
A bionic carbon framework activates the oxygen lattice sites of NiFe2O4 to enhance electrochemical water splitting†
Can
Wang‡
a,
Yue
Xiao‡
a,
Kailin
Li
*b,
Qing
Sun
c,
Bo
Yang
a and
Yuxin
Zhang
 *a
*a
aCollege of Materials Science and Engineering, Chongqing University, Chongqing 400043, P. R. China. E-mail: zhangyuxin@cqu.edu.cn
bState Key Laboratory of Molecular Engineering of Polymers, Department of Chemistry, Fudan University, Shanghai 200433, P. R. China. E-mail: likailin@fudan.edu.cn
cSchool of Chemistry and Chemical Engineering, Multi-Scale Porous Materials Center, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 400044, P. R. China
First published on 5th April 2025
Abstract
A well-designed carbon-supported material enables the promotion of efficient oxygen evolution. Herein, a NiFe2O4 catalyst supported by a bionic carbon framework (BCF) was synthesized, and the elements doped into the BCF facilitate electron transfer between oxygen and active metal centers. The as-prepared catalyst exhibits a low overpotential and Tafel slope, and the design approach presented here represents a promising method to design electrocatalysts for water splitting.
With the development of industry and the requirement for green energy, electrocatalytic water splitting for hydrogen production and carbon emissions reduction has become the best choice.1–3 However, the whole electrocatalytic system is restricted by the oxygen evolution reaction (OER), which is controlled by a four-electron transfer process.4–7 In recent years, NiFe2O4 has emerged as one of the most promising non-precious metal-based electrocatalysts for the OER. Despite the potential of NiFe2O4, it still has poor electrical conductivity and a limited number of active sites, and thus a lot of strategies, such as morphology control, modulation of the material composition and defect engineering, have been utilized to solve these issues with the aim of achieving a low cost and highly active electrocatalyst for water splitting.8–10
A favorable carrier not only disperses the catalyst better but also modulates the electronic structure and enhances the electrocatalytic performance. Carbon supports are frequently employed as conductive frameworks for catalysts. However, existing studies typically focus on doping with a single element, which at the same time has a limited effect on altering the reaction mechanism by which NiFe2O4 operates.11–14 This constraint significantly hinders the efficiency of most carbon-composite NiFe2O4 catalysts in the oxygen evolution reaction (OER). Additionally, the utilization of these carbon materials tends to be relatively insufficient.15–19 Constructing a bionic structure from diatomite is an effective method to address the issue above.20–22
There are two potential mechanisms for NiFe2O4 in the OER in alkaline electrolytes, and the lattice oxygen mechanism (LOM) is considered to be the most promising pathway.23–26 Compounding and doping are great solutions for improving the catalytic performance of NiFe2O4 by forcing the material to operate mainly under LOM control rather than a switchable OER route.27,28 However, these methods often require rare-earth or precious metals and involve complex synthesis methods, which limits their scalability for large-scale applications. Moreover, the ability to control the shift in catalytic mechanisms by doping the complex rather than NiFe2O4 itself remains an area that is yet to be fully explored.
Herein, diatomite (De) was used as a bionic hard template to construct a sulfur and nitrogen co-doped bionic carbon framework (SNC) on which the NiFe2O4 catalyst was loaded. Characterized by high conductivity and more active sites, in alkaline media, the NiFe2O4/SNC electrode delivered a small overpotential of 242 mV at 10 mA cm−2 and exhibited a lower Tafel slope of 91.42 mV dec−1 (RuO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :140.2 mV dec−1). Additionally, its excellent OER performance was confirmed through chemical probe studies and pH-dependent analysis, which revealed the activation of the LOM in NiFe2O4. The electrode also demonstrated outstanding durability, maintaining more than 98% of its initial performance at 10 mA cm−2 over 48 h of continuous electrocatalysis, significantly outperforming its NiFe2O4 counterpart.
:140.2 mV dec−1). Additionally, its excellent OER performance was confirmed through chemical probe studies and pH-dependent analysis, which revealed the activation of the LOM in NiFe2O4. The electrode also demonstrated outstanding durability, maintaining more than 98% of its initial performance at 10 mA cm−2 over 48 h of continuous electrocatalysis, significantly outperforming its NiFe2O4 counterpart.
The NiFe2O4/SNC material was synthesized in four steps (details provided in the ESI†), with the corresponding schematic of the process illustrated in Fig. 1a. As shown in Fig. S1 (ESI†), the sulfur and nitrogen-doped bionic carbon framework was synthesized via the polymerization of metanilic acid (MA) and p-toluenesulfonic acid (PA) on the surface of the diatomite. Subsequently, NiFe layered double hydroxides (LDH) were synthesized in situ on the polymer framework, and the composites were reduced to NiFe2O4/SNC@De by heating at 900 °C in nitrogen (Fig. S2, ESI†). Finally, the NiFe2O4/SNC was obtained after removing the diatomite hard templates using KOH (Fig. S3, ESI†). The multi-hollow, cake-like morphology of the diatomite template is clearly visible in scanning electron microscopy (SEM) image shown in Fig. 1b, confirming the successful synthesis of the bionic structure through this method. The sheet-like NiFe2O4 structure is uniformly distributed and retains the pores from the diatomite template, as observed in the higher magnification image in Fig. 1c. The pore size distribution and specific surface area (SSA) of NiFe2O4/SNC were measured using the Brunauer–Emmett–Teller (BET) method (Fig. S4, ESI†), which indicated that the pores on the surface are predominantly mesoporous (average pore size = 6.18 nm, SSA = 19.97 m2 g−1). The transmission electron microscopy (TEM) images in Fig. 1d reveal the sulfur and nitrogen-doped carbon structure beneath the NiFe2O4 nanosheets. The high-resolution TEM (HRTEM) image in Fig. 1e shows clear lattice fringes corresponding to the (220) crystal planes of NiFe2O4/SNC, demonstrating the high crystallinity of the material. In contrast, the disordered carbon structure of SNC does not exhibit lattice fringes due to its low crystallinity. The uniformity of the NiFe2O4/SNC synthesis and the successful doping of sulfur and nitrogen into the carbon structure were further confirmed by the Ni, Fe, O, S, and N elemental mapping results shown in Fig. 1f.
 | ||
| Fig. 1 (a) Synthesis of NiFe2O4/SNC; (b) and (c) SEM images of the as-prepared NiFe2O4/SNC; (d) TEM image of NiFe2O4/SNC and (e) HRTEM image and (f) elemental mapping of Ni, Fe, O, S and N from NiFe2O4/SNC. | ||
The X-ray diffraction (XRD) pattern of NiFe2O4/SNC exhibited a set of peaks corresponding to NiFe2O4 (JCPDF no. 74-2081), as shown in Fig. 2a. The specific peaks associated with diatomite (JCPDF no. 76-0935) were absent, indicating the complete etching of the diatomite structures from NiFe2O4/SNC, as well as in the other samples. The Fourier-transform infrared (FTIR) spectrum of NiFe2O4/SNC is presented in Fig. S5 (ESI†). Characteristic peaks for the O–H band, Fe–O stretching, and Ni–O stretching of NiFe2O4 were observed at approximately 3436.5 cm−1, 600.9 cm−1, and 436.8 cm−1, respectively.29 The peaks associated with SNC appeared at 1653.3 cm−1, 1408.4 cm−1, and 1006.6 cm−1, which were attributed to C–N, N–H, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S stretching vibrations, respectively.30
S stretching vibrations, respectively.30
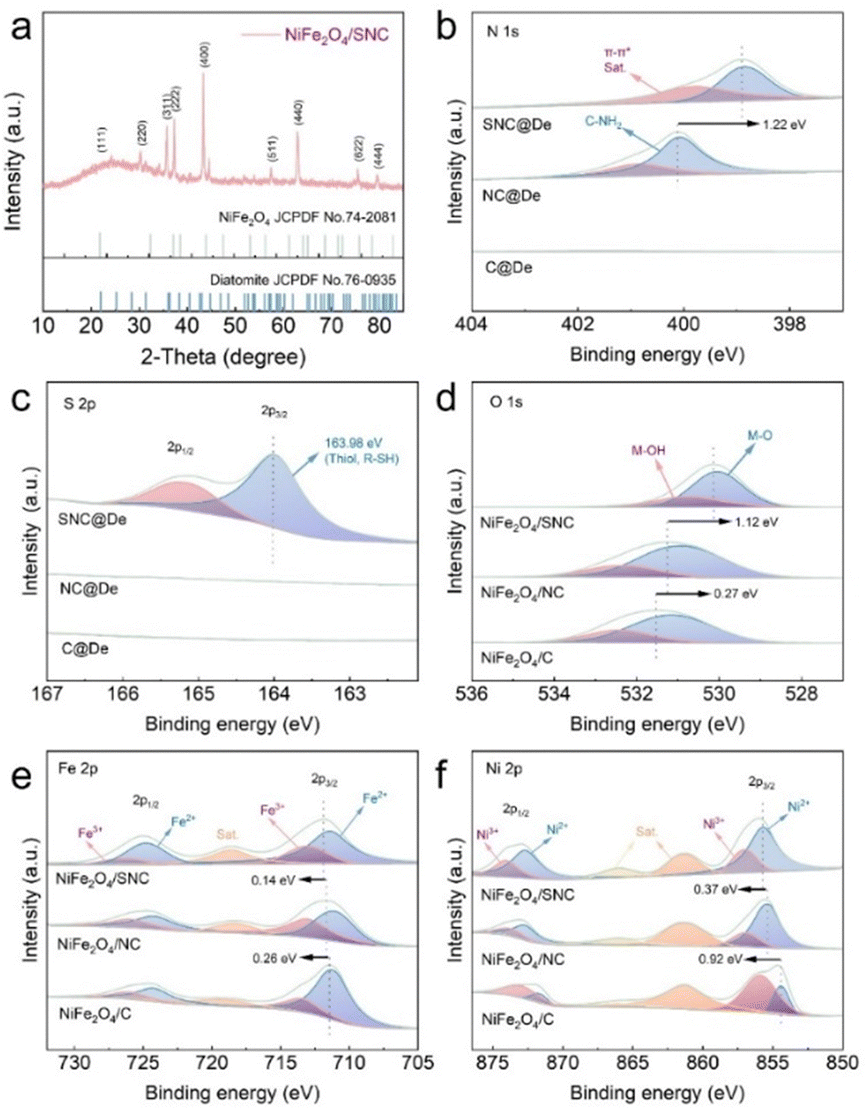 | ||
| Fig. 2 (a) XRD pattern of NiFe2O4/SNC; (b) N 2p XPS spectra and (c) S 2p XPS spectra of SNC, NC and C; (d) Ni 2p XPS spectra, (e) Fe 2p XPS spectra and (f) O 1s XPS spectra of NiFe2O4/SNC, NiFe2O4/NC and NiFe2O4/C. | ||
To confirm the success of the N and S doping into the carbon framework and to assess the electronic changes induced by doping, X-ray photoelectron spectroscopy (XPS) measurements were conducted on C@De, NC@De, and SNC@De for N and S, and on NiFe2O4/C, NiFe2O4/NC, and NiFe2O4/SNC for O, Fe, and Ni. The corresponding XPS spectra of N 1s (Fig. 2b) displayed two distinct peaks, confirming the successful doping of nitrogen into the lattice of NC@De and SNC@De. Moreover, the blue peak exhibited a positive shift (≈1.22 eV) from NC to SNC, demonstrating that the addition of sulfur enhances electron transfer between C and N. The XPS spectra of S 2p (Fig. 2c) confirm the successful doping of sulfur into the SNC@De lattice. A peak at 163.98 eV corresponds to S 2p3/2, indicating a strong interaction between sulfur and the carbon framework. In the XPS spectrum of O 1s (Fig. 2d), peaks at 530.04 eV and 530.75 eV correspond to oxygen–metal bonds and hydroxyl groups, respectively, consistent with previous reports for NiFe2O4.31 The XPS spectra of Fe 2p for NiFe2O4/SNC (Fig. 2e) displayed two main peaks at 711.44 eV and 724.80 eV, corresponding to Fe2+ 2p3/2 and Fe2+ 2p1/2, respectively, and two peaks at 713.08 eV and 726.34 eV, corresponding to Fe3+ 2p3/2 and Fe3+ 2p1/2. Compared to NiFe2O4/NC and NiFe2O4/C, the peak positions in the O 1s spectrum were shifted positively, while those in the Fe 2p spectrum were shifted negatively, indicating enhanced electron transfer from O to Fe after doping. Similarly, doping increased the valence of the Ni species (Fig. 2f), suggesting enhanced electron transfer from O to Ni. Moreover, comparing the N-doped (≈0.26 eV for Fe, ≈0.92 eV for Ni) and S, N co-doped (≈0.40 eV for Fe, ≈1.29 eV for Ni) samples with the undoped sample, it was found that co-doping more effectively enhanced electron transfer, improving the binding energy. These results demonstrate that co-doping significantly promotes electron transfer between lattice oxygen and the catalytically active centers, thereby enhancing oxygen evolution performance.
Using a typical three-electrode system, the OER activity of NiFe2O4/SNC was evaluated in 1 M KOH solution without IR compensation, using a platinum mesh as a counter electrode and an Hg/HgO electrode as the reference electrode. For comparison, NiFe2O4/NC, NiFe2O4/C, RuO2 and Ni foam were also tested as comparison working electrodes. The linear sweep voltammetry (LSV) curves in Fig. 3a show that NiFe2O4/SNC can drive the OER with a lower overpotential (η10) of 242 mV at 10 mA cm−2 than NiFe2O4/NC (337 mV), NiFe2O4/C (347 mV) and RuO2 (502 mV). In addition, the NiFe2O4/SNC electrode has a lower Tafel slope which indicates faster OER kinetics, as shown in Fig. 3b. The Tafel slope of 91.42 mV dec−1 is lower than that of 132.5 mV dec−1 for NiFe2O4/NC, 124.6 mV dec−1 for NiFe2O4/C and 140.2 mV dec−1 for RuO2. The results reveal that S doping of the carbon can increase the catalytic activity of the electrode and enhance the reconstruction process of the pre-catalyst to the real catalyst.10,27 Furthermore, the capabilities of NiFe2O4/NC are superior to those of NiFe2O4/C, indicating that the presence of N leads to some positive effects on the catalytic properties. Electrochemical impedance spectroscopy (EIS) plots were obtained for a better understanding of the charge transfer efficiency of the electrodes. Fig. S6 (ESI†) shows the EIS plots and the equivalent circuit diagram of the NiFe2O4/SNC, NiFe2O4/NC and NiFe2O4/C electrodes, which confirms that the N,S-doped carbon layer reduces the charge transfer resistance and effectively promotes electron transfer, leading to enhanced OER activity.
 | ||
| Fig. 3 (a) LSV curves of the as-prepared catalysts in KOH electrolyte (1.0 mol L−1) without internal resistance (IR) compensation, scan rate: 5 mV s−1; (b) corresponding Tafel plots at low potentials; (c) current density (ΔJ = Ja − Jc) versus scanning rate and the corresponding linear slopes for NiFe2O4/SNC, NiFe2O4/NC, NiFe2O4/C and RuO2; (d) comparison of the performance of NiFe2O4/SNC in alkaline electrolyte with other reported OER catalysts. | ||
To further investigate the factors affecting the OER activity, the double layer capacitance (Cdl) was estimated by cyclic voltammetry tests at different sweep rates in the non-Faraday region. As the conversion between the electrochemical active surface area (ECSA) and Cdl is positively correlated, a higher Cdl can justify a larger ECSA. As shown in Fig. 3c and Fig. S7 (ESI†), the Cdl values are detected to be 2.22 mF cm−2, 1.93 mF cm−2, 1.90 mF cm−2 and 1.16 mF cm−2 for NiFe2O4/SNC, NiFe2O4/NC, NiFe2O4/C and RuO2, respectively. A larger ECSA means more active sites. Compared with NiFe2O4/NC and NiFe2O4/C, the N and S co-doping of the carbon layers also effects the ECSA, giving NiFe2O4/SNC the highest value. In comparison with other OER catalysts from the last two years, NiFe2O4/SNC exhibits extraordinary performance, as shown in Fig. 3d and Table S1 (ESI†). Long-term stability is a key parameter that we used to evaluate the stability of NiFe2O4/SNC by continuously running the OER operation for 48 h at a current density of 10 mA cm−2. The small voltage decay (≈1.8%) that we observed indicated that NiFe2O4/SNC has excellent stability for the OER (Fig. S8, ESI†).
To gain deeper insights into the influence that the S and N doping in the carbon layer has on OER activity, the OER mechanisms of the samples were investigated by using a chemical probe to identify the oxidized oxygen intermediate. The LOM route involved peroxo-like (O22−) and superoxol-like (O2−) negative species, and the O22− species can serve as an indicator of whether the OER proceeds via the LOM route.28 Owing to its strong interaction with the O22− species, the tetramethylammonium cation (TMA+) was added to the electrolyte to probe the presence of O22− species.23,24 As shown in Fig. 4a, The OER activity of NiFe2O4/SNC decreased remarkably when the electrolyte was changed to TMAOH. In contrast, the OER activity of NiFe2O4/NC and NiFe2O4/C only decreased slightly (Fig. 4b), reflecting that the OER process on the NiFe2O4/SNC electrode proceeded via the LOM route. Moreover, the pH dependence of the OER activity for NiFe2O4/SNC, NiFe2O4/NC and NiFe2O4/C is shown in Fig. 4c and Fig. S9 (ESI†). The OER activity of NiFe2O4/SNC was highly dependent on the pH of the electrolyte, while the pH dependence was considerably weaker for NiFe2O4/NC and NiFe2O4/C. Furthermore, the current densities of NiFe2O4/SNC, NiFe2O4/NC and NiFe2O4/C at 1.6 V as a function of the pH were plotted, as shown in Fig. 4d. These results confirmed that the OER activity of NiFe2O4/SNC had a much higher pH dependence than that of NiFe2O4/NC and NiFe2O4/C. The higher pH dependence of NiFe2O4/SNC implied that S doping may trigger the LOM pathway with a non-concerted proton–electron transfer process.26,31 In other words, S,N doping of the carbon layer trigged the transition of the OER mechanism of NiFe2O4 from the AEM to the LOM.23
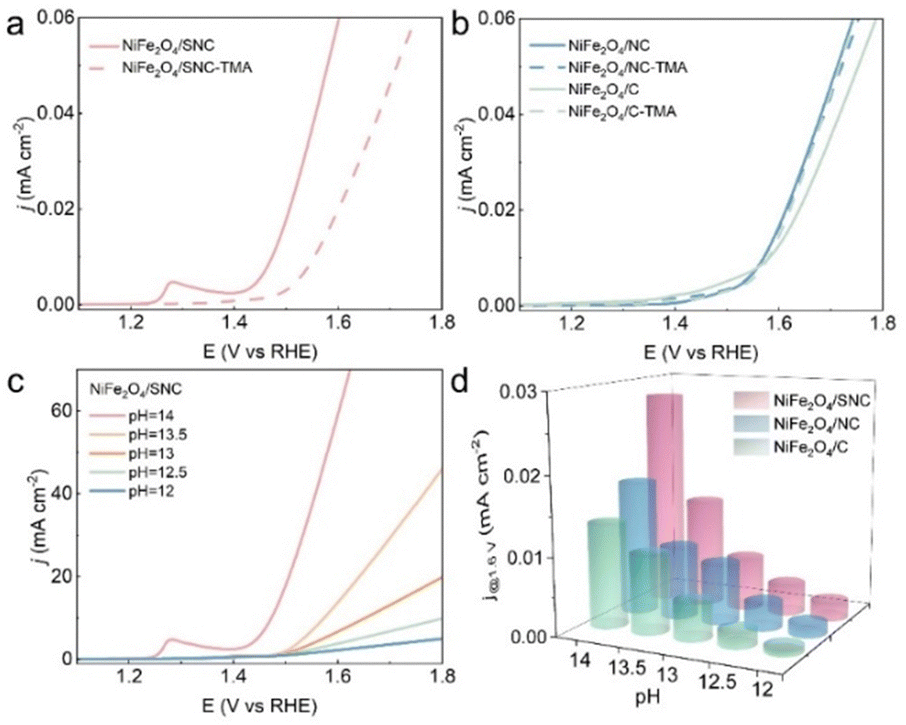 | ||
| Fig. 4 (a) and (b) Polarization curves for NiFe2O4/SNC, NiFe2O4/NC and NiFe2O4/C in 1.0 M KOH or 1.0 M TMAOH; (c) LSV tests for NiFe2O4/SNC in different concentration KOH solution; (d) current densities of NiFe2O4/SNC, NiFe2O4/NC and NiFe2O4/C at 1.6 V vs. RHE as a function of pH value. | ||
In summary, a series of highly efficient hybrid bionic NiFe2O4-based electrodes were developed using a multi-step sacrificial template method. NiFe2O4/SNC exhibits exceptionally high catalytic activity and remarkable durability. The outstanding OER performance of NiFe2O4/SNC can be attributed to the following factors: (i) the catalysts, synthesized via the described method, have the natural three-dimensional porous structure of diatoms, which provides a greater number of active sites; (ii) the carbon layer with defects (doping), formed through the polymerization of organic compounds followed by high-temperature calcination, enhances the electrical conductivity of the electrodes; and (iii) this is the first instance where sulfur and nitrogen co-doping in the carbon layer has effectively shifted the OER mechanism of NiFe2O4 from the AEM to the LOM pathway, significantly improving the catalytic performance of the catalyst. The well-designed NiFe2O4/SNC material provides novel insights into the transition of the OER mechanism in NiFe2O4-based catalysts induced by elemental doping in the supporting complexes rather than by directly modifying the catalysts themselves.
This work was supported by the financial support of the National Natural Science Foundation of China (grant no. 22405052 and 52378217), and the China Postdoctoral Science Foundation (grant no. 2024M750491). The project was supported (No. 2024 cdjqyjcyj−001) by the fundamental research funds for the central universities, and the Taishan Industry Leading Talents Project that provided valuable data and analyzed the prospect of water splitting electrocatalysts.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Norskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, Nat. Energy, 2021, 6, 462–474 CrossRef CAS.
- H. Li, Y. Lin, J. Duan, Q. Wen, Y. Liu and T. Zhai, Chem. Soc. Rev., 2024, 53, 10709–10740 RSC.
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174 CrossRef CAS PubMed.
- J.-T. Ren, L. Chen, H.-Y. Wang, W.-W. Tian and Z.-Y. Yuan, Energy Environ. Sci., 2024, 17, 49–113 RSC.
- Y. Zhao, D. P. A. Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Rev., 2023, 123, 6257–6358 CrossRef CAS PubMed.
- Z. Pei, H. Zhang, D. Luan and X. Lou, Matter, 2023, 6, 4128 CrossRef CAS.
- X. Wang, F. Liu, H. Qin, J. Li, X. Chen, K. Liu, T. Zhao, W. Yang, M. Yu, G. Fan and F. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202409628 Search PubMed.
- Z. Wu, H. Zhang, S. Zuo, Y. Wang, S. Zhang, J. Zhang, S. Zang and X. Lou, Adv. Mater., 2021, 33, 2103004 CrossRef CAS PubMed.
- Q. Chen, R. Wang, F. Lu, X. Kuang, Y. Tong and X. Lu, ACS Omega, 2019, 4, 3493–3499 Search PubMed.
- M. Fei, H. Shi, J. Zhao, N. Kang, W. He, H. Li and F. Yang, ChemCatChem, 2018, 10, 5174–5181 CrossRef CAS.
- Y. Chen, J. Xu, Y. Chen, L. Wang, S. Jiang, Z. Xie, T. Zhang, P. Munroe and S. Peng, Angew. Chem., Int. Ed., 2024, 63, e202405372 CrossRef CAS PubMed.
- L. Xu, S. A. Shah, H. Khan, R. Sayyar, X. Shen, I. Khan, A. Yuan, W. Yaseen, Z. A. Ghazi, A. Naeem, H. Ullah, X. Li and C. Wang, J. Colloid Interf. Sci., 2022, 617, 1–10 CrossRef CAS PubMed.
- Z. Liu, B. Tang, X. Gu, H. Liu and L. Feng, Chem. Eng. J., 2020, 395, 125500 CrossRef.
- T. N. Luong, T. L. L. Doan, P. M. Bacirhonde and C. H. Park, Int. J. Hydrogen Energy, 2025, 99, 1108–1118 Search PubMed.
- J. Zhang, Y. Jiang, Y. Wang, C. Yu, J. Cui, J. Wu, X. Shu, Y. Qin, J. Sun, J. Yan, H. Zheng, Y. Zhang and Y. Wu, Electrochim. Acta, 2019, 321, 134652 CrossRef CAS.
- X. Zhang, X. Zhang, X.-G. Wang, Z. Xie and Z. Zhou, J. Mater. Chem. A, 2016, 4, 9390–9393 RSC.
- W. Zong, D. Rao, H. Guo, Y. Ouyang, Y.-E. Miao, W. Wang, J. Wang, F. Lai and T. Liu, Nanoscale, 2020, 12, 10977–10986 RSC.
- Y. Yang, A. Li, X. Cao, F. Liu, S. Cheng and X. Chuan, RSC Adv., 2018, 8, 35672–35680 RSC.
- K. Li, X. Liu and Y. Zhang, Biogeotechnology, 2023, 1, 100037 Search PubMed.
- C. Wang, Y. Zhang, S. Liu and D. Wang, Trans. TJU, 2024, 30, 518–543 CAS.
- X. Wang, H. Zhong, S. Xi, W. S. V. Lee and J. Xue, Adv. Mater., 2022, 34, 2107956 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- Z. Wu, S. Zuo, Z. Pei, J. Zhang, L. Zhang, D. Luan, H. Zhang and X. Lou, Sci. Adv., 2025, 11, eadu5370 Search PubMed.
- X. Luo, H. Zhao, X. Tan, S. Lin, K. Yu, X. Mu, Z. Tao, P. Ji and S. Mu, Nat. Commun., 2024, 15, 8293 Search PubMed.
- Z. Abdi, P. A. Masouleh and A. M. Khachatourian, Inorg. Chem. Commun., 2023, 153, 110833 CrossRef CAS.
- C. Saka, I. Tegin and K. Kahvecioglu, Diam. Relat. Mater., 2023, 131, 109542 CrossRef CAS.
- X. Li, C. Deng, Y. Kong, Q. Huo, L. Mi, J. Sun, J. Cao, J. Shao, X. Chen, W. Zhou, M. Lv, X. Chai, H. Yang, Q. Hu and C. He, Angew. Chem., Int. Ed., 2023, 62, e202309732 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01015b |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |