
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical cavity enhancement of visible light-driven photochemical reaction in the crystalline state†
Ikuto
Sasaki
ab,
Kiyonori
Takahashi
 a,
Farsai
Taemaitree
ab,
Takayoshi
Nakamura
ac,
James A.
Hutchison
d,
Hiroshi
Uji-i
abef and
Kenji
Hirai
*ab
a,
Farsai
Taemaitree
ab,
Takayoshi
Nakamura
ac,
James A.
Hutchison
d,
Hiroshi
Uji-i
abef and
Kenji
Hirai
*ab
aResearch Institute for Electronic Science (RIES), Hokkaido University, N20W10, Kita ward, Hokkaido 001-0020, Japan. E-mail: hirai@es.hokudai.ac.jp
bGraduate School of Information Science and Technology, Hokkaido University, Sapporo, Hokkaido, Japan
cDepartment of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, Higashihiroshima 739-8526, Japan
dSchool of Chemistry and ARC Centre of Excellence in Exciton Science, The University of Melbourne, Masson Road, Parkville, Victoria 3052, Australia
eDepartment of Chemistry, Division of Molecular Imaging and Photonics, KU Leuven, Celestijnenlaan 200F, Leuven B-3001, Belgium
fInstitute for Integrated Cell-Material Science (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
First published on 9th January 2025
Abstract
Photochemical reactions enable the synthesis of energetically unfavorable compounds but often require irradiation with ultraviolet light, which potentially induces side reactions. Here, cavity strong coupling enhances the efficiency of an all-solid state photocyclization in crystals of 2,4-dimethoxy-β-nitrostyrene under irradiation with visible light. The exposure to visible light facilitates photocyclization by the transition to a lower polaritonic state, which is energetically lower than the original transition state.
Photochemical reactions enable the synthesis of compounds with large strain that can be thermally unstable, or thermally unattainable due to symmetry considerations.1–3 In particular, photocyclization represents a promising method for synthesizing cyclic compounds, which are crucial in the development of biological probes and pharmaceuticals.4–7 However, photochemical reactions often require irradiation with ultraviolet light (UV). This UV light exposure can potentially induce side reactions and photodegradation, resulting in poor yields of target compounds. Furthermore, the irradiance of sunlight is limited in the UV range and reactions that can run with visible light can be run renewably.
An optical cavity may be an excellent platform in which to drive photochemical reactions with lower photon energy. When molecules are placed in an optical cavity, the molecules can coherently exchange photonic energy with the vacuum fields of the optical cavity, reaching the state of strong coupling.8–11 Under strong coupling, photon and molecular excitons are hybridized, forming new energy states known as upper and lower polaritonic states. Both experimental and theoretical works demonstrate that the generation of the lower polaritonic state (P−) can enhance photochemical reaction rates driven by lower photonic energies, although a mechanistic understanding of this is still lacking.12–18 Although cavity strong coupling has demonstrated the ability to modify photochemical reactions, the systems studied to date have been limited to photoisomerization12,13,16,17 and photodegradation.15 To utilize cavity strong coupling for synthetic reactions, the expansion of cavity-modified reaction classes to include coupling reactions is required.
One way to examine coupling reactions under cavity strong coupling conditions is to utilize all solid-state crystalline phase reactions in optical cavities. In the crystalline phase, highly ordered molecular alignment allows for the controlled proximity of two molecules that can undergo coupling chemistry. For instance, nitrostyrene derivatives are crystallized into assembled structures that allow solid-state photocyclization.19,20 In the crystalline phase, [2+2] photocyclization of carbon–carbon double bonds of two proximal nitrostyrenes occurs. In this study, we introduce crystals of nitrostyrene derivatives into a Fabry–Perot (FP) cavity, demonstrating cavity strong coupling-enhanced photochemical synthetic reactions. In the absence of cavity effects, two nitrostyrene derivatives form a four-membered ring compound upon UV light irradiation. Visible radiation is inefficient due to tailing absorption beyond 400 nm. However, under strong coupling, there is a drastic redefining of the absorption spectrum, leading to two new peaks, the upper (P+) and lower (P−) polariton states peaking at ∼320 nm and ∼420 nm, respectively. Here we investigated if the emergence of the P− absorption in the visible region allows visible light to drive the nitrostyrene dimerization more efficiently (Fig. 1a).
 | ||
| Fig. 1 (a) Schematic illustration of photocyclization under strong coupling. The strong coupling of DN and an optical cavity mode generates polaritonic states P+ and P−. The irradiation of visible light at the P− absorption may facilitate the photocyclization of DN, whereas DN only absorbs UV light outside an optical cavity. (b) and (c) Single crystal structure of DN: the view from (b) the a axis and (c) the c axis. The carbon–carbon double bonds of adjacent DNs are in parallel and separated by ∼3.8 Å (green double arrows with dotted lines). | ||
We selected 2,4-dimethoxy-β-nitrostyrene (DN) as the model molecule. According to the Schmidt rule, the photocyclization occurs even in the crystalline phase if the pair of carbon–carbon double bonds is in a parallel configuration and its distance is less than 4.2 Å.21 To assess whether DN crystals meet the Schmidt rule, X-ray single crystal analysis of DN was conducted. The distance between carbon–carbon double bonds in a crystal of DN was determined to be 3.8 Å (Fig. 1b and c, Table S1, ESI†), satisfying the Schmidt rule condition. The X-ray structural analysis suggests that the photocyclization of DN occurs in the crystalline state. Notably, the [2+2] cyclization reaction is forbidden under thermal conditions.22 This allows us to solely focus on the effect of photoexcitation, eliminating the influence of photoinduced heating.
To examine the photocyclization of DN, we prepared DN crystals dispersed in a polymer film. To align the cavity and non-cavity experimental conditions as closely as possible, the polystyrene (PS) film containing DN crystals was sandwiched between polyvinyl alcohol (PVA) films, on a substrate coated with a distributed Bragg reflector (DBR) mirror, but without fabrication of a top Ag reflective mirror. DN crystals in the polymer films exhibited an absorption band around 365 nm (Fig. 2b and Fig. S1, ESI†), suggesting that DN undergoes a photocyclization upon UV light irradiation. The UV absorption gradually reduces as the reaction proceeds. The progression of the photocyclization can also be validated through Raman spectroscopy measurements with the excitation of a 785 nm laser, which is important as this spectral region is less affected by the optical cavity. In the Raman spectra, carbon–carbon double bonds typically exhibit signals around 1600 cm−1 (Fig. S2, ESI†). As the photocyclization of DN progresses, the carbon–carbon double bond transforms into a cyclic structure, leading to a decrease in the Raman signal intensity around 1600 cm−1. In contrast, the Raman signal due to the hydroxyl group from the PVA layers does not change in the photocyclization, allowing the signals to be used as the reference intensity for the Raman signals. Upon UV light irradiation (345–385 nm) on DN crystals, the relative intensity of the carbon–carbon double bond versus the hydroxyl group decreased, confirming the photocyclization of DN. The conversion after 10 minutes of UV irradiation was approximately 84%, as estimated by Raman spectroscopy.
 | ||
| Fig. 2 (a) Fabrication process of DN crystals into the FP cavity. (b) Absorbance of bare PS film containing DN crystals (black) and DN under strong coupling (blue). (c) Dispersion relation of DN crystals in an FP cavity. The black solid and dotted lines indicate the energy levels of the exciton and cavity photon, respectively. The white dotted lines of upper and lower polaritons are generated by a coupled oscillator model. | ||
The identical procedure was employed to validate the effect of visible light irradiation (405–415 nm) on the DN-dispersed polymer film, outside the cavity. The conversion after 10 minutes of visible light irradiation was much reduced, approximately 31% (Fig. S3, ESI†). Although the photocyclization of DN progressed under visible light irradiation due to the existence of an absorption shoulder in this wavelength region, the conversion efficiency (i.e., the conversion of monomers to cyclic compounds) under visible light irradiation was significantly lower compared to UV light irradiation. Although light irradiation at longer wavelengths (450 nm and above) was tested, a high conversion rate was not observed. This is likely because light at these wavelengths does not provide sufficient energy to overcome the barrier required for photocyclization (Fig. S4, ESI†).
To achieve a strong coupling condition for DN, we constructed an FP cavity using Ag and DBR mirrors. The DBR mirror, composed of alternate layers of Ta2O5 and SiO2, reflects UV light, serving as a reflective mirror to facilitate strong coupling of DN (Fig. S5, ESI†). Notably, the DBR mirror permits the transmission of visible light and a 785 nm laser, enabling the exposure of visible light irradiation for photocyclization and conducting Raman spectroscopy to assess the % conversion. In the fabrication of the FP cavities containing DN crystals (Fig. 2a), a DBR mirror was prepared on a quartz glass substrate. Subsequently, an aqueous solution of PVA was spin-coated on the DBR mirror, followed by spin-casting a toluene solution of PS containing DN molecules onto the PVA layer. By evaporation of toluene as the solvent, DN crystals were formed in the PS layer. An aqueous solution of PVA was further spin-coated on the PS-DN layer. Finally, Ag thin layers were sputtered onto the top PVA layer as a reflective surface. In this way, DN crystals were placed between the DBR and Ag mirrors where the UV absorption of DN (375 nm absorption maximum) could strongly couple with cavity photons. The thickness of the polymer layers was optimized to allow this resonance to occur at normal incidence to the cavity plane.23
UV-vis spectroscopy was conducted on the DN crystals in the FP cavity to validate the strong coupling. The DN crystals inside the FP cavity exhibited the emergence of polaritonic states around 325 nm and 420 nm, identified as upper and lower polaritonic states (P+ and P−, as shown in Fig. 2b). The absorption band of P− is stronger than the shoulder of the DN. This enhanced absorbance could increase the conversion efficiency of the photocyclization upon visible light irradiation. Strong light-matter coupling could be confirmed by the angular dispersion of UV-Vis transmission spectra for DN inside the FP cavity, which exhibited clear anti-crossing behavior of P+ and P− around 375 nm (3.31 eV) as the cavity photon and DN absorption come into resonance at normal incidence, 0 degrees (Fig. 2c). A coupled oscillator model reproduces the dispersion behaviour of P+ and P−24 (white dashed lines in Fig. 2c and ESI†), confirming that the DN inside the FP cavity is in a strong coupling state.
Visible light (405–415 nm) was irradiated from the DBR mirror side onto the DN crystals under strong coupling conditions (Fig. 2a). Before and after visible light irradiation, we performed Raman spectroscopy measurements to estimate the conversion (%) of DN photocyclization (Fig. 3). The obtained conversion (%) of the photocyclization was plotted as a function of time (Fig. 4a). The conversion (%) after 10 minutes of visible light irradiation on DN crystals under strong coupling was approximately 75%. The conversion was normalized by the spectral irradiance of the light source in the UV and in the visible region (Fig. S6, ESI†). It is noted that strong coupling enhances the conversion (%) under visible light irradiation (75% for strong coupling and 31% for non-strong coupling).
 | ||
| Fig. 3 Raman spectra of DN in an FP cavity. The exposure time of visible light varies from 0 to 10 minutes. The red and yellow shaded areas indicate Raman signals of carbon–carbon double bonds (indicating progressive cyclization of DN) and hydroxyl groups of the PVA film (reference signal), respectively. | ||
 | ||
| Fig. 4 Conversion (%) of DN crystals: UV light irradiation without strong coupling (black squares), visible light irradiation without strong coupling (black triangles), and visible light irradiation under strong coupling (black circles). The data is corrected for differing spectral irradiance of the light source. | ||
To confirm the impact of strong coupling on the reaction rate upon visible light irradiation, we fabricated a cavity with its mirror separation deliberately detuned from the exciton of DN. The mirror separation was set to be 558 nm, which is significantly detuned from the exciton of DN (Fig. S7, ESI†). After 10 minutes of visible light irradiation, the conversion of DN in the detuned FP cavities was estimated to be 34% (Fig. S8, ESI†), which is nearly the same as that of the DN crystals outside the FP cavities (31%). The DN crystals in the detuned cavity exhibit nearly no enhancement of conversion.
Photocyclization of DN crystals can be categorized as second order,19,20 where the relationship between the reaction rate constant and the molecular concentration is given by (see ESI†);
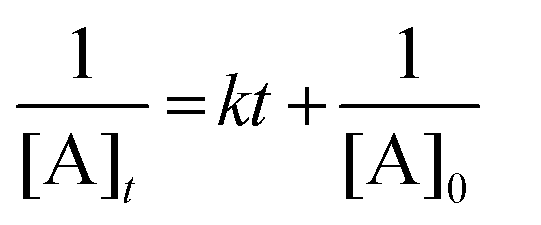
The mechanism behind the improved reaction kinetics under strong coupling is currently a subject of debate. Recently, discussions have focused on the contribution of cavity-modification of incident light fields affecting the light absorption rate, in addition to any intrinsic effect of polaritons and strong coupling on the reaction kinetics.16,18,25 Here we used transfer matrix method calculations to integrate the absorption in the DN layer for the non-cavity and FP cavity structures in the wavelength region 405–415 nm (Fig. S10, ESI†). These simulations suggest that the absorption rate is enhanced by 1.7 times for the FP cavity, which alone does not account for the 4.4-fold increase in reaction kinetics observed. It is necessary to consider, therefore, that strong coupling may also modify the potential energy curve for the photocyclizaton, contributing partially to the increased reaction kinetics. Evaluating the modification of the reaction barriers in photochemical reactions is difficult by experimental approaches and beyond the scope of this work, however these modification mechanisms have been treated in theoretical investigations.26–30
In summary, 2,4-dimethoxy-β-nitrostyrene (DN) under strong light-matter coupling in an optical cavity exhibits higher photocyclization kinetics compared to bare DN when irradiated with visible light. Enhanced kinetics are attributed to both enhanced visible light absorption, and changes to the reaction potential energy surface due to polariton formation. The conversion rate of [2+2] photocyclization is generally proportional to the number of irradiated photons, implying that higher irradiation intensity typically enhances the conversion rates. However, under strong coupling, the emergence of lower polaritonic states enables improved conversion efficiency even with light irradiation at longer wavelengths. Using strong light-matter coupling to drive photochemical reactions efficiently with lower energy photons could have important applications. For example, TiO2 is an excellent photocatalyst for water splitting, but the limiting of its absorption to UV light has long been a challenge as only 5% of the solar spectral irradiance falls in the UV. This method to drive typically UV photoreactions with lower photon energy could significantly improve the sustainability of important photocatalytic reactions.
IS carried out the experiment and wrote the manuscript. KT, TN, and KH carried out X-ray structural analysis. JAH carried out the simulation. KT, FT, JAH, and HU discussed the results and edited the manuscript. KH supervised the research project.
This work was supported by JSPS KAKENHI (24K01447, 23H04877, 23K17856, 21H04634), KU Leuven Internal Funds (C14/19/079), JST PRESTO (JPMJPR18TA), JST ACT-X (JPMJAX23DB) and Research Foundation of Flanders (FWO) research grant. We thank the Open Facility at Hokkaido University for allowing us to use Raman microscopy. This work was supported by the JSPS Core-to-Core Program. J. A. H. thanks the Australian Government for funding through the Australian Research Council Future Fellowship scheme (FT180100295) and Centre of Excellence in Exciton Science (CE170100026).
Data availability
The supporting data has been included in the ESI.†Conflicts of interest
There are no conflicts to declare.References
- V. Ramamurthy and K. Venkatesan, Chem. Rev., 1987, 87, 433–481 CrossRef.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef PubMed.
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef PubMed.
- U. C. Yoon, Y. X. Jin, S. W. Oh, C. H. Park, J. H. Park, C. F. Campana, X. Cai, E. N. Duesler and P. S. Mariano, J. Am. Chem. Soc., 2003, 125, 10664–10671 CrossRef PubMed.
- Y. Mifune, H. Nakamura and S. Fuse, Org. Biomol. Chem., 2016, 14, 11244–11249 RSC.
- Orville L. Chapman and Gilbert L. Eian, J. Am. Chem. Soc., 1968, 90, 5329–5330 CrossRef.
- S. G. Modha, A. Pöthig, A. Dreuw and T. Bach, J. Org. Chem., 2019, 84, 1139–1153 CrossRef PubMed.
- T. W. Ebbesen, Acc. Chem. Res., 2016, 49, 2403–2412 CrossRef PubMed.
- D. S. Dovzhenko, S. V. Ryabchuk, Y. P. Rakovich and I. R. Nabiev, Nanoscale, 2018, 10, 3589–3605 RSC.
- M. Hertzog, M. Wang, J. Mony and K. Börjesson, Chem. Soc. Rev., 2019, 48, 937–961 RSC.
- K. Hirai, J. A. Hutchison and H. Uji-i, Chem. Rev., 2023, 123, 8099–8126 CrossRef PubMed.
- J. A. Hutchison, T. Schwartz, C. Genet, E. Devaux and T. W. Ebbesen, Angew. Chem., Int. Ed., 2012, 51, 1592–1596 CrossRef PubMed.
- J. Mony, C. Climent, A. U. Petersen, K. Moth-Poulsen, J. Feist and K. Börjesson, Adv. Funct. Mater., 2021, 31, 2010737 CrossRef.
- J. Fregoni, G. Granucci, E. Coccia, M. Persico and S. Corni, Nat. Commun., 2018, 9, 4688 CrossRef PubMed.
- V. N. Peters, M. O. Faruk, R. Alexander, D. A. Peters and M. A. Noginov, Optica, 2019, 6, 318–325 CrossRef.
- P. A. Thomas, W. J. Tan, V. G. Kravets, A. N. Grigorenko and W. L. Barnes, Adv. Mater., 2024, 36, 2309393 CrossRef PubMed.
- H. Zeng, J. B. Pérez-Sánchez, C. T. Eckdahl, P. Liu, W. J. Chang, E. A. Weiss, J. A. Kalow, J. Yuen-Zhou and N. P. Stern, J. Am. Chem. Soc., 2023, 145, 19655–19661 CrossRef PubMed.
- I. Lee, S. R. Melton, D. Xu and M. Delor, J. Am. Chem. Soc., 2024, 146, 9544–9553 CrossRef PubMed.
- D. B. Miller, P. W. Flanagan and H. Shechter, J. Am. Chem. Soc., 1972, 18, 3912–3917 CrossRef.
- G. R. Desiraju and V. R. Pedireddi, J. Chem. Soc., Chem. Commun., 1989, 1112–1113 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef.
- S. Poplata, A. Tröster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef PubMed.
- K. Hirai, J. A. Hutchison and H. Uji-i, Chem. – Eur. J., 2023, 30, e202303110 CrossRef PubMed.
- J. Ren, Q. Liao, H. Huang, Y. Li, T. Gao, X. Ma, S. Schumacher, J. Yao, S. Bai and H. Fu, Nano Lett., 2020, 20, 7550–7557 CrossRef PubMed.
- T. Schwartz and J. A. Hutchison, arXiv, 2024, preprint, arXiv:2403.06001.
- J. Fregoni, G. Granucci, E. Coccia, M. Persico and S. Corni, Nat. Commun., 2018, 9, 4688 CrossRef PubMed.
- L. A. Martínez-Martínez, M. Du, R. F. Ribeiro, S. Kéna-Cohen and J. Yuen-Zhou, J. Phys. Chem. Lett., 2018, 9, 1951–1957 CrossRef PubMed.
- F. Herrera and F. C. Spano, Phys. Rev. Lett., 2016, 116, 238301 CrossRef PubMed.
- J. Galego, F. J. Garcia-Vidal and J. Feist, Phys. Rev. X, 2015, 5, 041022 Search PubMed.
- A. Mandal and P. Huo, J. Phys. Chem. Lett., 2019, 10, 5519–5529 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Raman spectra, UV-vis spectrum, and derivation of the formula. CCDC 2339682. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05598e |
| This journal is © The Royal Society of Chemistry 2025 |