
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational transition of a non-associative fluorinated amphiphile in aqueous solution. II. Conformational transition vs. supramolecular assembly†
Marc B. Taraban a,
Daniel J. Deredgea,
Margaret E. Smitha,
Katharine T. Briggsa,
Yue Feng‡
a,
Yu Lib,
Zhong-Xing Jiangb,
Patrick L. Wintrode*a and
Yihua Bruce Yu*a
a,
Daniel J. Deredgea,
Margaret E. Smitha,
Katharine T. Briggsa,
Yue Feng‡
a,
Yu Lib,
Zhong-Xing Jiangb,
Patrick L. Wintrode*a and
Yihua Bruce Yu*a
aDepartment of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, 20 Penn Street, Baltimore, MD 21201, USA. E-mail: byu@rx.umaryland.edu; pwintrod@rx.umaryland.edu; Fax: +1 410-706-5017; Tel: +1 410-706-7514 Tel: +1 410-706-6639
bSchool of Pharmaceutical Sciences, Wuhan University, Wuhan, Hubei 430071, China
First published on 14th January 2019
Abstract
Unlike many known amphiphiles, the fluorinated amphiphilic dendrimer studied in this work demonstrated a concentration-dependent conformational transition rather than micellization or assembly. Hydrophobic and hydrophilic interactions with water were suggested as the most probable driving force of this transition. This assumption was consistent with the observed 19F chemical shift changes of the dendrimer compared to a known micelle-forming fluorinated amphiphile. Since water is an important factor in the process, trends of the concentration-dependent changes in water proton transverse relaxation rate served as an indicator of structural changes and/or supramolecular assembly. The conformational transition process was also confirmed by ion-mobility mass-spectrometry. We suggested that structural features, namely, steric hindrances, prevented the micellization/assembly of the dendrimer of this study. This conclusion might inform the approach to develop novel unconventional amphiphiles.
Introduction
Amphiphiles are organic molecules formed by a hydrophobic moiety and a hydrophilic moiety. Many natural (e.g., phospholipids) and synthetic (e.g., detergents) amphiphiles are known, which play a variety of important roles in biology, medicine, engineering, and daily life. Therefore, the design of new amphiphiles with novel properties is of much interest to diverse areas of human activities. Structure and properties of amphiphiles in solution often depend on their concentration—a result of their amphiphilicity and a source of their function. The most commonly known concentration-dependent behavior of amphiphiles is, perhaps, micellization, where the individual amphiphilic molecules assemble together to form an aggregated supramolecular entity as the concentration increases. When the solvent is water, such an assembly is driven by the need to minimize the interaction between water and the hydrophobic moiety of the amphiphile. As a result, hydrophobic groups of an amphiphile are buried inside the supramolecular assembly (micelles), and thereby, sequestered from water, while the hydrophilic groups are exposed to interact with the surrounding water molecules. The concentration where half of the amphiphile population exists in the aggregated state (micelles) is called the critical micelle concentration (CMC).1Not all amphiphiles form micelles. In certain cases, the assembly of amphiphile monomers might be sterically hindered. For example, cyclodextrins (CDs), a class of cyclic oligosaccharides, have a hydrophilic outer surface and a hydrophobic inner cavity. The amphiphilic nature of CDs is exploited in drug formulation for solubilization of otherwise insoluble molecules via the formation of inclusion complexes.2 The exterior of CDs provides solubility in water while their hydrophobic cavity accommodates hydrophobic drug molecules.3 Their amphiphilicity notwithstanding, the structure of CDs is incompatible with micellization and unmodified CDs do not assemble into micelles. In fact, self-association of unmodified CDs is negligible and transient in aqueous solutions.4 CDs assemble into stable aggregated forms when they either form noncovalent inclusion complexes with hydrophobic guest molecules, where hydrophobic interactions between these molecules drive the assembly process,5 or when CDs are covalently modified by hydrophobic pendant groups.6
We developed a family of fluorinated dendritic nonionic amphiphiles, each comprising a hydrophobic dendron and a hydrophilic dendron. The hydrophobic dendron contains multiple chemically identical trifluoromethyl (–CF3) groups, which act as the signal source for 19F NMR and MRI, while the hydrophilic dendron contains multiple chemically identical oligo-oxyethylene chains, which provide aqueous solubility. The hydrophobic dendron and the hydrophilic dendron are connected by amide bonds, which also contribute to aqueous solubility of the amphiphile. The dendrimers were developed as 19F Imaging Tracers and are denoted as 19FIT-n, with n referring to the number of chemically identical fluorine atoms per dendrimer (n = 32, 33, 34, 35…).7,8 Fig. 1 shows the structure of 19FIT-27. MRI studies in animals indicate that, as imaging agents, 19FIT-27 has many advantages over perfluorocarbons (e.g., perfluoro-octylbromide), including high water solubility (>150 mM), a singlet 19F signal from 27 magnetically equivalent fluorine atoms, low toxicity, and, most importantly, no excessive and prolonged accumulation in the liver and spleen.9 It is metabolically stable and is excreted primarily through the urine with an elimination t1/2 of ∼12 h. In contrast, perfluorocarbons accumulate excessively in the liver and spleen with an elimination t1/2 on the order of 6 months or longer,10 are exhaled mainly through the lungs, and may cause fatal chemical pneumonitis.11
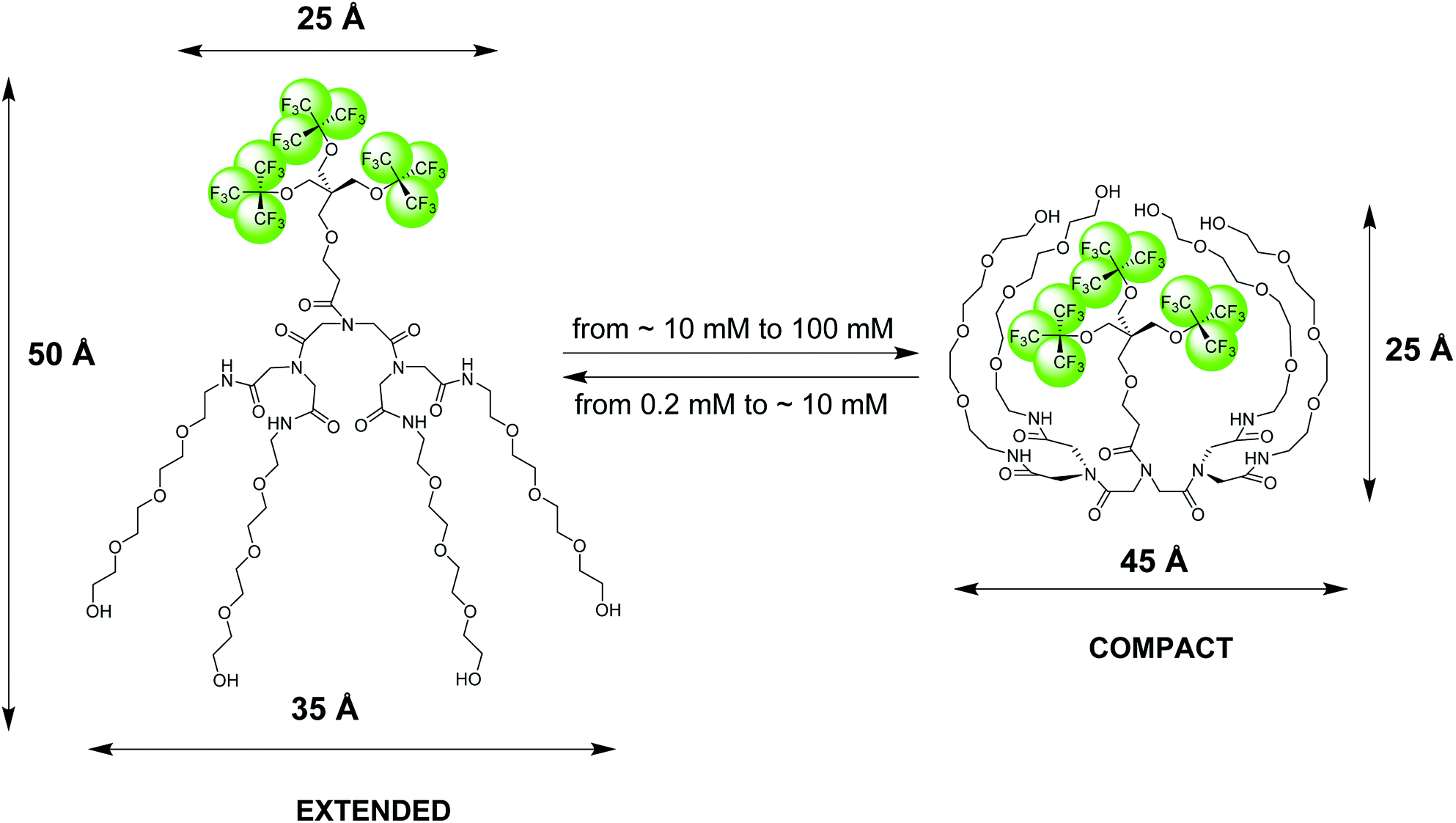 | ||
| Fig. 1 Pictorial presentation of the structure and conformational transition of an amphiphilic fluorinated dendrimer, 19FIT-27, confirmed by SAXS, SANS, DLS, and NMR diffusometry data.12 The extended conformation dominates at low dendrimer concentrations (<10 mM) while the compact conformation dominates at high dendrimer concentrations (>10 mM). The dimensional parameters of the extended and compact forms of 19FIT-27 are from the SAXS data.12 | ||
The NMR 19F chemical shift, δ(19F), of two versions of 19FIT-27, which differ only by one –CH2– group in the linker between the two dendrons, displays systematic up-field drift as their concentration C(19FIT-27) increases.7,12 Initially, based on the plot of δ(19F) vs. 1/C(19FIT-27) alone, a method often used to determine CMC of surfactants,1,13 we concluded that 19FIT-27 forms micelles with a CMC around 8 mM.7,9 However, the magnitude of the δ(19F) up-field shift is very small, ∼0.06 ppm, much smaller than that of conventional fluorinated amphiphiles, which are typically ∼1–2 ppm.13,14 To explore the reasons of such small magnitude of δ(19F) up-field shift of 19FIT-27, we conducted more detailed characterizations of 19FIT-27 at three concentrations, 1, 10 and 100 mM—respectively below, around, and above the observed transition point (∼8 mM). The assortment of structural and dynamic characterization techniques, including small-angle X-ray and neutron scattering (SAXS and SANS), dynamic light scattering (DLS) and NMR diffusometry (PFG NMR), have shown that 19FIT-27 remains monomeric as its concentration increases up to 100 mM. There is no aggregation.12
Based on our observations using the above listed methods, we proposed the following model for 19FIT-27.12 Instead of concentration-driven micellization/assembly, 19FIT-27 undergoes a concentration-driven conformational transition. At lower concentration, below ∼10 mM, 19FIT-27 adopts an extended conformation, while at higher concentration, above ∼10 mM, 19FIT-27 transitions to a compact conformation. Upon transition, the fluorocarbon moiety in 19FIT-27 is sequestered, at least to some extent, from water (Fig. 1). The inability of 19FIT-27 to form micelles/assemblies probably stems from steric hindrances. It comprises two conical dendrons connected by a short linker; assembling doubly conical objects into conventional micelles might be spatially hindered. There are indeed examples of nonionic dendritic amphiphiles forming micelles, but they comprise a single conical dendron and a linear tail, a shape compatible with tight packing for micellization.15
The conclusion that an amphiphile undergoes a concentration-dependent conformational transition instead of micellization is surprising. To our knowledge, it has not been reported before and, therefore, warrants further investigation. The nature of the transition can be clarified directly, if high-resolution structures of 19FIT-27 at various concentrations are available. This, however, proves an elusive goal to us thus far.
In this work, we aim to strengthen the conclusion regarding the conformational transition of 19FIT-27 based on a repertoire of techniques different from those used in our previous study.12 Here, we compared the concentration-dependent transition of 19FIT-27 with the behavior of another fluorinated amphiphile, sodium perfluorooctanoate (NaFC8), which is known to form micelles. We found that micellization/assembly results in orders of magnitude larger changes in 19F chemical shifts as compared to the transition of 19FIT-27, suggesting that 19FIT-27 does not undergo micellization.
We have previously shown that the transverse relaxation rate of water protons, R2(1H2O), is sensitive to a number of structural transformations of solutes, including the micellization of amphiphiles.16 Since R2(1H2O) is affected by the proton exchange rates between water and solute molecules, the assembly of solute molecules will affect their proton exchange with water, resulting in a subsequent change in R2(1H2O). Here, we compared the concentration-dependent profile of R2(1H2O) of 19FIT-27 with those of sodium octanoate (NaC8) and α-CD. NaC8 is known to form micelle as its concentration increases while α-CD is known to undergo neither micellization nor conformational transition as its concentration increases. We observed that R2(1H2O) of both 19FIT-27 and NaC8 display nonlinear dependence on solute concentration with clear transition points while that of α-CD display linear dependence on solute concentration with no transition point. However, R2(1H2O) of 19FIT-27 and NaC8 follow opposite trends, suggesting they undergo different types of concentration-dependent transitions.
We also monitored the concentration-dependent behavior of 19FIT-27 using ion-mobility mass-spectrometry (IM-MS). Since the drift time in IM-MS is defined by the collision cross-section of an ion in the gas phase, IM-MS data can provide comparative information on the relative dimensional and shape changes of ions. Indeed, e.g., for proteins, it has been demonstrated that under gentle ionization, many proteins typically retain their shape and compactness also in the gas phase.17 We found that during ionization, 19FIT-27 similarly retains its solution phase shape with no indications of the supramolecular assembly which would have led to longer drift time at higher concentrations. Instead, the opposite occurred—the drift time of 19FIT-27 becomes shorter at higher concentrations, suggesting compaction, rather than aggregation, as the dendrimer concentration increases.
Thus, the combination of results from multiple analytical techniques clearly points to the concentration-dependent conformational transition of the fluorinated dendrimer 19FIT-27 rather than micellization and/or other supramolecular assembly. In the compact form of 19FIT-27, the hydrophobic CF3-groups are sequestered, albeit incompletely, from water.
Results and discussion
Summary of structural characterizations of 19FIT-27
From our previous work,12 we made three related conclusions about the concentration-dependent behavior of 19FIT-27: (i) it does not undergo micellization/assembly; (ii) its structure becomes more compact as its concentration increases; and (iii) it experiences significant inter-molecular interactions at high concentration. The experimental observations corroborating each of the above conclusions are briefly summarized below.(i) Our conclusion that 19FIT-27 does not form micelles/assemblies was based on the following evidence. First, SAX(N)S (small-angle X-ray or neutron scattering) scattering profiles12 showed no signs of micellization/assembly. Indeed, the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) I(Q) vs. logQ plots for 1 mM, 10 mM, and 100 mM concentrations of 19FIT-27 were flat in the low region of the scattering vector Q—in case of assembly, I(Q) is known to show steep growth in the low Q region.18 Moreover, the low-resolution 3D-shape of 19FIT-27 molecule reconstructed from SAXS data showed comparable dimensional characteristics12 of 19FIT-27 at 1 mM, 10 mM, and 100 mM, also suggesting no assembly. Second, DLS (dynamic light scattering) data suggested that 19FIT-27 had very close values of hydrodynamic radii Rh at 1 mM, 10 mM, and 100 mM (31 Å, 33 Å, and 34 Å, respectively), suggesting there is no assembly.12 The slight increase in Rh at higher concentrations might reflect somewhat slower motion of the individual 19FIT-27 molecules. Third, the self-diffusion coefficient of 19FIT-27 normalized by water diffusion to account for viscosity effects, D(19FIT-27)/D(1H2O)—both were measured by PFG (pulsed field gradient) NMR—decreases slightly from 0.036 at 1 mM to 0.034 at 10 mM.12 If 19FIT-27 molecules assemble around 10 mM, one would expect a much larger decrease of Ds(19FIT-27)/Ds(1H2O). At 100 mM, Ds(19FIT-27)/Ds(1H2O) indeed decreases more significantly, to 0.020.12 This is more likely due to the crowding effect at 100 mM rather than assembly, which is discussed in the details below.
I(Q) vs. logQ plots for 1 mM, 10 mM, and 100 mM concentrations of 19FIT-27 were flat in the low region of the scattering vector Q—in case of assembly, I(Q) is known to show steep growth in the low Q region.18 Moreover, the low-resolution 3D-shape of 19FIT-27 molecule reconstructed from SAXS data showed comparable dimensional characteristics12 of 19FIT-27 at 1 mM, 10 mM, and 100 mM, also suggesting no assembly. Second, DLS (dynamic light scattering) data suggested that 19FIT-27 had very close values of hydrodynamic radii Rh at 1 mM, 10 mM, and 100 mM (31 Å, 33 Å, and 34 Å, respectively), suggesting there is no assembly.12 The slight increase in Rh at higher concentrations might reflect somewhat slower motion of the individual 19FIT-27 molecules. Third, the self-diffusion coefficient of 19FIT-27 normalized by water diffusion to account for viscosity effects, D(19FIT-27)/D(1H2O)—both were measured by PFG (pulsed field gradient) NMR—decreases slightly from 0.036 at 1 mM to 0.034 at 10 mM.12 If 19FIT-27 molecules assemble around 10 mM, one would expect a much larger decrease of Ds(19FIT-27)/Ds(1H2O). At 100 mM, Ds(19FIT-27)/Ds(1H2O) indeed decreases more significantly, to 0.020.12 This is more likely due to the crowding effect at 100 mM rather than assembly, which is discussed in the details below.
(ii) Two observations suggested that the 19FIT-27 molecule is more compact at 10 mM and 100 mM than at 1 mM. First, SAXS data showed that 19FIT-27 has a smaller radius of gyration Rg at 10 mM (17.2 Å) than at 1 mM (17.8 Å).12 Second, particle size distributions from DLS showed that 19FIT-27 has a broader distribution of Rh at 1 mM than at 10 and 100 mM, suggesting 19FIT-27 is more flexible and could sample more conformations at 1 mM than at 10 mM and 100 mM. This is consistent with 19FIT-27 existing in an extended more flexible conformation at 1 mM, but in a more compact conformation at 10 and 100 mM.12
(iii) Finally, we concluded that inter-molecular interactions between 19FIT-27 molecules are negligible at 1 mM and 10 mM, but become pronounced at 100 mM. First, in both SAXS and SANS scattering profiles,12 a broad peak corresponding to the distances between 19FIT-27 molecules ∼70 Å, was observed at 100 mM, but absent at 1 and 10 mM, and is a clear evidence of strong interparticle interference. Second, at all three concentrations, DLS data of 19FIT-27 display a peak in the μs range. However, at 100 mM, DLS data display an additional peak that is absent at 1 and 10 mM. This peak is in the ms time range, which suggests slow cooperative motion of 19FIT-27 molecules at 100 mM.12 Third, the normalized self-diffusion coefficient of 19FIT-27, Ds(19FIT-27)/Ds(1H2O), decreased from 0.034 at 10 mM to 0.020 at 100 mM, suggesting the diffusion of 19FIT-27 is hindered at 100 mM. Fourth, the ratio between the collective diffusion coefficient of 19FIT-27, Dc(19FIT-27), determined by DLS, and the self-diffusion coefficient of 19FIT-27, Ds(19FIT-27), determined by PFG NMR, increased from ca. 1.2 at 1 mM and 10 mM to ca. 2.3 at 100 mM. Such an increase in the ratio between two diffusion coefficients is known19 to be an indication of much stronger inter-particle interactions, as observed when the concentration of 19FIT-27 increases from 10 mM to 100 mM.12
In this work, we address additional details of the concentration-dependent conformational transition of 19FIT-27. Particular attention is on the extent of shielding of the fluorocarbon moiety from water upon conformational transition and on the driving force of the transition. Also, mass spectrometry is employed to verify conformational transition vs. supramolecular assembly as the concentration of 19FIT-27 increases.
Concentration-dependent transition of 19FIT-27 vs. assembly of NaFC8
To clarify the differences between conformational transition and micellization/assembly, we compared the concentration-dependent behavior of the fluorinated amphiphilic dendrimer, 19FIT-27, with a well-characterized fluorinated amphiphile, sodium perfluorooctanoate (NaFC8). As a reference molecule, NaFC8 can shed light on two aspects of 19FIT-27. First, how shielded are the fluorocarbons in 19FIT-27 from water in the compact state? The aliphatic fluorocarbon chain of NaFC8, 19F3C8–19F2C7–19F2C6–19F2C5–19F2C4–19F2C3–19F2C2–, is buried to a differing extent in the micelle core, with the 19F3C8-group the deepest and the –19F2C2-group the shallowest. Only the –19F2C2-group, adjacent to the carboxylate head group, has contact with water according to NMR relaxation data.14,20 According to SANS data, no water is present in the center of NaFC8 micelles.21 In other words, sequestration of water from the micellar core of NaFC8 is rather complete. This feature makes NaFC8 an ideal reference point to assess the sequestration of fluorocarbon groups from water in 19FIT-27. Second, NaFC8 forms micelles with a CMC of 30–32 mM at 25 °C,13 and increasing to 36 mM at 8 °C.21 Its aggregation number, N, referring to number of monomers per micelle, increases with concentration and reaches 23 at its CMC at 25 °C.22 Concentration-dependent assembly of 23 NaFC8 molecules is a much more cooperative process as compared to the conformational transition of a single 19FIT-27 molecule.The concentration-dependent transitions for 19FIT-27 and NaFC8 were monitored using the 19F chemical shift, δ(19F). Previously, the micellization of NaFC8 was characterized using its δ(19F) changes at 35 °C and 1.41 T (56.4 MHz for 19F).13 We measured δ(19F) of 19FIT-27 and NaFC8 at 22 °C and 9.4 T (376.5 MHz for 19F). All 27 fluorine atoms in 19FIT-27 are magnetically equivalent and thereby emit a single un-split 19F signal. In contrast, 15 fluorine atoms in the perfluorocarbon chain of NaFC8 are not magnetically equivalent, and thereby emit multiple 19F signals. Of the seven fluorocarbon groups in NaFC8, 19F3C8- and –19F2C2-groups are respectively distal and proximal to the polar carboxylic group, wherein the former is closest to the center of micellar core, while the latter is farthest from the core. We, therefore, chose δ(19F3C8–) and δ(–19F2C2–) chemical shifts to monitor the micellization of NaFC8. Fig. 2 shows concentration-dependent changes of chemical shifts δ(19F3C–) for 19FIT-27, and δ(19F3C8–) and δ(–19F2C2–) for NaFC8. The data are plotted in two ways. One is δ(19F) vs. 1/C(solute), which is a commonly used method to determine CMC of amphiphiles,23 and has been previously applied to NaFC8.13 Another is δ(19F) vs. C(solute), which is used in more recent work to determine CMC of amphiphiles.24
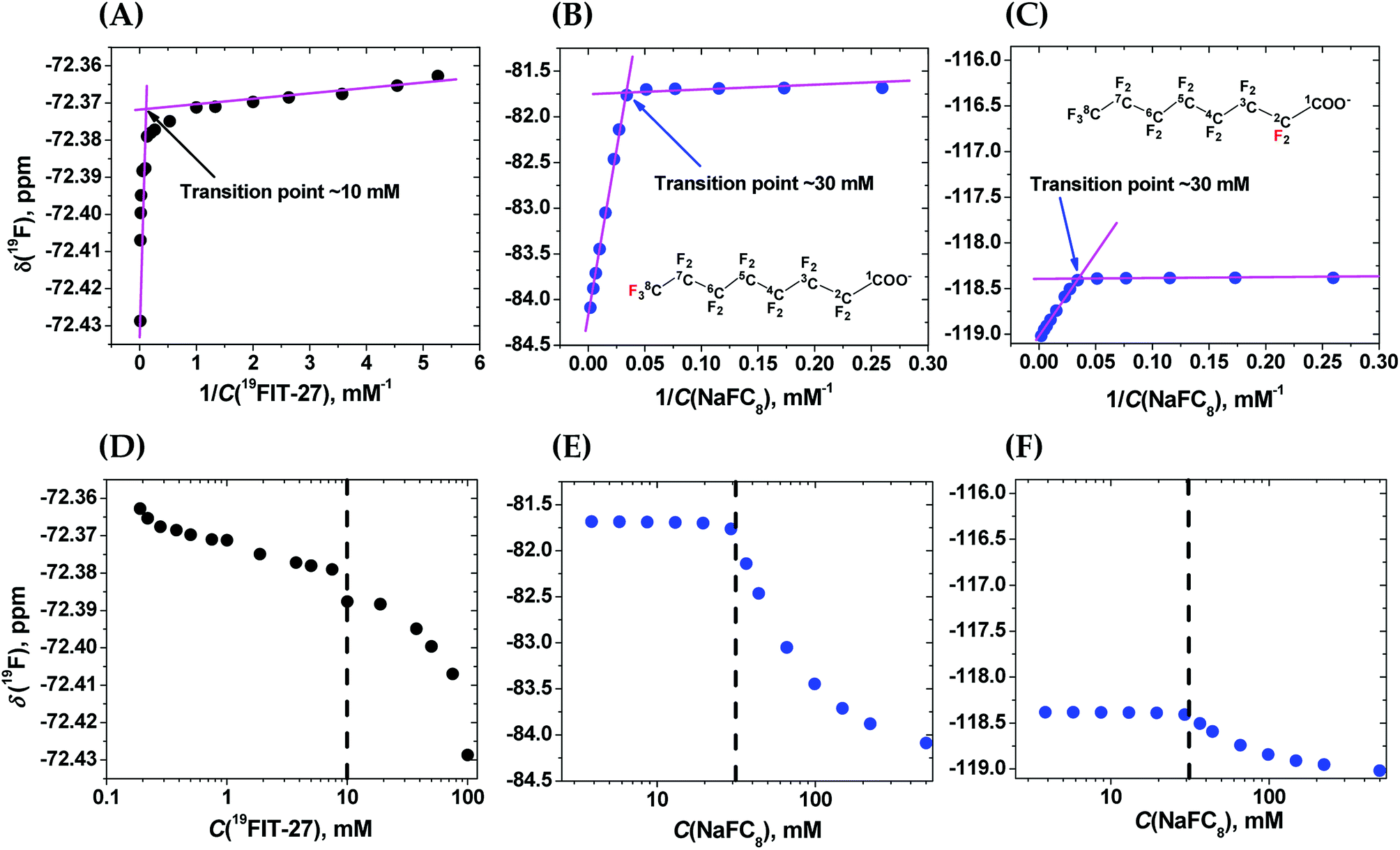 | ||
| Fig. 2 Comparison of the concentration-dependent transitions of 19FIT-27 and NaFC8 monitored by the 19F chemical shift δ(19F) measured at 9.4 T and 22 °C. Top row—δ(19F) vs. 1/C(solute). (A) δ(19F3C–) for 19FIT-27; (B) δ(19F3C8–) for NaFC8 (red label); (C) δ(–19F2C2–) for NaFC8 (red label). The two segments in each plot are fitted to linear lines (magenta) and the crossing point of the two lines gives the transition point (shown in each panel). Bottom row—δ(19F) vs. C(solute). (D) δ(19F3C–) for 19FIT-27; (E) δ(19F3C8–) for NaFC8; (F) δ(–19F2C2–) for NaFC8. C(solute) is shown in the logarithmic scale so that the transition point for each process is better revealed. The concentration range for 19FIT-27 is 0.2–100 mM (0.4–190 mg mL−1); that for NaFC8 is 3.9–500 mM (1.8–231.5 mg mL−1). | ||
In the δ(19F) vs. 1/C(solute) plot, the transition, for both 19FIT-27 and NaFC8, appears more salient than in the δ(19F) vs. C(solute) plot. This difference between these two ways to plot data on amphiphiles has been observed before,23 and perhaps accounts for the prevalence of plotting a physical chemical parameter of the amphiphile vs. 1/C(amphiphile) to extract CMC of amphiphiles. We initially concluded that two versions of 19FIT-27 form micelles with CMC of ca. 8 mM based on δ(19F) vs. 1/C(solute) plots.7,9 Absent from our previous work was a side-by-side comparison with a known micelle-forming amphiphile. Here, with the δ(19F) vs. 1/C(solute) plots of 19FIT-27 and NaFC8 presented together, it can be seen that 19FIT-27 displays a much less cooperative transition compared to NaFC8 (Fig. 2(A)–(C)). In the δ(19F) vs. C(solute) plot, the transition of 19FIT-27 appears even more diffusive (Fig. 2(D)–(F)). These results suggest that the concentration-dependent transition of 19FIT-27 is much less cooperative than that of NaFC8, which is known to involve 23 molecules. Although this does not prove that the transition of 19FIT-27 is monomeric, it does suggest that it involves much less than 23 molecules.
In all three δ(19F) vs. C(solute) profiles, transition results in the upfield shift of δ(19F). Greater shielding of 19F nuclei from external magnetic field is known to result in the upfield shift of δ(19F), when the environment of fluorocarbon groups become less polar.25 Thus, in all cases, the upfield shift of δ(19F) of the fluorocarbon groups is consistent with their transition from a more polar environment to a less polar environment. In other words, the concentration-dependent transitions in both 19FIT-27 and NaFC8 result in a sequestration of the fluorocarbon groups from water to some extent.
As we have already mentioned, the micellization of NaFC8 results in complete sequestration of its 19F3C8-groups from water.21 The tight packing of the core of NaFC8 micelles also excludes the effects of Na+-counterion on the chemical shifts of deeply buried 19F3C8-groups. Therefore, had the fluorocarbon groups in 19FIT-27 been completely sequestered from water and Na+-ions upon its concentration-dependent transition, the extent of the upfield shift of δ(19F) of these groups would have been comparable to that of NaFC8. However, the magnitude of the upfield shift of δ(19F) is very different all three cases (Fig. 2). At 22 °C, the magnitude of δ(19F) upfield shift is ∼0.06 ppm for 19FIT-27 (Fig. 2(A) and (D)), ∼2.5 ppm for 19F3C8– (Fig. 2(B) and (E)), and ∼0.6 ppm for –19F2C2– (Fig. 2(C) and (F)). The smaller upfield shift of –19F2C2-groups compared to 19F3C8-groups in NaFC8 is due to the location of the former closer to the micelle surface and their partial exposure to water. The 19F3C8-groups which are completely sequestered from water upon micellization,14,20 demonstrate a much larger upfield shift (cf., e.g., Fig. 2(B) and (C)). Therefore, the much smaller upfield shift of δ(19F3C–) of 19FIT-27 upon transition suggests that the sequestration of its fluorocarbon groups from water is very incomplete compared to NaFC8 micellization.
Comparison with NaFC8 confirms that 19FIT-27 indeed undergoes a concentration-dependent transition, but this transition process is much less pronounced than in the case of NaFC8 micellization, and the water molecules still interact with the fluorocarbon groups of 19FIT-27 in its compact state. Although the sequestration is rather incomplete, one might still expect that water will interact with the extended and compact forms of 19FIT-27 differently, i.e., water might differentiate different conformations of the dendrimer. In light of our previous work, where we demonstrated that the water proton transverse relaxation rate, R2(1H2O), is sensitive to solute–water interaction,16 we explored whether the concentration-dependent transition of 19FIT-27 can be monitored by R2(1H2O).
Water proton transverse relaxation rate R2(1H2O) as a probe of transition vs. assembly
As we have already mentioned, the sensitivity of water proton transverse rate, R2(1H2O), to the efficiency of proton exchange between solute and water molecules, makes it a potent probe of concentration-dependent behavior of solutes.16 Indeed, the conformational transitions and/or micellization/assembly of amphiphiles are expected to affect their proton exchange with water resulting in the changes of R2(1H2O).We, therefore, compared concentration-dependent changes of R2(1H2O) for 19FIT-27 (in the range 0.2–100 mM (0.4–190 mg mL−1)) and a known micelle-forming amphiphile, sodium octanoate (NaC8) (in the range 9–2400 mM (1.5–400 mg mL−1)). NaC8 forms micelles with two CMCs, one in the range 0.3–0.4 M and another in the range 0.9–1.2 M. Its aggregation number N is not fixed, but rather increases from 10–15 to 20–25 with increasing concentration of NaC8.26 Here, we also used a third amphiphile, α-CD, as a benchmark for R2(1H2O) concentration-dependent response, since α-CD is known not to form micelles and is not prone to any conformational transitions within its studied concentration range (0.2–105 mM (0.2–102 mg mL−1)).27 In the essence, α-CD serves as a negative control for both micellization and conformational transition.
For the purpose of comparing the three amphiphiles, we normalized both R2(1H2O) and C(solute), using the following formula to obtain corresponding Xnormalized values
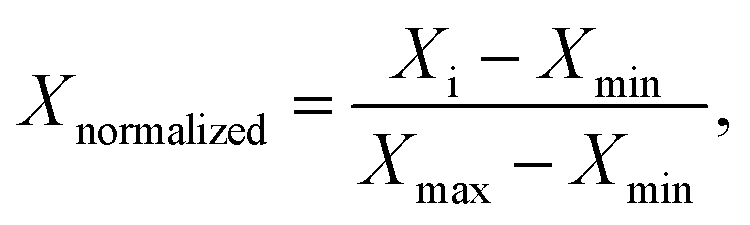 | (1) |
As seen from Fig. 3(A), in the absence of micellization/assembly and any conformational transitions affecting solute–water interactions, such as in the case of α-CD, the normalized dependence of R2(1H2O) vs. C(solute) is linear without any transition point. A linear relationship between R2(1H2O) and the proton molar fraction of α-CD has been reported,28 and is consistent with our observations. However, the plots R2(1H2O) vs. C(solute) for two other amphiphiles deviate from linearity and show evident transition points around a CMC value (NaC8) or a conformational transition (19FIT-27). The linear behavior of α-CD, the negative control, demonstrates that the observed nonlinear transition of NaC8 and 19FIT-27 is not simply due to increasing fraction of water molecules in the hydration shell of the solute as C(solute) increases.
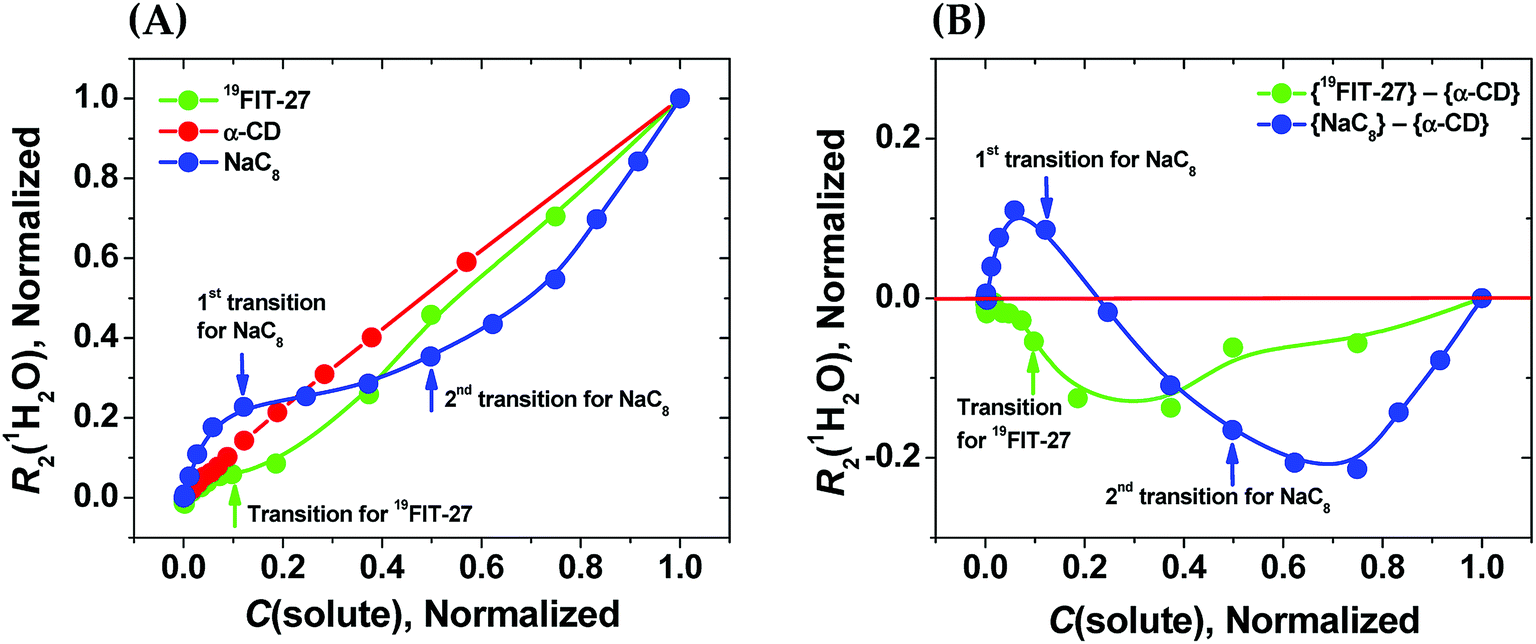 | ||
Fig. 3 Sensitivity of water proton transverse relaxation rate, R2(1H2O), towards conformational transitions and/or assembly of amphiphiles. (A) Comparative normalization plots of R2(1H2O) vs. C(solute) for three amphiphiles ( 19FIT-27, 19FIT-27,  α-CD, α-CD,  NaC8); (B) difference plots using data in (A) showing opposite deviations from linearity for transition vs. assembly ( NaC8); (B) difference plots using data in (A) showing opposite deviations from linearity for transition vs. assembly ( {19FIT-27}–{α-CD}, {19FIT-27}–{α-CD},  {NaC8}–{α-CD}). Arrows show corresponding transition points for NaC8 and 19FIT-27. The absolute values for the 1st and 2nd transition points for NaC8 are respectively ∼300–400 mM and ∼1000–1200 mM (ref. 26) while that for 19FIT-27 is ∼10 mM (see also ESI Tables S1 and S2† for the detailed data before and after normalization). {NaC8}–{α-CD}). Arrows show corresponding transition points for NaC8 and 19FIT-27. The absolute values for the 1st and 2nd transition points for NaC8 are respectively ∼300–400 mM and ∼1000–1200 mM (ref. 26) while that for 19FIT-27 is ∼10 mM (see also ESI Tables S1 and S2† for the detailed data before and after normalization). | ||
In Fig. 3(A), opposite concentration-dependent trends of R2(1H2O) were observed for micellization/assembly (NaC8) and conformational transition (19FIT-27). Indeed, R2(1H2O) of NaC8 shows initially positive deviation and then negative deviation from the linear trend of α-CD, while R2(1H2O) of 19FIT-27 shows negative deviation in the entire concentration range (Fig. 3(A)). This contrast between the three amphiphiles is better visualized in the difference plot shown in Fig. 3(B), where the linear α-CD dependence is subtracted from all three R2(1H2O) vs. C(solute) plots. The positive deviation of NaC8 from linearity occurs near its 1st transition point, which is attributed to micelle formation, while the negative deviation occurs near its 2nd transition point, which is attributed to the structural reorganization29 of the already assembled micelles to reduce the crowding at such high concentrations.30 In essence, the 2nd transition of NaC8 is a conformational transition at the micellar level, analogous to the conformational transition of 19FIT-27 at the molecular level. The common feature for both is that the normalized R2(1H2O) displays negative deviation from the linear behavior of α-CD.
These observations show that sequestration of hydrophobic groups from water indeed alters R2(1H2O), whether the sequestration is complete (as in NaC8) or incomplete (as in 19FIT-27). Distinctive behavior of normalized R2(1H2O) vs. C(solute) for micellization/assembly and conformational transitions makes it possible to distinguish between these two types of concentration-dependent transitions. However, the generality of the observation that supramolecular assembly leads to positive deviation of normalized R2(1H2O) from linearity and monomeric conformational transition leads to negative deviation of normalized R2(1H2O) from linearity remains to be seen.
Sequestration of the –CF3 groups in 19FIT-27 upon transition is presumably due to compaction of the dendrimer. To verify, we used ion-mobility mass-spectrometry (IM-MS) to monitor the structural dimension of 19FIT-27 as its concentration increases.
Transition vs. assembly of 19FIT-27 from the perspective of mass-spectrometry
Positive mode ESI-MS spectra of 19FIT-27 were collected at various concentrations, from 0.38 mM to 100 mM. Fig. 4 shows the spectrum at the highest concentration of 19FIT-27 (100 mM). No concentration-dependent changes in ESI-MS spectra for 19FIT-27 were observed in the experimental concentration range (see ESI, Fig. S2†). Whereas very minor populations of dimeric 19FIT-27 were persistently observed and potential degradation products were occasionally observed, the various mass spectral peaks corresponding to the monomeric 19FIT-27 were overwhelmingly abundant (Fig. 4 and ESI, Fig. S2†). Furthermore, any putative concentration-dependent accumulation of either higher order oligomers or degradation products were largely absent. The absence of concentration-dependent behavior suggests that any presence of dimers and degradation products are largely artefactual (in-source dimerization and fragmentation) and the abundance of monomeric mass peaks confirms the previously determined12 monomeric nature of 19FIT-27 in solution.
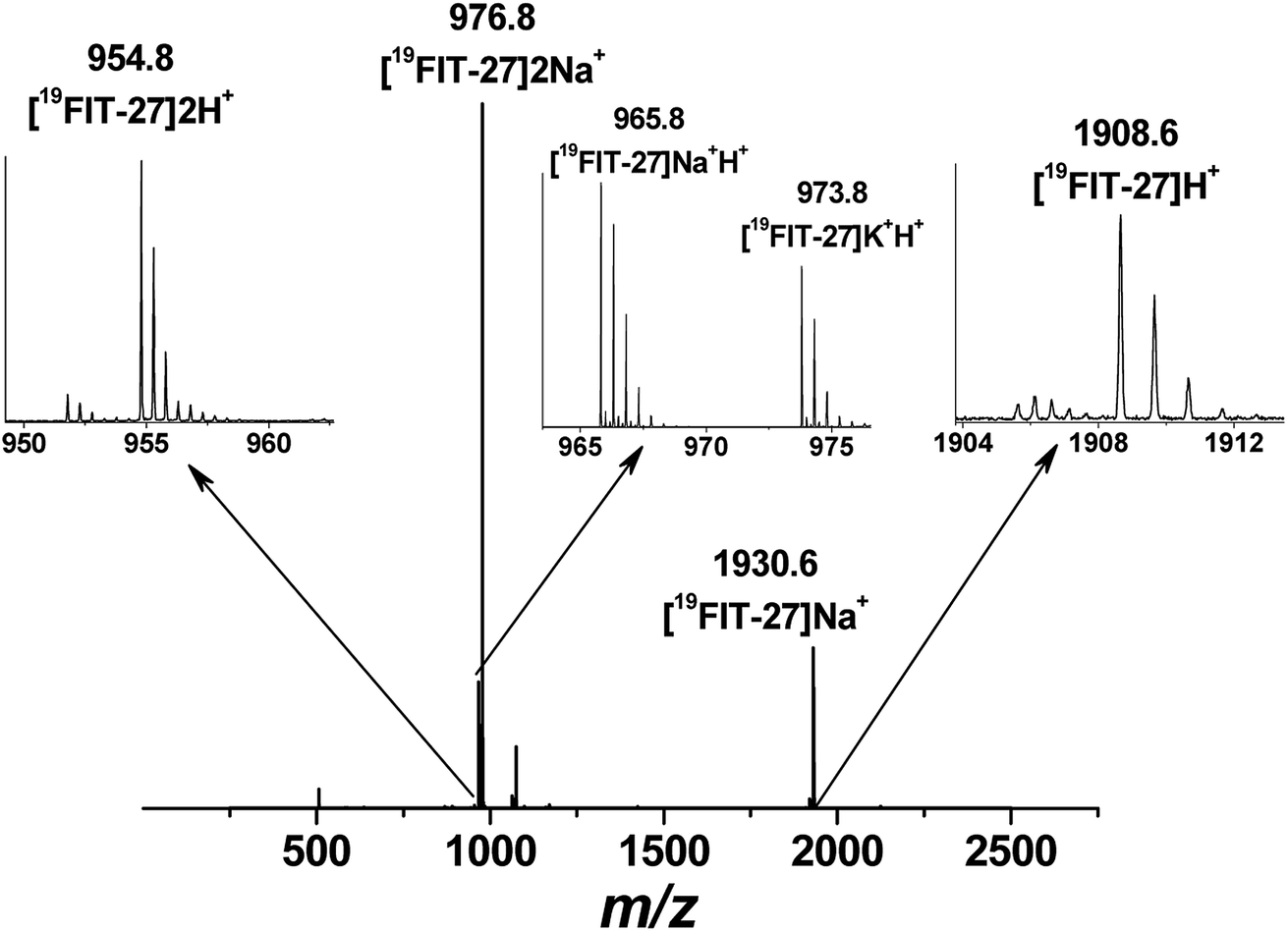 | ||
| Fig. 4 Positive mode electrospray ionization mass-spectrum (ESI-MS) of 19FIT-27 at 100 mM with peak assignment, showing no assemblies of the fluorinated dendrimer at highest concentration within the studied range. The molecular weight of monomeric 19FIT-27 is 1908 Da. | ||
Within the experimental concentration range of 19FIT-27, we detected subtle changes in the drift times of the single peak corresponding to the positively charged ion [19FIT-27]Na+ (Fig. 5(A)). At lower concentrations, the peak can be adequately fitted to a single Gaussian curve with the center of the peak giving the drift time, which is 17.9 ms. Meanwhile, as C(19FIT-27) increases, the observed peak (arrival time distribution) gradually broadens and can no longer be adequately fitted by a single Gaussian curve (Fig. 5(A)). Most telling, the center of the broadened peak moves to shorter drift time, consistent with more compact structure at high C(19FIT-27).
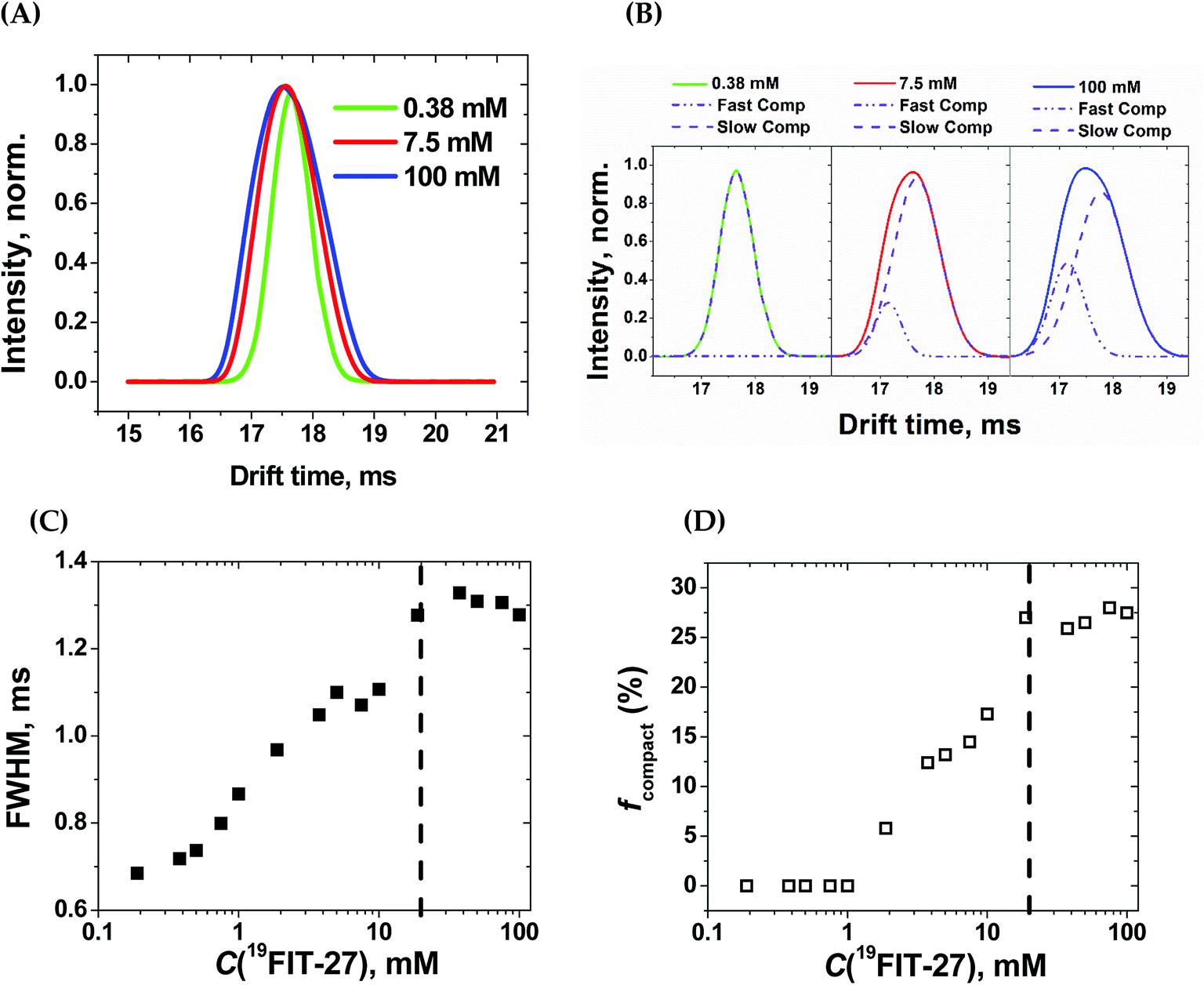 | ||
| Fig. 5 Ion-mobility mass-spectrometry (IM-MS) characterization of 19FIT-27 at different concentrations. (A) Concentration-dependent changes in spectral peak width for [19FIT-27]Na+; (B) single- and two-Gaussian deconvolution of the ion peaks shown in (A). The slow component has drift time of 17.9 ms; the fast component, 17.2 ms; (C) dependence of the full width at half-maximum (FWHM) of the ion peaks shown in (A) vs. C(19FIT-27); and (D) fraction of faster drifting compact form, fcompact, of [19FIT-27]Na+ vs. C(19FIT-27). The vertical dashed line in (C) and (D) highlights the transition point of ∼20 mM in the gas phase. | ||
Quantitatively, the broadened spectral peak could be reliably described by a two-Gaussian fit (Fig. 5(B)) with deconvolution into a slower drifting component (17.9 ms, the same as with lower C(19FIT-27)), and an emerging faster drifting component (17.2 ms). This result points to two dendrimer populations detected by IM-MS—one with a slower drift time of 17.9 ms corresponding to the extended form of the dendrimer, and another with a faster drift time of 17.2 ms corresponding to the compact form of the dendrimer. At first glance, such small changes in drift time distribution (3.9% reduction) may not seem to correlate well with the above mentioned significant volume reduction (25%) upon transition. However, drift time in IM-MS is determined by collision cross-section, not volume. We, therefore, used previously generated12 low-resolution SAXS 3D models at 1 mM and 10 mM 19FIT-27 to calculate collision cross-section by means of IMPACT projection approximation routine,35 and determined that, upon transition, there is only ∼1.5% reduction in collision cross section (∼1767.6 Å2 at 1 mM vs. ∼1741.3 Å2 at 10 mM). This explains the small decrease of drift time observed upon conformational transition.
The appearance of the faster drifting component broadens the IM-MS peak of the [19FIT-27]Na+ ion. The full width of the arrival time distribution at half-maximum (FWHM) demonstrates steady growth until the plateau, at which point no further changes were observed (Fig. 5(C)). Based on the two-Gaussian fits of the drift peak at higher concentrations, one might estimate the fraction of the compact form (fcompact) calculated from the areas of deconvoluted peaks. Fig. 5(D) shows the gradual growth of fcompact, which, similar to FWHM, also demonstrates plateauing at higher concentrations.
The plateau point in Fig. 5(C) and (D) give an estimate of the 19FIT-27 conformational transition point in the gas phase. As the data shows, the conformational transition point is delayed from ∼10 mM in aqueous solution to ∼20 mM in the gas phase. Also, similar to the pre-transition gradual changes of δ(19F) (Fig. 2(D)), both FWHM and fcompact display gradual pre-transition changes (Fig. 5(C) and (D)). Such pre-transition changes demonstrate the sensitivity of IM-MS towards the increasing presence of the compact form of 19FIT-27 similar to 19F NMR above.
The conformational transition of 19FIT-27 is not only delayed to a higher concentration, but also incomplete; fcompact is less than 30% completely transitioned even at 100 mM. From our previous SAXS and SANS experiments in aqueous solutions,12 one might conclude that at 100 mM the transition of 19FIT-27 into its compact form should be almost complete. Such discrepancies between liquid and gas state results are consistent with the weakened driving force for folding. The unavoidable partial dehydration in the vacuum chamber of the MS instrument could lead to a shift in the equilibrium and/or kinetics of interconversion between the extended and compact forms. The observed delay and incomplete conformational transition of 19FIT-27 in the gas phase supports the proposition that the transition is driven, at least in part, by the need to shield the 19F3C-groups from water.
In sum, no supramolecular assemblies of 19FIT-27 were detected by mass-spectrometry within the concentration range from 0.38 mM to 100 mM. IM-MS found evidence of the conformational transition of 19FIT-27 at higher concentrations. These observations confirm that shielding of fluorocarbon groups from water is part of the driving force of the conformational transition of 19FIT-27.
Conclusions
This work and its predecessor12 provide evidence of an asymmetric dendritic amphiphile, 19FIT-27, that undergoes conformational transition rather than micellization when its concentration exceeds certain level. This conclusion is supported by direct characterization of 19FIT-27 (SAXS, SANS, DLS, 19F NMR and MS), indirect characterization of 19FIT-27 (1H2O NMR), and side-by-side comparison of 19FIT-27 with both micelle-forming (NaFC8, NaC8) and non-micelle-forming (α-CD) amphiphiles. The origin of conformational transition in lieu of micellization likely lies in the shape of the amphiphile, which is incompatible with micellization. This points to a potential strategy to design novel amphiphiles with unconventional properties.Experimental section
Materials
Synthesis of the fluorinated amphiphilic dendrimer, 19FIT-27, followed the earlier described procedure,9 modified in order to introduce an additional methylene group (–CH2–) separating the hydrophobic fluorocarbon head and the hydrophilic tetraoxyethylene tails (Fig. 1).12 Sodium perfluorooctanoate (NaFC8, Sigma-Aldrich), sodium octanoate (NaC8, Sigma-Aldrich), and α-cyclodextrin (α-CD, TCI-GR) were used without further purification.Sample preparation
NMR experiments
All 1H and 19F NMR experiments were carried out using a Varian INOVA 400 NMR spectrometer (Varian, Inc., 399.75 MHz for 1H and 376.11 MHz for 19F) using a broadband detection probe. In all NMR experiments the temperature of sample solutions always was 22 °C (±0.1 °C).| I(t) = I0 × exp(−t/T2) | (2) |
Mass spectrometry experiments
All mass spectrometry data were acquired on a Waters Synapt G2 (Waters Co.) in electrospray ionization (ESI-MS) mode with direct infusion. All data acquisitions were performed in positive resolution mode at 3 kV capillary voltage, 30 V sample cone voltage, 5 V extraction cone voltage, 80 °C cone temperature, 175 °C desolvation temperature and 10 eV transfer collision energy. Data acquired were integrated over acquisition time in Masslynx software and exported for further analysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Early phase of this work was supported by an NSF grant (CBET 1133908). Mass-spectrometry experiments were supported in part by the University of Maryland Baltimore, School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014). Financial support from the University of Maryland, Baltimore and the University of Maryland, College Park seed grant is gratefully acknowledged.References
- N. Muller and R. H. Burkhahn, J. Phys. Chem., 1967, 71, 957–962 CrossRef CAS.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025 CrossRef CAS PubMed.
- P. Jansook, N. Ogawa and T. Loftsson, Int. J. Pharm., 2018, 535, 272–284 CrossRef CAS PubMed.
- (a) A. J. M. Valente, R. A. Carvalho and O. Södermann, Langmuir, 2015, 31, 6314–6320 CrossRef CAS PubMed; (b) A. J. M. Valente, R. A. Carvalho, D. Martinho and O. Södermann, Langmuir, 2017, 33, 8233–8238 CrossRef CAS PubMed.
- A. Ryzhakov, T. Do Thi, J. Stappaerts, L. Bertoletti, K. Kimpe, A. R. Sá Kouto, P. Saokham, G. Van den Mooter, P. Augustijns, G. W. Somsen, S. Kurkov, S. Inghelbrecht, A. Arien, M. I. Jimidar, K. Schrijnemakers and T. Loftsson, J. Pharm. Sci., 2016, 105, 2556–2569 CrossRef CAS PubMed.
- F. Salas and R. Darcy, Eur. J. Org. Chem., 2008, 957–969 CrossRef.
- Z.-X. Jiang and Y. B. Yu, J. Org. Chem., 2010, 75, 2044–2049 CrossRef CAS PubMed.
- X. Yue, M. B. Taraban, L. L. Hyland and Y. B. Yu, J. Org. Chem., 2012, 77, 8879–8887 CrossRef CAS PubMed.
- Z.-X. Jiang, X. Liu, E. K. Jeong and Y. B. Yu, Angew. Chem., Int. Ed., 2009, 48, 4755–4758 CrossRef CAS PubMed.
- K. L. Meyer, M. J. Carvlin, B. Mukherji, H. A. Sloviter and P. M. Joseph, Invest. Radiol., 1992, 27, 620–627 CrossRef CAS PubMed.
- Y. Nosé, Artif. Organs, 2004, 28, 807–812 CrossRef PubMed.
- M. B. Taraban, Y. Li, Y. Feng, E. V. Jouravleva, M. A. Anisimov, Z.-X. Jiang and Y. B. Yu, RSC Adv., 2014, 4, 54565–54575 RSC.
- N. Muller and H. Simsohn, J. Phys. Chem., 1971, 75, 942–945 CrossRef CAS.
- L. Nordstierna, P. V. Yushmanov and I. Furó, J. Phys. Chem. B, 2006, 110, 25775–25781 CrossRef CAS PubMed.
- (a) A. Richter, A. Wiedekind, M. Krause, T. Kissel, R. Haag and C. Olbrich, Eur. J. Pharm. Sci., 2010, 40, 48–55 CrossRef CAS PubMed; (b) R. Tyagi, S. Malhotra, A. F. Thünemann, A. Sedighi, M. Weber, A. Schäfer and R. Haag, J. Phys. Chem. C, 2013, 117, 12307–12317 CrossRef CAS; (c) D. R. Sikwal, R. S. Kalhapure, M. Jadhav, S. Rambharose, C. Mocktar and T. Govender, RSC Adv., 2017, 7, 14233–14246 RSC.
- Y. Feng, M. B. Taraban and Y. B. Yu, Chem. Commun., 2015, 51, 6804–6807 RSC.
- (a) B. T. Ruotolo, J. L. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed; (b) Y. Zhong, S.-J. Hyung and B. T. Ruotolo, Expert Rev. Proteomics, 2012, 9, 47–58 CrossRef CAS PubMed.
- M. B. Taraban, Y. Feng, B. Hammouda, L. L. Hyland and Y. B. Yu, Chem. Mater., 2012, 24, 2299–2310 CrossRef CAS PubMed.
- S. J. Law and M. M. Britton, Langmuir, 2012, 28, 11699–11706 CrossRef CAS PubMed.
- J. Ulmius and B. Lindman, J. Phys. Chem., 1981, 85, 4131–4135 CrossRef CAS.
- K. Shinoda, M. Hato and T. Hayashi, J. Phys. Chem., 1972, 76, 909–914 CrossRef CAS.
- S. S. Berr and R. R. M. Jones, J. Phys. Chem., 1989, 93, 2555–2558 CrossRef CAS.
- H. Gustavsson and B. Lindman, J. Am. Chem. Soc., 1978, 100, 4647–4654 CrossRef CAS.
- (a) W. Al-Soufi, L. Piñeiro and M. Novo, J. Colloid Interface Sci., 2012, 370, 102–110 CrossRef CAS PubMed; (b) M. V. C. Cardoso and E. Sabadini, Langmuir, 2013, 29, 15778–157786 CrossRef CAS PubMed.
- J. Sloop, Rep. Org. Chem., 2013, 3, 1–12 Search PubMed.
- (a) J. B. Hayter and T. Zemb, Chem. Phys. Lett., 1982, 93, 91–94 CrossRef CAS; (b) B. O. Persson, T. Drakenberg and B. Lindman, J. Phys. Chem., 1979, 83, 3011–3015 CrossRef CAS; (c) T. Zemb, M. Drifford, M. Hayoun and A. Jehanno, J. Phys. Chem., 1983, 87, 4524–4528 CrossRef CAS; (d) D. K. Chokappa and S. Das, Indian J. Chem., 1994, 33A, 795–800 CAS.
- W. Sliwa and T. Girek, Cyclodextrins: Properties and Applications, Wiley-VCH, Weinheim, 2017 Search PubMed.
- E. Sabadini, F. d. C. Egidio, F. Y. Fujiwara and T. Cosgrove, J. Phys. Chem. B, 2008, 112, 3328–3332 CrossRef CAS PubMed.
- P. Ekwall, in Chemistry, Physics and Application of Surface Active Substances, ed. J. Th. G. Overbeek, Gordon and Breach, New York, 1967, vol. 2, pp. 651–658 Search PubMed.
- P. Ekwall and L. Mandell, J. Colloid Interface Sci., 1979, 69, 384–397 CrossRef CAS.
- (a) C. A. Scarff, A. E. Ashcroft and S. E. Redford, Methods Mol. Biol., 2016, 1345, 115–132 CrossRef CAS PubMed; (b) G. Wang, A. J. Johnson and I. A. Kaltashov, Anal. Chem., 2012, 84, 1718–1724 CrossRef CAS PubMed.
- C. Bleiholder, N. F. Dupuis, T. Wyttenbach and M. T. Bowers, Nat. Chem., 2011, 3, 172–177 CrossRef CAS PubMed.
- L. Ceraulo, G. Giorgi, V. T. Liveri, D. Bongiorno, S. Indelicato, F. D. Gaudio and S. Indelicato, Eur. J. Mass Spectrom., 2011, 17, 525–541 CrossRef CAS PubMed.
- W. Hoffmann, G. von Helden and K. Pagel, Curr. Opin. Struct. Biol., 2017, 46, 7–15 CrossRef CAS PubMed.
- E. G. Marklund, M. T. Degiacomi, C. V. Robinson, A. J. Baldwin and J. L. P. Benesch, Structure, 2015, 23, 791–799 CrossRef CAS PubMed.
- M. J. O'Nell, P. E. Heckelman, C. B. Kock and K. J. Roman, The Merck Index, an Encyclopedia of Chemicals, Drugs, and Biologics, Merck, Whitehouse Station, 2006 Search PubMed.
- (a) H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630–638 CrossRef CAS; (b) S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Water proton transverse relaxation rates vs. concentration of fluorinated dendrimer, sodium octanoate and α-cyclodextrin; mass-spectra of fluorinated dendrimer at different concentrations. See DOI: 10.1039/c8ra08795d |
| ‡ Current address: Pfizer, Inc., Groton, CT 06340, USA. |
| This journal is © The Royal Society of Chemistry 2019 |