Transition-metal-catalyzed direct β-functionalization of simple carbonyl compounds
Guobing
Yan
* and
Arun Jyoti
Borah
Department of Chemistry, Lishui University, No. 1, Xueyuan Road, Lishui, 323000, Zhejiang Province, China. E-mail: 0gbyan@tongji.edu.cn
First published on 19th June 2014
Abstract
Chemical transformations via catalytic C–H bond activation have been established as one of the most powerful tools in organic synthetic chemistry. Transition-metal-catalyzed direct functionalization of β-C(sp3)–H bonds of carbonyl compounds has been developed in recent years. This highlight will focus on recent advances in this active area and their mechanisms are also discussed.
 Guobing Yan | Guobing Yan was born in Jiangxi, China, in 1975. He obtained his B.Sc. degree from Jinggangshan Normal University and his M.Sc. degree from Suzhou University, and in 2010 he obtained his Ph.D. degree from Tongji University. He spent two years, 2008 and 2009, as a visiting student in Professor Jianbo Wang's group at Peking University. He has been at Lishui University since 2010 as an associate professor. His current research interests focus on transition-metal-catalyzed activation of inert chemical bonds and green synthetic chemistry. |
1. Introduction
Over the past several decades, transition-metal-catalyzed direct functionalization of C–H bonds has emerged as a powerful tool in synthetic organic chemistry, due to its economical and environmental advantages.1 Although remarkable progress has been made in the sp2 C–H bond activation, the catalytic functionalization of sp3 C–H bonds remains an important fundamental challenge. This is primarily due to intrinsic inertness of sp3 C–H bonds and difficulty in controlling the regioselectivity.2 Carbonyl compounds are important intermediates in organic synthesis. Functionalization of carbonyl compounds at the α-position is well known for decades, which is usually achieved through enolate chemistry.3 However, the corresponding β-C–H bonds are typically unreactive. In the recent past, few synthetically attractive strategies for transition-metal-catalyzed functionalization of β-C(sp3)–H bonds have been developed, nevertheless these are successful only under the influence of a suitable directing group such as linear amides and ester substrates.42. β-Amination of alkyl ketones
In 2009, Ueno and co-workers developed an efficient method for the formation of a carbon–nitrogen bond through nickel-catalyzed β-C–H bond activation of alkyl ketones (Scheme 1).5 This is the first example of a one-step catalytic functionalization at the β-carbon atom of saturated ketones. Both ethyl ketones and secondary aliphatic amines are suitable for this catalytic system and provide the corresponding enaminones. However, benzylamine and N-methylaniline could not be compatible with this transformation. More importantly, exclusive β-regioselectivity has been achieved in the reaction.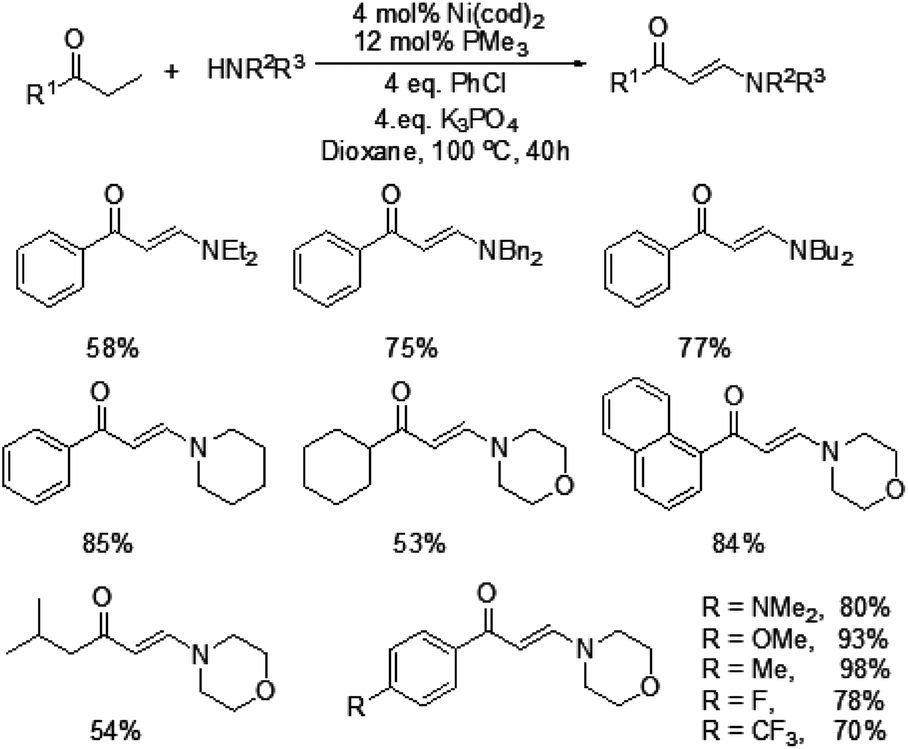 | ||
| Scheme 1 Nickel-catalyzed formation of C–N bond with alkyl ketones. | ||
A plausible pathway was proposed for this transformation (Fig. 1). The Ni(0) species was in situ generated from [Ni(cod)2] and PMe3, which underwent an oxidative addition with chlorobenzene. The resulting complex A reacted with an alkyl ketone to give the carbon-bound nickel enolate B, which could afford enone C through β-hydride elimination. Subsequently, the 1,4-addition of an amine to enone C took place to form a carbon–nitrogen bond at the β-position. The β-aminoketone D was converted into β-enaminone through formation of the nickel enolate E and subsequent β-hydride elimination regenerates the Ni(0) species to complete the catalytic cycle.
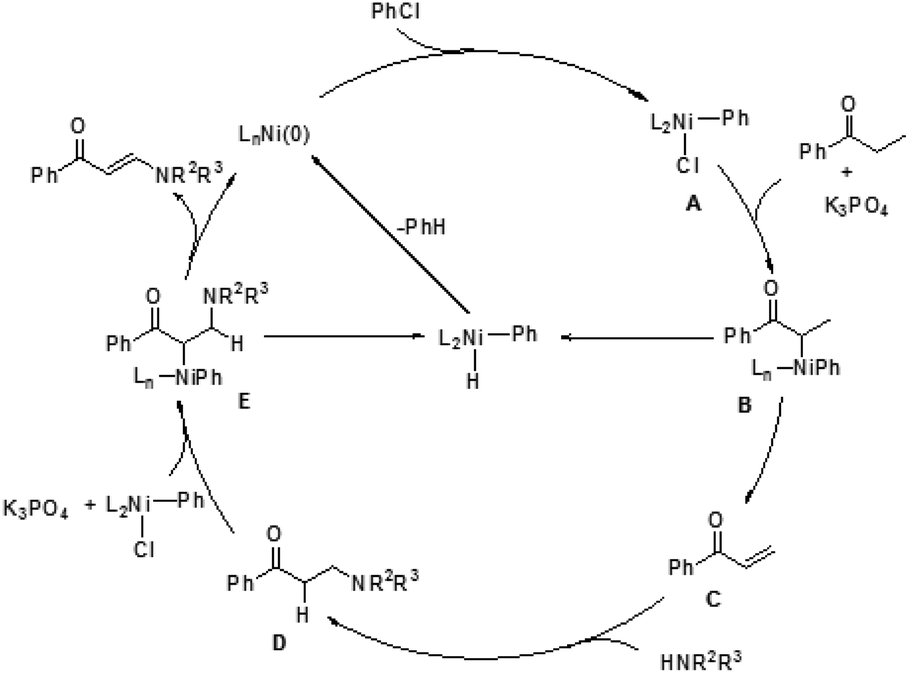 | ||
| Fig. 1 Possible mechanism. | ||
3. β-Arylation of carbonyl compounds
Baudoin and co-workers reported a mild and efficient intermolecular β-arylation of an unactivated sp3 C–H bond in carboxylic esters (Scheme 2).6 A broad range of useful functionalized carboxylic esters could be synthesized by this method. The advantages of this reaction are the mild conditions, even at room temperature, no polyarylation products formation and use of simple carboxylic esters as substrates. Moreover, β-arylated products have been obtained with enantiomeric ratios up to 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23 by changing Davephos to a chiral ligand. This novel protocol is complementary to directing-group-controlled β-arylation reactions. Although the mechanism of this reaction is not yet clear, their computational studies suggested that the mechanism could involve β-hydride elimination/Pd-η2 (C
:23 by changing Davephos to a chiral ligand. This novel protocol is complementary to directing-group-controlled β-arylation reactions. Although the mechanism of this reaction is not yet clear, their computational studies suggested that the mechanism could involve β-hydride elimination/Pd-η2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) bond rotation/hydride insertion/reductive elimination manifolds.
C) bond rotation/hydride insertion/reductive elimination manifolds.
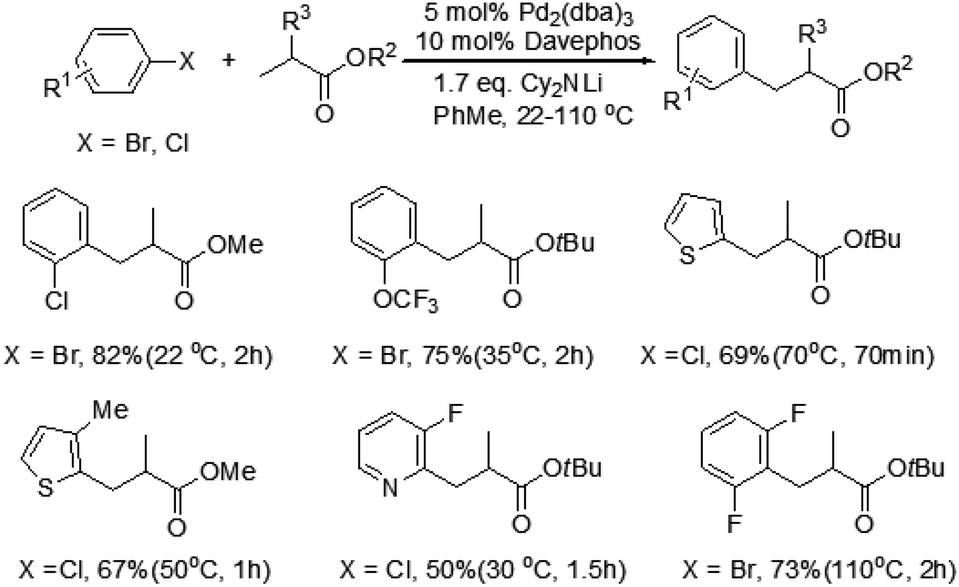 | ||
| Scheme 2 Palladium-catalyzed β-arylation of carboxylic esters. | ||
Recently, MacMillan and co-workers reported efficient and user-friendly reaction conditions using a combination of a photoredox catalyst and an amine catalyst to effect highly selective β-arylation of aldehydes and ketones with electron-deficient arylnitriles as the coupling partners.7 In comparison with Buchwald–Hartwig–Miura α-arylation of carbonyl compounds, this new approach achieves an important breakthrough in controlling β-regioselectivity. In the presence of a 26 W fluorescent light bulb and 1,4-diazobicyclo[2.2.2]nonane (DABCO) as a base, the use of Ir(ppy)3 in combination with N-isopropylbenzylamine (i-PrBnNH) or azepane can provide the corresponding β-arylation of aldehydes or ketones in good yields, without formation of any α-amine arylation adducts (Scheme 3). The catalytic system exhibits a broad range of substrate scope, including an array of alkanals and cyclohexanone derivatives. With respect to aromatic and heteroaromatic partners, a broad range of cyanosubstituted arenes with both electron-donating and electron-withdrawing substituents can undergo β-arylation coupling with good levels of efficiency. In addition, this strategy is applicable in the presence of a wide range of functional groups, such as ethers, esters, sulfones, alkenes, alkynes, arenes, and carbamates.
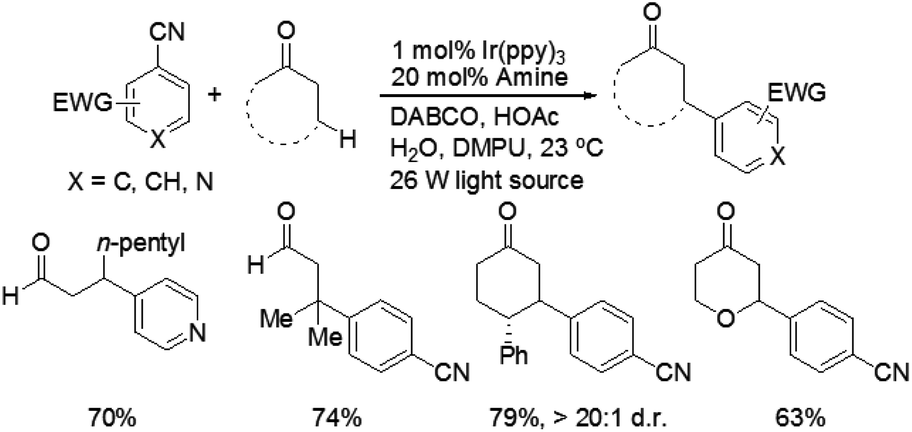 | ||
| Scheme 3 Direct β-arylation of aldehydes and ketones with electron-deficient arylnitriles. | ||
A prospective mechanism was proposed for the direct β-arylation of aldehydes or ketones through a photoinduced electron transfer pathway (Fig. 2). The reaction proceeds via formation of an arene radical anion A by single electron transfer (SET) from the excited state of *Ir(III)(ppy)3 to 1,4-dicyanobenzene, along with the oxidized photocatalyst Ir(IV)(ppy)3. Concurrent with this photoredox step, the amine catalyst (i.e., pyrrolidine) gets condensed with aldehyde to form the electron-rich enamine B. The electron-deficient Ir(IV)(ppy)3 can readily accept an electron via SET from the electron-rich enamine B to generate the corresponding radical cation C, which in turn isomerizes leading to the formation of β-enamine radical D. Intermolecular coupling between arene radical anion A and β-enamine radical D affords cyclohexadienyl anion E, which can undergo rapid β,δ-elimination of cyanide to regain aromaticity. Hydrolysis of the resulting enamine delivers the desired product to complete the organocatalytic cycle.
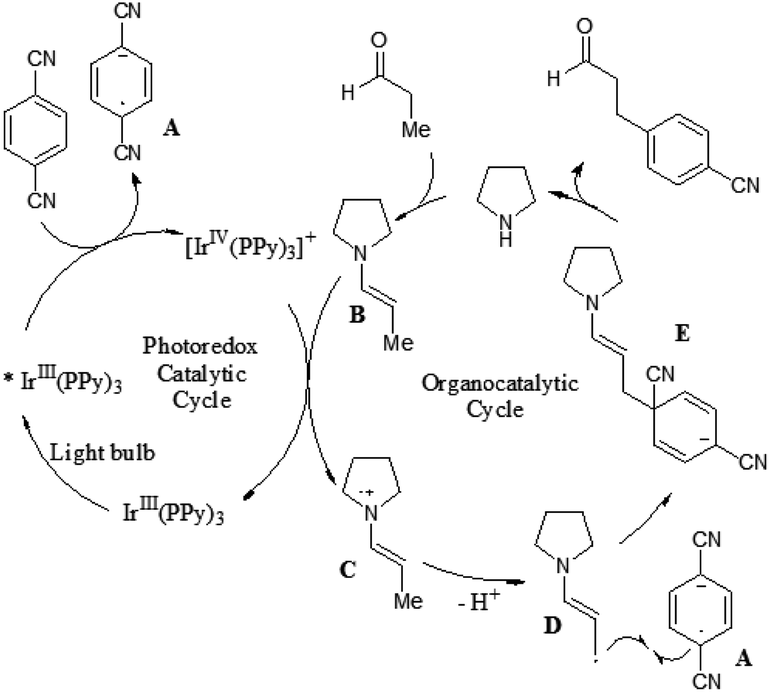 | ||
| Fig. 2 Possible mechanism. | ||
Although this photoredox-based strategy has been successfully developed for the direct β-arylation of carbonyl compounds, cyano-substitution appears to be essential for the arene coupling partner. More recently, Dong and co-workers disclosed a catalytic coupling that arises from the combination of electron-rich phosphine–Pd complexes and widely available aryl halides to enable the direct arylation of simple cyclic and acyclic ketones at the β-position (Scheme 4).8 This reaction exhibits a completely different site-selectivity under acidic conditions, whereas the use of stoichiometric bases in Buchwald–Hartwig–Miura α-arylation can generate the corresponding enolates and the reaction finally affords α-arylation products. Compared to the conventional conjugate addition of aryl nucleophiles to α,β-unsaturated compounds, some distinct advantages of this procedure are the operational simplicity, excellent functional group tolerance and no need of conjugated enones and aryl nucleophiles, which need several steps and redox procedures to prepare. This protocol is also applicable to the gram-scale synthesis of β-arylation of ketones with excellent yields under the optimized conditions. In addition, this new methodology is important and complementary to the previous directing group chelation-assisted C–H bonds activation4 and photoredox-based strategy.7
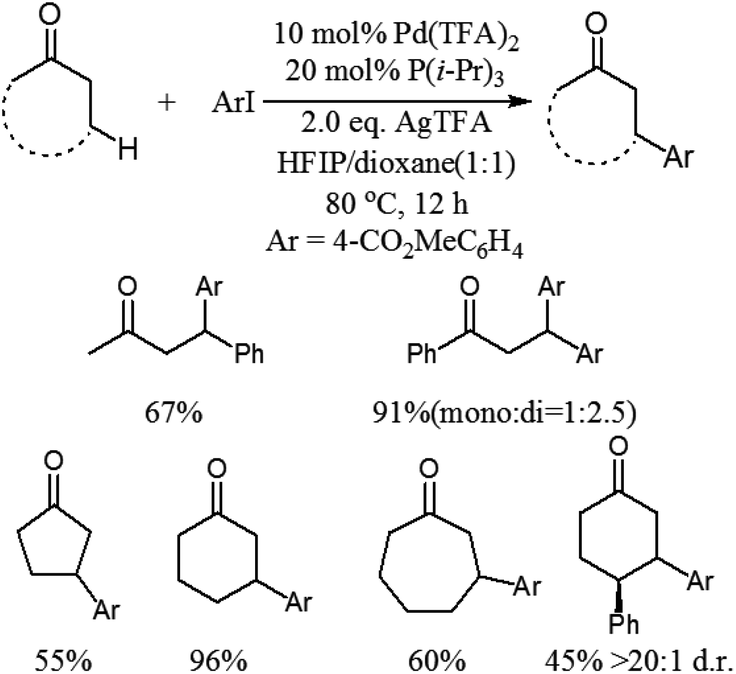 | ||
| Scheme 4 Palladium-catalyzed β-arylation of simple ketones. | ||
Mechanistically the reaction proceeds via the ketone oxidation, aryl halide activation and conjugate addition through a single catalytic cycle, in which an electrophilic palladium complex is generally required for ketone oxidation and oxidative addition into carbon–halide bonds which prefer electron-rich catalysts. Thus, the development of a suitable catalyst system, which is able to accommodate both needs during the catalytic cycle, is an important challenge of this strategy. Ultimately, the highly selective β-arylation of ketone has been achieved using electron-deficient Pd(TFA)2 in combination with electron-rich phosphine. A plausible mechanism is proposed (Fig. 3). Firstly, an enone and a Pd(0) species can be in situ generated from a sequence of Pd(II)–enolate formation, β-H elimination and reductive elimination of an acid (HX). And then the oxidative addition of Pd(0) into arylhalide bonds gives Aryl-Pd(II) species, which can undergo migratory insertion into the conjugated enone olefin to provide a Pd(II)–enolate intermediate. Protonation occurs from a Pd(II)–enolate intermediate by HX to give the β-arylated product along with a Pd(II) species.
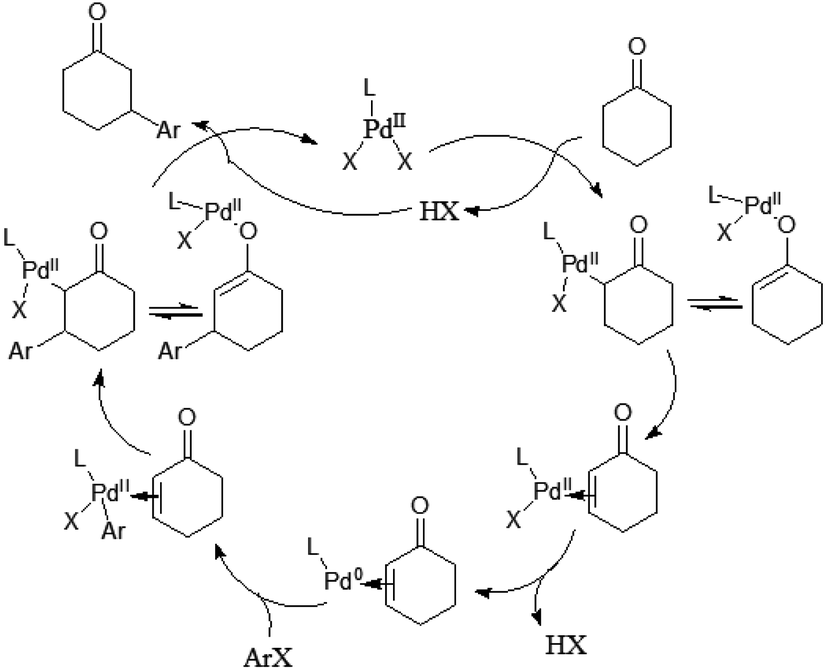 | ||
| Fig. 3 Possible mechanism. | ||
4. β-Aldol coupling of cyclic ketones
Although the α-aldol reaction of carbonyl compounds with electrophilic ketones or aldehydes has been widely exploited in organic chemistry, the corresponding “homo-aldol” transformation remains an important unsolved problem for direct β-functionalization of carbonyls.9 More recently, MacMillan and co-workers also explored the direct β-aldol coupling of cyclic ketones with aryl ketones based on a photoredox strategy (Scheme 5).10 A series of differentially substituted cyclohexanone derivatives and cyclopentanone were readily coupled with benzophenone under the analogous conditions for their previous β-arylation research, whereas seven-membered ketones gave low yields of the desired products. For aryl–alkyl ketone reaction partners, the excited state of the photocatalyst Ir(ppy)3 is not sufficiently reducing to induce ketyl radical formation. Fortunately, both electron-deficient and electron-rich acetophenone derivatives can readily couple with cyclohexanone to generate γ-hydroxyketone adducts using an analogous photocatalyst Ir(p-MeO-ppy)3, in which incorporation of electron-donating substituents into the aryl ligand would enhance the reduction potential of the IrL3 excited state. These results suggested that the photoredox cycle of this reaction can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. The specific catalytic mechanism is similar to that of their previous β-arylation of ketones and aldehydes (Fig. 4).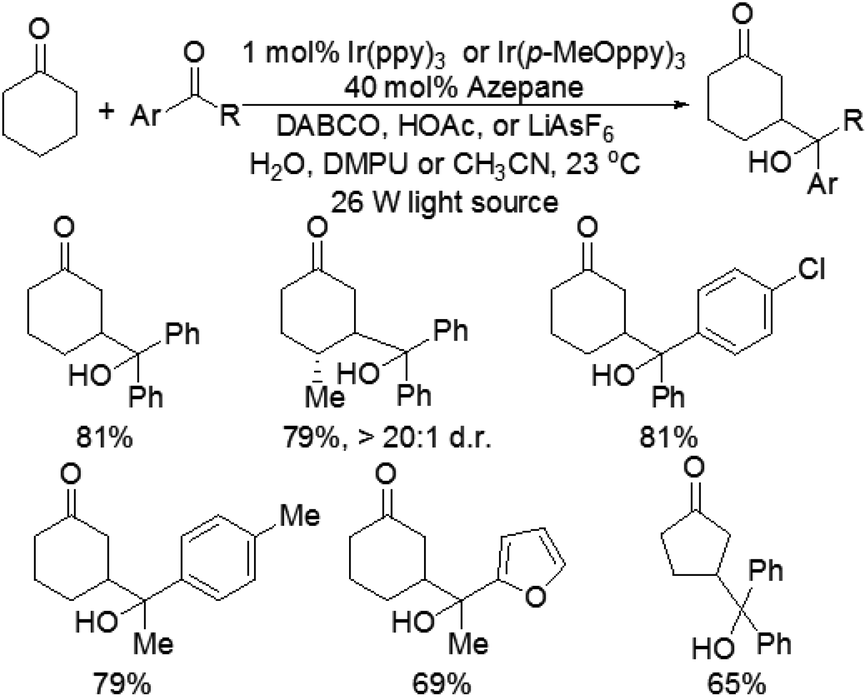 | ||
| Scheme 5 Direct β-coupling of cyclic ketones with aryl ketones. | ||
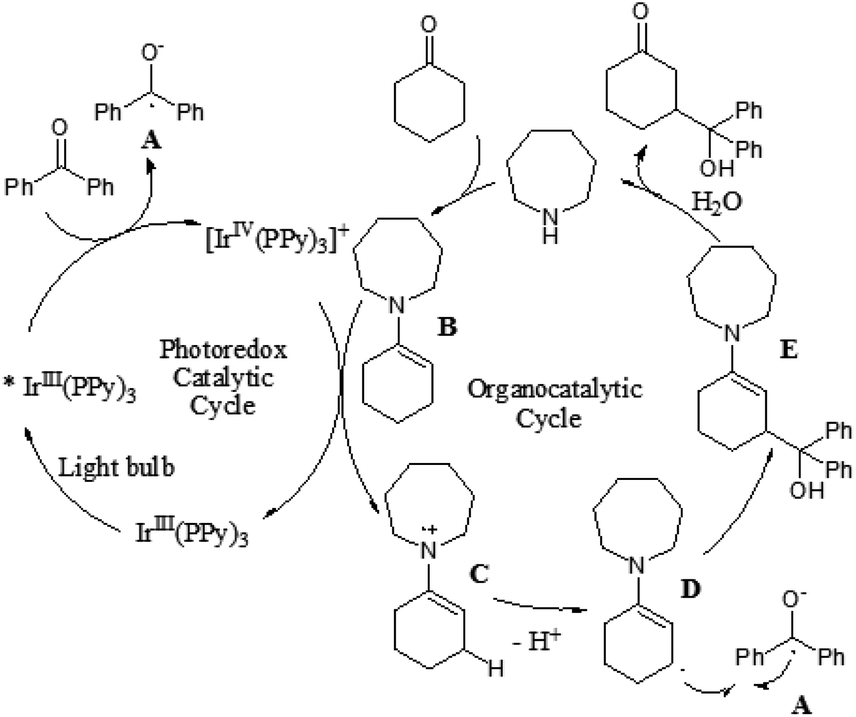 | ||
| Fig. 4 Possible mechanism. | ||
5. Conclusions
To summarize, we have presented a highlight of the area of direct functionalization of β-C(sp3)–H bonds of carbonyl compounds. From a synthetic point of view, these reactions provide novel and efficient tools for constructing various useful compounds. Gathering mechanistic insights could help us to understand the nature of these reactions and lead to the discovery of unique reactivities of metal catalysts. However, there are still some major challenges that need to be addressed in this rapidly developing area. Some reactions still suffer from some issues, such as the limitation of substrates scope and harsh conditions.The development of cheap and robust catalytic systems for the β-functionalization of carbonyl compounds is more attractive. The important challenge will be to develop an efficient protocol for construction of such carbon–heteroatom bonds and to extend the substrate scope. Although still in infancy, these seminal works have opened an entirely new dimension in the field of carbon–hydrogen bonds activation. As a consequence, new achievements in this field are expected to appear in the near future.
References
- (a) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (b) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- (a) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146 CrossRef CAS PubMed; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed.
- (a) D. A. Evans, G. Helmchen and M. Rüping, in Asymmetric Synthesis. The Essentials, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- For selected references, see: (a) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed; (b) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984–12986 CrossRef CAS PubMed; (c) M. Wasa, K. S. L. Chan, X.-G. Zhang, J. He, M. Miura and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570–18572 CrossRef CAS PubMed; (d) M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750–5753 CrossRef CAS PubMed; (e) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030–6032 CrossRef CAS PubMed.
- S. Ueno, R. Shimizu and R. Kuwano, Angew. Chem., Int. Ed., 2009, 48, 4543–4545 CrossRef CAS PubMed.
- A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261–7265 CrossRef CAS PubMed.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed.
- Z. X. Huang and G. B. Dong, J. Am. Chem. Soc., 2013, 135, 17747–17750 CrossRef CAS PubMed.
- For reviews on aldol reactions, see: (a) D. A. Evans, J. V. Nelson and T. R. Taber, Stereoselective Aldol Condensations, in Topics in Stereochemistry, ed. N. L. Allinger and E. L. Eliel, Wiley, New York, 1982, vol. 13 Search PubMed; (b) S. G. Nelson, Tetrahedron: Asymmetry, 1998, 9, 357–389 CrossRef CAS; (c) S. E. Denmark and R. A. Stavenger, Acc. Chem. Res., 2000, 33, 432–440 CrossRef CAS PubMed.
- F. R. Petronijević, M. Nappi and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323–18326 CrossRef PubMed.
| This journal is © the Partner Organisations 2014 |