
Interaction strength in molecular junctions consisting of π-stacked antiaromatic molecules†
Shintaro
Fujii
 *a,
Ryoya
Tomida
b,
Aoshi
Yamane
a,
Kazuki
Nabeyama
a,
Harunari
Ohkura
a,
Hiroshi
Shinokubo
*b and
Tomoaki
Nishino
*a
*a,
Ryoya
Tomida
b,
Aoshi
Yamane
a,
Kazuki
Nabeyama
a,
Harunari
Ohkura
a,
Hiroshi
Shinokubo
*b and
Tomoaki
Nishino
*a
aDepartment of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1 W4-10 Ookayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: fujii.s.af@m.titech.ac.jp; tnishino@chem.titech.ac.jp
bDepartment of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan. E-mail: hshino@chembio.nagoya-u.ac.jp
First published on 19th December 2023
Abstract
Elucidation of intermolecular interactions is essential for understanding charge transport properties in organic materials and organic electronic devices. While interactions between aromatic molecules have been extensively studied, little is known about interactions between antiaromatic molecules. Theoretical considerations predict that when two antiaromatic porphyrin molecules are superimposed, the antiaromatic molecules should be stabilized by attractive intermolecular interactions. In this study, we used atomic force microscopy to evaluate intermolecular interactions originating from the π–π stacking of the two antiaromatic porphyrin moieties at the molecular level. We evaluated substantial interaction between the antiaromatic porphyrin moieties as an adhesion force of the antiaromatic porphyrin molecular pairs using force spectroscopy. The present result supports the formation of π–π stacking due to the attractive interaction between the two antiaromatic π-systems at the single-molecule level.
Introduction
The importance of intermolecular π–π stacking interactions in enhancing charge transport has been widely studied in organic electronics.1,2 Effective overlap of molecular orbitals along the π–π stacking has contributed to the enhancement of charge transport in organic electronic and bioelectronic materials. Recent developments in molecular electronics have made it possible to probe the π–π stacking at the single molecule level, revealing unique charge transport properties2 such as quantum interference effects,3,4 length-independent charge transport,5 and high structural responsiveness to the external electric field.6 To understand the charge transport properties of π–π stacking, it is of crucial importance to clarify the interactions between π-stacked molecules. Indeed, extensive studies devoted to the interactions between aromatic molecules have shed light on their effects on electron transport. In terms of the π–π stacking interactions, recent studies have shown that antiaromatic compounds, characterized by planar rings of sp2 carbon atoms sharing multiples of four π-electrons, form unique π–π stacking structures in solution and in the solid state that spatially delocalize the π-electrons of the two antiaromatic systems and that the molecular pairs of antiaromatics exhibit stacked-ring aromaticity.7,8 Moreover, excellent charge transport properties were found for such antiaromatic compounds with a small HOMO–LUMO gap.9 Detailed knowledge of the π–π interactions is required to fully exploit the superior properties of the antiaromatics. However, unlike the aromatic counterpart, little is known about interactions between antiaromatic molecules.7,10,11 In this study, atomic force microscopy (AFM) was used to assess the intermolecular interactions between antiaromatic systems, norcorrole Ni(II) (denoted as Ni(nor)). We found substantial interaction between two antiaromatic Ni(nor) systems using force spectroscopy. This result supports the formation of π–π stacking due to the attractive interaction between the two antiaromatic π-systems. The present work reveals the intermolecular interactions of the antiaromatic systems at the single-molecule level and provides a better understanding of the interactions that make antiaromatic π-stacks suitable for building blocks in organic electronics.Results and discussion
The antiaromatic intermolecular interaction was evaluated by measuring the interaction force between antiaromatic molecules using AFM. We used a molecular matrix of a hexanethiol (C6) monolayer as a host to support and isolate single dodecanethiol (C12) chains appended with an antiaromatic moiety of Ni(nor) as the tail group (Fig. 1a). Similar experiments were performed for C12 with an aromatic moiety of Ni(porph) as the tail group (Fig. 1a), which is expected to form the usual aromatic π-stack. Fig. 1b–d show scanning tunneling microscopy (STM) topography of the sample surfaces, in which isolated individual molecules with either the antiaromatic (C12–Ni(nor)) or the aromatic moiety (C12–Ni(porph)) were visualized as bright spots. Such bright spots were not observed in the STM images of C6 monolayers without C12–Ni(nor) and C12–Ni(porph) (Fig. 1c). The higher topography observed for C12–Ni(nor) and C12–Ni(porph) reflects the higher physical height of C12–Ni(nor) and C12–Ni(porph) than C6. By counting the number of bright spots in the STM image in Fig. 1b, the surface density of C12–Ni(nor) molecules isolated in the C6 matrix on Au(111) can be estimated to be 0.02 nm−2. | ||
| Fig. 1 (a) The chemical structures of hexanethiol (C6) and acetyl-protected dodecanethiol with the norcorrole Ni(II) moiety (C12–Ni(nor)) and with the porphyrin Ni(II) moiety (C12–Ni(porph)). The acetyl group of C12–Ni(nor) and C12–Ni(porph) is desorbed during adsorption to the Au surface, allowing the formation of a C12–Ni(nor)Au–S or a C12–Ni(porph)Au–S bond.12 (b)–(d) Scanning tunneling microscopy topography of (b) and (c) the self-assembled monolayer (SAM) matrix with isolated C12–Ni(nor) and C12–Ni(porph) on Au(111), and (d) the SAM of C6 on Au(111). Imaging conditions: tunneling current (It) = 0.3 nA and sample bias voltage (Vs) = +1.0 V. | ||
Following the characterization of the isolated target molecules in the C6 matrix, an intermolecular interaction force between Ni(nor) molecules was evaluated by AFM force spectroscopy (Fig. 2a). Au-coated AFM cantilever tips were functionalized with the C12–Ni(nor) molecules isolated in the C6 matrix, in a similar fashion to the Au(111) substrate (see Fig. 1b), to facilitate the formation of the π–π stacking interactions between the sample molecules on the tip and the substrate surfaces. After bringing the AFM tip in contact with the sample surface of the C6 matrix, the AFM tip was pulled away from the surface to measure the adhesive force between the AFM tip and the sample surface.13 The measured adhesion force includes the contribution of the interaction force of π–π stacking of the antiaromatic moieties (Fig. 2b). To extract the contribution of the π–π stacking, we also performed AFM force spectroscopy on a C6 monolayer sample without C12–Ni(nor), using the same AFM tip used in AFM spectroscopy in Fig. 2a. Fig. 2c shows the AFM force spectroscopy results for the C6 monolayer without C12–Ni(nor), which clearly shows weaker adhesion than for the C6 matrix with C12–Ni(nor). By subtracting the adhesive forces measured for the C6 matrix with C12–Ni(nor) and the C6 monolayer without C12–Ni(nor), the contribution of the π–π stacking interaction between antiaromatics can be estimated (see ΔF in Fig. 3a). The interaction between antiaromatics arises from the π–π stacking of multiple antiaromatic pairs formed between the sample and the AFM probe. The number of antiaromatic pairs formed in the force spectroscopy measurements can be estimated by considering the contact area between the sample and the AFM tip as well as the surface density of the antiaromatic C12–Ni(nor) molecules. Based on the surface density calculated from the STM image (0.02 nm−2) (Fig. 1b), the nominal radius of curvature of the AFM tip (∼50 nm),14 and the contact area corresponding to that nominal radius of curvature, the interaction force of individual antiaromatic pairs is calculated to be on the order of 150 pN, which is on the same order of rapture force reported for the hydrogen bonding between two 2-ureido-4[1H]-pyrimidinone (UPy) moieties (ca. 180 pN).15 Force measurements of C12–Ni(porph) aromatic pairs suggest that the π–π stacking interactions of aromatics are of the same order as that of antiaromatics (Fig. 3b and Fig. S1, ESI†) and that the forces for aromatic and antiaromatic interactions are in the order of 150–220 pN. The result is consistent with our previous observation based on 1H NMR analysis.10 This argument was supported by density functional theory calculations.10
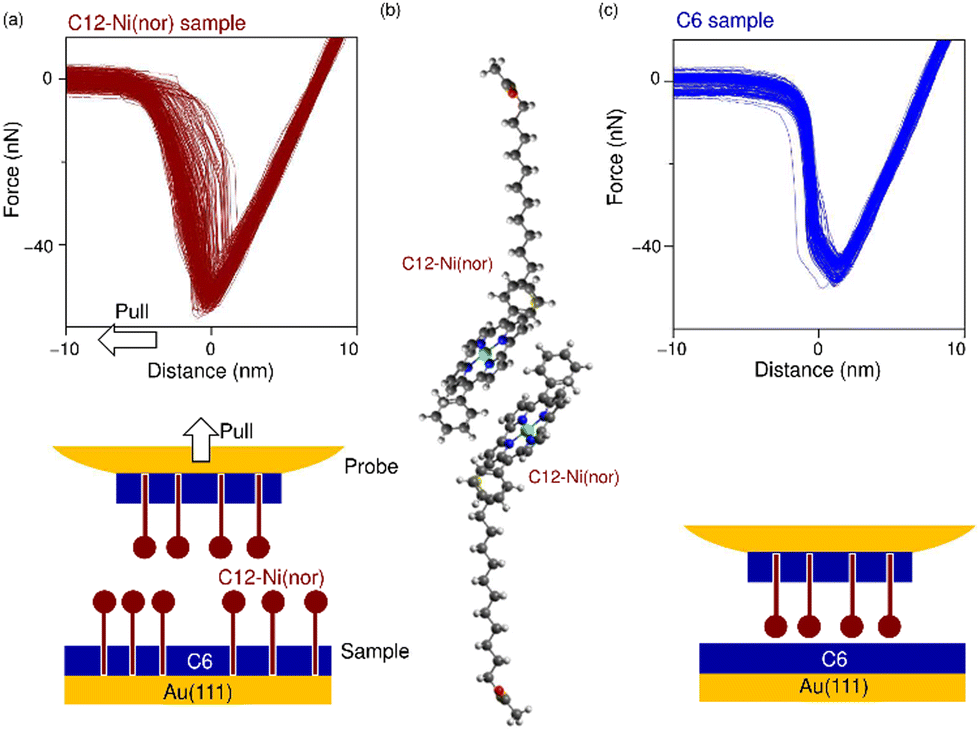 | ||
| Fig. 2 (a) and (c) Force curves measured on the C6 matrix with and without C12–Ni(nor). Adhesion force was defined as the maximum attraction (most negative force signal value) measured by the force curve. Schematics of the experimental systems are shown at the bottom. (b) Schematic illustration of the π-stack formation between C12–Ni(nor) molecules. Grey, white, blue, red, yellow, and green balls correspond to C, H, N, O, S and Ni atoms, respectively. | ||
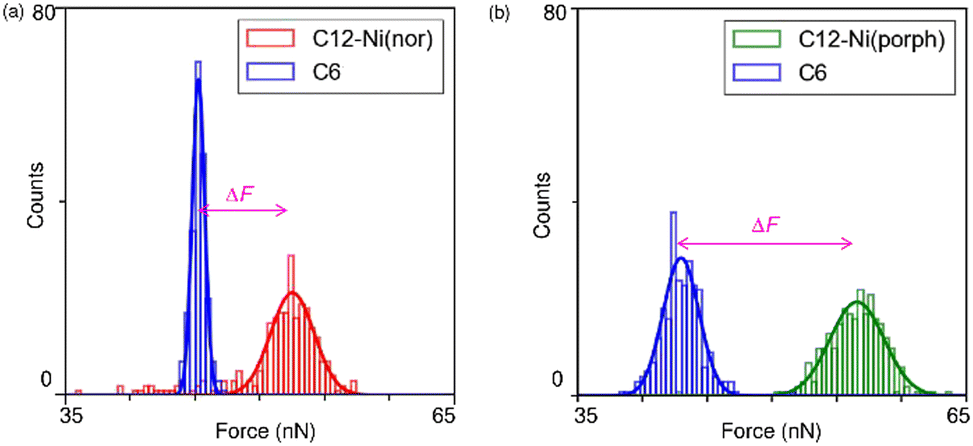 | ||
| Fig. 3 Histograms of the observed adhesive force constructed from the 256 force curves for (a) the C6 matrix with and without C12–Ni(nor) and (b) the C6 matrix with and without C12–Ni(porph). The bold lines are the result of the Gaussian fitting of the histograms. ΔF is the difference between peak adhesion forces for C6 and C12–Ni(nor) or C6 and C12–Ni(porph). | ||
Face-to-face stacking of antiaromatics leads to energetic stabilization, which is referred to as stacked-ring aromaticity.8,16 This energetic stabilization is due to the small HOMO–LUMO gap of the antiaromatics and the interaction between the frontier orbitals when the antiaromatics are stacked face-to-face.17 Meanwhile, energy decomposition analyses of the π–π stacking structures of norcorrole and porphyrins demonstrate that the dispersion interaction is more favourable for the antiaromatic system than the aromatic one. The analyses also show stronger repulsive interaction for the former than the latter, due to large exchange repulsion in the stacked norcorrole with short stacking distance. The overall interactions in the stacking structures of the antiaromatic and aromatic compounds are counterbalanced by attractive and repulsive interactions. Consequently, the interaction energies and rapture forces of π-stacked aromatics and π-stacked antiaromatics can be comparable in the force spectroscopy measurements. Theoretical calculations predict interaction energies of 1.34 eV and 1.58 eV for aromatic and antiaromatic pairs, respectively (Fig. S2 and ESI†). To accurately determine the interaction force at the single molecule scale, it is necessary to clarify the surface density of antiaromatic molecules adsorbed on the probe surface, the actual contact area between the AFM probe and the sample, and the dynamics of the breaking process of the interacting molecular pairs, which are far beyond the current standard experimental level and will be addressed in the future. This study supports the formation of π-stacks through an attractive interaction between two antiaromatic π-systems at the single-molecule scale to demonstrate the potential of π-stacks of antiaromatics as building blocks for the construction of organic frameworks in molecular electronics.
Experimental
Materials and sample preparation
Nickel(II) antiaromatic and aromatic compounds (C12–Ni(nor) and C12–Ni(porph), Fig. 1a) were prepared according to previously reported procedures.18 Au(111) substrates were prepared by the thermal vacuum deposition of Au on mica substrates. STM tips were mechanically cut from an Au wire (diameter 0.3 mm, purity >99.9%; Nilaco). Samples are prepared by dipping the Au(111) substrate into a 1 mM ethanol solution of hexanethiol (C6) for 1 hour. Then, the substrate was immersed in 0.5–1 mg ml−1 toluene solution of C12–Ni(nor) or 12-Ni(porph) (Fig. 1a) for 10 min. After immersion, the substrate was washed thoroughly with toluene and dried under a flow of inert gas. For control experiments, samples were prepared by dipping the Au(111) substrate into a 1 mM ethanol solution of C6 for 1 h. After immersion, the substrate was washed thoroughly with ethanol and dried under a flow of inert gas.STM and AFM measurements
STM measurements were performed using an MS-10 STM (Bruker), controlled by a NanoScope V (Bruker) under ambient conditions. All the STM images presented were recorded in constant current mode using mechanically cut Au tips.Force spectroscopy measurements were performed using a multimode AFM (Bruker) controlled by a NanoScope V (Bruker) under ambient conditions. Au-coated AFM cantilevers (PPP-NCSTAu) were purchased from NANOSENSORS. The nominal spring constant of the cantilever and radius of curvature of the cantilever probe are 7.6 N m−1 and 50 nm, respectively. Prior to the force spectroscopy measurements, the surface of the AFM cantilever tip was modified with C6 and C12–Ni(nor) or C6 and C12–Ni(porph) in the same manner as the sample preparation.
Conclusions
The interaction between the π-systems of norcorrole Ni(II) was investigated at the single molecule scale using scanning probe microscopy. The interaction was measured as the adhesive force acting between norcorrole molecules anchored to the probe surface and the sample surface. This study reveals substantial interactions in antiaromatic π-systems and provides a better understanding of the interactions that make π-stacks of antiaromatics suitable for building blocks in organic electronics.Author contributions
All the authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by JSPS KAKENHI (No. JP22H04974, JP23K04517, and JP21H01959) and JST SICORP (JPMJSC22C2).Notes and references
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- X. Li, W. Ge, S. Guo, J. Bai and W. Hong, Angew. Chem., Int. Ed., 2023, 62, e202216819 CrossRef CAS PubMed.
- R. Frisenda, V. A. E. C. Janssen, F. C. Grozema, H. S. J. van der Zant and N. Renaud, Nat. Chem., 2016, 8, 1099–1104 CrossRef CAS PubMed.
- M. Carlotti, A. Kovalchuk, T. Wächter, X. Qiu, M. Zharnikov and R. C. Chiechi, Nat. Commun., 2016, 7, 13904 CrossRef CAS PubMed.
- X. Li, Q. Wu, J. Bai, S. Hou, W. Jiang, C. Tang, H. Song, X. Huang, J. Zheng, Y. Yang, J. Liu, Y. Hu, J. Shi, Z. Liu, C. J. Lambert, D. Zhang and W. Hong, Angew. Chem., Int. Ed., 2020, 59, 3280–3286 CrossRef CAS PubMed.
- Y. Tang, Y. Zhou, D. Zhou, Y. Chen, Z. Xiao, J. Shi, J. Liu and W. Hong, J. Am. Chem. Soc., 2020, 142, 19101–19109 CrossRef CAS PubMed.
- R. Nozawa, H. Tanaka, W.-Y. Cha, Y. Hong, I. Hisaki, S. Shimizu, J.-Y. Shin, T. Kowalczyk, S. Irle, D. Kim and H. Shinokubo, Nat. Commun., 2016, 7, 13620 CrossRef CAS PubMed.
- J. Aihara, J. Phys. Chem. A, 2009, 113, 7945–7952 CrossRef CAS PubMed.
- S. Fujii, S. Marques-Gonzalez, J.-Y. Shin, H. Shinokubo, T. Masuda, T. Nishino, N. P. Arasu, H. Vázquez and M. Kiguchi, Nat. Commun., 2017, 8, 15984 CrossRef CAS PubMed.
- H. Kawashima, S. Ukai, R. Nozawa, N. Fukui, G. Fitzsimmons, T. Kowalczyk, H. Fliegl and H. Shinokubo, J. Am. Chem. Soc., 2021, 143, 10676–10685 CrossRef CAS PubMed.
- R. Nozawa, J. Kim, J. Oh, A. Lamping, Y. Wang, S. Shimizu, I. Hisaki, T. Kowalczyk, H. Fliegl, D. Kim and H. Shinokubo, Nat. Commun., 2019, 10, 3576 CrossRef PubMed.
- J. M. Tour, L. Jones, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh and S. Atre, J. Am. Chem. Soc., 1995, 117, 9529–9534 CrossRef CAS.
- O. H. Olubowale, S. Biswas, G. Azom, B. L. Prather, S. D. Owoso, K. C. Rinee, K. Marroquin, K. A. Gates, M. B. Chambers, A. Xu and J. C. Garno, ACS Omega, 2021, 6, 25860–25875 CrossRef CAS PubMed.
- https://www.nanosensors.com/pointprobe-plus-non-contact-soft-tapping-mode-au-coating-afm-tip-PPP-NCSTAu .
- S. Zou, H. Schönherr and G. J. Vancso, Angew. Chem., Int. Ed., 2005, 44, 956–959 CrossRef CAS PubMed.
- Y. Tsuji, K. Okazawa and K. Yoshizawa, J. Org. Chem., 2023, 88, 14887–14898 CrossRef CAS PubMed.
- K. Okazawa, Y. Tsuji and K. Yoshizawa, J. Phys. Chem. A, 2023, 127, 4780–4786 CrossRef CAS PubMed.
- (a) R. Nozawa, K. Yamamoto, J.-Y. Shin, S. Hiroto and H. Shinokubo, Angew. Chem., Int. Ed., 2015, 54, 8454–8457 CrossRef CAS PubMed; (b) G.-Y. Gao, A. J. Colvin, Y. Chen and X. P. Zhang, J. Org. Chem., 2004, 69, 8886–8892 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04166b |
| This journal is © The Royal Society of Chemistry 2024 |