
Construction of an intimately riveted Li/garnet interface with ultra-low interfacial resistance for solid-state batteries†
Jie
Wang‡
 ab,
Saisai
Zhang‡
a,
Hailei
Zhao
*ab,
Jintao
Liu
a,
Min-An
Yang
a,
Zhaolin
Li
a and
Konrad
Świerczek
cd
ab,
Saisai
Zhang‡
a,
Hailei
Zhao
*ab,
Jintao
Liu
a,
Min-An
Yang
a,
Zhaolin
Li
a and
Konrad
Świerczek
cd
aSchool of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: hlzhao@ustb.edu.cn
bBeijing Municipal Key Laboratory of New Energy Materials and Technologies, Beijing 100083, China
cFaculty of Energy and Fuels, Department of Hydrogen Energy, AGH University of Krakow, Mickiewicza 30, 30-059 Krakow, Poland
dAGH Centre of Energy, AGH University of Krakow, Czarnowiejska 36, 30-054 Krakow, Poland
First published on 23rd January 2024
Abstract
The application of ceramic garnet-type Li7La3Zr2O12 electrolytes is restricted by the challenge of poor contact with metallic lithium, which results in high interfacial resistance, uneven current distribution, and severe lithium dendrite penetration. Herein, a remarkably ultra-low interfacial impedance (∼2.7 Ω cm2) is achieved by employing Si addition into molten Li, because of the realized intimate interface contact resulting from the decreased surface tension of molten Li and, most importantly, the reinforced interfacial connection as a result of the minor reaction between the Li4.4Si and Li6.4La3Zr1.4Ta0.6O12 (LLZTO), further improving charge transfer across the Li/LLZTO interface. The generated Li4.4Si at the interface (I-Li4.4Si), showing strong combination with garnet, can function as a “rivet” for the stable interface upon cycling. Moreover, the I-Li4.4Si species with Li+ diffusivity favor the reduced local current density and thus the decreased over-potential to some extent. Thus, the resulting symmetric Li–Li4.4Si|LLZTO|Li–Li4.4Si cell delivers a stable Li plating–stripping performance for over 1200 h at a current density of 0.1 mA cm−2 (1000 h)/0.2 mA cm−2 (200 h) and a temperature of 60 °C with a small over-potential (5.3 mV@0.1 mA cm−2; 10.2 mV@0.2 mA cm−2). The fabricated full cell with a LiFePO4 cathode delivers a high reversible capacity of ∼169 mA h g−1 at 0.05C and excellent cycling stability with a capacity decay rate of only 0.038% per cycle.
1 Introduction
Rechargeable lithium-ion battery (LIB) technology, applied in portable electronics and electric vehicles (EVs), has enabled significant advances in human life over the past few decades. The increased demand for high energy density LIBs makes developing high-energy cathodes and high-capacity anodes crucial.1–3 For the anode, the lithium metal, considered the ultimate choice, shows the characteristics of ultrahigh theoretical specific capacity (∼3860 mA h g−1), low density, and the lowest redox potential (−3.04 V vs. the standard hydrogen electrode).4,5 Unfortunately, when using a lithium metal anode in LIBs with flammable organic-liquid electrolytes, safety concerns, caused mainly by lithium dendrite growth and thus the induced short-circuit, significantly hinder its practical application. Replacing combustible organic-liquid electrolytes with solid-state electrolytes (SSEs), which have high mechanical strength, is believed to be a suitable approach to address the critical issue of lithium dendrite penetration. The so-called solid-state lithium metal battery (SSLB) offers a preferable solution to realize the lithium metal anode into practical application.6–8SSEs should meet the requirements of high ionic conduction, good chemical compatibility with electrodes, and good electrochemical stability, with all factors being crucial for ideal electrolytes. Among the various proposed inorganic SSEs, garnet-type Li7La3Zr2O12 (LLZO) with a cubic structure has been recognized as one of the most attractive electrolytes, owing to its intrinsic advantages of high ionic conductivity (10−4–10−3 S cm−1 at room temperature), high Li+ transference number (∼1), excellent compatibility with lithium metal, and wide electrochemical window that enables paring with high-voltage cathodes.9–13 However, despite the above-mentioned merits, the successful application of garnet-type LLZO electrolyte in SSLBs is strongly restricted by the big challenge of large interfacial resistance originating from the noncompact contact between the Li metal anode and garnet SSEs due to the rigid ceramic nature of LLZO, which is further worsened by the uncontrollable formation of lithiophobic Li2CO3 and LiOH passivation layers on the LLZO surface, because of the instability of LLZO during air exposure.14–16 The insufficient physical contact is not only harmful for the charge transfer at the Li/LLZO interface, but also results in inhomogeneous current distribution and an uneven ion flow and thus the promoted Li dendrite formation/growth, especially at high currents.17,18 When the Li dendrites rapidly penetrate through the SSEs along grain boundaries and voids in LLZO, a short-circuit occurs.
To realize an improved interfacial contact between LLZO and the Li anode, various approaches have been proposed, including applying the mechanical pressure,19 eliminating surface Li2CO3 contaminants (polishing, high temperature annealing with reductants or acid treating),20–23 introducing a lithiophilic interlayer (Au, Sn, Mg, Al, Si, Ge, C, ZnO, etc.),24–31 adding a soft polymer/gel buffer layer,32–34 or compositing the Li anode with a lithiophilic phase to adjust surface energy of molten Li (Li@graphite, Li@Na, Li@Si3N4, Li@g-C3N4, Li@Mg3N2, Li@Zn, Li@Sn, Li@S, etc.).18,35–41 Among the strategies mentioned above, tuning the wettability of molten Li by constructing an alloy anode shows great promise because of its easy operability and manufacturing scalability. Benefitting from the improved wettability of molten Li with LLZO, an intimate interface contact and thus a much reduced interfacial resistance is achieved. For example, with the additive of Sn and graphite, the resultant molten Li–Sn and Li–graphite composites can be spread out easily on the LLZO surface, achieving an intimate Li–Sn/garnet and Li–graphite/garnet interface with a dramatically reduced interfacial impedance as low as ∼7 Ω cm2 and 11 Ω cm2, respectively.35,39 However, it should be pointed out that such a composite approach still has its own drawbacks. Firstly, the introduction of lithiophilic additives could have an adverse effect on the energy density of the whole battery. Secondly, the lithiated phases at the interface will bring the newly introduced solid/solid (lithiated phase/LLZO) interfaces, which also affect ion transportation to some extent, even if such a lithiated phase/LLZO interface is not continuous. Moreover, it is noteworthy that although intimate physical contact is achieved, the reported interfacial resistances vary greatly, and in some reports, the lithium dendrite-induced short-circuit still occurs at a relatively low current density. Therefore, under the premise of an intimate physical connection between the anode and garnet SSEs, how to further reduce the interfacial resistance and promote the charge transfer process at the interface, as well as simultaneously suppress the lithium dendrite formation still faces an enormous challenge and is of great importance.
In this work, we report a strategy of introducing a micro-sized Si powder additive into molten Li to perform multiple functions in not only improving the wettability of molten Li with Li6.4La3Zr1.4Ta0.6O12 (LLZTO) electrolyte, because of the spontaneous reaction between Si and Li, to ensure an intimate Li/LLZTO interface, but also reinforcing the interface connection owing to the minor reaction characteristics between the lithiated product (Li4.4Si) and garnet for further improving charge transfer across the Li|LLZTO interface. Moreover, the strong combination of Li4.4Si at the interface (I-Li4.4Si) with garnet can enable the Li4.4Si phase to serve as a “rivet” and a “bridge” to maintain the interfacial structure integrity to a certain degree, as well as provide additional ion transport pathways to mitigate the interface void effect upon repeated Li plating–stripping processes. Owing to these advantages, a remarkably ultra-low interfacial impedance of ∼2.7 Ω cm2 at room temperature is achieved, and the fabricated symmetric cell (Li–Li4.4Si|LLZTO|Li–Li4.4Si) delivers long-term cycling exceeding 1200 h with a small overpotential (5.3 mV at 0.1 mA cm−2 and 10.2 mV at 0.2 mA cm−2) at 60 °C. Besides, the constructed (Li–Li4.4Si|LLZTO|LiFePO4) full cell shows a more stable cycling performance with a notably high coulombic efficiency close to 100% than the Li|LLZTO|LiFePO4 full cell, demonstrating the potential application of Li–Li4.4Si in interface optimization for all-solid-state full batteries.
2 Experimental
2.1 Preparation of LLZTO electrolyte and Li–Li4.4Si composite anodes
The Li6.4La3Zr1.4Ta0.6O12 solid electrolyte was prepared via a solid-state reaction method. Typically, stoichiometric amounts of Li2CO3 (Sinopharm, AR, with 10 wt% excess Li2CO3 to compensate for the lithium loss at elevated temperatures), La2O3 (Sinopharm, 4N), ZrO2 (Sinopharm, AR), and Ta2O5 (Aladdin, 99.5%) were ball-milled for 12 h at a rotation speed of 300 rpm. After calcining at 900 °C for 12 h, the as-prepared precursor was ball-milled again, mixed with 10 mol% Al2O3 sintering aid, uniaxially pressed into pellets, and then sintered at 1175 °C for 6 h in air. The pressed pellets were covered with the Li6.4La3Zr1.4Ta0.6O12 precursor powder to prevent the lithium loss during the high-temperature sintering process. Before assembling cells, the obtained LLZTO solid electrolyte was mechanically polished on both sides using sand papers in an Ar-filled glove box to ensure a Li2CO3-free flat surface.The Li–Li4.4Si composites were prepared by a simple fusion method. After completely melting the pure Li metal at 250 °C in an Ar-filled glove box, different amounts of micron-sized Si powders (5 wt%, 10 wt%, 15 wt%, and 30 wt%) were added and kept under continuous mechanical stirring for 30 minutes. For simplicity, the Li–Li4.4Si composite with different mass ratios of Si powder addition was labeled as LSi05 (5 wt% Si), LSi10 (10 wt% Si), LSi15 (15 wt% Si), and LSi30 (30 wt% Si), respectively.
2.2 Materials characterization
X-ray diffraction (XRD) data were collected using a Rigaku D/max-A X-ray diffractometer (Cu Kα radiation source, λ = 1.5406 Å). During the XRD measurements, the Li–Li4.4Si composites were sealed with polyimide tape to avoid air exposure. The morphology observations were performed on a LEO-1450 scanning electron microscope (SEM). The elemental mapping images were obtained using SEM coupled with an energy dispersive X-ray spectroscopy (EDX) detector. The interface reaction energy between LLZTO and Li or Li4.4Si was examined using density functional theory (DFT) calculations, which are performed based on the Materials Project database that is available on the website (https://materialsproject.org/).422.3 Cell fabrication
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) was infiltrated in the LFP cathode. The Li–Li4.4Si|LLZTO|LFP full cell was assembled by employing the liquid electrolyte infiltrated LFP, LLZTO, and the Li–Li4.4Si composite as the cathode, electrolyte, and anode, respectively. The thickness of the LLZTO electrolyte used for full cell fabrication is about 0.3 mm.
:1:1) was infiltrated in the LFP cathode. The Li–Li4.4Si|LLZTO|LFP full cell was assembled by employing the liquid electrolyte infiltrated LFP, LLZTO, and the Li–Li4.4Si composite as the cathode, electrolyte, and anode, respectively. The thickness of the LLZTO electrolyte used for full cell fabrication is about 0.3 mm.
2.4 DFT calculations
The calculations are performed using the Cambridge sequential total-energy package (CASTEP) module in Materials Studio software. The generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) functional was used for the exchange–correlation energy. Lattice parameters and atomic positions are optimized before calculations. The plane-wave energy cutoff was set to 300 eV. The convergence thresholds for the energy, maximum force, maximum stress, and maximum displacement were set as 2.0 × 10−5 eV per atom, 0.05 eV Å−1, 0.1 GPa, and 2.0 × 10−3 Å, respectively. The interface formation energies were obtained according to the equation Ef = (Einterface − Ea − Eb)/S,28,36,43 where Einterface, Ea, and Eb is the total energy of the Li(001)|LLZO(001) or Li22Si5(001)|LLZO(001) interface, the energy of separated LLZO(001), and the energy of separated Li(001) or Li22Si5(001), respectively, and S is the interfacial area.2.5 Electrochemical measurements
The galvanostatic Li plating–stripping of the Li–Li4.4Si|LLZTO|Li–Li4.4Si symmetric cells and the cycling performance tests of the Li–Li4.4Si|LLZTO|LFP full cells were performed with a LAND CT2001A battery test system. The cells, fabricated by employing the pasted Ag layers as Li-blocking electrodes, on both sides of the polished LLZTO, were used for the conductivity test. The EIS measurements were performed with a Solartron 1260 frequency response analyzer coupled with a Solartron 1287 electrochemical interface, in a frequency range from 0.1 Hz to 106 Hz with a voltage amplitude of 50 mV, for the Ag|LLZTO|Ag cells and the Li–Li4.4Si|LLZTO|Li–Li4.4Si cells, respectively.3 Results and discussion
The garnet-type solid-state electrolyte with a nominal composition of Li6.4La3Zr1.4Ta0.6O12 (LLZTO) was selected in this work because of its high ionic conductivity.44 The material was prepared by a conventional solid-state method, and the corresponding XRD result shown in Fig. S1a† reveals a pure phase without any detectable impurities, corresponding to a highly crystalline cubic structure (JCPDS Card No. 80-0457) with a space group of Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d. The as-prepared LLZTO pellet has a dense microstructure (relative density: ∼96%), in which tightly connected grains (average size: ∼6 μm) and only a few isolated pores or voids at the grain boundaries are observed (Fig. S1b†). The high total Li-ion conductivity (∼4.1 × 10−4 S cm−1 at 25 °C) of the LLZTO pellet, estimated from the typically fitted Nyquist plot (Fig. S1c†), with an activation energy of 0.355 eV (Fig. S1d†), demonstrates the high quality of the synthesized garnet-type LLZTO, which provides the basic electrolyte support for this work. The Li–Si composite melt was prepared by simply adding the commercial Si power (particles size: 0.5–2 μm (Fig. S2a†)) into the molten Li at 250 °C in an Ar-filled glove box. Fig. 1a shows the digital images of the molten Li and Si powder before and after contact. It is observed that once the molten Li comes into contact with the Si particles, the molten lithium will rapidly permeate into Si powder, driven by the spontaneous reaction between Li and Si (22Li + 5Si → Li22Si5, Gibbs free energy: ΔG < 0, Fig. S2b†), to fulfill the continuous lithiation reaction, as demonstrated in Video S1.† To ensure the uniformity of the lithiated Si particles in molten Li, the resultant composite melt was further kept under continuous stirring for 30 minutes, and the XRD result of the as-cooled Li–Si composite is shown in Fig. 1b. The peaks related to the pure Li (JCPDS Card No. 15-0401) and Li22Si5 (JCPDS Card No. 73-2049) phases can be clearly distinguished. The absence of the peaks attributable to Si and the newly detected peaks belonging to the Li22Si5 phase demonstrate that the Si has thoroughly reacted with molten Li during the high-temperature (250 °C) mixing process. The generation of the lithium-rich alloy phase (Li4.4Si) can also be verified according to the Li–Si binary phase diagram (Fig. S3†). Notably, there is a broader peak appearing at a low angle, which originates from the polyimide tape that is used for sample sealing to avoid oxidization when exposed to air during sample transfer and characterization.
d. The as-prepared LLZTO pellet has a dense microstructure (relative density: ∼96%), in which tightly connected grains (average size: ∼6 μm) and only a few isolated pores or voids at the grain boundaries are observed (Fig. S1b†). The high total Li-ion conductivity (∼4.1 × 10−4 S cm−1 at 25 °C) of the LLZTO pellet, estimated from the typically fitted Nyquist plot (Fig. S1c†), with an activation energy of 0.355 eV (Fig. S1d†), demonstrates the high quality of the synthesized garnet-type LLZTO, which provides the basic electrolyte support for this work. The Li–Si composite melt was prepared by simply adding the commercial Si power (particles size: 0.5–2 μm (Fig. S2a†)) into the molten Li at 250 °C in an Ar-filled glove box. Fig. 1a shows the digital images of the molten Li and Si powder before and after contact. It is observed that once the molten Li comes into contact with the Si particles, the molten lithium will rapidly permeate into Si powder, driven by the spontaneous reaction between Li and Si (22Li + 5Si → Li22Si5, Gibbs free energy: ΔG < 0, Fig. S2b†), to fulfill the continuous lithiation reaction, as demonstrated in Video S1.† To ensure the uniformity of the lithiated Si particles in molten Li, the resultant composite melt was further kept under continuous stirring for 30 minutes, and the XRD result of the as-cooled Li–Si composite is shown in Fig. 1b. The peaks related to the pure Li (JCPDS Card No. 15-0401) and Li22Si5 (JCPDS Card No. 73-2049) phases can be clearly distinguished. The absence of the peaks attributable to Si and the newly detected peaks belonging to the Li22Si5 phase demonstrate that the Si has thoroughly reacted with molten Li during the high-temperature (250 °C) mixing process. The generation of the lithium-rich alloy phase (Li4.4Si) can also be verified according to the Li–Si binary phase diagram (Fig. S3†). Notably, there is a broader peak appearing at a low angle, which originates from the polyimide tape that is used for sample sealing to avoid oxidization when exposed to air during sample transfer and characterization.
 | ||
| Fig. 1 (a) The digital images of the molten Li and Si powder before and after contact at 250 °C. (b) The XRD pattern of the Li–Li4.4Si composite with 15 wt% Si addition (LSi15). The surface SEM images of (c) pure Li and Li–Li4.4Si composite with (d) 5 wt% Si addition (LSi05), (e) 10 wt% Si addition (LSi10), (f) 15 wt% Si addition (LSi15), and (g and h) 30 wt% Si addition (LSi30). (i) The EDS elemental mapping image of the surface of the LSi30 composite. (j) The SEM and (k) corresponding EDS elemental mapping images of the cross-section of the LSi30 composite. | ||
To characterize the dispersivity of the lithiated Si (Li4.4Si) in the composite, SEM coupled with EDS analysis was conducted, and the results are shown in Fig. 1c–k and S4.† Unlike pure metallic Li with a clean and smooth surface (Fig. 1c), for all Li–Li4.4Si composites (LSi05, LSi10, LSi15, and LSi30), significant dispersion of Li4.4Si particles in the solidified Li matrix is observed. The generated Li4.4Si phase, tightly embedded into the Li matrix, shows a relatively good uniformity overall, except for particle aggregation in small local areas, as confirmed by the EDS elemental mapping result (Fig. 1i and S4b†). The cross-sectional SEM (Fig. 1j) combined with the related EDS analysis result (Fig. 1k) of sample LSi30 further indicates that the lithiated Li4.4Si phase presents a uniform distribution in the composite both in the transverse and longitudinal directions. It is noteworthy that since a trace amount of Si can dissolve in molten Li at 250 °C, the precipitated nano-sized Li4.4Si during the cooling process could contribute to the Si element distribution in the whole sample to some extent.
The polished LLZTO (Fig. 2a) was employed to investigate the wettability of the developed Li–Li4.4Si composites with the solid electrolyte. As shown in Fig. 2b and c, the molten Li and the LSi15 melt exhibit entirely different wetting behaviors in a stainless-steel crucible, with a spherical droplet for molten Li and a flat thin layer for the LSi15 melt. This difference in contact mode has also been verified on the LLZTO surface. In contrast to the pure molten Li on the garnet that shows a lithiophobic nature with a contact angle (CA) of >130°, the LSi15 melt can spread easily onto the LLZTO pellet with a significantly reduced CA down to circa 28° after being heated at 250 °C for 5 minutes (Fig. 2d and e). Such improved interfacial wettability is associated with the decreased surface tension of the liquid melt with the introduction of Si, which is probably ascribed to the generated Li4.4Si phase that destroys the interior metallic bonding network in molten Li, as well as the liquid melt dissolved Si element in a trace amount that weakens the interior atomic interactions to a certain degree. The improved interfacial contact of the modified melt with LLZTO electrolyte has also been evidenced in the cross-sectional SEM results, as illustrated in Fig. 2f–j. It is observed that even though the surface Li metal was treated by an additional heating and external pressing, large voids still exist at the Li/LLZTO interface (Fig. 2f). By comparison, a close adherence of the Li–Li4.4Si composites to the LLZTO surface without interface gaps is achieved, demonstrating the effectiveness of Si addition in improving the interfacial contact. The sufficient coverage of the composite melt on the garnet surface could not only benefit the Li+ ion transfer across the interface, but also homogenize the current distribution and thus suppress the lithium dendrite nucleation and growth.
 | ||
| Fig. 2 Digital photos showing (a) the LLZTO electrolyte and the liquid melt in a stainless-steel crucible: (b) pure molten Li, and (c) LSi15 melt. Comparison of wettability of melted (d) Li and (e) LSi15 on the surface of the LLZTO pellet. Cross-sectional SEM images of the (f) Li/LLZTO, (g) LSi05/LLZTO, (h) LSi10/LLZTO, (i) LSi15/LLZTO, and (j) LSi30/LLZTO interfaces. | ||
Note that apart from the original Li/LLZTO interface, the generated Li-rich alloy in the composite inevitably introduces the Li4.4Si/LLZTO interface, as marked in Fig. 2j. To better understand the function of Li4.4Si particles for improved wettability between the Li–Li4.4Si composite and LLZTO, DFT calculations were performed to compare the interfacial formation energies of Li/LLZTO and Li4.4Si/LLZTO (Fig. 3a). The calculated value of the formation energy (−1.22 J m−2) of the Li4.4Si/LLZTO interface is more negative than that of the Li/LLZTO interface (−0.94 J m−2), signifying the positive role of the Li4.4Si species in favoring interfacial contact between the Li–Li4.4Si composite and LLZTO. Besides, from the perspective of interfacial chemical compatibility, we found that compared to the Li/Li7La3Zr2O12 interface system, a more favorable reaction between the Li22Si5 and Li7La3Zr2O12 with a mutual reaction energy of −17 to −51 eV per atom is delivered (Fig. 3b and Table S1†). It is undeniable that the minor interfacial reaction with the generation of thermodynamically stable phases probably affects the charge transfer at the Li4.4Si/LLZTO interface, but it could benefit the adhesion of Li4.4Si species on the LLZTO surface and thus further improve the interfacial wettability of the Li–Li4.4Si melt towards garnet, as well as guarantee the “rivet” effect of I-Li4.4Si in reinforcing the interface connection and thus favoring charge transfer and interface stability during electrochemical cycling.
 | ||
| Fig. 3 (a) DFT calculations of the formation energies of the Li(001)/Li7La3Zr2O12(001) and Li22Si5(001)/Li7La3Zr2O12(001) interfaces. (b) Calculated mutual reaction energy between garnet-type Li7La3Zr2O12 and Li or lithiated silicon phases. | ||
The efficacy of the Si addition for decreasing the interfacial resistance was evaluated by the EIS studies of the Li–Li4.4Si|LLZTO|Li–Li4.4Si and Li|LLZTO|Li symmetric cells. As shown in Fig. 4a, the symmetric cell with pure Li electrodes shows a much higher area specific resistance (ASR) of 273.1 Ω cm2 (referring to the one-sided electrode–electrolyte interface),41 while a considerable impedance decrease is observed for the Li–Li4.4Si|LLZTO|Li–Li4.4Si symmetric cells, which is mainly attributed to the better interface connection between the Li–Li4.4Si composites and the LLZTO pellet. Specifically, as the mass ratio of Si addition increases, the arc impedance displays an initially decreasing and then increasing trend. When the Si ratio increases from 5 to 15 wt%, the calculated ASR gradually decreases from ∼121 Ω cm2 down to as low as ∼2.7 Ω cm2, which is smaller than other reported values.21–31,33–35,37,39,40,43 Notably, a further increase in the Si ratio (30 wt%) does not result in an additional decrease in ASR, but rather a significant increase in interfacial impedance (∼41 Ω cm2), even though the LS30 composite shows a tight physical connection with LLZTO. It is believed the Li4.4Si species at the interface can be recognized as the topological structure of the electrolyte because of its Li-ion transport characteristic. However, the loss of the direct Li/LLZTO interface contact area, as a result of the excessively accumulated Li4.4Si at the interface, could contribute to the deteriorated charge transfer at the interface and thus the ASR increase due to the lower ionic conductivity of Li4.4Si than that of LLZTO. Moreover, the possibly generated interfacial reaction products could also have a potential effect on Li-ion transfer across the Li4.4Si/LLZTO interface and are responsible for the change in ASR to some extent.
 | ||
| Fig. 4 (a) Nyquist plot of the symmetric cells with LLZTO solid electrolyte and Li–Li4.4Si composites (LSi05, LSi10, LSi15, and LSi30) or pure Li electrodes. The inset shows enlarged impedance curves. (b) Galvanostatic cycling of the LSi15|LLZTO|LSi15 symmetric cell under stepped increasing the current density from 0.02 to 1.2 mA cm−2. (c) Reversible Li plating–stripping curves of the LSi15|LLZTO|LSi15 symmetric cell at 0.1 mA cm−2 and a temperature of 25 °C. Inset: the corresponding Li plating–stripping curves at different cycling stages. (d) Galvanostatic Li plating–stripping curves of the LSi15|LLZTO|LSi15 and Li|LLZTO|Li symmetric cells at 60 °C, and (e) the corresponding magnified voltage curve at 0.1 and 0.2 mA cm−2. (f) SEM and corresponding EDS elemental mapping images of a dark spot on the LLZTO surface after long-term cycling in the LSi15|LLZTO|LSi15 symmetric cell. Schematic illustrations of charge transfer differences at the electrode/electrolyte interface in the (g) LSi15|LLZTO|LSi15 and (h) Li|LLZTO|Li symmetric cells. | ||
Galvanostatic cycling tests of the symmetric cells directly revealed the performance-improving effects of the introduction of Si. The critical current density (CCD), as defined as the maximum applied current density where a short circuit occurs in the cell, was used to measure the capability for Li dendrite suppression. As shown in Fig. 4b, the symmetric cell with the LSi15 electrode exhibits an exacerbating voltage hysteresis as the current density step increases, and soft breakdown (sudden voltage decrease), reflecting the mixed ion/electron transportation caused by the formed small lithium filaments in the SSE,45–47 is observed when the current density reaches 1 mA cm−2. In contrast, the Li|LLZTO|Li cell shows a CCD of only 0.2 mA cm−2 (Fig. S5†) because of the poor Li/LLZTO interface contact. Additionally, the LSi15|LLZTO|LSi15 symmetric cell displays a stable cycling performance for as long as 400 h without short circuiting (Fig. 4c) under the test condition of 0.1 mA cm−2/0.05 mA h cm−2 (25 °C). The delivered small overpotential of 7.4 mV further confirms the low interfacial impedance. Besides, it only presents a slight increase upon long-term cycling (7.4 mV → 9 mV), as evidenced by the zoomed-in charge/discharge curves at different cycling stages showing almost overlapped ultraflat Li plating–stripping voltage profiles (inset of Fig. 4c), which indicates the dendrite-suppressing ability of the LSi15 electrode and superior interface stability during Li cycling. In comparison, the control cell with the pristine Li electrode, reported in our previous work,41 shows a noisy potential with large voltage polarization and serious voltage fluctuations upon cycling. The cell fails to cycle in a few hours owing to the lithium dendrite-induced short-circuit, as confirmed by the post-morphological observation on the cross section of the LLZTO pellet (Fig. S6†) that gives evident Li dendrite proliferation through the grain boundaries as a result of the poor interface contact, the uneven electric field and lithium-ion flux distribution, the presence of localized hot spots, and thus the promoted lithium dendrite nucleation and propagation.
The interface stability of the symmetric cells (LSi15|LLZTO|LSi15 and Li|LLZTO|Li) is also investigated and compared visually by galvanostatic cycling at 60 °C. As shown in Fig. 4d, the LSi15|LLZTO|LSi15 symmetric cell exhibits a stable voltage profile in the consecutive plating–stripping cycles for over 1200 hours (1000 h at 0.1 mA cm−2/0.1 mA h cm−2 and 200 h at 0.2 mA cm−2/0.2 mA h cm−2) without Li infiltration, while the polarization voltage for the control Li|LLZTO|Li cell soars during cycling (0.1 mA cm−2/0.1 mA h cm−2). A smooth and stable curve with a small over-potential of 5.3 and 10.2 mV is achieved at a current density of 0.1 and 0.2 mA cm−2, respectively. The voltage hysteresis at each current density shows a negligible increase with cycling, and even after long-term cycling, the voltage curve still possesses a well-maintained plateau-like characteristic (Fig. 4e), suggesting the remained intimate interface structure without any obviously generated defects as a consequence of the uniform Li plating–stripping enabled by the charge homogenization.
After dissembling the long-term cycled LSi15|LLZTO|LSi15 symmetric cell and removing the LSi15 electrode with water, the dried LLZTO surface with some dark spots, which are very difficult to remove mechanically, is revealed (Fig. S7a†). Further EDS analysis on the LLZTO surface (Fig. 4f, S7b and S8†) demonstrates a distinguished elemental distribution difference between the dark spot region and other regions. Note that the Ta element can directly interfere with the determination of Si distribution because of the closer Si Kα/Ta Mα peak values (Si Kα: 1.740 keV and Ta Mα: 1.710 keV). The brighter Si element and almost undetected Ta element in the dark spot indicate the enrichment of Si in this region, which should be attributed to the products generated by the minor reaction occurring at the Li4.4Si/LLZTO interface. Besides, we noticed that the Si distribution in this region is inhomogeneous and the La/Zr/Ta element distribution is almost absence. Such a phenomenon probably results from the presence of the formed Li2O species on the surface, as evidenced by the significant O distribution in this area. Certainly, it should be pointed out that the elemental mapping results do not indicate that there are no interfacial minor reactions on the LLZTO surface except for the dark spot area, as the observable reaction area is related to the aggregation degree of the I-Li4.4Si particles.
Based on the results above, the much-improved Li plating–stripping properties of the LSi15|LLZTO|LSi15 symmetric cell can be attributed to the following aspects (Fig. 4g): (1) in contrast to the pure Li with poor physical contact with LLZTO and the resultant huge interfacial resistance (Fig. 4h), the intimate LSi15/LLZTO interface significantly lowers the interfacial impedance, homogenizes the current distribution and thus avoids selective Li deposition and extends cell's cycling life. (2) The minor reaction at the introduced Li4.4Si/LLZTO interface can provide the good chemical contact of I-Li4.4Si towards LLZTO, and the good bonding characteristics not only reinforce the interface connection and thus further decrease the interfacial charge transfer resistance, but also enable the I-Li4.4Si to function as a “rivet” for the stable interface during repeated Li plating–stripping processes. (3) The Li4.4Si with Li+ transport characteristic can enlarge the charge transfer area and thus not only reduce the local current density and the over-potential but also mitigate the interface voids effect on the cycling performance to some extent. It is undeniable that when cycling the symmetric cell at higher current densities, since there is limited lithium diffusivity in metal, defects still inevitably form and accumulate at the interface, leading to uneven current distribution, thereby promoting lithium dendrite nucleation/growth and inducing short-circuit failure.
Apart from electrochemical characterization on symmetric cells, the LSi15|LLZTO|LFP full cell was assembled to verify the feasibility of the LSi15/LLZTO interface. As shown in Fig. 5a, the LSi15|LLZTO|LFP full cell delivers a reversible capacity of around 169 mA h g−1 at 0.05C and more than 96% capacity retention after 90 cycles. The high coulombic efficiency was kept close to 100% throughout cycling. Besides, the corresponding charge–discharge curves shown in Fig. 5b display a well-defined flat voltage plateau at ∼3.4 V, comparable to that of the LFP-based cell with liquid electrolyte, and the maintained voltage polarization upon cycling implies the almost non-deteriorated electrode reaction kinetics and further suggests the interfacial stability. By contrast, the fabricated full cell with a pristine Li anode only presents an initial discharge capacity of ∼68 mA h g−1 along with subsequently rapid capacity decay, especially after 10 cycles. Fig. 5c shows the cycling performance of the LSi15|LLZTO|LFP cell with the current density step increasing from 0.05C to 1C. An average discharge capacity of 154.5, 139.4, 125.4, 107.4, and 82.6 mA h g−1 is achieved at 0.1C, 0.2C, 0.3C, 0.5C, and 1C, respectively. Even at a high current rate, the two-phase reaction characteristic with a flat voltage plateau of LiFePO4 is well-maintained (Fig. 5d). Note that only 3 μL liquid electrolyte was infiltrated into the LFP cathode for wetting the LFP/LLZTO interface and constructing ion transport paths in the cathode as well. Further optimization of the charge transfer at the cathode side could be capable of realizing a more reduced voltage polarization and improved rate-capability. When the current rate suddenly decreases from 1C to 0.05C, the specific capacity can return to its original value with a recovered cell polarization of 67.9 mV, further indicating the stable LSi15/LLZTO interface. All the above-presented results demonstrate the effectiveness of incorporating Si powder in molten Li in achieving a perfect interface contact with garnet electrolyte and provide certain technique support for the practical development of SSLBs.
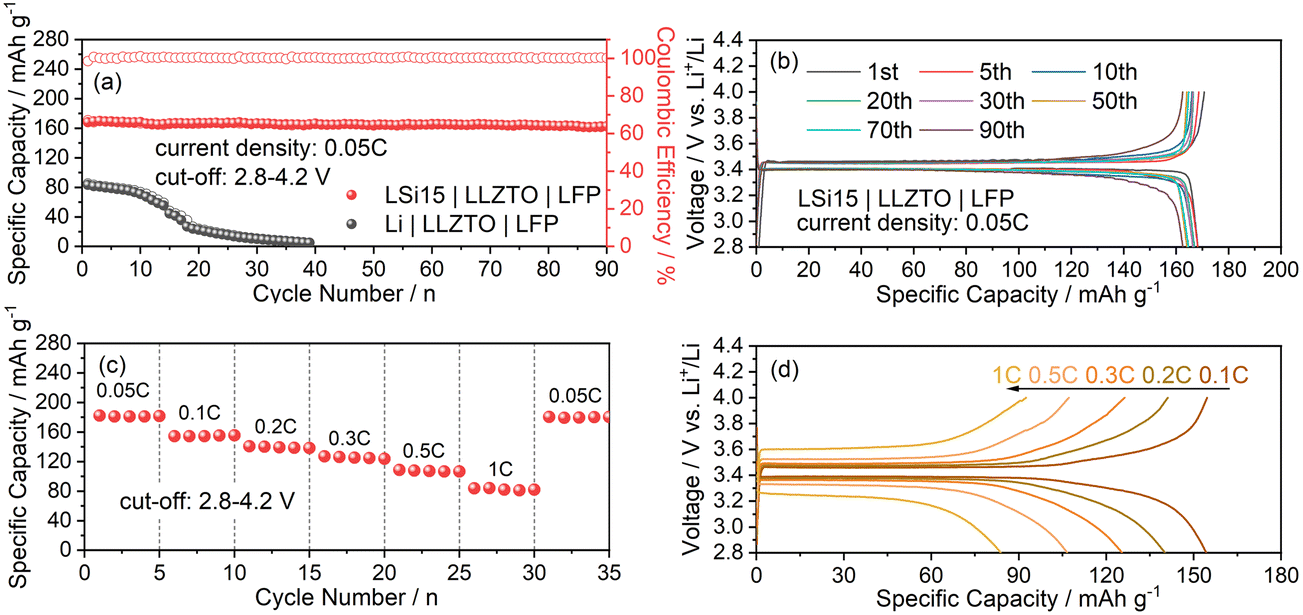 | ||
| Fig. 5 (a) Cycling performances of the assembled LSi15|LLZTO|LFP and Li|LLZTO|LFP cells at a current density of 0.05C at 25 °C, and (b) the typically corresponding discharge/charge capacity–voltage curves at different cycles. (c) Stepped rate performance and (d) corresponding discharge/charge voltage profiles of the LSi15|LLZTO|LFP cell at different current rates. | ||
4 Conclusions
In summary, we have demonstrated the utility of Si powder as an efficient additive to tune the surface tension of molten Li and thus significantly resolve the Li/LLZTO interfacial contact issue. The added Si powder spontaneously reacts with lithium, generating a Li–Si alloy with the Li4.4Si composition tightly embedded in the Li matrix. The generated Li4.4Si phases can decrease the interface formation energy of Li–Li4.4Si|LLZTO and thus simultaneously favor improved interfacial contact. Moreover, the minor interfacial reaction between I-Li4.4Si and LLZTO can endow the I-Li4.4Si species with good bonding characteristics towards garnet, benefitting the reinforced interface connection and the maintained stable interface structure (“rivet” effect) upon repeated cycling. In addition, I-Li4.4Si with Li+ diffusivity can lead to an enlarged charge transfer area and thus a decreased local current density, favoring reduced over-potential to a certain degree. As a result, compared to the control cells with pure Li electrodes, the symmetric cells with optimized Li–Li4.4Si composite electrodes (LSi15: 15 wt% Si addition) show a remarkably decreased interfacial resistance (∼2.7 Ω cm2), an enhanced CCD (∼1 mA cm−2), a lower over-potential (7.4 mV@0.1 mA cm−2, 25 °C; 5.3 mV @0.1 mA cm−2, 60 °C; 10.2 mV@0.2 mA cm−2, 60 °C), and a significantly improved cycling stability (over 1200 h: 1000 h@0.1 mA cm−2/0.1 mA h cm−2 + 200 h@0.2 mA cm−2/0.2 mA h cm−2). A superior cycling performance with high discharge capacity (∼169 mA h g−1 at 0.05C) and good cycling stability (capacity fading rate: 0.038% per cycle) is also achieved in the LSi15|LLZTO|LFP full cell. This work sheds new light on the research of solid-state Li metal batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52074023 and 52102205), the Beijing Natural Science Foundation (2222062), and the Interdisciplinary Research Project for Young Teachers of USTB (Fundamental Research Funds for the Central Universities) (FRF-IDRY-21-023).Notes and references
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS
.
- W. Lee, S. Muhammad, C. Sergey, H. Lee, J. Yoon, Y. M. Kang and W. S. Yoon, Angew. Chem., Int. Ed., 2020, 59, 2578–2605 CrossRef CAS PubMed
- C. Wang, C. Yang and Z. Zheng, Adv. Sci., 2022, 9, 2105213 CrossRef CAS PubMed
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed
- Y. Guo, H. Li and T. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed
- H. Wang, L. Sheng, G. Yasin, L. Wang, H. Xu and X. He, Energy Storage Mater., 2020, 33, 188–215 CrossRef
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed
- J. Liang, J. Luo, Q. Sun, X. Yang, R. Li and X. Sun, Energy Storage Mater., 2019, 21, 308–334 CrossRef
- H. Buschmann, J. Dölle, S. Berendts, A. Kuhn, P. Bottke, M. Wilkening, P. Heitjans, A. Senyshyn, H. Ehrenberg, A. Lotnyk, V. Duppel, L. Kienlee and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 19378–19392 RSC
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef
- Y. Zhu, J. G. Connell, S. Tepavcevic, P. Zapol, R. Garcia-Mendez, N. J. Taylor, J. Sakamoto, B. J. Ingram, L. A. Curtiss, J. W. Freeland, D. D. Fong and N. M. Markovic, Adv. Energy Mater., 2019, 9, 1803440 CrossRef
- J. Wolfenstine, J. L. Allen, J. Read and J. Sakamoto, J. Mater. Sci., 2013, 48, 5846–5851 CrossRef CAS
- R. Ye, M. Ihrig, N. Imanishi, M. Finsterbusch and E. Figgemeier, ChemSusChem, 2021, 14, 4397–4407 CrossRef CAS PubMed
- C. Galven, J.-L. Fourquet, M.-P. Crosnier-Lopez and F. Le Berre, Chem. Mater., 2011, 23, 1892–1900 CrossRef CAS
- S. Zhang, H. Zhao, J. Wang, T. Xu, K. Zhang and Z. Du, Chem. Eng. J., 2020, 393, 124797 CrossRef CAS
- Y. Song, L. Yang, W. Zhao, Z. Wang, Y. Zhao, Z. Wang, Q. Zhao, H. Liu and F. Pan, Adv. Energy Mater., 2019, 9, 1900671 CrossRef
- Y. Zhang, J. Meng, K. Chen, H. Wu, J. Hu and C. Li, ACS Energy Lett., 2020, 5, 1167–1176 CrossRef CAS
- T. Krauskopf, H. Hartmann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2019, 11, 14463–14477 CrossRef CAS PubMed
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS
- Y. Li, X. Chen, A. Dolocan, Z. Cui, S. Xin, L. Xue, H. Xu, K. Park and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 6448–6455 CrossRef CAS PubMed
- H. Xu, Y. Li, A. Zhou, N. Wu, S. Xin, Z. Li and J. B. Goodenough, Nano Lett., 2018, 18, 7414–7418 CrossRef CAS PubMed
- H. Huo, Y. Chen, N. Zhao, X. Lin, J. Luo, X. Yang, Y. Liu, X. Guo and X. Sun, Nano Energy, 2019, 61, 119–125 CrossRef CAS
- J. Wakasugi, H. Munakata and K. Kanamura, J. Electrochem. Soc., 2017, 164, A1022–A1025 CrossRef CAS
- M. He, Z. Cui, C. Chen, Y. Li and X. Guo, J. Mater. Chem. A, 2018, 6, 11463–11470 RSC
- K. K. Fu, Y. Gong, Z. Fu, H. Xie, Y. Yao, B. Liu, M. Carter, E. Wachsman and L. Hu, Angew. Chem., Int. Ed., 2017, 56, 14942–14947 CrossRef CAS PubMed
- K. K. Fu, Y. Gong, B. Liu, Y. Zhu, S. Xu, Y. Yao, W. Luo, C. Wang, S. D. Lacey, J. Dai, Y. Chen, Y. Mo, E. Wachsman and L. Hu, Sci. Adv., 2017, 3, e1601659 CrossRef PubMed
- W. Luo, Y. Gong, Y. Zhu, K. K. Fu, J. Dai, S. D. Lacey, C. Wang, B. Liu, X. Han, Y. Mo, E. D. Wachsman and L. Hu, J. Am. Chem. Soc., 2016, 138, 12258–12262 CrossRef CAS PubMed
- W. Luo, Y. Gong, Y. Zhu, Y. Li, Y. Yao, Y. Zhang, K. K. Fu, G. Pastel, C. F. Lin, Y. Mo, E. D. Wachsman and L. Hu, Adv. Mater., 2017, 29, 1606042 CrossRef PubMed
- Y. Shao, H. Wang, Z. Gong, D. Wang, B. Zheng, J. Zhu, Y. Lu, Y. S. Hu, X. Guo, H. Li, X. Huang, Y. Yang, C. W. Nan and L. Chen, ACS Energy Lett., 2018, 3, 1212–1218 CrossRef CAS
- C. Wang, Y. Gong, B. Liu, K. Fu, Y. Yao, E. Hitz, Y. Li, J. Dai, S. Xu, W. Luo, E. D. Wachsman and L. Hu, Nano Lett., 2017, 17, 565–571 CrossRef CAS PubMed
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS PubMed
- H. Huo, J. Gao, N. Zhao, D. Zhang, N. G. Holmes, X. Li, Y. Sun, J. Fu, R. Li, X. Guo and X. Sun, Nat. Commun., 2021, 12, 176 CrossRef CAS PubMed
- B. Liu, Y. Gong, K. Fu, X. Han, Y. Yao, G. Pastel, C. Yang, H. Xie, E. D. Wachsman and L. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 18809–18815 CrossRef CAS PubMed
- J. Duan, W. Wu, A. M. Nolan, T. Wang, J. Wen, C. Hu, Y. Mo, W. Luo and Y. Huang, Adv. Mater., 2019, 31, 1807243 CrossRef PubMed
- M. Du, Y. Sun, B. Liu, B. Chen, K. Liao, R. Ran, R. Cai, W. Zhou and Z. Shao, Adv. Funct. Mater., 2021, 31, 2101556 CrossRef CAS
- Y. Huang, B. Chen, J. Duan, F. Yang, T. Wang, Z. Wang, W. Yang, C. Hu, W. Luo and Y. Huang, Angew. Chem., Int. Ed., 2020, 59, 3699–3704 CrossRef CAS PubMed
- L. Chen, R.-A. Tong, J. Zhang, H. Wang, G. Shao, Y. Dong and C.-A. Wang, Angew. Chem., Int. Ed., 2023, 62, e2023050 Search PubMed
- G. V. Alexander, O. V. Sreejith, M. S. Indu and R. Murugan, ACS Appl. Energy Mater., 2020, 3, 9010–9017 CrossRef CAS
- C. Wang, H. Xie, L. Zhang, Y. Gong, G. Pastel, J. Dai, B. Liu, E. D. Wachsman and L. Hu, Adv. Energy Mater., 2018, 8, 1701963 CrossRef
- J. Wang, S. Zhang, S. Song, J. Liu, Z. Li and H. Zhao, J. Mater. Chem. A, 2023, 11, 251–258 RSC
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef
- C. Cao, Y. Zhong, K. C. Wasalathilake, M. O. Tadé, X. Xu, H. Rabiee, M. Roknuzzaman, R. Rahman and Z. Shao, J. Mater. Chem. A, 2022, 10, 2519–2527 RSC
- Y. Li, J.-T. Han, C.-A. Wang, H. Xie and J. B. Goodenough, J. Mater. Chem., 2012, 22, 15357–15361 RSC
- C. Wang, T. Deng, X. Fan, M. Zheng, R. Yu, Q. Lu, H. Duan, H. Huang, C. Wang and X. Sun, Joule, 2022, 6, 1–12 CrossRef
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS PubMed
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06883h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |