
Zinc(II)-catalysed cyclocondensation of oxime ethers triggered by polarity inversion: construction of fused pyrroles†
Norihiko
Takeda
 ,
Sora
Fujita
,
Kaho
Murakami
,
Chihiro
Honda
,
Hina
Tomoda
,
Motohiro
Yasui
,
Takahiro
Yamada
and
Masafumi
Ueda
*
,
Sora
Fujita
,
Kaho
Murakami
,
Chihiro
Honda
,
Hina
Tomoda
,
Motohiro
Yasui
,
Takahiro
Yamada
and
Masafumi
Ueda
*
Kobe Pharmaceutical University, Motoyamakita, Higashinada, Kobe 658-8558, Japan. E-mail: masa-u@kobepharma-u.ac.jp
First published on 23rd April 2025
Abstract
An efficient approach for the synthesis of fused pyrroles such as indoles, thieno[3,2-b]pyrroles, and benzothieno[3,2-b]pyrroles possessing an N-alkoxy group was developed via the Zn(II)-catalysed cyclocondensation of α-hydroxy oxime ethers. Important features include the scalable synthesis with high yield achieved (up to 99%), environmentally benign, low-cost catalysts, and less hazardous reaction conditions.
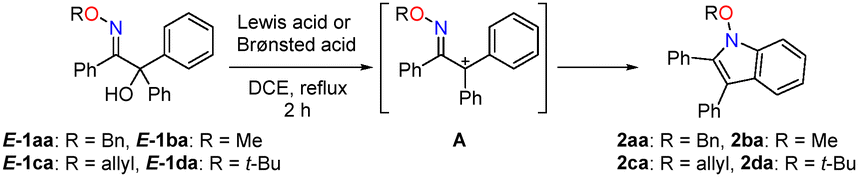
Fused pyrroles are crucial core moieties that are present in many bioactive natural products and pharmaceuticals.1 Benzene-fused pyrrole (indole) is considered a privileged motif in medicinal chemistry and natural pruducts.2 Interestingly, an N-alkoxyindole bearing an alkoxy group on the nitrogen atom of the indole unit is occasionally found in natural and biologically active products such as (R)-paniculidine B3a and N-butoxyindole-3-carbinol.3b This N-alkoxyindole moiety is also useful as a synthetic intermediate in total synthesis.4 Therefore, various strategies for synthesizing this motif have been developed, including (i) alkylation of N-hydroxyindoles,5 (ii) alkylative cycloaddition of nitrosoarenes with alkynes,6 (iii) modified Cadogan–Sundberg reductive cyclisation of o-nitro ethylbenzene carrying α,β-dialkoxycarbonyl groups,7 (iv) base-mediated cyclisation of o-nitrostyrenes,8 and (v) oxidative cyclisation of oxime ethers.9 However, these reactions often do not afford high yields. Although intramolecular cyclisation utilising oxime ethers is a reliable and practical synthetic method, not only is a cyano group at the Cα position of the oxime ethers essential, but stoichiometric amounts of a Lewis acid (FeCl3)9a or hypervalent iodine(III) reagent (PhICl2)9b are also required for successful transformations, resulting in excessive waste (Scheme 1(a)). Therefore, from the viewpoint of sustainability, the development of effective catalysed cyclisation reactions is still in high demand. Umpolung strategies are of considerable interest to organic chemists because they provide alternative synthetic routes to those imposed by the natural polarity of classical synthons.10 For example, the umpolung of an imino carbon enables the reaction with electrophiles and the umpolung of the α-carbon atom enables the reaction with nucleophiles.11 Furthermore, the regioselectivity of various cycloadditions can be inverted by polarity inversion.12 Various highly reactive intermediates such as azoalkene and nitrosoalkenes have been utilised to achieve the umpolung reactions of imine derivatives at the Cα-position.13 These intermediates can be generated in situ from the corresponding precursors, which are hydrazones and oximes with a leaving group at the Cα-position. Furthermore, these reactions are confined to functionalized hydrazones and oximes, or are accompanied by the liberation of HX, requiring more than two equivalents of the Grignard reagent or base for successful transformation (Scheme 1(b)). To address these limitations and expand our continuous endeavors in umpolung chemistry,14 we envisaged that the use of α-hydroxy oxime ether with a Lewis or Brønsted acid in the catalytic cyclodehydration triggered by polarity inversion at the Cα-position should furnish fused pyrrole scaffolds in a single step. This acid catalysed-cyclodehydration would proceed smoothly because the introduction of an alkoxy group on the imino nitrogen would effectively stabilize the in situ generated α-carbocation by the resonance effect. Moreover, the hydroxy group, as a leaving group at the Cα position, suppresses spontaneous degradation of the substrates. To the best of our knowledge, there is only one example of the synthesis of N-methoxyindole via the cyclisation of α-hydroxy oxime ether (67 mol% of TFA in CDCl3).15 However, a long reaction time (four days) was required and the substrate scope was not investigated. We herein describe the catalytic cyclisation of α-hydroxy oxime ethers by polarity inversion to produce fused pyrroles such as N-alkoxyindoles and thieno[3,2-b]pyrroles. The synthetic utility is highlighted by gram-scale reactions, the low catalyst loading, and low waste generation.
 | ||
| Scheme 1 (a) Representative examples of synthesis of N-alkoxyindoles, (b) umpolung reaction at the Cα-position, (c) this work. | ||
The umpolung cyclisation of α-hydroxy oxime ether 1aa, prepared from ethyl benzoylformate in 2 steps (for detail see, ESI†), as a model substrate was initially investigated (Table 1). A series of Lewis acid was screened as catalysts for the synthesis of N-alkoxyindole (Table 1). When E-1aa was treated with 20 mol% Sc(OTf)3 in 1,2-dichloroethane (DCE) under reflux, the desired N-benzyloxyindole 2aa was obtained in 32% yield (entry 1). The use of Yb(OTf)3 or Al(OTf)3 led to a slight increase in the yield, whereas Mg(OTf)2 or Cu(OTf)2 failed to afford the desired products (entries 2–6). Zn(OTf)2 as a catalyst increased the yield to 67% (entry 7). Other Lewis and Brønsted acids were not effective for this cyclisation (entries 8 and 9).16 The effects of the substituent on the oxygen atom of the oxime on the cyclisation reaction were then examined. Although the cyclisation of 1ba (R = Me) afforded the corresponding 2ba in a yield similar to that of 2aa, 1ca (R = allyl) afforded a lower yield (entries 10 and 11). In contrast, a significant increase in the yield to 85% was observed when 1da carrying a tert-butyl group (R = t-Bu: CCDC 2423379†) was used (entry 12). This suggests that the reaction strongly depends on the substituent effect of the alkoxy group. Furthermore, a lower catalyst loading (1 mol%) improved in the yield of 2da to quantitative (entry 13). Therefore, the optimum reaction conditions were determined to be 1 mol% Zn(OTf)2 in DCE under reflux.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Catalyst (mol%) | Yielda (%) |
| a Yield of the chromatographically pure product. b Not detected. c Reaction time: 13 h. | |||
| 1 | E-1aa | Sc(OTf)3 (20) | 2aa: 32 |
| 2 | E-1aa | Yb(OTf)3 (20) | 2aa: 41 |
| 3 | E-1aa | Al(OTf)3 (20) | 2aa: 49 |
| 4 | E-1aa | Fe(OTf)3 (20) | 2aa: 32 |
| 5 | E-1aa | Mg(OTf)2 (20) | 2aa: NDb |
| 6 | E-1aa | Cu(OTf)2 (20) | 2aa: NDb |
| 7 | E-1aa | Zn(OTf)2 (20) | 2aa: 67 |
| 8 | E-1aa | BF3·OEt2 (20) | 2aa: 13 |
| 9 | E-1aa | MsOH (20) | 2aa: 26 |
| 10 | E-1ba | Zn(OTf)2 (20) | 2ba: 63 |
| 11 | E-1ca | Zn(OTf)2 (20) | 2ca: 30c |
| 12 | E-1da | Zn(OTf)2 (20) | 2da: 85 |
| 13 | E-1da | Zn(OTf)2 (1) | 2da: 99 |
Various oxime ethers were screened under the optimal conditions (Scheme 2). First, oxime ethers 1db–1dg with various substituents on the benzene ring were investigated for the construction of N-alkoxyindoles. The desired indoles 2db–df were obtained in good-to-high yields, and reactions with electron-donating groups (EDG: methoxy, methyl, tert-butyl), as well as phenyl and chloro groups, at the para-position were feasible. In contrast, indole 2dg bearing the p-trifluoromethoxy group as the electron-withdrawing group (EWG) was obtained in moderate yield, even with a longer reaction time. This result suggests that an EWG at the para-position could not stabilize the proposed carbocation intermediate A; therefore, the generation of intermediate A would be prevented. Indole 2dh, bearing an m-methoxy group on the benzene ring, was also regioselectively obtained in a good yield. The reaction with 1di bearing a 2,3-dihydro-1,4-benzodioxine unit proceeded smoothly (2di: 86%). Subsequently, oxime ethers bearing aryl and heteroaryl groups on the imino carbon were investigated. EDG or EWG at the para-, meta-, or ortho-position, as well as heteroaryl groups, such as thiophene and benzothiophene, afforded the corresponding indoles (2dj–2do: 90–99%). Furthermore, oxime ethers bearing alkyl groups on the imino carbon were investigated. All the oxime ethers studied, including those with primary, secondary, and cyclic alkyl groups, were smoothly converted to the corresponding 2-alkyl indoles (2dp–2du: 71–99%).
 | ||
Scheme 2 Substrate scope. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reaction time: 18 h. b1dg (45%) was recovered. cReaction time: 8.5 h. d1dh (47%) was recovered. eReaction time: 13.5 h. fReaction time: 8 h. Reaction time: 18 h. b1dg (45%) was recovered. cReaction time: 8.5 h. d1dh (47%) was recovered. eReaction time: 13.5 h. fReaction time: 8 h. | ||
The reactions of oxime ethers with aryl groups having different electron densities at the Cα position were examined (Scheme 3). The reaction of E-1dv carrying p-methoxyphenyl and phenyl groups at the Cα position afforded methoxybenzene-fused indole 2dv without formation of the benzene-fused indole 2dv′. When Z-1dv was used under the optimal conditions, the methoxybenzene-fused indole 2dv was obtained as the sole product. The geometry of the oxime ether did not significantly affect the reaction. Additionally, E-1dw carrying m-methoxyphenyl and phenyl groups showed a similar trend, and methoxybenzene-fused indole 2dw was obtained in a regioselective manner, although a longer reaction time was required for successful transformation. These results suggest that in this cyclisation, an electron-rich aromatic ring was preferentially incorporated to construct fused pyrroles.
 | ||
| Scheme 3 Regioselective cyclisation. | ||
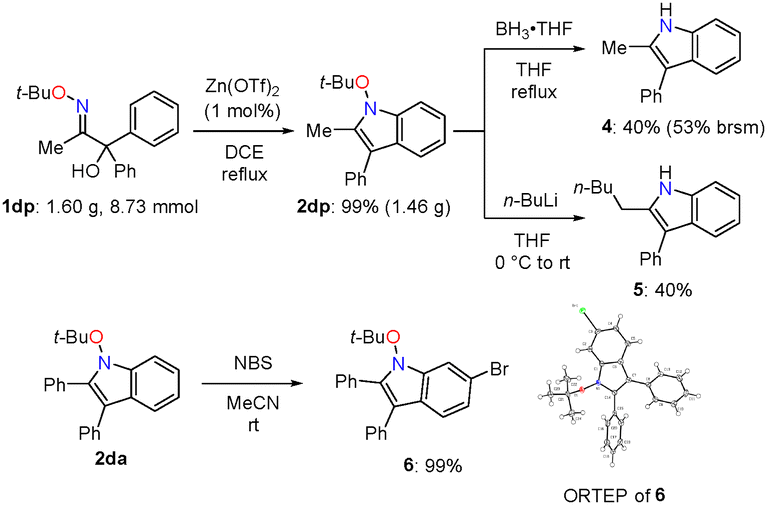
This umpolung cyclisation was scaled-up to the gram-scale, where N-alkoxyindole 2dp was obtained in 99% yield (1.46 g, Scheme 4). Chemical transformations were performed to demonstrate the synthetic utility of this methodology (Scheme 4). The tert-butyloxy group was removed by reductive N–O bond cleavage to afford NH indole 4 in moderate yield.17 Interestingly, reductive alkylation of 2dp with n-BuLi afforded NH indole 5 carrying an n-pentyl group at the 2-position.16 Moreover, with NBS in MeCN, N-alkoxyindole 2da was converted to brominated indole 6, which was structurally characterized by X-ray crystallography (CCDC 2423395†).
 | ||
| Scheme 4 Scalable synthesis and transformations. | ||
Several control experiments were performed to elucidate the reaction mechanism (Scheme 5). Considering the possibility that TfOH might be the actual catalyst, the reaction was performed with 1da and 1 mol% TfOH, yielding complex mixtures (eqn (1)). Additionally, in the absence of Zn(OTf)2, the reaction did not proceed at all (eqn (2)). These results tentatively indicate that Zn(OTf)2 acts as a catalyst in the umpolung cyclisation. To demonstrate the necessity of a second aryl group at the Cα position, 1dx bearing phenyl and methyl groups was utilised, leading to the formation of dehydrated 7 in low yield, along with the absence of the cyclised product (eqn (3)). This result indicates that the two aryl groups at the Cα position are critical for the umpolung cyclisation.
 | ||
| Scheme 5 Control experiments. | ||
Based on the above results and the literature,15,18 a plausible reaction pathway was proposed, as illustrated in Scheme 6. The coordination of Zn(OTf)2 with the hydroxy group of 1 (C-1), followed by dehydration, generates the tertiary carbocation D-1 at the Cα position, with resonance structures of nitrenium ion D-2, oxonium ion D-2′, and D-3. Three types of reaction pathways are possible for this cyclisation, depending on the resonance structure. (1) The 4π-electronic cyclisation (Aza-Nazarov cyclisation) of D-1 followed by aromatization of E affords N-alkoxyindole 2 (path a, 4π-electrocyclisation). (2) From the resonance structures D-2 and D-2′, electrophilic attack on the nitrenium ion by the benzene ring would give E (path b, SEAr cyclisation). (3) Alternatively, C–N bond formation could also occur via a nucleophilic amination pathway involving intermediate D-3 (path c, SNAr cyclisation). The tert-butyl group of α-hydroxy oxime ether 1da would efficiently prevent (i) the coordination of Zn(OTf)2 with the sterically congested tert-butyl ether moiety and (ii) degradation of the starting material 1 by Beckmann fragmentation from C-2.19 Therefore, the tert-butyl group as a substituent on the oxime ether 1da plays an important role in the umpolung cyclisation reaction.
 | ||
| Scheme 6 Proposed reaction pathway. | ||
Having defined an efficient protocol for the synthesis of fused pyrroles, the umpolung cyclisation of α-hydroxy oxime ethers containing thiophene and benzo[b]thiophens was next investigated (Scheme 7). The reaction of 8a–c having several different alkyl chains, such as n-hexyl, (benzyloxy)ethyl, and (tert-butylsilyloxy)ethyl, under the optimized conditions furnished the desired thieno[3,2-b]pyrroles 3a–c in good yields. Although thieno[3,2-b]pyrroles are present in π-functional materials and several biologically active compounds,20 reported synthetic methods of thieno[3,2-b]pyrroles depended on thermal- and metal-catalysed cyclisation of diazo compounds which are associated with potential explosion hazards.21 Therefore, from the viewpoint of safety, the developed synthetic approach is practical and enables diazo-free synthesis of thieno[3,2-b]pyrroles. Moreover, umpolung cyclisation of 8d–f carrying benzo[b]thiophene afforded benzothieno[3,2-b]pyrroles 3d–f in good to high yields.
 | ||
| Scheme 7 Synthesis of thieno[3,2-b]pyrroles and benzothieno[3,2-b]pyrroles. | ||
In summary, zinc-catalysed umpolung cyclisation of readily available α-hydroxy oxime ethers was developed for the synthesis of fused pyrroles such as N-alkoxyindoles, thieno[3,2-b]pyrroles, and benzothieno[3,2-b]pyrroles. This protocol is practical because of its simple operation, low catalyst loading, and low waste, as well as its relatively high yield and atom economy. Further studies to demonstrate the synthetic utility of the developed methodology, including in the synthesis of biologically active compounds, are underway.
This work was supported by JSPS KAKENHI (Grant Numbers JP24K09740, N. T. and JP21K06465, M. U.) and the Hyogo Science and Technology Association (N. T.).
Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. S. Gholap, Eur. J. Med. Chem., 2016, 110, 13–31 CrossRef CAS PubMed; (b) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC.
- (a) N. J. Pravin, R. S. Kavalapure, S. G. Alegaon, S. Gharge and S. D. Ranade, Bioorg. Chem., 2025, 154, 108092 CrossRef CAS PubMed; (b) A. Kumari and R. K. Singh, Bioorg. Chem., 2019, 89, 103021 CrossRef CAS PubMed; (c) C. Zheng and S.-L. You, Nat. Prod. Rep., 2019, 36, 1589–1605 RSC.
- (a) T. Kinoshita, S. Tatara and U. Sankawa, Chem. Pharm. Bull., 1985, 33, 1770–1773 CrossRef CAS; (b) S. M. Jump, J. Kung, R. Staub, M. A. Kinseth, E. J. Cram and L. N. Yudina, Biochem. Pharmacol., 2008, 75, 713–724 CrossRef CAS PubMed.
- (a) M. G. Ricardo, M. Schwark, D. Llanes, T. H. J. Niedermeyer and B. Westermann, Chem. – Eur. J., 2021, 27, 12032–12035 CrossRef CAS PubMed; (b) A. M. Levinson, Org. Lett., 2014, 16, 4904–4907 CrossRef CAS PubMed; (c) P. Wang, Y. Gao and D. Ma, J. Am. Chem. Soc., 2018, 140, 11608–11612 CrossRef CAS PubMed.
- Selected examples, see: (a) M. Somei, F. Yamada, T. Kurauchi, Y. Nagahama, M. Hasegawa, K. Yamada, S. Teranishi, H. Sato and C. Kaneko, Chem. Pharm. Bull., 2001, 49, 87–96 CrossRef CAS PubMed; (b) M. Somei, S. Inoue, S. Tokutake, F. Yamada and C. Kaneko, Chem. Pharm. Bull., 1981, 29, 726–738 CrossRef CAS.
- (a) N. Selvakumar, Y. Reddy, A. M. Azhagan, M. K. Khera, J. M. Babu and J. A. Iqbal, Tetrahedron Lett., 2003, 44, 7065–7069 CrossRef CAS; (b) N. Selvakumar and G. G. Rajulu, J. Org. Chem., 2004, 69, 4429–4432 CrossRef CAS PubMed.
- A. Penoni, G. Palmisano, G. Broggini, A. Kadowaki and K. M. Nicholas, J. Org. Chem., 2006, 71, 823–825 CrossRef CAS PubMed.
- B. T. McClay, K. E. Lambson, S. R. Banini, T. N. Akhmedov and B. C. G. Söderberg, Tetrahedron, 2023, 144, 133578 CrossRef CAS PubMed.
- (a) Y. Du, J. Chang, J. Reiner and K. Zhao, J. Org. Chem., 2008, 73, 2007–2010 CrossRef CAS PubMed; (b) Z. Yun, R. Cheng, J. Sun, D. Zhang-Negrerie and Y. Du, Adv. Synth. Catal., 2018, 360, 250–254 CrossRef CAS.
- (a) P. Spieß, S. Shaaban, D. Kaiser and N. Maulide, Acc. Chem. Res., 2023, 56, 1634–1644 CrossRef PubMed; (b) A. Y. Sukhorukov, Synlett, 2020, 439–449 CAS; (c) N. M. Rezayee, J. N. Lamhauge and K. A. Jørgensen, Acc. Chem. Res., 2022, 55, 1703–1717 CrossRef CAS PubMed.
- (a) Y. Hussain and P. Chauhan, Org. Biomol. Chem., 2021, 19, 4193–4212 RSC; (b) Y. Wu, L. Hu, Z. Li and L. Deng, Nature, 2015, 523, 445–450 CrossRef CAS PubMed; (c) J. Liu, C.-G. Cao, H.-B. Sun, X. Zhang and D. Niu, J. Am. Chem. Soc., 2016, 138, 13103–13106 CrossRef CAS PubMed.
- Selected examples, see: (a) L. Duan, X. Wang, Y. Gu, Y. Hou and P. Gong, Org. Chem. Front., 2020, 7, 2307–2312 RSC; (b) A. J. S. Alves, S. M. M. Lopes, M. S. C. Henriques, J. A. Paixao, T. M. V. D. Pinho and E. Melo, Eur. J. Org. Chem., 2017, 4011–4025 CrossRef CAS; (c) M. Xiang, C.-Y. Li, J. Zhang, Y. Zou, W.-S. Li, F. Tian, W.-J. Wan and L.-X. Wang, Org. Chem. Front., 2021, 6215–6219 RSC.
- Selected examples, see: (a) C. E. Sacks and P. L. Fuchs, J. Am. Chem. Soc., 1975, 97, 7372–7374 CrossRef CAS; (b) J. M. Hatcher and D. M. Coltart, J. Am. Chem. Soc., 2010, 132, 4546–4547 CrossRef CAS PubMed; (c) R. Sengupta, J. A. Witek and S. M. Weinreb, Tetrahedron, 2011, 67, 8229–8234 CrossRef CAS PubMed; (d) J. A. Witek and S. M. Weinreb, Org. Lett., 2011, 13, 1258–1260 CrossRef CAS PubMed; (e) I. Korboukh, P. Kumar and S. M. Weinreb, J. Am. Chem. Soc., 2007, 129, 10342–10343 CrossRef CAS PubMed.
- (a) N. Takeda, E. Futaki, Y. Kobori, M. Ueda and O. Miyata, Angew. Chem., Int. Ed., 2017, 56, 16342–16346 CrossRef CAS PubMed; (b) N. Takeda, M. Furuishi, Y. Nishijima, E. Futaki, M. Ueda, T. Shinada and O. Miyata, Org. Biomol. Chem., 2018, 16, 8940–8943 RSC; (c) E. Futaki, N. Takeda, M. Yasui, T. Shinada, O. Miyata and M. Ueda, Org. Biomol. Chem., 2020, 18, 1563–1566 RSC.
- X. Creary, E. A. Burtch and Z. Jiang, J. Org. Chem., 2003, 68, 1117–1127 CrossRef CAS PubMed.
- See ESI†.
- The reaction conditions for N–O bond cleavage, such as SmI2 and TfOH, led to the formation of complex mixtures.
- X. Creary, Y.-X. Wang and Z. Jiang, J. Am. Chem. Soc., 1995, 117, 3044–3053 CrossRef CAS.
- (a) J. H. M. Hill, D. P. Gilbert and A. Feldsott, J. Org. Chem., 1975, 40, 3735–3740 CrossRef CAS; (b) A. F. Ferris, J. Org. Chem., 1960, 25, 12–18 CrossRef CAS.
- (a) S. Stecko and D. T. Gryko, JACS Au, 2022, 2, 1290–1305 CrossRef CAS PubMed; (b) B. Fang, C. Hu, Y. Ding, H. Qin, Y. Luo, Z. Xu, J. Meng and Z. Chen, J. Heterocycl. Chem., 2021, 58, 1610–1627 CrossRef CAS; (c) K.-C. Ching, T. N. Q. Tran, S. N. Amrun, Y.-W. Kam, L. F. P. Ng and C. L. L. Chai, J. Med. Chem., 2017, 60, 3165–3186 CrossRef CAS PubMed; (d) Q. Ji, J. Gao, J. Wang, C. Yang, X. Hui, X. Yan, X. Wu, Y. Xie and M.-W. Wang, Bioorg. Med. Chem. Lett., 2005, 15, 2891–2893 CrossRef CAS PubMed.
- (a) B. J. Stokes, H. Dong, B. E. Leslie, A. L. Pumphrey and T. G. Driver, J. Am. Chem. Soc., 2007, 129, 7500–7501 CrossRef CAS PubMed; (b) S. Srinivasan and G. B. A. Schuster, Org. Lett., 2008, 10, 3657–3660 CrossRef CAS PubMed; (c) A. Vogt, F. Henne, C. Wetzel, E. Mena-Osteritz and P. Bäuerle, Beilstein J. Org. Chem., 2020, 16, 2636–2644 CrossRef CAS PubMed; (d) A. L. Samsonenko, A. S. Kostyuchenko, T. Y. Zheleznova, V. Y. Shuvalov, I. S. Vlasov and A. S. Flsyuk, Synthesis, 2022, 3227–3238 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2423379 and 2423395. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01362c |
| This journal is © The Royal Society of Chemistry 2025 |