
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalised polyaniline-based porous organic polymers for catalytic conversion of CO2 into cyclic carbonates†
Ulzhalgas Karatayeva and
Charl F. J. Faul*
and
Charl F. J. Faul*
School of Chemistry, University of Bristol, Bristol BS8 1TS, UK. E-mail: charl.faul@bristol.ac.uk
First published on 29th July 2025
Abstract
This study presents a sustainable CO2 capture and conversion strategy using functionalised polyaniline-based porous organic polymers. The catalyst, PTPA-COOH, enables efficient cyclic carbonate production, achieving high conversion and recyclability in the absence of solvents and co-catalysts. This route supports the transition to sustainable conversion of CO2 to green and economically attractive cyclic carbonates.
Global anthropogenic greenhouse gas (GHG) emissions predominantly originate from five sectors: electricity and heat production, agriculture, forestry, and other land use (AFOLU), industry, transportation, and buildings.1 According to the US Environmental Protection Agency (EPA), CO2 emissions represent the largest share, with the cement industry alone contributing approximately 7% (at least 2.1 billion tons annually).2 As shown in Fig. 1a, total global emissions reach 38.52 GtCO2eq,3 underscoring the need for coordinated mitigation strategies. Achieving net-zero targets will require stringent emission reduction policies, including the adoption of advanced technologies such as carbon capture, utilisation, and storage (CCUS), which is projected to reach commercial scale in the cement industry by 20304,5 and potentially reduce CO2 emissions by 0.43 tons per ton of cement by 2050.6
 | ||
| Fig. 1 (a) Sectoral contributions to GHG emissions. (b) Global market size of cyclic carbonates. Adapted with permission.3,17 Copyright 2024, Elsevier. | ||
CO2's increasing abundance, non-toxicity, and role as a sustainable carbon source render its fixation and conversion into chemicals, fuels, and polymers a promising strategy for atmospheric recycling. A notable utilisation pathway is the cycloaddition of CO2 to epoxides to form five-membered cyclic carbonates. This reaction exhibits high atom economy, generates no side products, and adheres to green chemistry principles.7,8 It simultaneously mitigates anthropogenic GHG emissions and produces high-value products, including electrolyte components for secondary batteries,9 monomers for polycarbonates and polyurethanes,9,10 plastics precursors,11 fuel additives,10,12 environmentally benign solvents,7,9,10 and intermediates for pharmaceutical and fine chemical synthesis.9,13 Cyclic carbonates, characterised by high solubility, elevated boiling points, low toxicity, and biodegradability, represent a valuable class of chemicals.14
The growing demand for cyclic carbonates, such as dimethyl, propylene, ethylene, glycerol, and styrene carbonates, is driving market expansion. The propylene carbonate segment is projected to grow at a compound annual growth rate (CAGR) of 5.57%, reaching US$752.3 million by 2030,15 while the overall market is expected to exceed US$7.1 billion by 2030 at a CAGR of 7.2% (Fig. 1b).16,17 Notably, current annual production of 100 kilotons of cyclic carbonates directly utilises approximately 40 kilotons of CO2.18
Over recent decades, numerous homogeneous catalysts, including organocatalysts,19 metal salen,20 and metalloporphyrin complexes,21 have been developed for CO2 conversion into cyclic carbonates. Although industrialised, these processes require high temperatures (T > 200 °C) and pressures (P > 6 MPa) and encounter challenges in product separation and catalyst reuse.22 In contrast, heterogeneous systems, despite issues of cost and efficiency, offer potential advantages. Among heterogeneous catalysts, such as metal complexes,23 metal–organic frameworks,24 and porous organic polymers (POPs),13,25 the latter have demonstrated superior performance owing to their flexible molecular design, tuneable pore parameters, and excellent chemical and mechanical stability. Moreover, POPs exhibit negligible volatility, facile regeneration, and robust covalent linkages, rendering them highly competitive for optimising CO2 adsorption and conversion via cost-effective synthesis routes.8
The targeted introduction of nitrogen active sites into POPs significantly enhances their CO2 binding capacity by promoting Lewis acid–base interactions.13,26 Functionalisation with amines and carboxylic acids further augments CO2 adsorption and separation through polar interactions and dipole–quadruple forces. Moreover, the abundant mesopores in these structures facilitate rapid reactant diffusion, thereby increasing the efficiency of CO2 conversion.8
Many effective heterogeneous catalysts require co-catalysts (e.g., tetrabutylammonium bromide) or rely on rare-earth or toxic heavy metals, raising sustainability and leaching concerns.9,23,24 Consequently, research is increasingly focused on developing metal-free nanoporous organic polymers with high activity and stability for ambient CO2 fixation, offering safer and more sustainable alternatives.
Here we report three porous polymers that incorporate both amines and carboxylic acids as desirable features using a Buchwald–Hartwig cross-coupling reaction (Scheme 1). Their chemical composition and morphology were confirmed by various characterisation techniques (Fig. S3–S5, ESI†). The polymers are insoluble in common organic solvents and water, facilitating recovery. We further evaluated the impact of introducing –COOH groups on conversion efficiency, along with practical factors such as recyclability, and performance with simulated flue gases.
 | ||
| Scheme 1 Synthesis of extended PTPA, PTPA-COOH and PTPA-2COOH. | ||
The synthesis procedure for generating these polymers, as shown in Scheme 1, is outlined in the ESI.†
N2 adsorption–desorption measurements indicate type II isotherms for the synthesised polymers (Fig. 2a), indicative of mesoporous materials. Specific surface areas (SSAs) of extended PTPA, PTPA-COOH and PTPA-2COOH are 118 m2 g−1, 117 m2 g−1 and 93 m2 g−1, respectively. A broad pore size distribution (PSD) (Fig. 2b) was observed within the mesoporous region, with some degree of microporosity. Gas uptake measurements indicate that extended PTPA, PTPA-COOH and PTPA-2COOH exhibit a CO2 capture capacity of 2.16 wt%, 3.44 wt% and 2.71 wt%, respectively, at 273 K and 1.37 wt%, 2.32 wt% and 1.73 wt%, respectively, at 298 K, up to 1 bar (Fig. 2c and d). These values are relatively high given their low SBET, implying strong interactions of CO2 molecules with the pore surfaces. The heats of adsorption (Qst) for extended PTPA, PTPA-COOH and PTPA-2COOH are 28.96 kJ mol−1, 27.63 kJ mol−1 and 28.07 kJ mol−1, respectively, and are typical values found for related materials as reported by our group before.27 Such values below 40 kJ mol−1 are indicative of physisorption, which offers an energy efficient means for catalytic surface regeneration compared to chemisorption (which would require significant more energy for desorption processes). These values therefore suggest that the polymers’ structural features are sufficient to promote CO2 enrichment near catalytic sites.
 | ||
| Fig. 2 (a) N2 sorption isotherms at 77 K and (b) PSD for the synthesised materials. CO2 isotherms of the polymers (c) at 273 K and (d) at 298 K. | ||
Table 1 provides information about materials’ porosity properties, CO2 uptakes and Qst values. It is noteworthy that despite higher pore volume and heat of adsorption of the extended PTPA, its ability for CO2 uptake is lower because of the high degree of mesoporosity in comparison with the other materials. In contrast, the extended PTPA-COOH not only has a higher proportion of micropores, which enhances CO2 adsorption, but also outperforms PTPA-2COOH, benefiting from both larger SSA and total pore volumes (Fig. 2b).
| Polymer | SBETa (m2 g−1) | Pore volume (cm3 g−1) | CO2 uptake at 1 bar, (wt%) | Qst (kJ mol−1) | |
|---|---|---|---|---|---|
| 273 K | 298 K | ||||
| a Calculated from N2 adsorption isotherms collected at 77 K (the uncertainty of N2 uptake is calculated by the standard deviation over three repeat experiments). | |||||
| PTPA | 118 ± 7 | 0.362 | 2.16 | 1.37 | 28.96 |
| PTPA-COOH | 117 ± 6 | 0.214 | 3.44 | 2.32 | 27.63 |
| PTPA-2COOH | 93 ± 9 | 0.211 | 2.71 | 1.73 | 28.07 |
To explore the functionality of these materials beyond simple CO2 capture, they were used for CO2 fixation in cycloaddition reactions with epoxides. This well-known reaction results in the production of cyclic carbonates, typically in the presence of solvents and co-catalysts.7 Previous work from our group showed successful CO2 fixation without these additives.28 Our initial investigations therefore used the well-studied and widely reported cycloaddition of CO2 with neat epichlorohydrin (ECH) with no co-catalyst to form the corresponding cyclic carbonate to benchmark the performance of our materials.
As anticipated, our bifunctional porous materials effectively converted CO2 and ECH into the corresponding cyclic carbonate with high yields, all without the use of solvents or co-catalysts (Table 2, Entries 1–3). Their broad PSDs facilitate efficient mass transfer for substrate molecules. Non-functionalised PTPA showed lower yields (63%) for this cycloaddition reaction, compared with >90% for the two COOH-functionalised materials. The activity of the two functionalised PTPAs can be attributed to the inclusion of carboxyl groups within the hierarchical cross-linked network structure. Interestingly, the material with two carboxylic groups per repeat unit showed identical results to the material with one –COOH group. Based on previous reports,9,29,30 a possible catalytic pathway for PTPA-COOH is proposed (see Fig. S16, ESI†). It is noteworthy that on carbonisation of these materials surface area increases significantly (>300 m2 g−1), but catalytic activity all but disappears (3% yields). See Table S2, ESI† for these details.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Substrate | t (h) | PCO2 (MPa) | Yieldc (%) |
| a After 5 cycles.b CO2/N2 = 15/85.c Values are derived from two repeat experiments. | |||||
| 1 | PTPA | ECH | 72 | 0.125 | 63 |
| 2 | PTPA-COOH | ECH | 72 | 0.125 | 93 |
| 3 | PTPA-2COOH | ECH | 72 | 0.125 | 92 |
| 4 | PTPA-COOH | ECH | 48 | 3 | 97 |
| 5a | PTPA-COOH | ECH | 48 | 3 | 88a |
| 6b | PTPA-COOH | ECH | 48 | 3 | 99b |
| 7 | PTPA | SO | 72 | 0.125 | 4 |
| 8 | PTPA-COOH | SO | 72 | 0.125 | 17 |
| 9 | PTPA-2COOH | SO | 72 | 0.125 | 8 |
Considering these results, studies to optimise the catalytic performance for the cycloaddition of ECH and CO2 were conducted using the best-performing extended PTPA-COOH. To further illustrate the advantages of the heterogeneous PTPA-COOH catalyst and explore wider use of our materials, we conducted reactions under elevated CO2 pressures (3.0 MPa) while maintaining the same temperature (100 °C) as previous trials. After 48 hours, a 97% yield for the conversion of ECH to (chloromethyl)ethylene carbonate was obtained (Table 2, entry 4). A crucial feature for any practical catalyst is its ability to be reused without a significant decrease in catalytic activity. PTPA-COOH was therefore tested for five cycles under identical reaction conditions (100 °C, 3 MPa, 48 hours) using ECH as the model substrate. The results demonstrated that the bifunctional PTPA-COOH catalyst can be efficiently recycled, with a slight decrease in performance after five cycles (Fig. 4 and Table 2, Entry 5) to 88%. Additionally, the FTIR spectrum (Fig. S17, ESI†) confirmed that the main peaks of PTPA-COOH were preserved.
The high catalytic performance of PTPA-COOH led us to explore its effectiveness under conditions with relatively low CO2 concentrations (15% CO2 in N2), which mimic industrial CO2 emissions (i.e., simulated flue gas). PTPA-COOH demonstrated exceptional catalytic activity, achieving a yield of 99% after 48 hours at 100 °C and 3.0 MPa (Table 2, Entry 6).
Another commonly studied terminal epoxide, styrene oxide (SO), was then used to further evaluate the catalytic abilities of our materials. Converting SO to its corresponding cyclic carbonate is challenging due to the less reactive β-carbon atom.31 Recent published results report typical low conversion values of trace amounts (120 °C, no co-catalyst),12 16% (100 °C, no co-catalyst),30 and 21% (25 °C, with TBAB).32 This lower reactivity is also reflected in the results we obtained (co-catalyst and solvent free), with the conversion yields under mild conditions for all three materials below 20%, as shown in Table 2, Entries 7–9. Among these materials, extended PTPA-COOH demonstrated the highest conversion (17%), followed by the extended PTPA-2COOH (8%). The non-functionalised PTPA exhibited the lowest capability towards this specific cycloaddition reaction, with a yield of only 4% recorded. These results clearly showed that harsher conditions, such as those used for the ECH cycling experiment, would be needed to explore useful catalytic activity for the more challenging epoxide substrate.
We therefore examined the effectiveness of PTPA-COOH with SO, as well as a further 7 epoxide substrates under the conditions used to obtain the highest ECH conversion (i.e., 100 °C, 48 hours and 3.0 MPa CO2, see Fig. 3). When using larger substrates under the same conditions, we noted reduced yields, which can be attributed either to the inherent activity of the substrates or to the blocking of the active sites, hindering access and decreasing their activity and potential for conversion. Additionally, product yields decreased from 99% to 71% as the kinetic diameter of the substrates increased. Despite these challenges, all the corresponding carbonates were produced with good to excellent yields. These results demonstrate the high versatility of PTPA-COOH in the cycloaddition of CO2.
 | ||
| Fig. 3 Catalytic performance of PTPA-COOH in the reaction of various epoxides with CO2 (values are derived from two repeat experiments). | ||
In a final exploration, we also tested the recyclability of PTPA-COOH with the challenging substrate, SO, with results shown in Fig. 4 alongside the results from the ECH cycling study. Keeping in mind that SO conversion yields are typically very low, it is noteworthy that after an initial 15% drop in conversion, this system continues to produce styrene carbonate at yields >50% after 5 cycles. A comparison of PTPA-COOH with other catalysts for the CO2 cycloaddition with SO is shown in Table S3 (ESI†).
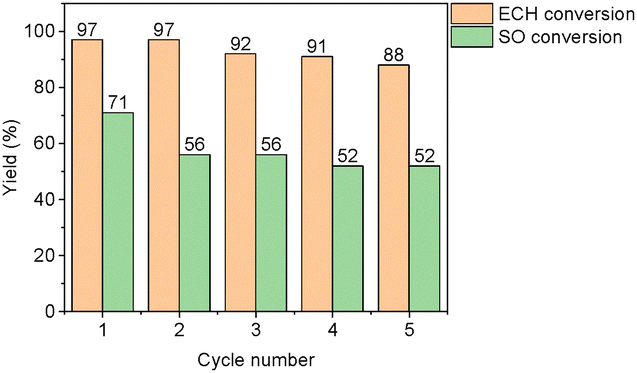 | ||
| Fig. 4 The recycling stability of PTPA-COOH (100 °C and 3.0 MPa CO2 pressure) over 5 cycles for ECH (orange) and SO (green). | ||
In conclusion, incorporating carboxyl groups into porous polymers enhances gas sorption and catalytic performance, although additional carboxylation did not consistently improve activity. The heterogeneous catalyst PTPA-COOH exhibits good CO2 adsorption capacity, easy recoverability, and good reusability, retaining catalytic activity over at least five cycles. These features not only reduce operational costs but also offer significant advantages over traditional homogeneous catalysts, which often suffer from limited reusability and high recovery costs. Given its bifunctional nature, structural robustness, substrate compatibility, and ability to catalyse reactions under industrially relevant conditions, we believe that PTPA-COOH is a promising candidate for practical applications in CO2 capture and conversion. These findings provide a promising strategy for the sustainable conversion of CO2 into cyclic carbonates of economic and technological interest, contributing to CO2 emission reduction and providing real routes to a green transition to address global challenges.
U. K. is grateful to the Bolashak International Scholarship program. The authors also appreciate the support provided by the Electron Microscopy Facility, Glassblowing Workshop, Mechanical Workshop, NMR Facility, and X-ray Crystallography Facility at the School of Chemistry, University of Bristol. The authors acknowledge use of the University of Bristol NanoESCA Laboratory.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- IPCC, 2014: Climate Change 2014: Synthesis Report, IPCC, Geneva, Switzerland, p. 151.
- K. Korczak, M. Kochański and T. Skoczkowski, J. Clean. Prod., 2022, 358, 132006 CrossRef CAS.
- M. Filonchyk, M. P. Peterson, L. Zhang, V. Hurynovich and Y. He, Sci. Total Environ., 2024, 935, 173359 CrossRef CAS PubMed.
- N. Thonemann, L. Zacharopoulos, F. Fromme and J. Nühlen, J. Clean. Prod., 2022, 332, 130067 CrossRef CAS.
- M. Wasim, A. Abadel, B. H. Abu Bakar and I. M. H. Alshaikh, Case Stud. Constr. Mater., 2022, 17, e01318 Search PubMed.
- M. Hanifa, R. Agarwal, U. Sharma, P. C. Thapliyal and L. P. Singh, J. CO2 Util., 2023, 67, 102292 CrossRef CAS.
- S. Bhunia, R. A. Molla, V. Kumari, S. M. Islam and A. Bhaumik, Chem. Commun., 2015, 51, 15732–15735 RSC.
- Q. Wu, K. Huang, F. Liu, P. Zhang and L. Jiang, Ind. Eng. Chem. Res., 2017, 56, 15008–15016 CrossRef CAS.
- X. Zhang, H. Zhang, B. Qiu, D. Zhu, S. Zhang, Y. Bian, J. Wang, D. Li, S. Wang, W. Mai, J. Chen and T. Li, Fuel, 2023, 331, 125828 CrossRef CAS.
- Y. Lu, Z. Chang, S. Zhang, S. Wang, Q. Chen, L. Feng and Z. Sui, J. Mater. Sci., 2020, 55, 11856–11869 CrossRef CAS.
- Y. Xie, T. T. Wang, R. X. Yang, N. Y. Huang, K. Zou and W. Q. Deng, ChemSusChem, 2014, 7, 2110–2114 CrossRef CAS PubMed.
- X. Meng, Y. Liu, S. Wang, J. Du, Y. Ye, X. Song and Z. Liang, ACS Appl. Polym. Mater., 2020, 2, 189–197 CrossRef CAS.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- F. Zhou, Q. Deng, N. Huang, W. Zhou and W. Deng, ChemistrySelect, 2020, 5, 10516–10520 CrossRef CAS.
- Propylene Carbonate Market Report Size, Share & Growth, https://www.marketresearchfuture.com/reports/propylene-carbonate-market-8089.
- Carbonate Market Statistics, Trends, https://www.alliedmarketresearch.com/carbonates-market.
- V. Mishra and S. C. Peter, Chem Catal., 2024, 4, 100796 CAS.
- P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS.
- Y. D. Li, D. X. Cui, J. C. Zhu, P. Huang, Z. Tian, Y. Y. Jia and P. A. Wang, Green Chem., 2019, 21, 5231–5237 RSC.
- X. Wu and M. North, ChemSusChem, 2017, 10, 74–78 CrossRef CAS PubMed.
- M. Yang, J. Deng, D. Guo, J. Zhang, L. Yang and F. Wu, Org. Biomol. Chem., 2019, 17, 5367–5374 RSC.
- Z. Lu, J. He, B. Guo, Y. Zhao, J. Cai, L. Xiao and L. Hou, Chin. J. Chem. Eng., 2022, 43, 110–115 CrossRef CAS.
- D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029–2035 CrossRef CAS.
- J. Zheng, M. Wu, F. Jiang, W. Su and M. Hong, Chem. Sci., 2015, 6, 3466–3470 RSC.
- P. B. Chen, J. W. Yang, Z. X. Rao, Q. Wang, H. T. Tang, Y. M. Pan and Y. Liang, J. Colloid Interface Sci., 2023, 652, 866–877 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- Y. Liao, J. Weber and C. F. J. Faul, Chem. Commun., 2014, 50, 8002–8005 RSC.
- B. B. Narzary, U. Karatayeva, J. Mintah, M. Villeda-Hernandez and C. F. J. Faul, Mater. Chem. Front., 2023, 7, 4473 RSC.
- U. Karatayeva, S. A. Al Siyabi, B. Brahma Narzary, B. C. Baker and C. F. J. Faul, Adv. Sci., 2024, 11, 2308228 CrossRef CAS PubMed.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- K. Qiao, F. Ono, Q. Bao, D. Tomida and C. Yokoyama, J. Mol. Catal. A: Chem., 2009, 303, 30–34 CrossRef CAS.
- D. Guo, C. Li, J. Zhang, G. Liu, X. Luo and F. Wu, J. Solid State Chem., 2021, 293, 121770 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc04043d |
| This journal is © The Royal Society of Chemistry 2025 |