applications of MP and MPc as model systems in electrocatalytic reactions. None of the catalytic systems discussed in this review have been prepared by thermo-pyrolysis. The well-defined chemical structures of these model systems benefit understanding the structure–performance relationship of the catalytic processes and mechanisms. First, the electrocatalytic properties of MP and MPc as soluble and insoluble catalysts are summarized. Then, the effects of substituent groups, axial ligands and substrates on electrocatalytic reactions are discussed. The applications of MP- and MPc-based framework materials in electrocatalysis are introduced. Next, we focus on the applications of advanced in situ characterization techniques in revealing the interfacial catalytic processes and mechanisms of MP and MPc. Finally, challenges and future directions for this field are discussed.
2 MP and MPc in molecular electrocatalysis
2.1 Molecular electrocatalysts
Molecular catalysts can be divided into two categories, i.e., soluble catalysts which can dissolve in the electrolyte, and insoluble catalysts supported on electrodes.30,31 Although some of the soluble electrocatalysts are not efficient in electron transfer during the catalytic process, they can provide important insights into understanding the basic catalytic processes and mechanisms in the reaction.32Kuwana and co-workers developed a series of water-soluble iron(III) 5,10,15,20-tetrakis(N-methylpyridyl)porphyrins (FeTMPyP) (Fig. 2a, 1–3) for the ORR in acidic solution. Derivatives 1 and 2 were demonstrated to be active for the ORR, and the generated FeII species can react with O2 rapidly in the reaction. For derivative 3, since the four positive charges are closer to the porphyrin ring, the FeIII/FeII reduction potential of 3 is 0.15 V higher than that of 1 and 2, which is more favourable for the ORR.33
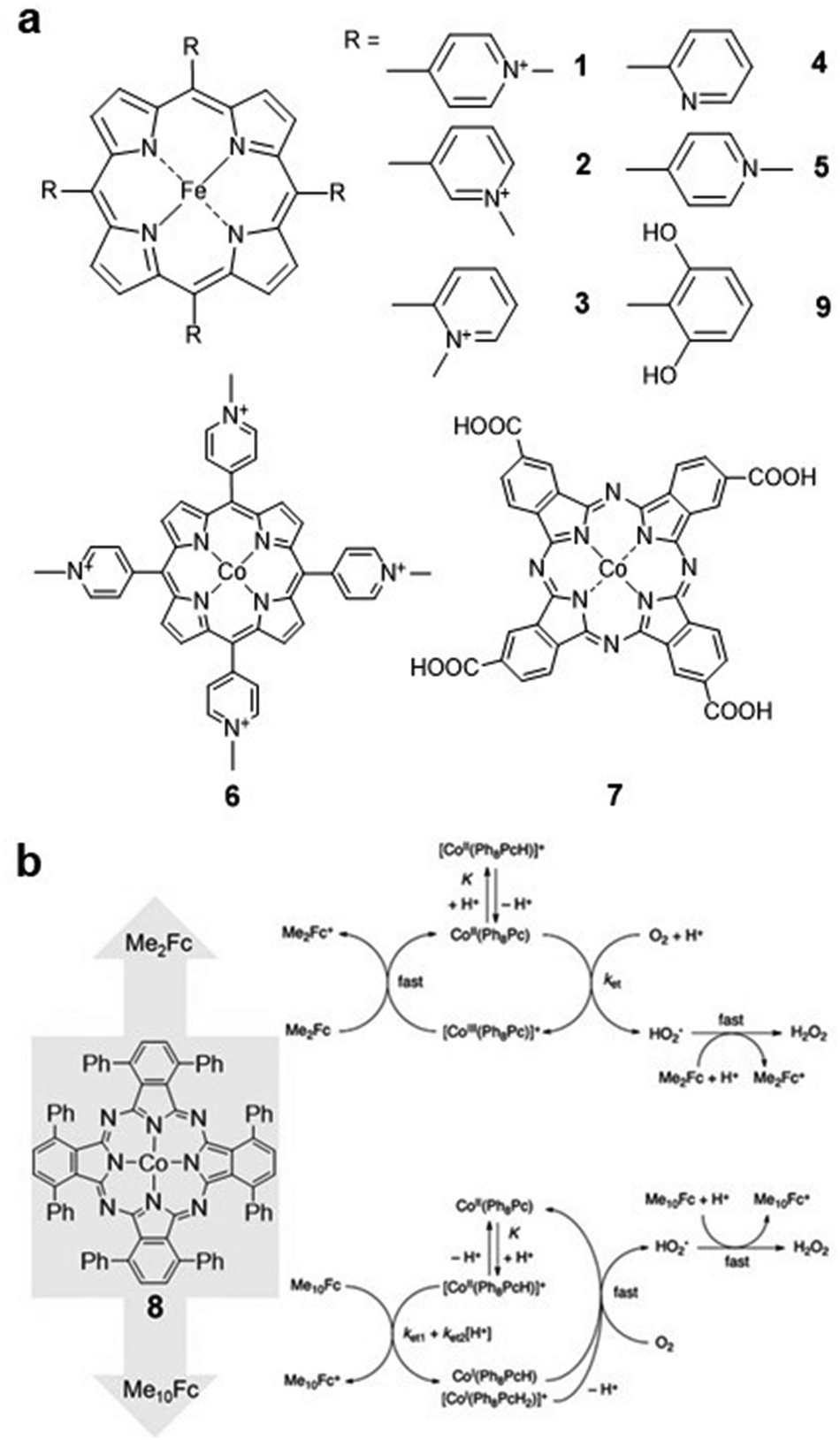 | ||
| Fig. 2 (a) Molecular structures of 1–7 and 9. (b) The possible ORR pathway of compound 8 after the addition of dimethylferrocene (Me2Fc) and decamethylferrocene (Me10Fc). Reprinted with permission from ref. 36. Copyright 2012 American Chemical Society. | ||
Matson et al. investigated the ORR selectivity of the 2-pyridyl (Fig. 2a, compound 4) and 4-pyridyl (Fig. 2a, compound 5) substituted-FeTMPyP derivatives in an acidic solution (pH = 0.3). The cyclic voltammetry (CV) results showed that the ORR onset potential of catalyst 4 was 0.4 V (vs. normal hydrogen electrode, NHE). The overpotential of complex 5 was approximately 100 mV higher than that of complex 4. Compared with 1–3, 4 and 5 are more favorable for H2O production in the ORR (<5% and <15% H2O2, respectively).16
Cobalt porphyrins are also commonly used as soluble electrocatalysts. Chan and co-workers investigated the ORR catalysed by cobalt(III) 5,10,15,20-tetrakis(4-N-methylpyridyl)porphyrin (CoTMPyP) (Fig. 2a, compound 6) in 0.1 M trifluoromethanesulfonic acid (TFMSA) solution. The catalyst reduces O2 to H2O2via a 2e− pathway with a selectivity of 90%.34 Cobalt phthalocyanine (CoPc) and its derivatives have also been reported for the ORR. For instance, compound 7 (Fig. 2a) reduces O2 to H2O2 with an onset potential of −0.4 V (vs. saturated calomel electrode, SCE), and further reduction of H2O2 to H2O can be observed at more negative reduction potentials.35
Recent works have tended to perform the electrocatalytic ORR in organic solvents to improve the solubility of the catalysts. Catalyst 8 (Fig. 2b) was investigated using various chemical reductants in benzonitrile and formic acid, and the catalytic mechanism was found to be related to the reducing strength of the reductants. As shown in Fig. 2b, when Me2Fc was used as a reducing agent, the rate-limiting step was found to be the formation of the *HO2 complex. With the increasing amount of Me10Fc, the rate-limiting step changes to the protonation of 8. In addition, whether Me2Fc or Me10Fc is used, the selectivity for H2O2 is very high (>74%).36
Similar to the ORR, the CO2RR has been studied in organic solvents. Fe porphyrin 9 (Fig. 2a) was reported to catalyse the CO2RR with high selectivity to CO in N,N-dimethylformamide (DMF)/0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) electrolyte in the presence of 2 M H2O. In addition, modifying iron tetraphenylporphyrin (FeTPP) with phenolic hydroxyl groups at all ortho-sites of the phenyl groups improves its activity and stability in reducing CO2 to CO, which is due to the high local proton concentration after the introduction of phenolic hydroxyl groups.19
Insoluble catalysts loaded on the electrode exhibit higher electron transport efficiency during the catalytic process, and can be recovered from the substrate after the reaction, which can effectively reduce the cost of industrial production. The commonly used strategies for immobilizing MP and MPc molecules on electrodes involve non-covalent or covalent interactions between catalysts and substrates.37
Noncovalent immobilization via π–π interactions has been shown to be a facile strategy for immobilizing MPs and MPc on electrodes. For example, cobalt tetraphenylporphyrin (CoTPP) supported on carbon nanotubes (CNTs) showed a CO Faradaic efficiency (FECO) of more than 90%, a low overpotential and long-term stability.38 Similarly, CoPc anchored on CNTs has been proven to exhibit higher activity towards the CO2RR, and the catalytic stability and product selectivity are better than those of CoPc anchored on carbon fiber paper.39
Noncovalent immobilization of molecular catalysts can also be achieved through electrostatic interactions. Reduced graphene oxide frameworks (FePGF), synthesized by utilizing the electrostatic interaction between the ammonium ion of FeIII porphyrin (FeMAP) and the carboxylate anion of reduced liquid crystalline graphene oxide (Fig. 3a), were used to catalyse the CO2RR. As shown in Fig. 3b, FePGF exhibited a better onset potential of −0.59 V (vs. reversible hydrogen electrode, RHE). The electrolysis test of FePGF at −0.59 V (vs. RHE) showed remarkable catalytic stability (Fig. 3c), and the CO selectivity was maintained at a high level after 24 h, with a FECO of 93% at an overpotential of 480 mV.40
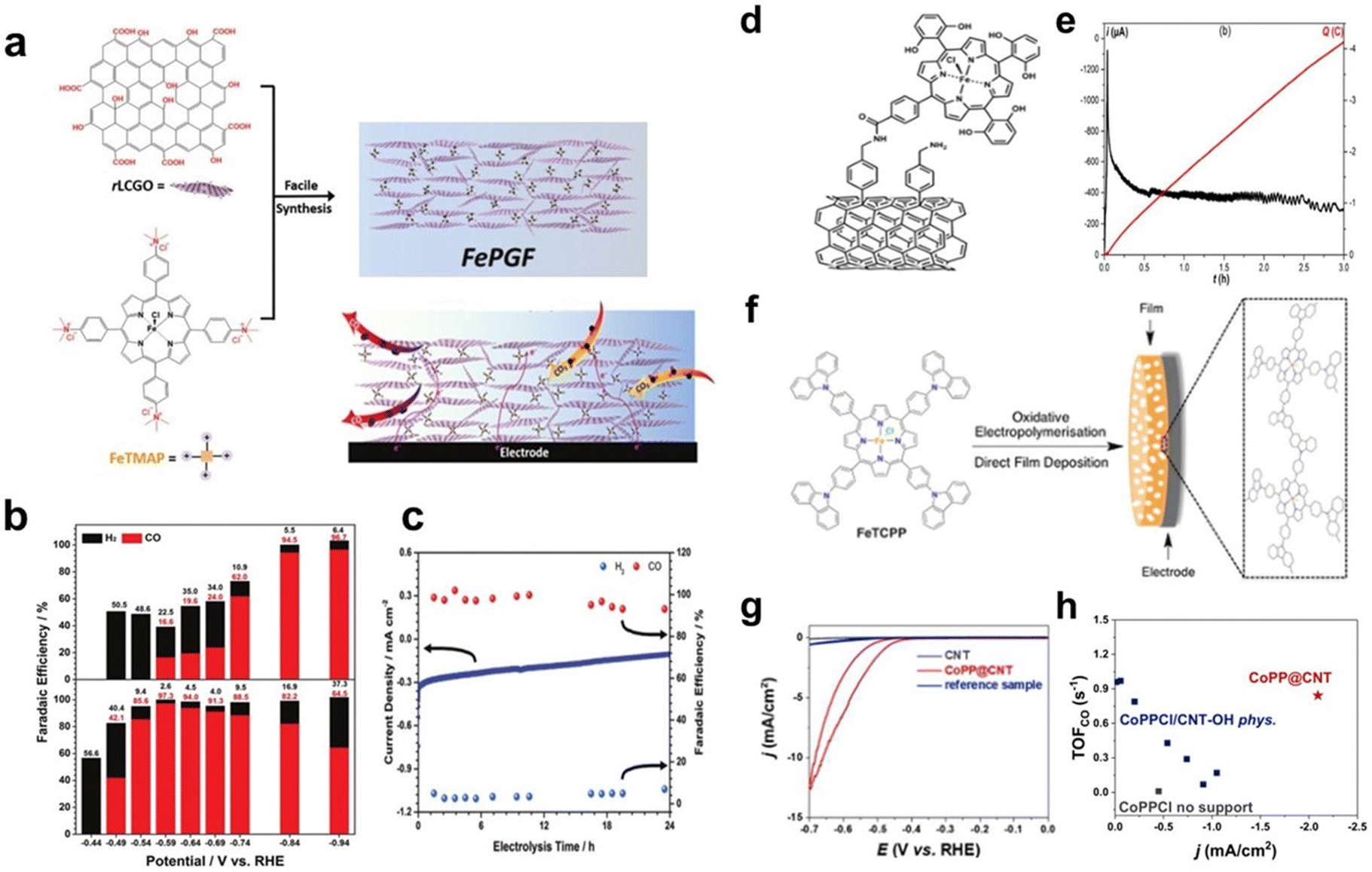 | ||
| Fig. 3 (a) Schematic illustration of the synthesis of FePGF. (b) FE of FePGF as an insoluble catalyst (bottom) for CO (red) and H2 (black) formation at various applied potentials. (c) Long-term stability with respect to current density (blue solid line) and FE (colored dots) of CO2 reduction with FePGF at −0.59 V (vs. RHE) for 24 h. Reprinted with permission from ref. 40. Copyright 2018 Wiley-VCH. (d) Covalent immobilization of the iron porphyrin catalyst on multiwalled carbon nanotubes (MWCNTs). (e) Current (black curve) and charge (red curve) during controlled potential electrolysis (E = −1.06 V vs. standard hydrogen electrode, SHE) with the catalyst covalently immobilized on MWCNTs. Reprinted with permission from ref. 41. Copyright 2016 Royal Society of Chemistry. (f) Illustration of film formation via oxidative electropolymerisation of tetrakis(4-carboxyphenyl)-porphyrin-Fe (FeTCPP). Reprinted with permission from ref. 42. Copyright 2016 Royal Society of Chemistry. (g) CVs recorded on CNT-OH, CoPP@CNT and the control sample. (h) Turnover frequency (TOF)CO as a function of current densities at −0.55 V (vs. RHE) for all tested electrodes. Reprinted with permission from ref. 43. Copyright 2019 Elsevier. | ||
Covalent immobilization of catalysts on substrates can be achieved through coupling reactions. Robert et al. constructed an amide bond between tetraphenyl iron porphyrin containing OH- and NH2-modified CNTs, and a composite catalyst with iron porphyrin anchored on CNTs was prepared successfully as a heterogeneous catalyst (Fig. 3d). The catalyst exhibits high catalytic activity for the CO2RR in a neutral solution with a high turnover frequency and excellent stability. As shown in Fig. 3e, CO was selectively produced with a FE close to 95% after 1 h of electrolysis in CO2-saturated 0.5 M NaHCO3 solution at −1.06 V (vs. SHE).41
Covalent immobilization can also be achieved through electrochemical reactions. Daasbjerg et al. used FeTCPP to form a catalyst film on an indium–tin oxide substrate through electropolymerization (Fig. 3f). The prepared film showed good electrocatalytic activity for CO2 reduction. The stability test showed 12% activity loss after the first voltammetric cycle, followed by a 12% loss in the next four cycles, which indicates that the film is not chemically stable enough for long-term use.42
In addition, there are other strategies for covalently immobilizing catalysts onto substrates. Han et al. covalently linked protoporphyrin IX cobalt chloride (CoPPCl) to hydroxyl-functionalized carbon nanotubes (CNT-OH) through a metal-centred substitution reaction to obtain the functionalized material CoPP@CNT. In Fig. 3g, the CV curves show significant CO2 reduction currents at −0.4 V (vs. RHE). During the steady-state electrolysis process, the catalyst has a high CO selectivity. The FECO reaches 90% at −0.65 V (vs. RHE). Through covalent anchoring, the amount of catalyst on the electrode can be optimized for a higher current density (Fig. 3h).43
2.2 Tailoring model catalysts by molecular fabrication
Influence of the electronic effect of functional groups. The electronic effects of substituents have been extensively considered an important factor in molecular fabrication. With the introduction of different substituents, the electron density of the metal centre changes, and the charge transfer between the active site and the substrate is adjusted. In addition, the interactions between the intermediates and active sites vary, thus influencing the catalytic behaviour.
Cao et al. reported Co porphyrins 10 and 11 (Fig. 4a) with different substituents as model systems to investigate the electronic effect of substituents on the ORR.44 The results showed that the ORR activity of 10 was higher than that of 11 with fluorine substitutions in an alkaline solution. Density functional theory (DFT) calculations showed that the electron density of the Co site in compound 11 is significantly lower than that in 10, which is due to the strong electron-withdrawing ability of the pentafluorophenyl substituent (Fig. 4b). Karami et al. found that the introduction of electron-donating methyl and methoxy substituents on the porphyrin ring is beneficial for the catalytic activity of cobalt porphyrin, which is due to the stronger binding between O2 and the Co centre and the promoted electron transfer.25 Therefore, for the ORR, electron-donating substituents can increase the electron density of the metal centre, thereby increasing the binding to O2 and electron transfer, which is crucial for the increase in the ORR activity.
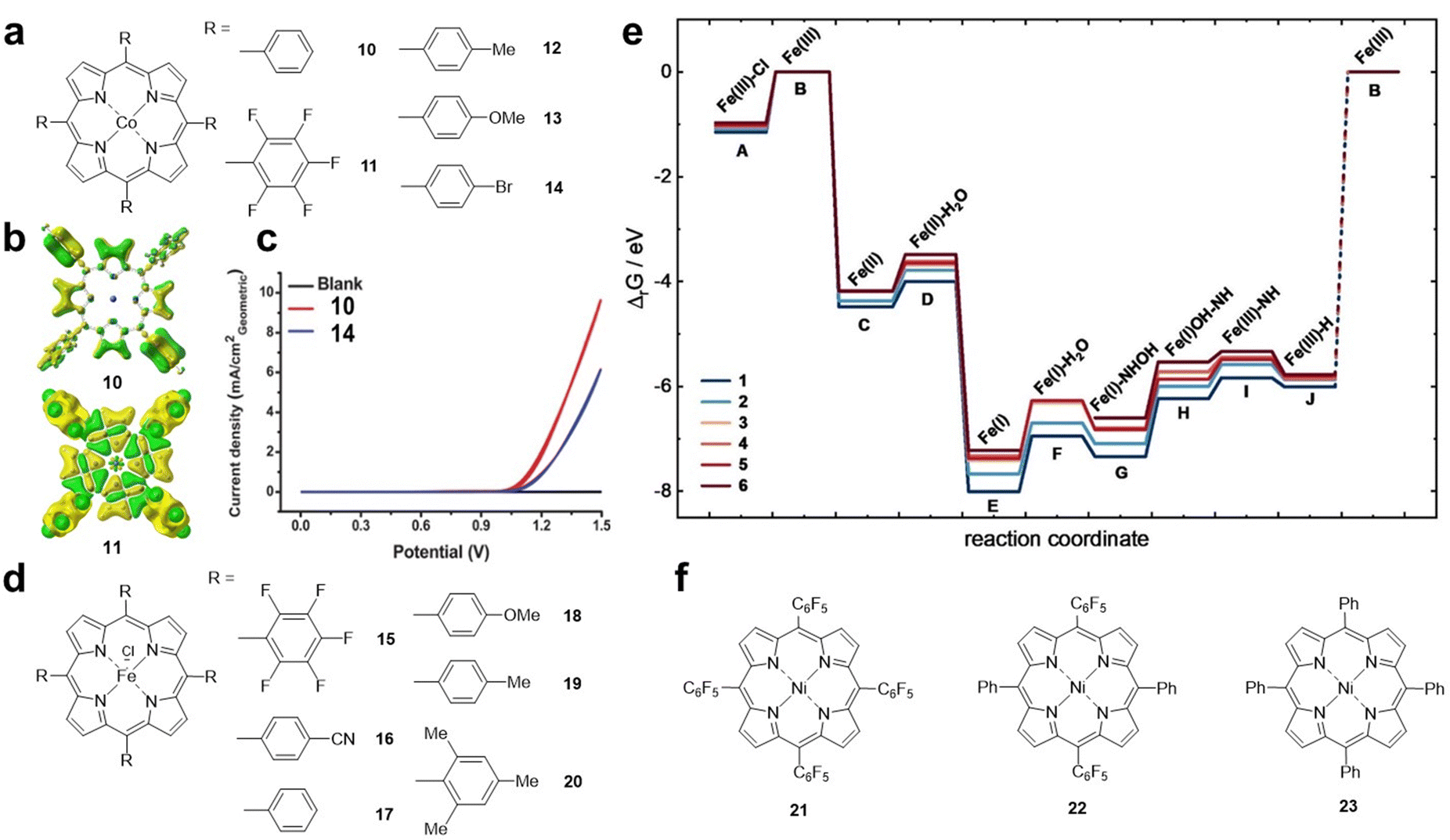 | ||
| Fig. 4 (a) Molecular structures of Co porphyrins 10–14. (b) The corresponding calculated electron density of 10 and 11. Reprinted with permission from ref. 25. Copyright 2007 Wiley-VH. (c) Cyclic voltammograms obtained using fluorine doped indium tin oxide coated with 20 nmol cm−2 cobalt porphyrin complexes as the working electrodes in a 0.5 M borate buffer solution. Red plot: 10 and blue plot: 14. Reprinted with permission from ref. 45. Copyright 2014 Royal Soc Chemistry. (d) Molecular structures of Fe porphyrins 15–20. (e) Calculated Gibbs free enthalpies of HER intermediates relative to the FeIII species. Reprinted with permission from ref. 46. Copyright 2023 Wiley-VH. (f) Molecular structures of Ni porphyrins 21–23. | ||
The electronic effects of substituents on the OER have also been studied. As shown in Fig. 4c, compound 10 has better OER activity than the electron-withdrawing group-substituted Co porphyrin 14 (Fig. 4a).45 The results show that, similar to the ORR, electron-withdrawing groups are not conducive to the promotion of OER activity.
To investigate the influence of substituents on the HER, Kramm et al. designed a series of iron porphyrin derivatives with different side chain groups (Fig. 4d).46 From 15 to 20, the electron density of the Fe–N4 centre gradually increases. Theoretical calculations indicated that the anionic radical *[FeII(N4)]− (that is, FeI) is important for the HER throughout the catalytic process (Fig. 4e). In the *[FeII(N4)]− reduced state, porphyrins with electron-withdrawing groups, which facilitate the electron density shift towards the ligand π system, are more active in the HER. Cao et al. synthesized a series of Ni porphyrins 21–23 (Fig. 4f) for the HER. They found that the HER activity of 22 and 23 was lower than that of 21 after replacing C6F5 with the substituent C6H5.22
From the above results, it can be inferred that MP with electron-withdrawing groups is beneficial for the HER. According to the literature, MI or M0 is necessary for the HER, and strong electron-withdrawing substituents promote a positive shift in the equilibrium potential of MI or M0, which makes it easier to trigger the reduction of protons.
Similar to the HER, some electron-withdrawing groups are also preferred for the CO2RR. Savéant et al. studied FeTPP 24 and its derivatives 25–28 (Fig. 5a), and the electron-withdrawing effect of the complexes increased sequentially. FeTPP derivative 28 with methoxy substituents on the benzene ring was provided as a reference because of its strong electron-donating effect. Electrochemical tests showed that the peak potential of the catalysts shifted positively from 24 (E = −1.428 V vs. SHE) to 25 (E = −1.365 V vs. SHE), 26 (E = −1.276 V vs. SHE) and 27 (E = −1.118 V vs. SHE). In contrast, the presence of electron-donating substituents on 28 results in a 340 mV shift of E in the negative direction (E = −1.725 V vs. SHE) and a higher TOF value (Fig. 5b).47
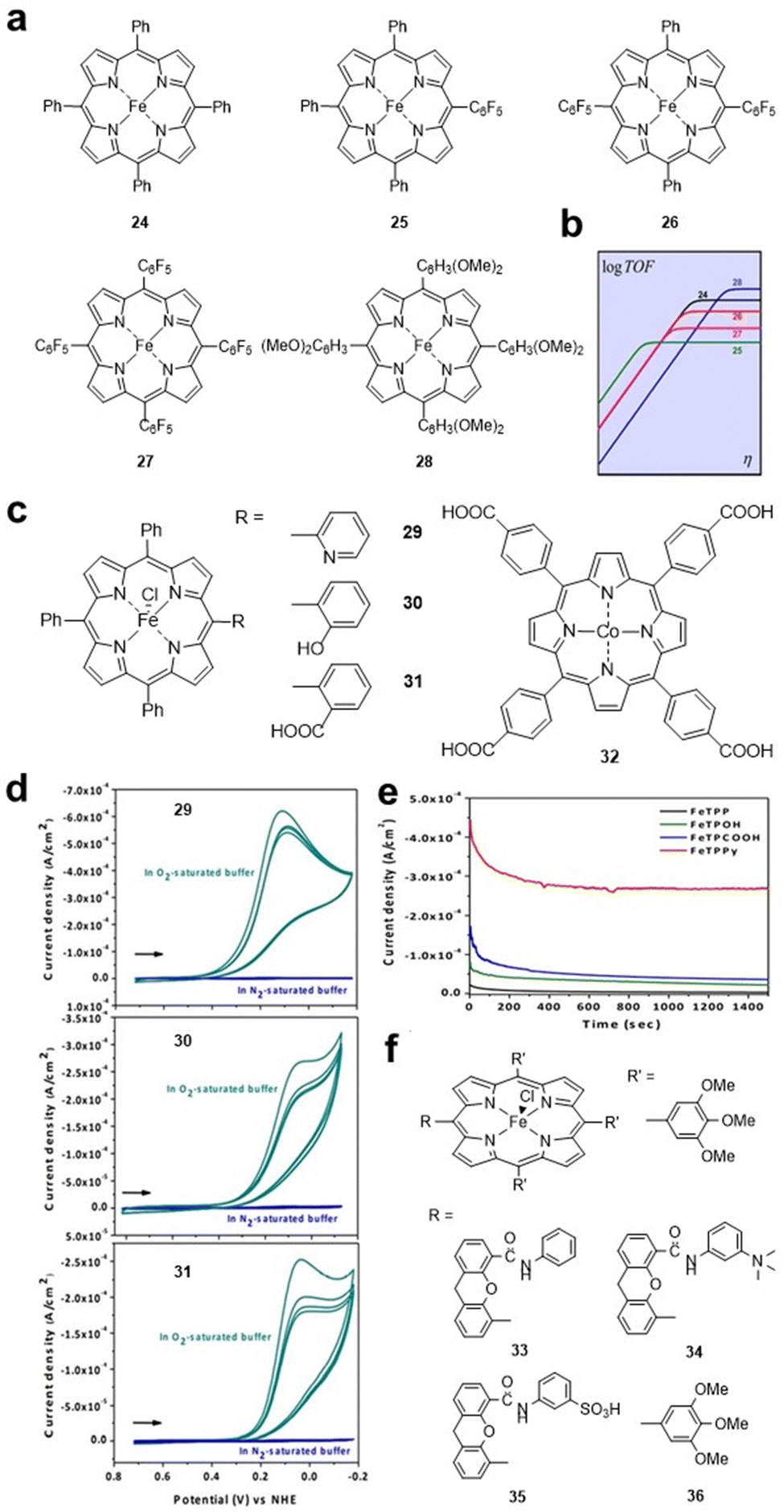 | ||
| Fig. 5 (a) Molecular structures of iron porphyrin catalysts 24–28. (b) Catalytic Tafel plots of catalysts 24–28. Reprinted with permission from ref. 47. Copyright 2016 American Chemical Society. (c) Molecular structures of 29–32. (d) CVs for 29–31 under an N2 and O2 atmosphere on basal plane graphite (BPG) electrodes. (e) Controlled potential (0.1 V (vs NHE)) experiments for Fe porphyrin catalysts on BPG electrodes. Reprinted with permission from ref. 48. Copyright 2015 Wiley-VH. (f) Molecular structures of hangman Fe porphyrins 33–36. | ||
Effect of proton relay groups. Substituents containing proton relays and multiple redox sites are important for electrocatalytic reactions, and introduced proton relays can promote H+ transfer in electrocatalytic processes. Warren et al. studied the ORR catalysed by iron porphyrins 29–31 (Fig. 5c) with Brønsted acid–base groups as the side chain. Among them, 29 with pyridine as the proton relay exhibited the best ORR catalytic activity with a peak current of 0.7 mA cm−2 in an acidic aqueous solution (Fig. 5d and e). For 30 and 31, the peak current is 0.3 mA cm−2, which is higher than that of FeTPP. The authors showed that FeTPPy 29 may be more acidic than FeTPPOH 30 or FeTPPCOOH 31 in the adsorbed catalyst layer. The authors believe that the strong acidity of the substituents is beneficial for functional groups (pyridine and carboxylic acid) to facilitate the transfer of protons to the reduced iron oxide intermediate, which could significantly enhance the catalytic ORR activity.48 Similarly, a carboxyl group was introduced into the CoTPP macrocycle to form a new compound cobalt meso-tetra(4-carboxyphenyl)porphyrin (CoTcPP) 32 (Fig. 5c), which also exhibited enhanced ORR activity.49
The introduction of protic groups can also facilitate the electrocatalytic reduction process by lowering the energy barrier for proton reactions. Nocera et al. synthesized a series of hangman iron porphyrins 33–36 (Fig. 5f) to investigate the influence of side chain protic groups on the electrocatalytic HER ability. The authors clarified that the presence of proton donors in iron porphyrin will enhance the catalytic activity of the HER. HPFe-3SA 35 with a sulfonic acid hanging group exhibits the best catalytic performance, which may be due to its ability for intramolecular rather than intermolecular H+ transfer from the sulfonic acid. Theoretical calculations showed that the energy barrier for proton transfer is lowered when protons are transferred within the molecule. Furthermore, when intramolecular proton transfer was delayed by increasing the hanging group pKa (HPDFe-DMA, 34), a large decrease in the catalytic rate compared with 35 was observed.50
The catalytic activity of MP with proton groups can be regulated by adjusting the microenvironment around the catalyst. Savéant et al. introduced phenolic groups at all ortho-positions of the phenyl group in FeTPP to improve the electrocatalytic efficiency of CO2 reduction to CO. 5,10,15,20-Tetrakis(20,60-dihydroxyphenyl) iron porphyrin (FeTDHPP, 9 in Fig. 2a) was electrolyzed at −1.16 V (vs. SHE) for 4 h, and no degradation was observed, showing a FECO of 94%. The enhanced catalytic activity of 9 was attributed to the higher local concentration of protons originating from the phenolic hydroxyl substituents.19
Steric effects of substituents. When large steric hindrance groups are introduced into the side chains of MP and MPc, the reaction intermediates can be stabilized by direct interaction, thus improving the activity. Hellman investigated the OER and ORR processes catalysed by metal hangman-porphyrins (MHPs) by DFT calculations. By comparing the Gibbs free energies of the reaction intermediates of MP and MHP, the influence of the hangman structure on the reaction process was revealed. The authors found that the hangman group has little effect on the energy of *OH and *OOH intermediates, but can stabilize *O intermediates. As shown in Fig. 6a, by transferring a proton from the acid group to the *O intermediate, the hangman porphyrin can shift the energy to a more favourable position.51
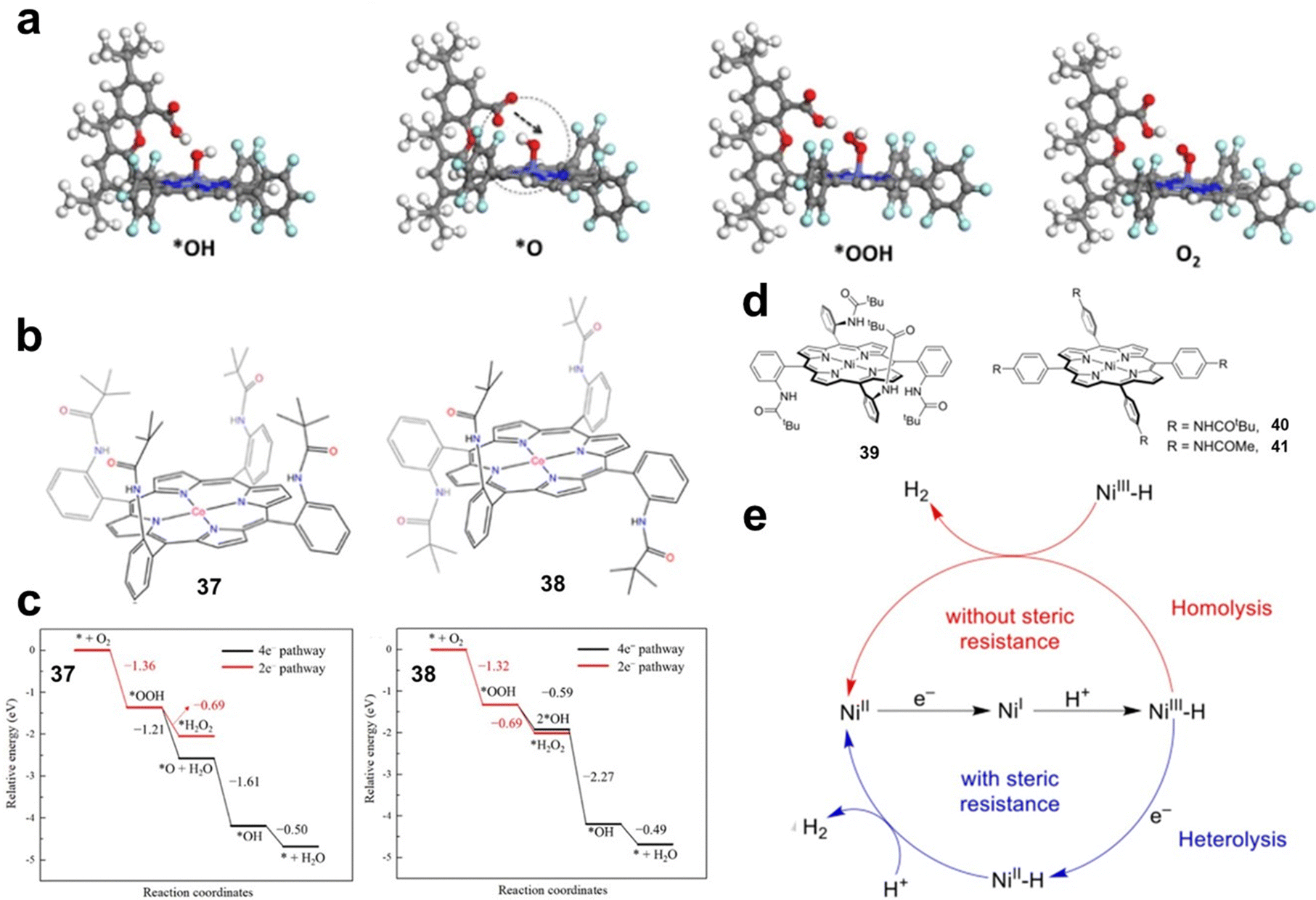 | ||
| Fig. 6 (a) The calculated structures of the *OH, *O, *OOH, and O2 intermediates on CoHP. Reprinted with permission from ref. 51. Copyright 2014 American Chemical Society. (b) Structures of 37 and 38. (c) The energies of key intermediate species in the basic steps in 2e− and 4e− ORR pathways for the catalysts 37 and 38. Reprinted with permission from ref. 52. Copyright 2023 Elsevier. (d) Molecular structures of Ni porphyrins 39–41. (e) Proposed catalytic HER mechanisms with 39 and 40. Reprinted with permission from ref. 53. Copyright 2020 Wiley-VH. | ||
The steric effect of the side chain may alter the reaction pathway by stabilizing the reaction intermediate. Zhao et al. constructed cobalt porphyrins 37 and 38 (Fig. 6b) with different substituents and used DFT calculations to study the ORR mechanism. The electronic structure and adsorption configuration analysis of O2 adsorbed on 37 indicated that *O2 was fully activated, which is beneficial for the ORR. The energies of key intermediate species in 2e− and 4e− ORR pathways were calculated respectively (Fig. 6c). The results showed that *OOH is more easily hydrogenated on 37 to generate *O and *OH with better selectivity for the 4e− ORR. In contrast, the 2e− pathway is more favoured for 38.52
The steric effect of side chain groups can directly affect the combination of reactants or the formation of intermediates, resulting in changes in the reaction pathway. Cao et al. designed and synthesized three nickel porphyrins (Fig. 6d, 39–41) with different steric effects and investigated their activity and mechanism for the HER. Compared with 40 and 41, 39 exhibits greater steric resistance on both sides of the porphyrin macrocycle. By studying the reduced states of 39 and 40 (39− and 40−) in the presence of trifluoroacetic acid (TFA), the authors demonstrated that 39− and 40− exhibit different H2 generation mechanisms when treated with TFA (Fig. 6e). For 39, the bimolecular homolytic mechanism of NiIII–H is blocked due to the steric hindrance effect of the pivalamide groups located on both sides of the porphyrin macrocycle of 39. NiIII–H requires an additional 1e− reduction to become more active to react with TFA. In the case of 40, due to the small steric hindrance in the direction perpendicular to the porphyrin macrocycle, the NiIII–H generated by 40− and TFA can undergo bimetallic homolysis to generate H2.53
Pyridine, as a nitrogen-containing group, is often studied as an axial ligand, which can change the coordination environment of the metal centre. Cho and co-workers reported a highly efficient ORR catalyst (FePc-Py-CNT) with pyridine axial ligands. Compared with the iron phthalocyanine (FePc)-CNT catalyst, the half-wave potential of pyridine-coordinated FePc is more positive. The effect of axial ligands on MP and MPc molecules is explained through electron-donating effects, that is, the axial coordination of the ligand group on the central metal increases the electron density on the macrocycle by donating electrons to the unoccupied d-orbital of the metal ion, thus improving its catalytic activity.54
Different axial ligands (thiosalts and imidazoles) can influence the rate-determining step (RDS) of the electrocatalytic reaction. Dey et al. investigated the electrocatalytic ORR of thiolate- and imidazole-coordinated iron porphyrin complexes. The results showed that for the sulfate complex, the RDS is the heterocleavage of the O–O bond in the FeIII–OOH intermediate. The electron-donating effect of sulfate increases the electron density of the Fe centre, thereby increasing the pKa of the FeIII–OOH species, which is beneficial for its protonation and the dissociation of the O–O bond. For compounds with imidazole ligands, the pKa value of FeIII–OOH is too low for protonation and O–O bond cleavage to occur. Therefore, it must be reduced to FeII–OOH first, followed by protonation and O–O bond cleavage.55
For MPs and MPcs with M–N4 sites, their electrocatalytic activity and catalytic reaction pathways are greatly affected by the electronic structure of the M–N4 site. The introduction of axial ligands can change the symmetry of the M–N4 structure and optimize its d orbital energy level, thereby regulating the electronic structure of the M–N4 sites. Li et al. selected a series of n-type metal chalcogenides as axial coordination groups and prepared p–n junctions with p-type FePc to achieve continuous and wide-range ORR activity control at the single-atom level.56 Especially, for gallium monosulfide (GaS), its introduction leads to a 2.5-fold increase in the ORR activity of FeN4. The rectification effect in the FePc/GaS junction spatially distorts the FeN4 fragment in the FeII center and induces a medium-high spin state transition from 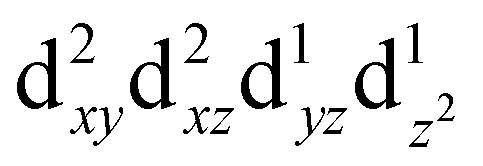 to
to 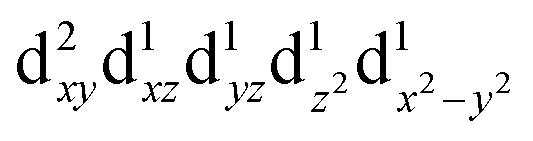 , making the FeN4 fragment more effective in adsorbing and dissociating O2 molecules. Liu et al. synthesized an N–Fe–N4 catalyst with N axial ligands for the ORR and used in situ Mössbauer spectroscopy to directly capture *O2− and *OH− to demonstrate the change in the electronic structure of the active site with changes in potential.57 When the ORR does not start, OH− adsorbs on the Fe center, and the formation of the *OH− intermediate slightly shortens the Fe–N bond length. Near the ORR onset potential (0.9 V vs. RHE), the generation of *O2− intermediates further shortens the Fe–N bond length, and the Fe2+ electronic state transitions from high spin (HS) to low spin (LS). Calculation proves that the interaction between the π orbital of the coordinated O2 and the dxz orbital of the Fe atom increases the splitting of the d orbital, leading to the transformation of the electronic configuration of Fe2+ when the *O2− intermediate is formed, and at the same time, it can significantly reduce the energy barrier formed by each intermediate in the ORR.
, making the FeN4 fragment more effective in adsorbing and dissociating O2 molecules. Liu et al. synthesized an N–Fe–N4 catalyst with N axial ligands for the ORR and used in situ Mössbauer spectroscopy to directly capture *O2− and *OH− to demonstrate the change in the electronic structure of the active site with changes in potential.57 When the ORR does not start, OH− adsorbs on the Fe center, and the formation of the *OH− intermediate slightly shortens the Fe–N bond length. Near the ORR onset potential (0.9 V vs. RHE), the generation of *O2− intermediates further shortens the Fe–N bond length, and the Fe2+ electronic state transitions from high spin (HS) to low spin (LS). Calculation proves that the interaction between the π orbital of the coordinated O2 and the dxz orbital of the Fe atom increases the splitting of the d orbital, leading to the transformation of the electronic configuration of Fe2+ when the *O2− intermediate is formed, and at the same time, it can significantly reduce the energy barrier formed by each intermediate in the ORR.
2.3 Combination of MP and MPc with low-dimensional nanomaterials
Low-dimensional nanomaterials such as graphene, CNTs, carbon black, and MoS2 have been widely used as conductive substrates due to their excellent electrical conductivity and large specific surface area. Immobilizing molecular catalysts on these substrates can increase the charge transfer between the catalyst molecules and substrates, enhancing catalytic performance.58Defect engineering means the introduction of well-defined defects into catalytic materials through heteroatom doping and removal, plasma irradiation, hydrogenation, and amorphization to provide more abundant sites for improving the intrinsic activity.60,61 Defect engineering has been proven to be an effective method to modify the surface properties and electronic structure of graphene. When MPs and MPcs are supported on graphene with defects, the defects change the electron density of the metal centre, thereby affecting its catalytic activity. For Fe–N4, the higher the electron density of the metal centre, the better its catalytic ORR activity. Yang and co-workers dispersed FePc on defective graphene (DG) via π–π stacking. In 0.1 M KOH, FePc/DG exhibited excellent catalytic activity and long-term stability as a cathode ORR catalyst. DFT calculations showed that DG with 585 defects (DG-585) is favourable for charge redistribution on FePc and the substrate. Electron transfer from the DG-585 defect to FePc leads to the formation of an electron-rich region on the Fe atom (Fig. 7a), which accelerates the ORR reaction kinetics and accelerates electron transfer from the Fe atom to the adsorption intermediate. This adjustment of the electronic structure of Fe atoms is beneficial for the adsorption and reaction of O2 molecules, resulting in a larger positive initial potential and higher current density for the ORR.62
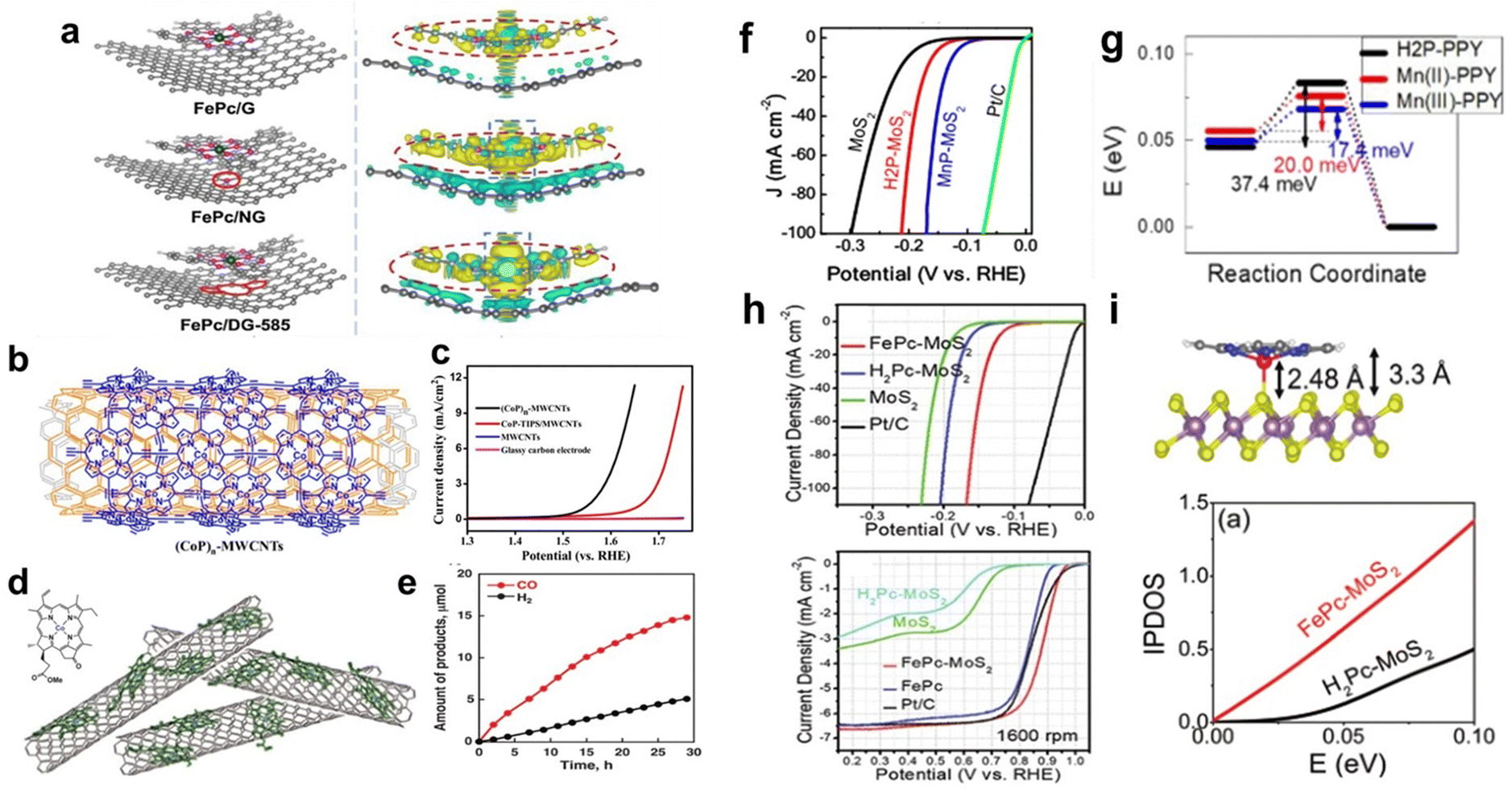 | ||
| Fig. 7 (a) Top views of th optimized FePc/DG-585 (FePc/G or FePc/NG) based hybrid interfaces, and side views of the 3D charge density difference plot for the interfaces between DG-585 (G or NG) sheet and FePc layer. Reprinted with permission from ref. 62. Copyright 2021 Elsevier. (b) The structure of (CoP)n-MWCNTs. (c) Linear sweep voltammetry (LSV) curves using different materials in 1.0 M KOH. Reprinted with permission from ref. 65. Copyright 2015 American Chemical Society. (d) Structure of CoII(Ch) and schematic image of CoII(Ch) on MWCNTs. (e) Time courses of evolution of CO and H2 in the CO2RR on the glassy carbon electrode modified with CoII(Ch) adsorbed on MWCNTs at an applied potential of −1.1 V (vs. NHE). Reprinted with permission from ref. 66. Copyright 2015 Royal Soc Chemistry. (f) LSV curves for MoS2, H2P-MoS2, MnP-MoS2, and commercial Pt/C as catalysts towards the HER in H2-saturated 0.5 M H2SO4. (g) Activation barrier of the Heyrovsky reaction calculated for the slab geometry. Reprinted with permission from ref. 67. Copyright 2019 Royal Soc Chemistry. (h) LSV curves of FePc-MoS2, FePc, Pt/C, H2Pc-MoS2, and MoS2 for the HER and ORR in 0.1 M KOH. (i) Top: Optimized structures of the 1T′ phase MoS2-FePc. Bottom: Integrated partial density of states (IPDOS) for MoS2 in FePc-MoS2 and H2Pc-MoS2. Reprinted with permission from ref. 68. Copyright 2019 Royal Soc Chemistry. | ||
Graphene has been extensively studied as a support for the HER. Chen et al. reported a facile method to prepare cobalt porphyrin-functionalized electrochemically reduced graphene oxide (CoTMPyP/ERGO) thin films. Oppositely charged CoTMPyP and ERGO nanosheets were directly assembled, followed by electrochemical reduction to synthesize CoTMPyP/ERGO composites. The composite exhibits excellent catalytic activity for the HER in alkaline media. CoTMPyP acts as an active centre in HER catalysis. The authors attributed the excellent catalytic performance of the composite to the efficient electron transport between ERGO and CoTMPyP; moreover, the wrinkled structure present inside and between the graphene flakes exposes more active sites, which is beneficial for the reaction of reactants and the transport of products.63
Du et al. reported the noncovalent bonding of (CoP)n to MWCNTs through in situ polymerization using MWCNTs as templates to prepare a highly efficient electrocatalyst ((CoP)n-MWCNTs) for water oxidation (Fig. 7b). (CoP)n-MWCNTs exhibit good OER activity in alkaline solution (pH ∼ 13.6) (Fig. 7b). At a potential of 1.52 V (vs. RHE), the catalytic current density was 1.0 mA cm−2, corresponding to a low overpotential of 0.29 V. These results demonstrate that (CoP)n-MWCNTs are efficient OER catalysts that can significantly reduce the overpotential for the water oxidation reaction (Fig. 7c). In addition, the excellent stability was confirmed through chronopotentiometry measurements, indicating that the (CoP)n-MWCNT catalyst has good OER durability.65
For the CO2RR, Fukuzumi et al. prepared a cobalt chloric acid complex CoII(Ch) on MWCNTs to form CoII(Ch)@MWCNT composites as CO2RR catalysts (Fig. 7d). Under acidic conditions, the CoII(Ch)@MWCNT complex selectively catalyses the reduction of CO2 to CO. At pH 4.6, the Faradaic yield of CO is as high as 89%, while that of H2 is only 11% (Fig. 7e). Compared with the 2D catalyst formed by CoII(Ch) supported on planar reduced graphene oxide (rGO), the authors proposed that the three-dimensional (3D) structure formed by MWCNTs and CoII(Ch) may play an important role in the selective electrocatalytic reduction of CO2 to CO.66
Kang et al. formed a unique 1T' intercalation complex through a one-step hydrothermal reaction between 2D MoS2 and Mn porphyrin (MnP) molecules. MnP-MoS2 exhibits excellent electrocatalytic activity for the HER with a current density of 10 mA cm−2 at an overpotential of 0.125 V (vs. RHE) (Fig. 7f). In addition, the authors synthesized MoS2 nanosheets by using MnCl2 instead of MnP, and the results showed that the enhanced catalytic efficiency of MnP-MoS2 was due to the porphyrin ring and Mn centre. On the basis of DFT calculations, the authors attributed the excellent catalytic performance to the high electron concentration and low activation barrier of the rate-determining Heyrovsky reaction (Fig. 7g).67
In addition to MnP, FePc has been supported on MoS2 as a bifunctional catalyst for the OER and ORR. Kang et al. also synthesized a 2D hybrid structure composed of FePc molecules and MoS2 (FePc–MoS2). FePc–MoS2 exhibits excellent catalytic activity for both the HER and ORR. As shown in Fig. 7h, the catalytic HER activity of FePc–MoS2 is higher than that of H2Pc–MoS2 and pristine MoS2. The IPDOS calculation (Fig. 7i, bottom) results showed that the electron concentration in FePc–MoS2 is higher than that in H2Pc–MoS2, and the high electron concentration helps improve the HER performance. For the ORR, the authors attributed the high catalytic activity of FePc–MoS2 to the nonplanar geometry of the Fe–N4 active sites in FePc adsorbed on MoS2 (Fig. 7i, top).68
In addition, two-dimensional materials like phosphorene,69 borophene,70 and silicene71 have also been widely used in electrocatalytic reactions with M–N4 sites due to the large surface area, high conductivity, and favourable charge transfer, which have shown great promise in electrocatalysis.
3 MP/MPc-based framework materials
3.1 Metal–organic frameworks (MOFs)
MP/MPc-based MOF materials are a specific kind of MOF, and porphyrin and phthalocyanine can be used as both the structural unit and the catalytic site. The adjustable pore size and various topologies of MOFs are responsible for their wide application in electrocatalytic reactions such as the ORR, OER, HER, and CO2RR.72In addition to high-valent metal ions/clusters, polyoxometalates, such as ε-PMo8VMo4VIO40Zn4 (Zn-ε-Keggin) clusters, are widely used as an electron-rich linker in 3D Por/Pc-MOFs that can easily transfer electrons in reversible redox reactions without changing their metal-oxo-cluster structures. Lan et al. used reduced Zn-ε-Keggin clusters and MP as building blocks to synthesize a series of MOFs (M-PMOF) with different metals for catalysing CO2 reduction (Fig. 8a). All of these MOFs exhibit excellent electrochemical CO2 reduction performance. (Fig. 8b). In particular, Co-PMOFs have the best catalytic performance, with a FECO of 99%, a high TOF of 1656 h−1 (at an overpotential of −0.8 V vs. RHE), and excellent stability over 36 h. Based on the experimental results and theoretical calculations, the author proposed the possible mechanism of the Co-PMOF catalysed CO2RR. The Co centre is reduced from CoII to CoI at first with the electron transferred from Zn-ε-Keggin. Then, CoI site interacts with CO2 to generate *COOH, and the electrocatalytic process is triggered. Reducing Zn-ε-Keggin clusters and MP can directly lead to directional electron transport in an electric field, which is beneficial for multielectron transfer in the electrocatalytic CO2RR.74
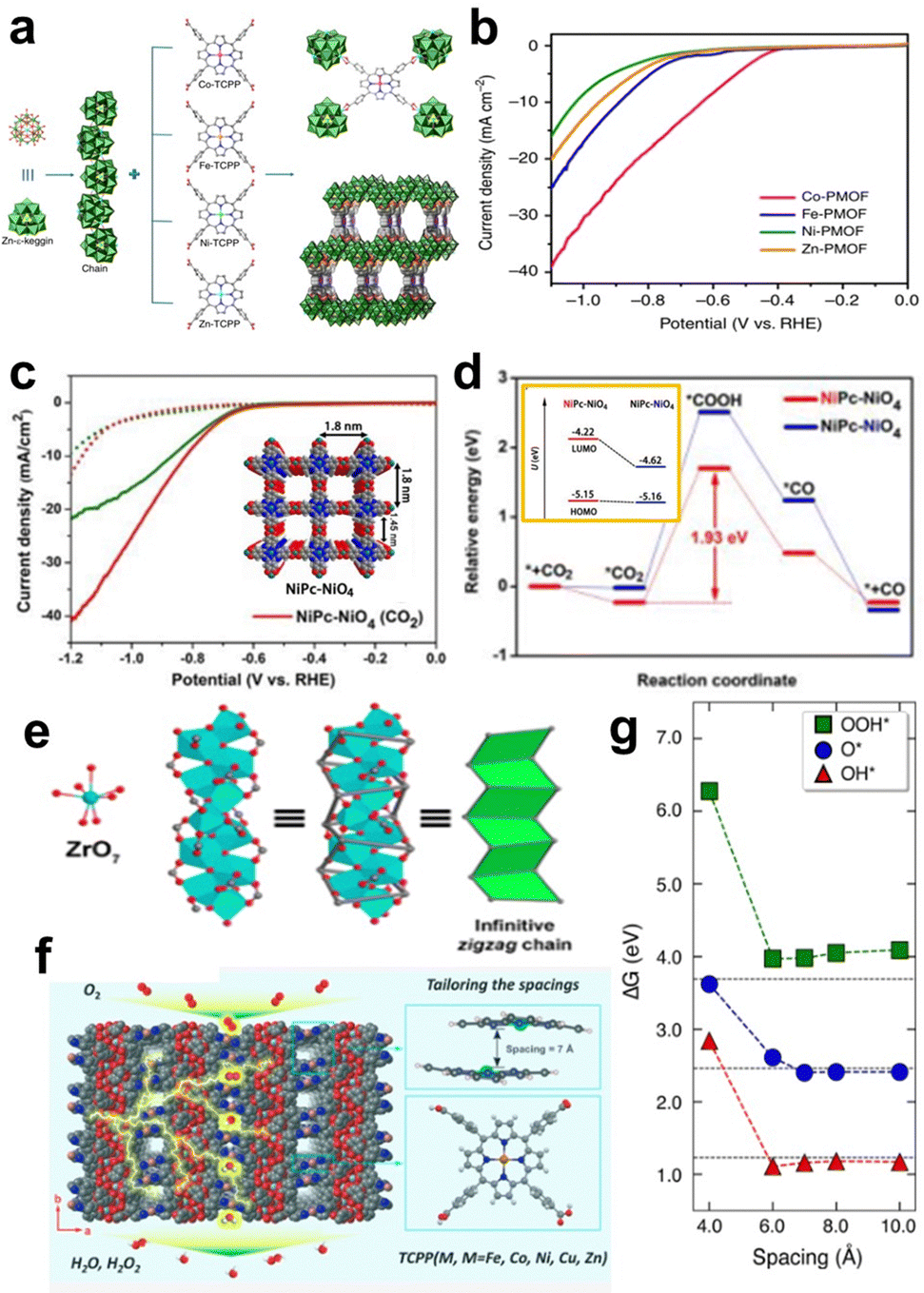 | ||
| Fig. 8 (a) Schematic illustration of the structures of M-PMOFs (M = Co, Fe, Ni, and Zn). (b) LSV curves of M-PMOFs. Reprinted with permission from ref. 74. Copyright 2018 Nature Publishing Group. (c) LSV curves of NiPc-NiO4 and NiPc-OH in CO2, and Ar-saturated 0.5 M KHCO3. (d) Calculated energy diagrams for CO2-to-CO conversion on two proposed active sites in NiPc-NiO4. Orange box: the energy level of the HOMO and LUMO of different Ni atoms in NiPc-NiO4 with the introduction of CO2. Reprinted with permission from ref. 76. Copyright 2021 Wiley-VH. (e) The structure of zigzag Zr chains. (f) Crystal structure of PCN-226 viewed along the c-axis. (g) Adsorption energy of ORR intermediates as a function of the spacing distances between the porphyrin units. Reprinted with permission from ref. 80. Copyright 2020 American Chemical Society. | ||
Copper-based MOFs were also constructed for the CO2RR. Gu et al. reported that 2D Cu-MOF nanosheet catalysts synthesized with Cu porphyrin as an organic linker and Cu2(COO)4 as nodes can efficiently and selectively reduce CO2 to formate and acetate. At −1.55 V (vs. Ag/Ag+) and a total current density of 4.5 mA cm−2, the highest Faradaic efficiencies of HCOO− and CH3COO− increase to 68.4% and 16.8%, respectively. The TOF of the catalyst for the production of HCOO− and CH3COO− can be as high as 2037 h−1 and 148 h−1, respectively.75
Ni–N4-based MOFs have also been explored for the CO2RR. Phthalocyanine-based 2D MOF (NiPc-NiO4) nanosheets are linked by NiPc-OH and nickel salt.76 NiPc-NiO4 exhibits remarkable CO2RR catalytic performance (Fig. 8c) with a CO selectivity of 98.4% at −1.2 V (vs. RHE) and a TOF of up to 2603 h−1. To further explore the active sites of NiPc-NiO4, the authors calculated the CO2RR reaction pathways of two different Ni active sites in the MOFs (Fig. 8d). As shown in the orange box of Fig. 8d, the formation energy of the *COOH intermediate at the NiPc site is 1.93 eV, which is lower than that at the NiO4 site (2.53 eV), suggesting that the CO2RR may preferentially occur at the Ni–N4 site. Mulliken charge analysis revealed that the Ni environment in NiPc sites is more electron-rich than that in NiO4 sites. Therefore, the strong CO2 adsorption capacity and electron-rich environment of Ni centres in phthalocyanines make them good active sites for the CO2RR.
In addition, the crystal structure of porphyrin-based MOFs can affect their electrocatalytic activity. For instance, MOF-525-Fe, PCN-223-Fe and PCN-222-Fe all use FeTCPP as the organic linker and a Zr6 cluster as the connecting node, but the crystal structures are different. Both MOF-525-Fe and PCN-222-Fe exhibited great electrocatalytic CO2RR activity, while PCN-223-Fe exhibited good electrocatalytic ORR activity.13
MTCPP can also be linked with Zr clusters, forming multiple 3D MP/MPc-MOFs. MOFs with 3D topology have a higher active site density, resulting in enhanced current density and high catalytic activity. Ghalkhani et al. synthesized 3D-MOFs with 3D nanochannels using stable Zr6 clusters and CoTCPP. Compared with commercial Pt/C catalysts, the composite exhibits a smaller overpotential and higher current density for both the ORR and OER. The high surface area and large pores of 3D MOFs promote the diffusion of the reactant H2O and O2 throughout the framework, which facilitates the electrocatalytic reaction. In addition, due to the good stability and fine dispersion of 3D MOFs in solvents, the cobalt porphyrin immobilized in the 3D structure can better exhibit its catalytic performance.78
The density of active sites in MOFs can be adjusted with different node structures. Huang et al. reported a new 3D MOF material (named PCN-226) linked by MTCPP (M = Cu, Fe, Co) and Zr4+ cations as an efficient electrocatalyst for the ORR. PCN-226 has a zigzag Zr-oxide chain structure (Fig. 8e), which enhances the overall stability of the MOF and forms a compact packing structure of porphyrin molecules. Moreover, CV and LSV measurements showed that PCN-226-Co exhibited excellent ORR activity, showing an ORR onset potential of 0.83 V (vs. RHE), and a half-wave potential of 0.75 V (vs. RHE). The crystal structure of PCN-226-Cu (Fig. 8f) indicates a pore size of 7.2 Å × 4.8 Å when viewed along the b-axis, and a pore size of 5.4 Å × 4.2 Å when viewed along the c-axis. Theoretical calculations show that a suitable packing distance (∼7 Å) is beneficial for the adsorption of *O, *OH, and *OOH intermediates (Fig. 8g). The authors attributed the good catalytic performance to the fact that the chain structure of PCN-226 increases the density of active sites while providing better pathways for electron transfer.80
3.2 Covalent–organic frameworks (COFs)
COFs are formed through covalent bonds between organic monomers such as MP and MPc, which have been widely used in electrocatalytic reactions.81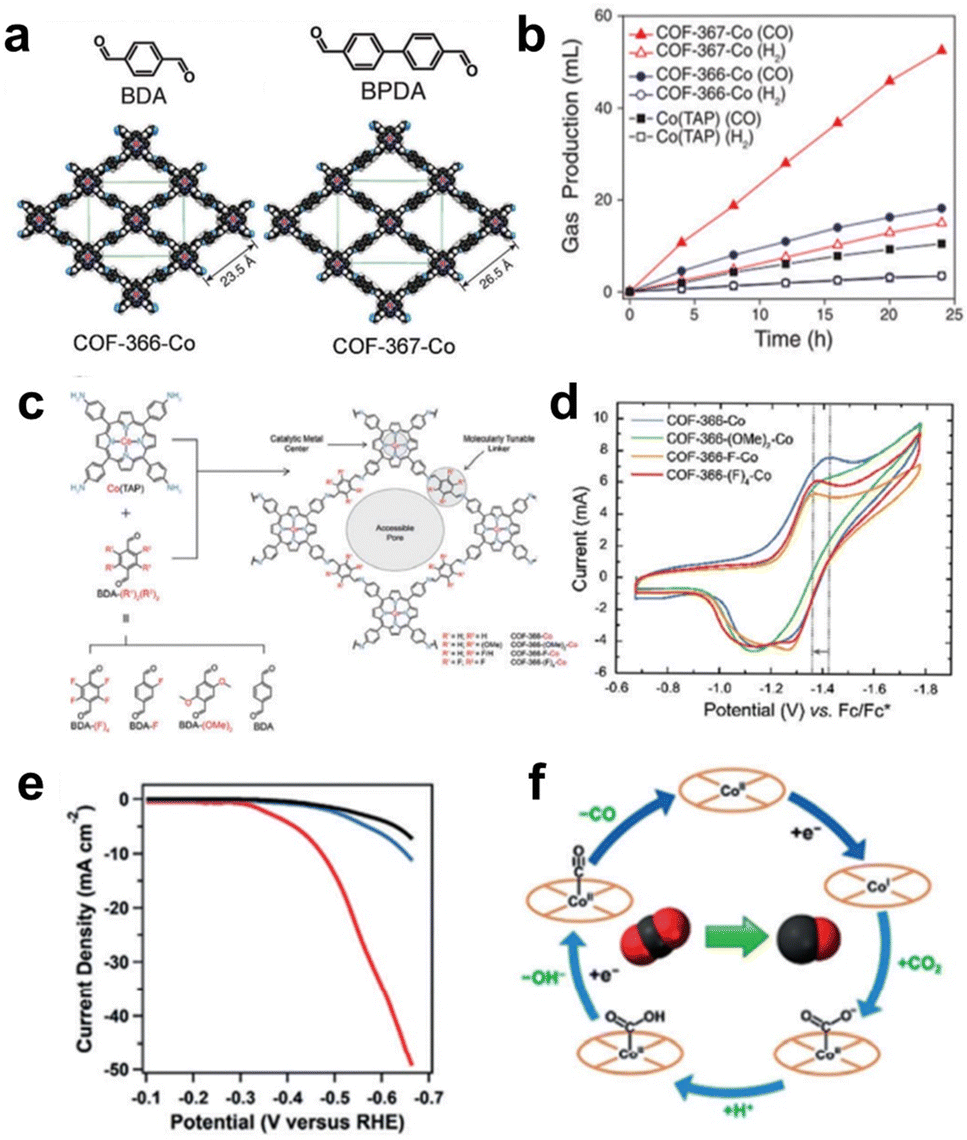 | ||
| Fig. 9 (a) Schematic of COF-366-Co and COF-367-Co. (b) The volume of CO produced by electrolysis. Reprinted with permission from ref. 82. Copyright 2015 American Association for the Advancement of Science. (c) Schematic illustration of the synthesis of COFs from CoPs with different substituents. (d) CV curves of CoP-COFs with different substituents. Reprinted with permission from ref. 83. Copyright 2018 American Chemical Society. (e) LSV curves of CoPc-PDQ-COF (red) and contrast catalysts. (f) The schematic diagram of the electrocatalytic CO2 reduction cycle by Co sites. Reprinted with permission from ref. 84. Copyright 2020 Wiley-VH. | ||
By modifying the network structure of COFs through electron absorbing/donating groups, the electronic properties of active sites can be adjusted. Yaghi et al. prepared a series of COFs with different BDA building block derivatives (BDA-(F)4, BDA-F, and BDA-(OMe)2) as organic linkers to study the structure–property relationship of COFs with substituent groups (Fig. 9c). The CV results in Fig. 9d show that the COF-366-F-Co catalyst modified with the strongest electron-withdrawing group shows the best CO2RR performance and the highest CO formation current density (65 mA mg−1) compared to other catalysts. However, COF-366(F)4-Co modified with the second strongest electron-withdrawing group (BDA-(F)4) exhibited the worst electrocatalytic performance. This may be due to the high hydrophobicity of the BDA-(F)4 group in COF-366-(F)4-Co, which can reduce the contact of the catalytic sites with the electrolyte. A series of aligned thin films of these COFs exhibited high CO selectivity (FECO = 87%) and high current density (65 mA mg−1) at −0.67 V (vs. RHE), far surpassing molecular catalysts in terms of selectivity and efficiency. The catalyst remained stable for 12 hours without obvious loss.83
By changing the structure of the organic building blocks connected with MP/MPc, the structure of COFs can be adjusted for optimizing the catalytic performance. A novel CoPc building block ((NH2)8CoPc) was designed to react with a robust phenazine linkage (4,5,9,10-pyrenediquinone, PDQ) for the construction of a 2D COF (CoPc-PDQ-COF).84 The FECO of CoPc-PDQ-COF-catalysed CO2 reduction was as high as 96% at −0.66 V (vs. RHE), and the TOF value was as high as 320![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, which is 32 times that of molecular CoPc (Fig. 9e). The excellent catalytic performance is attributed to the specific π structure of CoPc-PDQ-COF, which consists of complete π-conjugation along the x and y axes and π-conduction along the z-direction, so electrons are more easily transported to the catalytic sites. By studying the overall proton/electron transfer pathway at Co sites during the CO2RR (Fig. 9f), it can be concluded that the change from high-valence CoII to low-valence CoI is the first step of electron injection. Subsequently, electron transfer occurs from CoI to the adsorbed CO2 molecule, which then undergoes proton transfer to form the intermediate COOH*. Finally, CO is produced. Jiang et al. used 2,3,9,10,16,17,23,24-octacarboxyphthalocyanine tetraanhy-dride Co(TAPc) and 1,3,5,7-tetra(4-aminophenyl)adamantine (TAPA) to prepare a 3D COF (CoPc-PI-COF-3). CoPc-PI-COF-3 doped carbon black was used to fabricate an electrocatalytic cathode for the CO2RR in KHCO3 solution, and a current density of 31.7 mA cm−2 was obtained at −0.90 V (vs. RHE). According to powder X-ray diffraction and gas adsorption analysis, CoPc-PI-COF-3 has an 3D interpenetrating network structure. The unique 3D porous structure of CoPc-PI-COF-3 ensures that 32.7% of CoPc units serve as active centres.85
000, which is 32 times that of molecular CoPc (Fig. 9e). The excellent catalytic performance is attributed to the specific π structure of CoPc-PDQ-COF, which consists of complete π-conjugation along the x and y axes and π-conduction along the z-direction, so electrons are more easily transported to the catalytic sites. By studying the overall proton/electron transfer pathway at Co sites during the CO2RR (Fig. 9f), it can be concluded that the change from high-valence CoII to low-valence CoI is the first step of electron injection. Subsequently, electron transfer occurs from CoI to the adsorbed CO2 molecule, which then undergoes proton transfer to form the intermediate COOH*. Finally, CO is produced. Jiang et al. used 2,3,9,10,16,17,23,24-octacarboxyphthalocyanine tetraanhy-dride Co(TAPc) and 1,3,5,7-tetra(4-aminophenyl)adamantine (TAPA) to prepare a 3D COF (CoPc-PI-COF-3). CoPc-PI-COF-3 doped carbon black was used to fabricate an electrocatalytic cathode for the CO2RR in KHCO3 solution, and a current density of 31.7 mA cm−2 was obtained at −0.90 V (vs. RHE). According to powder X-ray diffraction and gas adsorption analysis, CoPc-PI-COF-3 has an 3D interpenetrating network structure. The unique 3D porous structure of CoPc-PI-COF-3 ensures that 32.7% of CoPc units serve as active centres.85
The density of active sites can also be optimized with different structures. Cao et al. synthesized a porous 3D cobalt porphyrin COF (3D-por(Co/H)COF) for catalytic CO2 reduction by using tetrakis(4-formylphenyl)methane (TFPM) mixed with 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (Co-TAPP) and 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAPP) through a Schiff base condensation reaction. Since the CoP building blocks are spatially separated in the porous 3D-Por(Co/H)-COF, all Co active sites in the framework can fully react and thus exhibit high activity for the CO2RR. The FECO at −0.6 V (vs. RHE) is 92.4%, and the TOF is 4610 h−1.86
OER and HER. Graphdiyne (GDY) with alkyne bonds has a conjugated structure, which makes it a suitable platform for bonding with MP and MPc to construct the complex catalysts. Chen et al. synthesized 2D COF nanosheets (CoPDY) as electrocatalysts for the OER and HER in alkaline solution using GDY and Co porphyrin. The stable Co–N4 centres, distributed uniformly on the CoPDY plane, serve as the active sites for the reaction. The commercial copper foam (CF) not only serves as a catalyst for generating Co-PDY but also serves as a robust porous conductive substrate that can further enhance the electrocatalytic activity for the OER. Co-PDY/CF exhibits excellent catalytic activity for the OER (an overpotential of 270 mV at 10 mA cm−2 and a low Tafel slope of 99 mV dec−1) with long-term stability and durability. In addition, Co-PDY/CF exhibits high electrocatalytic performance for the HER.87
Likewise, for the HER, a higher surface area, allowing easier access to catalytically active sites, is beneficial for catalytic reactions. Villagran and co-workers reported a CoTcPP-based COF for the electrocatalytic HER in strongly acidic media. A cathodic current density of 10 mA cm−2 was reached, and the overpotentials of COF and monomer molecule CoTcPP were 0.475 V and 0.666 V, respectively. Compared with the monomeric CoTcPP molecule (3.44 m3 g−1), the crystalline polymer has a larger surface area (441.74 m3 g−1). The increase in exposed active sites and porous channels enhances the catalytic HER performance. In addition, the authors proposed that increasing the conjugated system can enhance the electrocatalytic efficiency. The COF also showed good resistance to HER electrolysis in the presence of acid and remained active for 10 h.88
4 Unravelling the catalytic mechanism with MP and MPc model systems
The design of high-efficiency electrocatalysts requires an in-depth understanding of the catalytic mechanisms and structure–activity relationships of the electrocatalysts. To gain a comprehensive understanding of such catalytic processes, in situ characterization techniques have been performed to investigate model catalytic systems based on MP and MPc.894.1 Electrochemical Raman spectroscopy
Raman spectroscopy (RS), which is based on the changes in the interaction of incident radiation with the vibrations of molecular bonds, can provide valuable information on the adsorption states and vibrations of catalytic species on electrode surfaces, allowing for the identification of active sites and reaction intermediates involved in catalytic reactions.90RS relies on the inelastic scattering of a laser to detect the vibrations of molecules with varying polarizability in electrochemical experiments. RS can be performed in electrochemical cells to probe surface species at the electrode–electrolyte interface under operating conditions. To enhance the strength of the Raman signal of probed molecules, specific Raman techniques, including surface-enhanced Raman spectroscopy (SERS), tip-enhanced Raman spectroscopy (TERS) and shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS), have been developed.90
RS in vacuum can be used to identify adsorbed species. Nguyen et al. studied the adsorption of O2 on CoPc assembled on Ag(111) using ultra-high vacuum TERS (UHV-TERS). In situ UHV-TERS analysis combined with scanning tunnelling microscopy (STM) imaging, 18O2 isotope experiments, and DFT simulations revealed that there are two oxygen-adsorbed species on the electrode surface: O2/CoPc/Ag(111) and O/CoPc/Ag(111). O2/CoPc/Ag(111) shows a characteristic band at 1151 cm−1 (18O–18O), while O/CoPc/Ag(111) shows vibrations at 661 cm−1 (Co–16O) and 623 cm−1 (Co–18O).91
By combining RS and electrochemical measurements, the in situ observation of species evolution during catalysis can be achieved. Dey and colleagues investigated the iron porphyrin-catalysed ORR using rotating disk electrochemistry coupled to surface enhanced resonance Raman spectroscopy (SERRS-RDE). SERRS-RDE was performed with an electrode held at −0.5 V in an air-saturated pH 7 buffer. The collected data showed a clear increase in the intensity of the peaks at 1369 cm−1 and 1565 cm−1 compared to the data obtained without O2, indicating the formation of low-spin (LS) FeIII species. Peaks were observed at 1352 cm−1 and 1540 cm−1, which were attributed to reduced FeII species. Increased peak intensities were also observed at 1371 cm−1 and 1571 cm−1, suggesting the formation of high-valent FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. Therefore, FeII species, high-spin (HS) FeIII species, LS FeII–OOH species, and FeIVO species can be observed during the catalyst-catalysed steady-state ORR. Furthermore, the data at variable potentials showed that the populations of HS FeII species, LS FeII–OOH species, and FeIVO species increased and the populations of quiescent HS FeIII species decreased as the potential decreased. Therefore, during the catalytic process, HS FeIII would be reduced to the catalytically active FeII state.92
O species. Therefore, FeII species, high-spin (HS) FeIII species, LS FeII–OOH species, and FeIVO species can be observed during the catalyst-catalysed steady-state ORR. Furthermore, the data at variable potentials showed that the populations of HS FeII species, LS FeII–OOH species, and FeIVO species increased and the populations of quiescent HS FeIII species decreased as the potential decreased. Therefore, during the catalytic process, HS FeIII would be reduced to the catalytically active FeII state.92
TERS can also be used to study the behaviour of MPc catalysts during electrocatalytic processes. Chen et al. used in situ electrochemical TERS (EC-TERS) to monitor the structural changes of FePc on Au(111) during the ORR. The in situ EC-TERS results showed that the intensity of the 593 cm−1 (α carbon), 682 cm−1 (isoindole nitrogen) and 751 cm−1 (bridge nitrogen) bands increased significantly when the potential was decreased from 0.7 V to 0.4 V (vs. RHE), which suggests that the nonplanar geometry formed by FePc molecules under short-term cathodic polarization is recoverable. When the potential was kept at 0.4 V for a long time (15 min), the recoverable spectra became irreversible. The 593 cm−1 and 751 cm−1 bands became almost negligible, and two new bands appeared at 724 and 795 cm−1. There was also a spectral change in the band in the 1083–1542 cm−1 region. These changes are due to the demetallation of FePc to H2Pc during the ORR process. Electrochemical measurements revealed that the loss of Fe sites after permanent dementalization resulted in reduced ORR activity.93
4.2 Scanning probe microscopy (SPM)
SPM techniques provide atomic-scale surface structure information in vacuum, air, and solution, and enable the observation of surface/interface processes with high spatial or temporal resolution, making them suitable techniques for investigating electrocatalytic mechanisms. SPM techniques include STM, atomic force microscopy, scanning electrochemical microscopy, scanning ion conductance microscopy, and scanning electrochemical cell microscopy. Notably, EC-STM, as one of the commonly used SPM techniques, can provide atomic-resolution surface information under electrochemical conditions, making it ideal for studying the transformation of species during electrocatalysis and the structure–activity relationship of electrocatalysts.89,94The adsorption of reactants on catalytic active sites is the initial step in electrocatalytic reactions and affects the subsequent electrocatalytic processes. STM can be used to characterize the adsorption of reactants on active sites. Hipps et al. reported the adsorption of molecular oxygen on cobalt octaethylporphyrin (CoOEP) assembled on HOPG during the ORR. They found that the contrast of the molecules became darker after the CoOEP molecules adsorbed O2.95 Speller et al. demonstrated the behaviour of MnIIItetradecyl-chlorinated porphyrin on Au(111) using tetradecane solvent as the liquid medium.96 The authors conducted the experiments in O2-saturated tetradecane solvent. Compared to the case without O2 saturation, the number of bright molecules increases, which indicates that the appearance of bright spots is caused by O2 adsorption on the metal centre.
Wan et al. investigated the adsorption of OH− on CoTPP in the OER. The CoTPP catalyst showed better OER activity when the electrolyte alkalinity was increased in CV measurements.97 EC-STM measurements of CoTPP were performed in acidic, alkaline, and neutral solutions (Fig. 10a–c), and the cross-sectional profiles along the white line in Fig. 10a–c are shown in Fig. 10d. In an acidic electrolyte, each CoTPP molecule can be distinguished as a bright dot that corresponds to the cobalt centre (Fig. 10a and d). However, in an alkaline solution, CoTPP can be identified as a species with two-fold symmetry containing two bright spots (Fig. 10b and d). The coexistence of the two species was observed in a neutral solution (Fig. 10c and d). The morphological differences revealed that CoTPP-OH− species were more likely to form in higher pH solutions, which was further confirmed using UV-vis absorption spectra. At the same time, the transformation of the reaction species *OH− and the product species *O2 is only observed in alkaline environments. This result shows that in the catalytic OER, pH affects the adsorption of reactants, thereby affecting the entire reaction process.
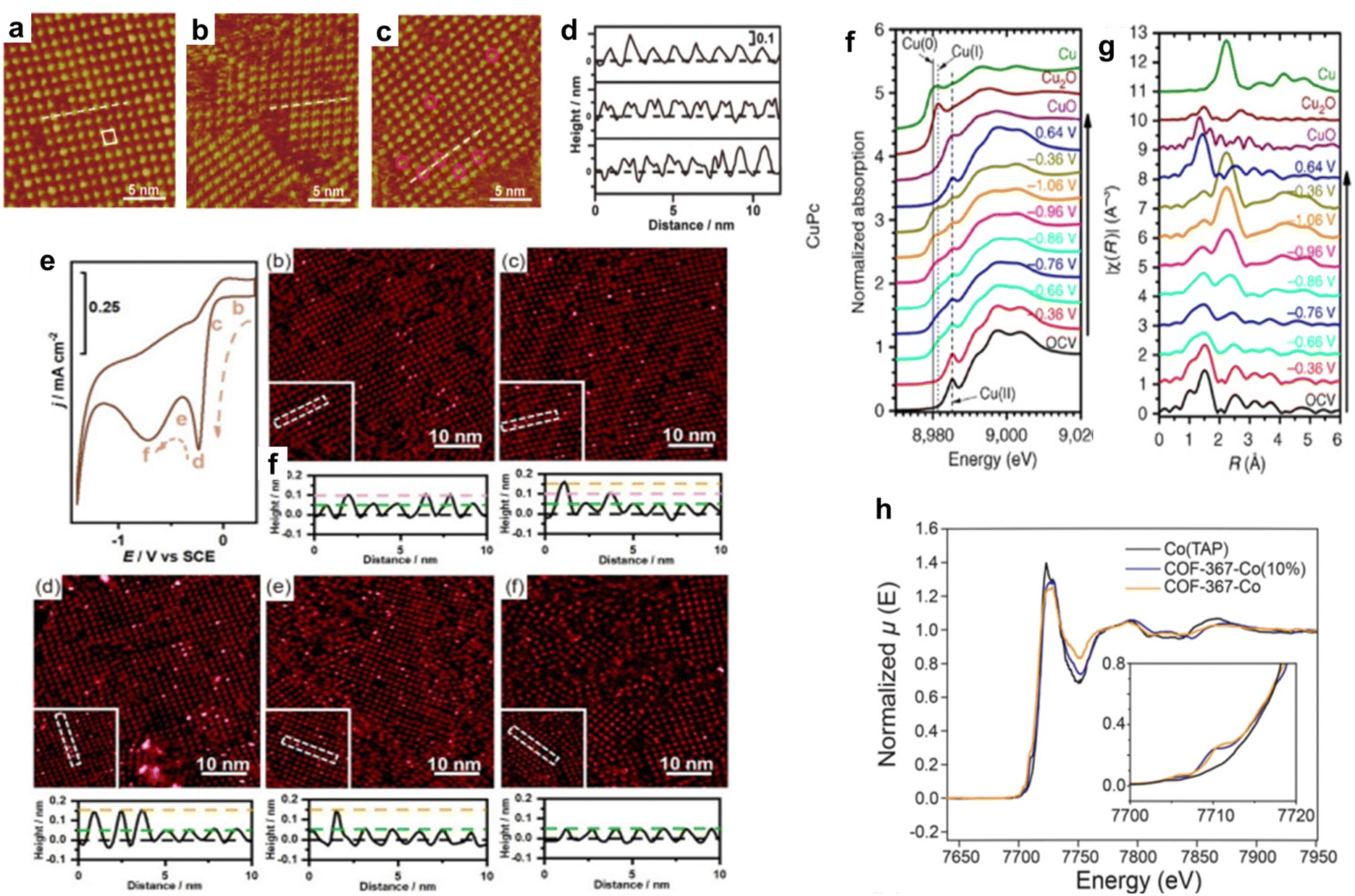 | ||
| Fig. 10 (a)–(c) STM images of the CoTPP adlayer on the Au(111) substrate in 0.1 mol L−1 HClO4 (a) 0.1 mol L−1 KOH (b) and 0.1 mol L−1 NaClO4 (c). (d) Cross-section profiles along the white lines in a to c (from top to bottom). Reprinted with permission from ref. 97. Copyright 2019 American Chemical Society. (e) CV curve of the CoTMPP-modified Au electrode-catalysed ORR and sequential STM images under different potentials. Reprinted with permission from ref. 101. Copyright 2023 Springer. (f) In situ Cu K-edge X-ray absorption near-edge structure (XANES) spectra of CuPc. (g) Fourier transformed Cu K-edge EXAFS spectra for CuPc. Reprinted with permission from ref. 104. Copyright 2018 Nature Publishing Group Springer. (h) X-ray absorption spectra of COF-367-Co (orange line), COF-367-Co (10%) (blue line), and Co(TAP) (black line). The inset shows the pre-edge regimes of the same spectra. Reprinted with permission from ref. 82. Copyright 2015 American Association for the Advancement of Science. | ||
Wan et al. also used EC-STM to observe the high-contrast CoPc-CO2 species formed by the adsorption of the reactant CO2 on CoPc in the CO2RR, and further confirmed the formation of CoPc-CO2 through theoretical simulation.98 It was further found that the adsorption ratio of CO2-adsorbed species increased when metal cations (Mg2+) were introduced into the solution, which is due to the synergistic adsorption of Mg2+ with CO2.99
In situ ECSTM can be used to investigate the electrocatalytic process at the molecular scale, which is beneficial for understanding the reaction mechanism. For instance, the FePc-catalysed ORR was investigated by ECSTM at the molecular scale. The adsorbed species exhibited high contrast in the O2-saturated electrolyte, corresponding to the FePc-O2 complex (E = 700 mV vs. SCE), and it could be reversibly transformed to FePc with low contrast during the ORR (E scans from 350 to 50 mV vs. SCE).100
In situ ECSTM was also used to study the electrocatalytic process and dynamic surface changes of catalyst molecules under different pH environments. Wang et al. used ECSTM to observe the process of cobalt porphyrin-catalyzed ORR in neutral and acidic electrolytes. The electrochemical results in a neutral environment revealed two stages of the ORR, including the formation of H2O2 and the further reduction of H2O2 at different potentials. In an acidic environment, the ORR only involves the generation process of H2O2. The authors observed the electrocatalytic process using in situ ECSTM experiments in a neutral environment, in which Co, Co–O2 and Co–OOH− species were interconverted at different potentials in the reaction (Fig. 10e).101In situ experiments in acidic environments can only observe the transformation process of Co, and Co–O2 species.
In situ ECSTM has been used to study the process of the CO2RR catalysed by MPc. An ordered adlayer of CoPc was prepared on a Au(111) substrate. In a CO2-saturated electrolyte, the proportion of high-contrast absorbed species (CoPc-CO2) was found to be larger than that in Ar and air environments using ECSTM. CoPc-CO2 formation was further confirmed by theoretical simulations. The transformation between high-contrast CoPc-CO2 (−0.8 V vs. SCE) and low-contrast CoPc (−1.25 V vs. SCE) was studied using ECSTM at different potentials. In potential step experiments, dynamic information about the initial stages of the reaction was obtained, including the reduction of CoPc and the CO2 binding step, and the RDS was determined to be the latter by calculating the rate constants.98
4.3 X-Ray absorption spectroscopy
X-Ray absorption spectroscopy (XAS) is a spectroscopic technique that uses signal changes before and after X-ray incidence to reveal the element composition, electronic state and microstructure of the analysed material. The intermediates in electrocatalytic reactions can be revealed by in situ XAS, which is beneficial for understanding the reaction mechanism.102In situ XAS is suitable for the identification of active sites in electrocatalysts. Wang et al. developed a transition metal-based zinc-porphyrin complex for the conversion of CO2 to CO with an FE of up to 95%. In situ XAS studies showed that the metal centre does not undergo redox reactions in the entire potential range. Combining XAS results and electrochemical measurements, the porphyrin ligand was found to be the active site for the CO2RR.103
In situ XAS is also useful for probing changes in the structure and oxidation state of electrocatalysts. Wang et al. studied the application of copper(II) phthalocyanine (CuPc) in the electrocatalytic CO2RR. At −1.06 V (vs. RHE), CuPc catalyses the CO2RR to methane with an FE of 66%. The results of X-ray absorption near-edge structure (XANES) (Fig. 10f) and extended X-ray absorption fine structure (EXAFS) (Fig. 10g) revealed the complex structural and oxidation state changes of CuPc during the CO2RR. Under the initial conditions of XANES, a characteristic peak of CuII appeared at ∼8985 eV. When the applied potential decreased to −0.86 V (vs. RHE), a small absorption peak corresponding to CuI appeared at ∼8981 eV and Cu0 started to dominate the XANES spectrum when the applied potential further decreased to −1.06 V (vs. RHE). This is consistent with the presence of the characteristic metallic Cu–Cu bond in the corresponding EXAFS spectrum. The CuII peak remained at all applied potentials in the XANES spectra, and peak features of CuPc persisted in the EXAFS spectra, indicating that CuII was not totally converted into lower oxidation states during the CO2RR. Interestingly, when the applied potential was switched back to positive 0.64 V (vs. RHE), the Cu0 peak of the CuPc electrocatalyst disappeared, and the XANES spectrum almost fully recovered, suggesting that the state changes of CuPc were reversible.104
In situ XAS was explored to study the influence of the coordination environment on the electronic structure of active sites. Chang et al. synthesized a cobalt porphyrin-based COF catalyst for the electrocatalytic CO2RR. The catalyst exhibited a high FE and TON and showed almost no degradation within 24 hours. The authors used XAS to assess the effect of the surrounding COF on the electronic structure of the catalytic active centre in cobalt porphyrin. The results showed that the cobalt K-edge XAS spectra of cobalt porphyrin and COF exhibited similar line sizes, shapes, and positions, consistent with the formal CoII oxidation states of all samples (Fig. 10h). The COF catalysts also exhibited additional pre-edge features not present in molecular cobalt porphyrins (Fig. 10h, inset). The additional pre-edge peaks indicated the direct electronic modulation of Co coupled into the extended lattice, which suggests that the chemical environment in the COF can influence the electronic properties of the active centre in porphyrin molecules.82
4.4 Mössbauer spectroscopy
Mössbauer spectroscopy is widely used to investigate the internal energy levels of atoms in samples. Due to its high chemical sensitivity, Mössbauer spectroscopy can provide information about the valence state, spin state and coordination environment of the active sites in electrocatalysis, which can offer precise information about the electronic structure of the metal centre of M–N4 SACs. It is mainly used to study three isotopes of 57Fe, 119Sn, and 151Eu, which can achieve the Mössbauer effect at room temperature rather than low temperatures.In situ Mössbauer spectroscopy can be used to monitor the spin state of the central metal in MPs during the catalytic process. Kramm et al. prepared iron porphyrin-based catalysts and investigated their catalytic ORR active sites by in situ Mössbauer spectroscopy.105 The authors showed that within the potential interval (from 0.9 V to 0.2 V) (vs. RHE) over which the ORR occurs, the iron signature of the active site changes with the applied potential. When the potential ranges from 0.9 V (vs. RHE) to 0.75 V (vs. RHE), the D1 peak intensity decreases in the fitted Mössbauer spectrum of the catalyst, while the D3 peak intensity appears or increases. D2 sites appear only at potentials of 0.6 V (vs. RHE) or less. The authors attributed the D3 peak to the iron high-spin configuration site responsible for the direct reduction of oxygen. Site D2 is associated with the further reduction of hydrogen peroxide, thereby contributing to the overall reduction of oxygen to water via a 4-electron reduction pathway. DFT calculation and thermodynamic properties indicate that the D3 site may be related to the iron-based fivefold FeN4 group with pyridine N4 coordination, while site D2 is similar to an iron-based intermediate spin FeN4-X, where X is a weakly binding anion.106
Conclusions and perspectives
In this review, we have systematically summarized the applications of MP, MPc and their derivatives as model systems in electrocatalytic reactions. We sequentially introduced the performance and catalytic behaviour of soluble and insoluble MP and MPc catalytic systems in electrocatalysis, the optimization and modification strategies of MP and MPc, and the application of MP and MPc motif-based framework materials in electrocatalysis. In addition, the application of advanced in situ characterization techniques for unravelling the interfacial catalytic processes and mechanisms of MP and MPc was discussed. This review provides important insights into understanding the catalytic mechanism of single-atom catalysts with M–N4 sites and is beneficial for bridging the material gap in the field of electrocatalysis.The research on the electrocatalysis with MP and MPc catalysts has attracted extensive attention and has great development prospects. First, the synthesis of some MP and MPc molecules with specific structural characteristics is highly desirable but challenging. For example, MPs/MPcs with different numbers and types of coordination atoms around the metal centre (in the first coordination sphere) can be used to reveal the influence of the coordination environment on active sites. In addition, the atoms in the second coordination sphere also have a significant impact on the catalytic performance of MP and MPc. The recent works have also shown the synergistic performance of multi-atomic catalysis systems. The design and synthesis of the corresponding molecular model catalysts could provide valuable information about the mechanisms and structure–performance relationship. Second, the development of in situ and operando characterization techniques for understanding the catalytic process of MP and MPc catalysts is another important direction. Different in situ techniques can reveal the dynamic chemical and structural evolution of the catalytic systems during the reaction process, which is very important for a comprehensive understanding of the reaction mechanism. The combination of spectroscopic techniques for obtaining chemical information, electrochemical techniques for obtaining catalytic properties, and imaging techniques for obtaining structural characterization is crucial. Meanwhile, improving the spatial resolution of the characterization techniques is also important for investigating the intrinsic catalytic behavior of single active sites in a local environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1501002), the National Natural Science Foundation of China (22132007, 21972147, and 22302208), and the National Postdoctoral Program for Innovative Talents (BX20220307) of the Chinese Postdoctoral Science Foundation.References
- C. Costentin, H. Dridi and J. M. Savéant, Molecular Catalysis of O2 Reduction by Iron Porphyrins in Water: Heterogeneous Versus Homogeneous Pathways, J. Am. Chem. Soc., 2015, 137, 13535–13544 CrossRef CAS PubMed.
- W. Schöfberger, F. Faschinger, S. Chattopadhyay, S. Bhakta, B. Mondal, J. A. A. W. Elemans, S. Müllegger, M. S. Stefano Tebi, R. Koch, F. Klappenberger, M. Paszkiewicz, J. V. Barth, E. Rauls, H. Aldahhak, W. Gero Schmidt and A. Deyet, A Bifunctional Electrocatalyst for Oxygen Evolution and Oxygen Reductio Reactions in Water, Angew. Chem., Int. Ed., 2016, 55, 2350–2355 CrossRef PubMed.
- Z. Liang, H. Y. Wang, H. Q. Zheng, W. Zhang and R. Cao, Porphyrin-Based Frameworks for Oxygen Electrocatalysis and Catalytic Reduction of Carbon Dioxide, Chem. Soc. Rev., 2021, 50, 2540 RSC.
- W. Zhang, W. Lai and R. Cao, Energy-Related Small Molecule Activation Reactions: Oxygen Reduction and Hydrogen and Oxygen Evolution Reactions Catalysed by Porphyrin-and Corrole-Based Systems, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS.
- B. Mondal, A. Rana, P. Sen and A. Dey, Intermediates Involved in The 2e−/2H+ Reduction of CO2 to CO by Iron(0) Porphyrin, J. Am. Chem. Soc., 2015, 137, 11214–11217 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- T. Zhang, C. Wang, S. B. Liu, J. L. Wang and W. B. Lin, A Biomimetic Copper Water Oxidation Catalyst with Low Over-Potential, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- B. B. Beyene and C. H. Hung, Recent Progress on Metalloporphyrin-Based Hydrogen Evolution Catalysis, Coord. Chem. Rev., 2020, 410, 213234 CrossRef CAS.
- Y. Han, H. Fang, H. Jing, H. Sun, H. Lei, W. Lai and R. Cao, Singly Versus Doubly Reduced Nickel Porphyrins for Proton Reduction: Experimental and Theoretical Evidence for a Homolytic Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2016, 55, 5457–5462 CrossRef CAS PubMed.
- S. Bhunia, A. Rana, S. Hematian, K. D. Karlin and A. Dey, Proton Relay in Iron Porphyrins for Hydrogen Evolution Reaction, Inorg. Chem., 2021, 60, 13876–13887 CrossRef CAS PubMed.
- M. B. Gawande, K. Ariga and Y. Yamauchi, Single-Atom Catalysts, Small, 2021, 17, 2101584 CrossRef CAS PubMed.
- Q. Zhang and J. Guan, Applications of single-atom catalysts, Nano Res., 2022, 15, 38–70 CrossRef CAS.
- J. Han, P. An, S. Liu, X. Zhang, D. Wang, Y. Yuan, J. Guo, X. Qiu, K. Hou, L. Shi, Y. Zhang, S. Zhao, C. Long and Z. Tang, Reordering d orbital energies of single-site catalysts for CO2 electroreduction, Angew. Chem., Int. Ed., 2019, 58, 12711–12716 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Heterogeneous Single-Atom Catalysts for Electrochemical CO2 Reduction Reaction, Adv. Mater., 2020, 32, 2001848 CrossRef CAS.
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbrüster, A. Knop-Gericke and R. Schlögl, Free-atom-like d states in Single-atom Alloy Catalysts, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed.
- S. Saini, J. H. Stenlid and F. Abild-Pedersen, Electronic Structure Factors and the Importance of Adsorbate Effects in Chemisorption on Surface Alloys, npj Comput. Mater., 2022, 8, 163 CrossRef CAS.
- L. Zhong and S. Li, Unconventional Oxygen Reduction Reaction Mechanism and Scaling Relation on Single-Atom Catalysts, ACS Catal., 2020, 10, 4313–4318 CrossRef CAS.
- Y. Chen, Z. Li, Y. Zhu, D. Sun, X. Liu, L. Xu and Y. Tang, Atomic Fe Dispersed on N-doped Carbon Hollow Nanospheres for High-Efficiency Electrocatalytic Oxygen Reduction, Adv. Mater., 2019, 31, 1806312 CrossRef.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, Electrochemical Reduction of CO2 Catalyzed by Fe-N-C Materials: a structure-selectivity study, ACS Catal., 2017, 7, 1520 CrossRef CAS.
- L. Feng, K. Y. Wang, E. Joseph and H. C. Zhou, Catalytic Porphyrin Framework Compounds, Trends Chem., 2020, 2, 555 CrossRef CAS.
- J. Peto, T. Ollar, P. Vancso, Z. I. Popov, G. Z. Magda, G. Dobrik, C. Hwang, P. B. Sorokin and L. Tapaszto, Spontaneous Doping of the Basal Plane of Mos2 Single Layers Through Oxygen Substitution Under Ambient Conditions, Nat. Chem., 2018, 10, 1246–1251 CrossRef CAS.
- S. Samanta, K. Mittra, K. Sengupta, S. Chatterjee and A. Dey, Second Sphere Control of Redox Catalysis: Selective Reduction of O2 To O2− or H2O by an Iron Porphyrin Catalyst, Inorg. Chem., 2013, 52, 1443–1453 CrossRef CAS PubMed.
- B. D. Matson, C. T. Carver, A. V. Ruden, J. Y. Yang, S. Raugei and J. M. Mayer, Distant Protonated Pyridine Groups in Water-Soluble Iron Porphyrin Electrocatalysts Promote Selective Oxygen Reduction to Water, Chem. Commun., 2012, 48, 11100–11102 RSC.
- X. P. Zhang, A. Chandra, Y. M. Lee, R. Cao, K. Ray and W. Nam, Transition Metal-Mediated O–O Bond Formation and Activation in Chemistry and Biology, Chem. Soc. Rev., 2021, 50, 4804–4811 RSC.
- M. Ardakani, P. Rahimi, H. Dehghani, P. Karami, H. Zare and S. Karami, Electrocatalytic Reduction of Dioxygen on the Surface of Glassy Carbon Electrodes Modified with Cobalt Porphyrin Complexes, Electroanalysis, 2007, 19, 2258–2263 CrossRef CAS.
- S. Amanullah, P. K. Das, S. Samanta and A. Dey, Tuning the Thermodynamic Onset Potential of Electrocatalytic O2 Reduction Reaction by Synthetic Iron–Porphyrin Complexes, Chem. Commun., 2015, 51, 10010–10013 RSC.
- L. Xie, X. P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U. P. Apfel and R. Cao, Enzyme-inspired Iron Porphyrins for Improved Electrocatalytic Oxygen Reduction and Evolution Reactions, Angew. Chem., Int. Ed., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- H. Jia, Y. Yao, Y. Gao, D. Lu and P. Du, Pyrolyzed Cobalt Porphyrin-Based Conjugated Mesoporous Polymers as Bifunctional Catalysts for Hydrogen Production and Oxygen Evolution in Water, Chem. Commun., 2016, 52, 13483–13486 RSC.
- S. Bhunia, K. Bhunia, B. C. Patra, S. K. Das, D. Pradhan, A. Bhaumik, A. Pradhan and S. Bhattacharya, Efficacious Electrochemical Oxygen Evolution From a Novel Co(II) Porphyrin/Pyrene-Based Conjugated Microporous Polymer, ACS Appl. Mater. Interfaces, 2019, 11, 1520–1528 CrossRef CAS.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Molecular Catalysts for Water Oxidation, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Molecular Electrocatalysts for the Oxygen Reduction Reaction, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS.
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts, Chem. Rev., 2018, 118, 2340–2391 CrossRef CAS PubMed.
- Y. O. Su, T. Kuwana and S. M. Chen, Electrocatalysis of Oxygen Reduction by Water-Soluble Iron Porphyrins. Thermodynamic and Kinetic Advantage Studies, J. Electroanal. Chem. Interfacial Electrochem., 1990, 288, 177–195 CrossRef CAS.
- R. J. H. Chan, Y. O. Su and T. Kuwana, Electrocatalysis of Oxygen Reduction to Hydrogen Peroxide Conversion, Inorg. Chem., 1985, 24, 3777–3784 CrossRef CAS.
- N. Kobayashi and Y. Nishiyama, Catalytic Electroreduction of Molecular Oxygen Using Iron or Cobalt 4,4′,4′,4′-Tetracarboxyphthalocyanine, J. Phys. Chem. C, 1985, 89, 1167–1170 CrossRef CAS.
- T. Honda, T. Kojima and S. Fukuzumi, Proton-Coupled Electron-Transfer Reduction of Dioxygen Catalysed by a Saddle-Distorted Cobalt Phthalocyanine, J. Am. Chem. Soc., 2012, 134, 4196–4206 CrossRef CAS.
- M. Abdinejad, K. Tang, C. Dao, S. Saedy and T. Burdyny, Immobilization Strategies for Porphyrin-Based Molecular Catalysts for the Electroreduction of CO2, J. Mater. Chem. A, 2022, 10, 7626 RSC.
- X. M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials, Angew. Chem., Int. Ed., 2017, 129, 6568–6572 CrossRef.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Highly Selective and Active CO2 Reduction Electrocatalysts Based on Cobalt Phthalocyanine/carbon Nanotube Hybrid Structures, Nat. Commun., 2017, 8, 14675 CrossRef.
- J. Choi, P. Wagner, R. Jalili, J. Kim, D. R. MacFarlane, G. G. Wallace and D. L. Officer, A Porphyrin/Graphene Framework: a Highly Efficient and Robust Electrocatalyst for Carbon Dioxide Reduction, Adv. Energy Mater., 2018, 8, 1801280 CrossRef.
- A. Maurin and M. Robert, Catalytic CO2-To-CO Conversion in Water by Covalently Functionalized Carbon Nanotubes with a Molecular Iron Catalyst, Chem. Commun., 2016, 52, 12084–12087 RSC.
- X. M. Hu, Z. Salmi, M. Lillethorup, E. B. Pedersen, M. Robert, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Controlled Electropolymerisation of a Carbazole-functionalised Iron Porphyrin Electrocatalyst for CO2 Reduction, Chem. Commun., 2016, 52, 5864–5867 RSC.
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y. F. Han, Cobalt Phthalocyanine Coordinated to Pyridine-Functionalized Carbon Nanotubes with Enhanced CO2 Electroreduction, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS.
- H. Qin, Y. Wang, B. Wang, X. Duan, H. Lei, X. Zhang, H. Zheng, W. Zhang and R. Cao, Cobalt Porphyrins Supported on Carbon Nanotubes as Model Catalysts of Metal-N4/C Sites for Oxygen Electrocatalysis, J. Energy Chem., 2021, 53, 77–81 CrossRef CAS.
- A. Han, H. Jia, H. Ma, S. Ye, H. Wu, H. Lei, Y. Han, R. Cao and P. Du, Cobalt Porphyrin Electrode Films for Electrocatalytic Water Oxidation, Phys. Chem. Chem. Phys., 2014, 16, 11209 Search PubMed.
- N. Heppe, C. Gallenkamp, S. Paul, N. Segura-Salas, N. von Rhein, B. Kaiser, W. Jaegermann, A. Jafari, I. Sergueev, V. Krewald and U. I. Kramm, Substituent Effects in Iron Porphyrin Catalysts for the Hydrogen Evolution Reaction, Chem. – Eur. J., 2023, 29, e202202465 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J. M. Savéant, Dissection of Electronic Substituent Effects in Multielectron-Multistep Molecular Catalysis. Electrochemical CO2-to-CO Conversion Catalysed by Iron Porphyrins, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- S. Sinha, M. S. Aaron, J. Blagojevic and J. J. Warren, Electrocatalytic Dioxygen Reduction by Carbon Electrodes Noncovalently Modified with Iron Porphyrin Complexes: Enhancements from a Single Proton Relay, Chem. – Eur. J., 2015, 21, 18072–18075 CrossRef CAS PubMed.
- Y. Yamada, Y. Fukunishi, S. Yamazakib and S. Fukuzumi, Hydrogen Peroxide as Sustainable Fuel: Electrocatalysts for Production with a Solar Cell and Decomposition with a Fuel Cell, Chem. Commun., 2010, 46, 7334–7336 RSC.
- D. J. Graham and D. G. Nocera, Electrocatalytic H2 Evolution by Proton-Gated Hangman Iron Porphyrins, Organometallics, 2014, 33, 4994–5001 CrossRef CAS.
- J. D. Baran, H. Grönbeck and A. Hellman, Analysis of Porphyrines as Catalysts for Electrochemical Reduction of O2 and Oxidation of H2O, J. Am. Chem. Soc., 2014, 136, 1320–1326 CrossRef CAS.
- X. Chen, Y. Dai, H. Zhang and X. Zhao, Revealing the Steric Effects of Cobalt Porphyrin on the Selectivity of Oxygen Reduction Reaction, Colloids Surf., A, 2023, 663, 131091 CrossRef CAS.
- X. Guo, N. Wang, X. Li, Z. Zhang, J. Zhao, W. Ren, S. Ding, G. Xu, J. Li, U. Apfel, W. Zhang and R. Cao, Homolytic Versus Heterolytic Hydrogen Evolution Reaction Steered by a Steric Effect, Angew. Chem., Int. Ed., 2020, 59, 8941–8946 CrossRef CAS PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. G. Kim, Q. Li, N. Park, M. Liu and J. Cho, Promotion of Oxygen Reduction by a Bio-Inspired Tethered Iron Phthalocyanine Carbon Nanotube-Based Catalyst, Nat. Commun., 2013, 4, 2076 CrossRef PubMed.
- S. Chatterjee, K. Sengupta, S. Samanta, P. Kumar Das and A. Dey, Concerted Proton–Electron Transfer in Electrocatalytic O2 Reduction by Iron Porphyrin Complexes: Axial Ligands Tuning H/D Isotope Effect, Inorg. Chem., 2015, 54, 2383–2392 CrossRef CAS PubMed.
- Z. Zhuang, L. Xia, J. Huang, P. Zhu, Y. Li, C. Ye, M. Xia, R. Yu, Z. Lang, J. Zhu, L. Zheng, Y. Wang, T. Zhai, Y. Zhao, S. Wei, J. Li, D. Wang and Y. Li, Continuous Modulation of Electrocatalytic Oxygen Reduction Activities of Single-Atom Catalysts through p-n Junction Rectification, Angew. Chem., Int. Ed., 2023, 62, e202212335 CrossRef CAS PubMed.
- X. Li, C. Cao, S. Hung, Y. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. Ercan Alp, W. Xu, T. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Identification of the Electronic and Structural Dynamics of Catalytic Centers in Single-Fe-Atom Material, Chem, 2020, 6, 3440–3454 CAS.
- X. Wang, L. Sun, V. Reddu and A. C. Fisher, Electrocatalytic Reduction of Carbon Dioxide: Opportunities with Heterogeneous Molecular Catalysts, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- Y. Jiang, Y. Lu, X. Lv, D. Han, Q. Zhang, L. Niu and W. Chen, Enhanced Catalytic Performance of Pt-Free Iron Phthalocyanine by Graphene Support for Efficient Oxygen Reduction Reaction, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS.
- X. Yan, L. Zhuang, Z. Zhu and X. Yao, Defect Engineering and Characterization of Active Sites for Efficient Electrocatalysis, Nanoscale, 2021, 13, 3327–3345 RSC.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, Nitrogen-Doped Graphene as Efficient Metal-Free Electrocatalyst for Oxygen Reduction in Fuel Cells, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS.
- X. Yu, S. Lai, S. Xin, S. Chen, X. Zhang, X. She, T. Zhan, X. Zhao and D. Yang, Coupling of Iron Phthalocyanine at Carbon Defect Site via Π-Π Stacking for Enhanced Oxygen Reduction Reaction, Appl. Catal., B, 2021, 280, 119437 CrossRef CAS.
- J. Ma, L. Liu, Q. Chen, M. Yang, D. Wang, Z. Tong and Z. Chen, A Facile Approach to Prepare Crumpled Cotmpyp/Electrochemically Reduced Graphene Oxide Nanohybrid as an Efficient Electrocatalyst for Hydrogen Evolution Reaction, Appl. Surf. Sci., 2017, 399, 535–541 CrossRef CAS.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, Carbon Nanotube-templated Synthesis of Covalent Porphyrin Network for Oxygen Reduction Reaction, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- H. Jia, Z. Sun, D. Jiang and P. Du, Covalent Cobalt Porphyrin Framework on Multiwalled Carbon Nanotubes for Efficient Water Oxidation at Low Overpotential, Chem. Mater., 2015, 27, 4586–4593 CrossRef CAS.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Selective Electrochemical Reduction of CO2 to CO With a Cobalt Chlorin Complex Adsorbed on Multi-Walled Carbon Nanotubes in Water, Chem. Commun., 2015, 51, 10226 RSC.
- I. S. Kwon, I. H. Kwak, H. G. Abbas, H. W. Seo, J. Seo, K. Park, J. Park and H. S. Kang, Two Dimensional Mos2 Meets Porphyrins via Intercalation to Enhance the Electrocatalytic Activity toward Hydrogen Evolution, Nanoscale, 2019, 11, 3780–3785 RSC.
- I. S. Kwon, I. H. Kwak, J. Y. Kim, H. G. Abbas, T. T. Debela, J. Seo, M. K. Cho, J. P. Ahn, J. Park and H. S. Kang, Two-Dimensional MoS2/Fe-Phthalocyanine Hybrid Nanostructures as Excellent Electrocatalysts for Hydrogen Evolution and Oxygen Reduction Reactions, Nanoscale, 2019, 11, 14266–14275 RSC.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. D. Tománek and P. D. Ye, Phosphorene: an Unexplored 2D Semiconductor with a High Hole Mobility, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS.
- M. Ou, X. Wang, L. Yu, C. Liu, W. Tao, X. Ji and L. Mei, The Emergence and Evolution of Borophene, Adv. Sci., 2021, 8, 2001801 CrossRef CAS.
- J. Liu, Y. Yang, P. Lyu, P. Nachtigall and Y. Xu, Few-Layer Silicene Nanosheets with Superior Lithium-Storage Properties, Adv. Mater., 2018, 30, 1800838 CrossRef.
- N. Lv, Q. Li, H. Zhu, S. Mu, X. Luo, X. Ren, X. Liu, S. Li, C. Cheng and T. Ma, Electrocatalytic Porphyrin/Phthalocyanine-Based Organic Frameworks: Building Blocks, Coordination Microenvironments, Structure-Performance Relationships, Adv. Sci., 2023, 2206239 CrossRef CAS.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, Metal–Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- Y. R. Wang, Q. Huang, C. T. He, Y. Chen, J. Liu, F. C. Shen and Y. Q. Lan, Oriented Electron Transmission in Polyoxometalate-Metalloporphyrin Organic Framework for Highly Selective Electroreduction of CO2, Nat. Commun., 2018, 9, 4466 CrossRef.
- J. X. Wu, S. Z. Hou, X. D. Zhang, M. Xu, H. F. Yang, P. S. Cao and Z. Y. Gu, Cathodized Copper Porphyrin Metal–Organic Framework Nanosheets for Selective Formate and Acetate Production from CO2 Electroreduction, Chem. Sci., 2019, 10, 2199 RSC.
- J. D. Yi, D. H. Si, R. Xie, Q. Yin, M. D. Zhang, Q. Wu, G. L. Chai, Y. B. Huang and R. Cao, Conductive Two-Dimensional Phthalocyanine-based Metal–Organic Framework Nanosheets for Efficient Electroreduction of CO2, Angew. Chem., Int. Ed., 2021, 133, 17245–17251 CrossRef.
- P. M. Usov, S. R. Ahrenholtz, W. A. Maza, B. Stratakes, C. C. Epley, M. C. Kessinger, J. Zhu and A. J. Morris, Cooperative Electrochemical Water Oxidation by Zr Nodes and Ni–Porphyrin Linkers of a PCN-224 MOF Thin Film, J. Mater. Chem. A, 2016, 4, 16818 RSC.
- S. Sohrabi, S. Dehghanpour and M. Ghalkhani, A Cobalt Porphyrin-Based Metal Organic Framework/Multi-Walled Carbon Nanotube Composite Electrocatalyst for Oxygen Reduction and Evolution Reactions, J. Mat. Sci., 2018, 53, 3624–3639 CrossRef CAS.
- P. M. Usov, B. Huffman, C. C. Epley, M. C. Kessinger, J. Zhu, W. A. Maza and A. J. Morris, Study of Electrocatalytic Properties of Metal–Organic Framework PCN-223 for The Oxygen Reduction Reaction, ACS Appl. Mater. Interfaces, 2017, 9, 33539–33543 CrossRef CAS.
- M. O. Cichocka, Z. Liang, D. Feng, S. Back, S. Siahrostami, X. Wang, L. Samperisi, Y. Sun, H. Xu, N. Hedin, H. Zheng, X. Zou, H. C. Zhou and Z. Huang, A Porphyrinic Zirconium Metal–Organic Framework for Oxygen Reduction Reaction: Tailoring The Spacing between Active-Sites through Chain-Based Inorganic Building Units, J. Am. Chem. Soc., 2020, 142, 15386–15395 CrossRef CAS PubMed.
- M. Chen, H. Li, C. Liu, J. Liu, Y. Feng, A. G. H. Wee and B. Zhang, Porphyrin-and Porphyrinoid-Based Covalent Organic Frameworks (COFs): From Design, Synthesis to Applications, Coord. Chem. Rev., 2021, 435, 213778 CrossRef CAS.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water, Science, 2015, 349, 1208–1213 CrossRef CAS.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, Reticular electronic tuning of porphyrin active sites in covalent organic frameworks for electrocatalytic carbon dioxide reduction, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, A Stable and Conductive Metallophthalocyanine Framework for Electrocatalytic Carbon Dioxide Reduction in Water, Angew. Chem., Int. Ed., 2020, 132, 16730–16736 CrossRef.
- B. Han, Y. Jin, B. Chen, W. Zhou, B. Yu, C. Wei, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, Maximizing Electroactive Sites in a Three-Dimensional Covalent Organic Framework for Significantly Improved Carbon Dioxide Reduction Electrocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202114244 CrossRef CAS.
- S. Y. Chi, Q. Chen, S. S. Zhao, D. H. Si, Q. J. Wu, Y. B. Huang and R. Cao, Three-dimensional Porphyrinic Covalent Organic Frameworks for Highly Efficient Electroreduction of Carbon Dioxide, J. Mater. Chem. A, 2022, 10, 4653 RSC.
- H. Huang, F. Li, Y. Zhang and Y. Chen, Two-dimensional Graphdiyne Analogue Co-Coordinated Porphyrin Covalent Organic Framework Nanosheets as a Stable Electrocatalyst for the Oxygen Evolution Reaction, J. Mater. Chem. A, 2019, 7, 5575–5582 RSC.
- Y. Wu, J. M. Veleta, D. Tang, A. D. Price, C. E. Botez and D. Villagrán, Efficient Electrocatalytic Hydrogen Gas Evolution by a Cobalt–Porphyrin-Based Crystalline Polymer, Dalton Trans., 2018, 47, 8801–8806 RSC.
- X. Wang, Y. Q. Wang, Y. C. Feng, D. Wang and L. J. Wan, Insights into Electrocatalysis by Scanning Tunnelling Microscopy, Chem. Soc. Rev., 2021, 50, 5832–5849 RSC.
- Z. Xu, Z. Liang, W. Guo and R. Zou, In Situ/Operando Vibrational Spectroscopy for The Investigation of Advanced Nanostructured Electrocatalysts, Coord. Chem. Rev., 2021, 436, 213824 CrossRef CAS.
- D. Nguyen, G. Kang, N. Chiang, X. Chen, T. Seideman, M. C. Hersam, G. C. Schatz and R. P. Van Duyne, Probing Molecular-Scale Catalytic Interactions Between Oxygen and Cobalt Phthalocyanine using Tip-Enhanced Raman Spectroscopy, J. Am. Chem. Soc., 2018, 140, 5948–5954 CrossRef CAS PubMed.
- K. Sengupta, S. Chatterjee and A. Dey, In Situ Mechanistic Investigation of O2 Reduction by Iron Porphyrin Electrocatalysts using Surface-Enhanced Resonance Raman Spectroscopy Coupled to Rotating Disk Electrode (SERRS-RDE) Setup, ACS Catal., 2016, 6, 6838–6852 CrossRef CAS.
- Z. Chen, S. Jiang, G. Kang, D. Nguyen, G. C. Schatz and R. P. Van, Duyne, Operando Characterization of Iron Phthalocyanine Deactivation during Oxygen Reduction Reaction using Electrochemical Tip-Enhanced Raman Spectroscopy, J. Am. Chem. Soc., 2019, 141, 15684–15692 CrossRef CAS.
- D. Liu, B. Zhang, G. Zhao, J. Chen, H. Pan and W. Sun, Advanced In-situ Electrochemical Scanning Probe Microscopies in Electrocatalysis, Chinese, J. Catal., 2023, 47, 93–120 CrossRef CAS.
- B. A. Friesen, A. Bhattarai, U. Mazur and K. W. Hipps, Single Molecule Imaging of Oxygenation of Cobalt Octaethylporphyrin at the Solution/solid Interface: Thermodynamics from Microscopy, J. Am. Chem. Soc., 2012, 134, 14897–14904 CrossRef CAS.
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan, R. J. M. Nolte, J. A. A. W. Elemans and S. Speller, Real-Time Single-Molecule Imaging of Oxidation Catalysis at a Liquid–solid Interface, Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS PubMed.
- X. Wang, Z. F. Cai, D. Wang and L. J. Wan, Molecular Evidence for the Catalytic Process of Cobalt Porphyrin Catalysed Oxygen Evolution Reaction in Alkaline Solution, J. Am. Chem. Soc., 2019, 141, 7665–7669 CrossRef CAS PubMed.
- X. Wang, Z. F. Cai, Y. Q. Wang, Y. C. Feng, H. J. Yan, D. Wang and L. J. Wan, In Situ Scanning Tunnelling Microscopy of Cobalt-Phthalocyanine-Catalysed CO2 Reduction Reaction, Angew. Chem., Int. Ed., 2020, 59, 16098–16103 CrossRef CAS.
- Y. Q. Wang, X. H. Dan, X. Wang, Z. Y. Yi, J. J. Fu, Y. C. Feng, J. S. Hu, D. Wang and L. J. Wan, Probing the Synergistic Effects of Mg2+ on CO2 Reduction Reaction on CoPc by In Situ Electrochemical Scanning Tunnelling Microscopy, J. Am. Chem. Soc., 2022, 144, 20126–20133 CrossRef CAS PubMed.
- J. Y. Gu, Z. F. Cai, D. Wang and L. J. Wan, Single-Molecule Imaging of Iron-Phthalocyanine-catalysed Oxygen Reduction Reaction by in Situ Scanning Tunnelling Microscopy, ACS Nano, 2016, 10, 8746–8750 CrossRef CAS PubMed.
- Y. C. Feng, X. Wang, Z. Y. Yi, Y. Q. Wang, H. J. Yan and D. Wang, In-situ ECSTM Investigation of H2O2 Production in Cobalt—Porphyrin-catalysed Oxygen Reduction Reaction, Sci. China: Chem., 2023, 66, 273–278 CrossRef CAS.
- M. Wang, L. Árnadóttir, Z. J. Xu and Z. Feng, In Situ X-Ray Absorption Spectroscopy Studies of Nanoscale Electrocatalysts, Nano-Micro Lett., 2019, 11, 1–18 CrossRef PubMed.
- Y. Wu, J. Jiang, Z. Weng, M. Wang, D. L. J. Broere, Y. Zhong, G. W. Brudvig, Z. Feng and H. Wang, Electroreduction Of CO2 Catalysed by a Heterogenized Zn−Porphyrin Complex With a Redox-innocent Metal Center, ACS Cent. Sci., 2017, 3, 847–852 CrossRef CAS.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Active Sites of Copper-Complex Catalytic Materials for Electrochemical Carbon Dioxide Reduction, Nat. Commun., 2018, 9, 415 CrossRef.
- L. Ni, P. Theis, S. Paul, R. W. Stark and U. I. Kramm, In Situ 57Fe Mössbauer Study of a Porphyrin Based Fenc Catalyst for ORR, Electrochim. Acta, 2021, 395, 139200 CrossRef CAS.
- L. Ni, C. Gallenkamp, S. Paul, M. Kubler, P. Theis, S. Chabbra, K. Hofmann, E. Bill, A. Schnegg, B. Albert, V. Krewald and U. I. Kramm, Active Site Identification in Fenc Catalysts and their Assignment to the Oxygen Reduction Reaction Pathway by in situ 57Fe Mössbauer Spectroscopy, Adv. Energy Sustainability Res., 2021, 2, 2000064 CrossRef CAS.
| This journal is © the Partner Organisations 2024 |