
Engaging hydrazine hydrate as a hydrogen source for cobalt(II)-catalysed transfer hydrogenation of nitroaromatics†
Ravichandran
Manikandan
a,
Ramasamy
Shanmugam
b and
Annamalai
Pratheepkumar
 *a
*a
aDepartment of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore, Tamil Nadu 632014, India. E-mail: pratheepkumar.a@vit.ac.in; kumarpratheep0@gmail.com
bCO2 Research and Green Technologies Centre, Vellore Institute of Technology, Vellore, Tamil Nadu, 632014, India
First published on 15th May 2025
Abstract
Herein, we report earth-abundant cobalt(II)-catalysed hydrogenation of nitroaromatics to amines by utilizing hydrazine hydrate as the liquid hydrogen carrier. The significant highlight of this method is the production of greener byproducts. Remarkably, the developed method is promising in accessing drugs like butamben and benzocaine and other potential pharmaceutical precursors. The current protocol is highly functional group tolerant and also scalable to gram-scale synthesis. Density functional theory calculations have been performed with control studies to comprehend the mechanistic pathways.
Amines are ubiquitous motifs prevalent in countless natural products, pharmaceuticals, agrochemicals and functional materials. Therefore, efficient approaches to their synthesis are in high demand (Scheme 1a), thus finding a spot in the global market with a volume of 9.9 million tons in 2023 and is estimated to reach 15.2 million tons by 2032.1 Owing to their significance in industry and academia, it is highly desirable to develop new methodologies to access these motifs.2 The dry distillation of indigo led to the first discovery of aniline by Otto Unverdorben in 1826. However, after a decade Zinin et al. developed a protocol employing stoichiometric amounts of sodium sulfides for the reduction of nitroaromatics. Other seminal work by Béchamp demonstrated the synthesis of anilines by reducing nitro compounds using metallic iron in acidic media (Scheme 1b).1b,3 Thus, the most accessible and routinely employed route to anilines is reduction of nitro compounds due to their easy access, availability and low cost, which greatly improve the practicability and influence of this chemistry.4 In recent times, transfer hydrogenation and direct hydrogenation using hydrogen gas have filled the major synthetic gaps in reduction of nitro compounds.5
 | ||
| Scheme 1 (a) Pharmacologically active compounds of our work. (b) Traditional stoichiometric reduction route to anilines. (c) Exploitation of transition-metal-catalyzed reduction of nitro compounds. (d) Sustainable cobalt-catalyzed transfer hydrogenation of nitroaromatics. | ||
However, hydrogenation reactions depend on catalytic or stoichiometric usage of noble metals such as Rh,6 Ir,7 Pt,8 Pd,9 and Ru10 and hydrogen gas, operating at high pressure and temperature which are existing drawbacks, enticing chemists to bypass and develop new methods. In recent years, the hydrogenation of nitroaromatics has extended its range from organometallic reagents and harsh reaction conditions employing hydrogen gas to sustainable alcohols,11 abundant silanes,12 hydrazine hydrate,13 formic acid,14 ammonia boranes15 and Hantzch esters16 (Scheme 1c). Among them, hydrazine hydrate is advantageous due to its atom economy, low cost and zero-waste nature. Moreover, it is considered as a promising liquid-phase chemical hydrogen storage material with a hydrogen content of w = 8.0%.17 Most of the developed methodologies utilize the above-mentioned sustainable hydrogen sources in large amounts or as bulk solvents.18 Development of stoichiometric loading of hydrogen sources in reduction of nitroaromatics by employing earth-abundant metal catalysts is still in early stages and yet to be developed.19 In addition, chemo-selective hydrogenation of nitro compounds is achieved under noble metal catalysis.20 In recent times, heterogeneous catalysts have been developed towards chemo-selective hydrogenation of nitro compounds.21–24 Despite there being advances in hydrogenation of nitro compounds, chemo-selective hydrogenation using earth-abundant catalytic systems still needs to be addressed. In this regard, here we hypothesized using a cobalt complex in chemo-selective hydrogenation in the absence of ligand and stoichiometric base. A significant improvement of nitro reduction has been developed using tailor-made cobalt complexes.25 With the above-mentioned impressive and impactful advances in utilization of hydrazine hydrate as hydrogen source, we envisioned employing an earth-abundant cobalt catalyst in the transfer hydrogenation of nitroaromatics. Herein, we report a cobalt-catalyzed hydrogenation of nitroaromatics using hydrazine hydrate as a liquid hydrogen carrier. A few remarkable features of this reaction are that the developed method (i) is sustainable and atom economical, (ii) requires a low catalyst loading, (iii) utilizes a stoichiometric amount of hydrogen source, (iv) allows synthesis of potential synthons for drug development and synthesis of drugs like benzocaine and butamben which is an important highlight of this work (Scheme 1d).
At the outset, we treated nitrobenzene 1a with hydrazine hydrate in the presence of Co(acac)2 as the catalyst and KOtBu as a base in ethanol at 100 °C for 12 h (see ESI,† for further details; Table S1). To our delight, during our initial investigation we observed the desired product aniline 2a in 17% yield (entry 1). On varying the base K2CO3 a slight increase in yield was observed (entry 2). Remarkably, the reaction proceeded smoothly in the absence of base delivering the product in 31% yield (entry 3). Interestingly, the reaction yielded 49% on increasing the equivalents of hydrazine hydrate (entry 4). Other non-polar solvents such as toluene, heptane, and m-xylene yielded the desired compound 2a in 53–68% yields (entries 5–8). Furthermore, screening of solvents (entries 9–12) resulted in the maximum yield of 91% for CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (entry 10). Inspired by this result, next we screened other catalysts; however, a diminished yield was observed for Co(OAc)2·4H2O (entry 13). Unsurprisingly, [CoCp*(CO)I2] yielded the desired product without notable reduction in the yield (entry 14). From an economic viewpoint and commercial availability, we chose Co(acac)2 to explore the substrate scope. Further lowering the catalyst loading resulted in poor yield (entry 15). Conversely, an increase in the catalyst loading does not alter the yield of the product (entry 16). Undoubtedly, the reaction halted in the absence of catalyst and hydrazine hydrate (entries 17 and 18). On varying the equivalents of hydrazine hydrate, the desired product was afforded with comparable yield using 6 equiv. (entry 21). Moreover, shorter reaction time and increase in temperature failed to improve the yield of 2a (entries 22 and 23). Hence, we opted for entry 21 as representing the optimized conditions for the transfer hydrogenation of nitroaromatics.
:H2O (entry 10). Inspired by this result, next we screened other catalysts; however, a diminished yield was observed for Co(OAc)2·4H2O (entry 13). Unsurprisingly, [CoCp*(CO)I2] yielded the desired product without notable reduction in the yield (entry 14). From an economic viewpoint and commercial availability, we chose Co(acac)2 to explore the substrate scope. Further lowering the catalyst loading resulted in poor yield (entry 15). Conversely, an increase in the catalyst loading does not alter the yield of the product (entry 16). Undoubtedly, the reaction halted in the absence of catalyst and hydrazine hydrate (entries 17 and 18). On varying the equivalents of hydrazine hydrate, the desired product was afforded with comparable yield using 6 equiv. (entry 21). Moreover, shorter reaction time and increase in temperature failed to improve the yield of 2a (entries 22 and 23). Hence, we opted for entry 21 as representing the optimized conditions for the transfer hydrogenation of nitroaromatics.
Having the suitable optimized conditions in hand, further to test the practical applicability of our developed methodology, we evaluated a wide spectrum of nitroaromatics in terms of substrate scope and the results are tabulated in Table 1. Gratifyingly, the reaction is amenable to electron-donating groups, with 4-Me and 4-OMe of nitroaromatics yielding the corresponding products in 76–89% yields (2a–2c). Interestingly, we further extended the scope to halides flanked on ortho and para positions under the standard reaction conditions and obtained the desired product in very good yields (2d–2g). Remarkably, iodo and bromo derivatives often undergo oxidative addition tolerated under these conditions to yield the corresponding products. Most importantly, the developed protocol is compatible with hydroxyl functional group in delivering the desired products with excellent yield of 90% (2h). Besides, nitroaromatics bearing amine substituted at the ortho position smoothly yielded the corresponding products 2i in 85% yield. The reaction was also possible with dual nitro substituents flanked on the aromatic ring (2j and 2k). To prove the effectiveness of this reaction, benign and widespread substituents such as alkyne 2l, nitriles (2m–2n), aldehyde 2o, ketone 2p, ester 2q and amides (2r–2t) smoothly underwent the reaction chemoselectively to furnish the desired products in moderate to good yields without any side reactions. However, 4-nitrobenzaldehyde and 3-nitroacetophenone delivered corresponding hydrazone products 2o and 2p in synthetically viable yield. Encouraged by the above results, then we investigated heterocyclic nitroaromatics under the standard reaction conditions. Surprisingly, the reaction was compatible and smooth, rendering the desired heterocyclic amines in 69–85% yields (2u–2z). However, treatment of 5-nitroisatin in the standard conditions yielded the desired amine and along with the reduction of ketone functional group afforded 5-aminoindolin-2-one. An important highlight of this methodology is the preparation of pharmaceutically important intermediates like BMS-986236 2aa, boscalid 2ab, tizanidine 2ac and paracetamol 2ad in good yields of 73–87%. Late-stage reduction of nitroaromatics led to the synthesis of synthons for linezolid 2ae and drugs like benzocaine 2af and butamben 2ag and emotional health hormone like 2-phenylethylamine 2ah which greatly improved the practicability and influence of this chemistry. Importantly the current protocol is scalable to gram-scale synthesis in delivering 2a in 84% yield (see ESI†).

To gain further insight into the reaction mechanism, we carried out a series of reactions to propose a plausible mechanism (see ESI,† for further details; Scheme S1). Initially, the mercury poisoning test was carried out under standard conditions with a drop of mercury, resulting in the desired product 2a without a notable reduction in the yield (Scheme S1a, ESI†) confirming that the system is indeed homogenous. Generally, the reduction of nitrobenzene occurs in two pathways, i.e., path a: formation of nitroso intermediate followed by hydroxylamine reduction to yield aniline. In the other case, path b, it undergoes reduction through formation of azoxy, azo and hydrazo intermediates. To further prove the reaction mechanism, we treated nitroso A under standard conditions. Gratifyingly, it delivered the aniline in 76% yield. Encouraged by this result, we further treated hydroxylamine B under standard conditions, predictably delivering aniline in 74% yield. Further to examine whether azoxy C, azo D and hydrazo E intermediates were involved, we treated azoxy, azo and hydrazo separately under standard conditions. Unfortunately, the azo intermediate resulted in aniline with a drop in yield to 12%. However, no product formation is observed in the case of azoxy and hydrazobenzene. Additionally, we carried out density functional theory (DFT) calculations at the M06-L/Def2-SVP level to gain mechanistic insight into the cobalt-catalyzed transfer hydrogenation of nitroaromatics (see ESI†).26 Initially, reduced CoI(acac)2 complex IN1 and the formation of active Co-H27 species occurs with free energy of +8.36 kcal mol−1, followed by nitrobenzene 1a coordination led to IN2. Further, first hydride transfer at TS1 with +36.44 kcal mol−1 and subsequent formation of intermediate IN3 −9.52 kcal mol−1 with the oxidation state of Co(III). Further, the nitroso A coordinated in IN4 Co(I) −10.44 kcal mol−1 underwent a second hydride transfer, rendering IN5 Co(III) −39.08 kcal mol−1. Further, third hydride transfer to hydroxylamine B at IN7 with Co(III) −26.03 kcal mol−1 produced aniline 2a and IN8 −77.32 kcal mol−1 followed by proton transfer, regenerated the Co(acac)2 catalyst. Above all, TS1 shows an energy barrier of 24.16 kcal mol−1. Hence, the hydride transfer is considered a rate-determining step. Based on the experimental and DFT calculations, the transfer hydrogenation of nitro to amine via direct route is considered to be a favourable pathway (Scheme 2).1b,25a
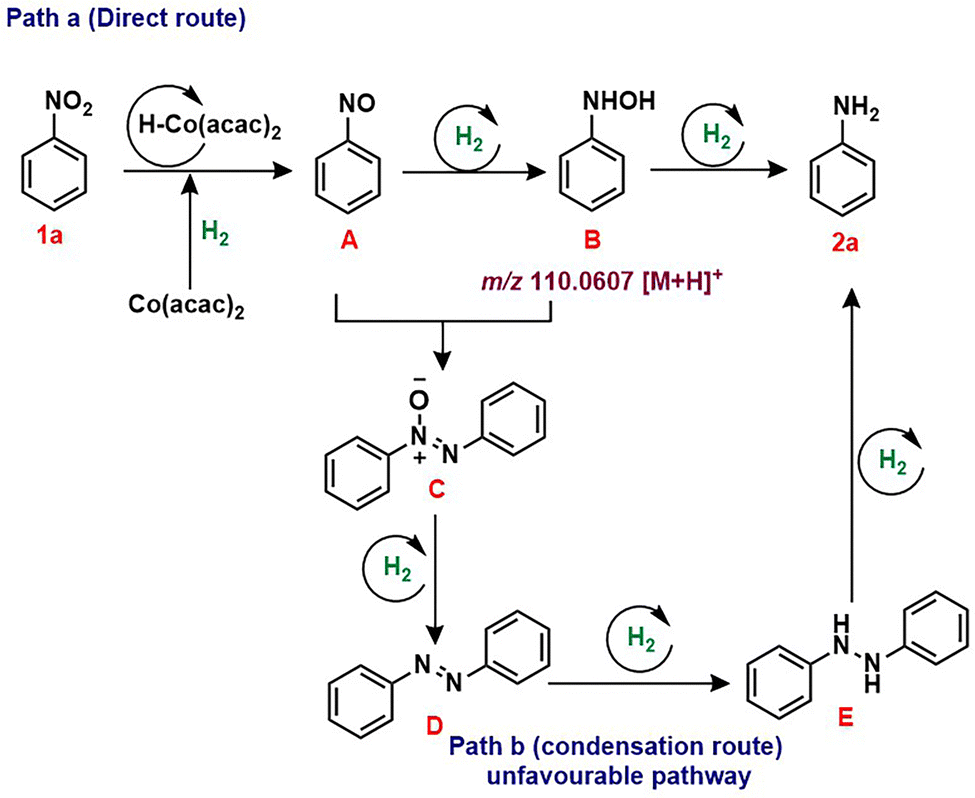 | ||
| Scheme 2 Plausible reaction mechanism for the transfer hydrogenation of nitroaromatics. | ||
In summary, we have presented an efficient method for the cobalt(II)-catalyzed selective hydrogenation of nitro compounds to amines, utilizing inexpensive and environmentally friendly hydrazine hydrate as the hydrogen carrier. Key highlights of this approach include chemoselective reduction, functional group tolerance, the synthesis of drug intermediates, and the late-stage hydrogenation of nitro compounds to potential drugs. Usage of stoichiometric amount of hydrazine hydrate as the hydrogen source enhances the value of this method. Importantly, being ligand and base free, using a commercially available and cost-effective cobalt catalyst, and affording greener byproducts augment its applicability in both academia and industry. We anticipate that this current method will be adopted into a modern organic chemist's toolbox in accessing biologically important amines by reducing commercially available and low-cost nitro compounds.
We gratefully acknowledge the financial support from the DST-SERB, India, Project File No. SRG/2023/002161 and Vellore Institute of Technology, Vellore for providing the seed grant – SG20220089 to conduct the research. We thank VIT-SIF for instrument facilities. R. M. acknowledges VIT for providing a fellowship. R. M. further thanks Dr. Charles Beromeo Bheeter for support during his initial period of doctoral degree.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed; (b) D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed; (c) V. Goyal, N. Sarki, B. Singh, A. Ray, M. Poddar, A. Bordoloi, A. Narani and K. Natte, ACS Appl. Nano Mater., 2020, 3, 11070–11079 CrossRef CAS.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
- (a) Y. Liu, Y. Lu, M. Prashad, O. Repič and T. J. Blacklock, Adv. Synth. Catal., 2005, 347, 217–219 CrossRef CAS; (b) F. A. Khan, J. Dash, C. Sudheer and R. K. Gupta, Tetrahedron Lett., 2003, 44, 7783–7787 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS.
- (a) R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed; (b) V. Zubar, A. Dewanji and M. Rueping, Org. Lett., 2021, 23, 2742–2747 CrossRef CAS PubMed; (c) N. Garg, A. H. Chowdhury and B. Sundararaju, Tetrahedron Green Chem., 2024, 3, 100043 CrossRef; (d) C. Dewangan, S. Kumawat, T. Bhatt and K. Natte, Chem. Comm., 2023, 59, 14709–14712 RSC.
- (a) V. Chugh, B. Chatterjee, W.-C. Chang, H. H. Cramer, C. Hindemith, H. Randel, T. Weyhermüller, C. Farès and C. Werlé, Angew. Chem., Int. Ed., 2022, 61, e202205515 CrossRef CAS PubMed; (b) Y. Wei, J. Wu, D. Xue, C. Wang, Z. Liu, Z. Zhang, G. Chen and J. Xiao, Synlett, 2014, 25, 1295 CrossRef.
- (a) S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376–7385 CrossRef CAS; (b) S. Chen, G. Lu and C. Cai, New J. Chem., 2015, 39, 5360–5365 RSC.
- (a) P. Lara, A. Suárez, V. Collière, K. Philippot and B. Chaudret, ChemCatChem, 2014, 6, 87–90 CrossRef CAS; (b) P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445–2465 RSC.
- (a) R. J. Rahaim and R. E. Maleczka, Org. Lett., 2005, 7, 5087 CrossRef CAS PubMed; (b) M.-U. Hung, S.-T. Yang, M. Ramanathan and S.-T. Liu, Appl. Organomet. Chem., 2018, 32, e3976 CrossRef.
- (a) R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A.-E. Surkus, H. Junge, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 14375–14379 CrossRef CAS PubMed; (b) K. Naveenkumar, P. A. Ravindra, B. Archana, N. Kannan, G. Pavithran and A. Sinha, J. Mol. Struct., 2023, 1294, 136302 CrossRef; (c) S. Revathi and T. Ghatak, J. Catal., 2024, 429, 115207 CrossRef CAS.
- (a) X. Liu, S. Ye, H.-Q. Li, Y.-M. Liu, Y. Cao and K.-N. Fan, Catal. Sci. Technol., 2013, 3, 3200–3206 RSC; (b) V. Goyal, N. Sarki, K. Natte and A. Ray, J. Indian Chem. Soc., 2021, 98, 100014 CrossRef CAS.
- (a) K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769–1771 RSC; (b) M. Orlandi, F. Tosi, M. Bonsignore and M. Benaglia, Org. Lett., 2015, 17, 3941–3943 CrossRef CAS PubMed; (c) S. Panda, A. Nanda, R. R. Behera, R. Ghosh and B. Bagh, Chem. Commun., 2023, 59, 4527–4530 RSC.
- (a) D. Cantillo, M. Baghbanzadeh and C. O. Kappe, Angew. Chem. Int. Ed., 2012, 51, 10190–10193 CrossRef CAS; (b) D. Cantillo, M. M. Moghaddam and C. O. Kappe, J. Org. Chem., 2013, 78, 4530–4542 CrossRef CAS PubMed; (c) H.-Z. Zhu, Y.-M. Lu, F.-J. Fan and S.-H. Yu, Nanoscale, 2013, 5, 7219–7223 RSC; (d) S.-C. A. Lin, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Molecular Catalysis, 2019, 466, 46–51 CrossRef CAS.
- A. H. Romero, ChemistrySelect, 2020, 5, 13054–13075 CrossRef CAS.
- (a) J. Du, J. Chen, H. Xia, Y. Zhao, F. Wang, H. Liu, W. Zhou and B. Wang, ChemCatChem, 2020, 12, 2426–2430 CrossRef CAS; (b) S. Kumari, S. Roy, K. Ranjan Saha and S. Kundu, ChemCatChem, 2024, 16, e202400901 CrossRef CAS.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- (a) A. Furst, R. C. Berlo and S. Hooton, Chem. Rev., 1965, 65, 51–68 CrossRef CAS; (b) D. Motta, I. Barlocco, S. Bellomi, A. Villa and N. Dimitratos, Nanomaterials, 2021, 11, 1340 CrossRef CAS PubMed; (c) V. A. Matyshak and O. N. Silchenkova, Kinet. Catal., 2022, 63, 339–350 CrossRef CAS; (d) S. Bellomi, D. Motta, M. Stucchi, L. Prati, N. Dimitratos and A. Villa, Catalysts, 2024, 14, 119 CrossRef CAS.
- (a) H. Lu, Z. Geng, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Lett., 2016, 18, 2774–2776 CrossRef CAS PubMed; (b) M. F. Ansari, Anshika, J.-B. Sortais and S. Elangovan, Eur. J. Org. Chem., 2024, e202301278 CrossRef CAS.
- V. Goyal, T. Bhatt, C. Dewangan, A. Narani, G. Naik, E. Balaraman, K. Natte and R. V. Jagadeesh, J. Org. Chem., 2023, 88, 2245–2259 CrossRef CAS PubMed.
- H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- H. K. Kadam and S. G. Tilve, RSC Adv., 2015, 5, 83391–83407 RSC.
- S. Dey, D. Panja, A. Sau, S. D. Thakur and S. Kundu, J. Org. Chem., 2023, 88, 10048–10057 CrossRef CAS PubMed.
- D. Sharma, P. Choudhary, S. Kumar and V. Krishnan, J. Colloid Interface Sci., 2024, 657, 449–462 CrossRef CAS PubMed.
- R. Lakshmi Priya and S. Ganesh Babu, Langmuir, 2024, 40, 10293–10304 CrossRef CAS PubMed.
- (a) U. Sharma, P. Kumar, N. Kumar, V. Kumar and B. Singh, Adv. Synth. Catal., 2010, 352, 1834–1840 CrossRef CAS; (b) S. Kumar and R. Gupta, ChemistrySelect, 2017, 2, 8197–8206 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (c) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC; (d) A. Kumar, S. Pattanaik, G. Joshi, M. K. Sahu, E. D. Jemmis and C. Gunanathan, ACS Catal., 2024, 14, 4249–4264 CrossRef CAS.
- (a) S. P. Semproni, C. Milsmann and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 9211–9224 CrossRef CAS PubMed; (b) P. Daw, S. Chakraborty, G. Leitus, Y. Diskin-Ponser, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 2500–2504 CrossRef CAS; (c) D. Mahapatra, A. Sau, T. Ghosh, A. Roy and S. Kundu, Org. Lett., 2024, 26, 6001–6005 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedure, characterization data and 1H, 13C, and HRMS spectral data. See DOI: https://doi.org/10.1039/d5cc01160d |
| This journal is © The Royal Society of Chemistry 2025 |