
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual cross-linked cellulose based hydrogel films†
Neethu
Thomas
,
Saphia
Moussaoui
,
Braulio
Reyes-Suárez
,
Olivier
Lafon
 and
G. N. Manjunatha
Reddy
*
and
G. N. Manjunatha
Reddy
*
University of Lille, CNRS, Centrale Lille Institut, Univ. Artois, UMR 8181–UCCS–Unité de Catalyse et Chimie du Solide, F-59000, Lille, France. E-mail: gnm.reddy@univ.lille.fr
First published on 22nd October 2024
Abstract
Polymeric hydrogels and the associated structural assemblies are endowed with exceptional capabilities for applications in biomedicine, chemical biology, molecular electronics and wider energy paradigm. Cross-linking chemistry adds extra handles to tailor the gelation process and functional properties, surpassing those of traditional hydrogels. Here, we present molecularly tethered gelation of a cellulose (C) derivative by taking advantage of covalent and non-covalent interactions using organic and ionic linkers, respectively. The dual-cross-linked C-based hydrogels can be synthesized at a moderate temperature (∼70 °C) and processed into thin films using a programmable dip-coater at room temperature. The hydrogel films exhibited enhanced pH stability compared to the mono-cross-linked gels, were long-lived (over 180 days) and showed excellent ion-exchange properties. The gelation mechanism, local structures, and ion-exchange properties were corroborated by high-field (28.2 T, 1H = 1200 MHz) solid-state NMR spectroscopy. A facile gelation process enabled by covalent linkages, metal coordination, and multimodal characterization demonstrated here is expected to provide opportunities for a number of unexplored applications.
Introduction
Gels derived from natural products are currently of great interest for a multitude of applications ranging from biomedicine, artificial tissues, wood engineering to a wider energy paradigm.1–5 The ability of hydrophilic polymer networks to swell in and retain significant fractions of water causes gelation, leading to soft and self-standing three-dimensional materials referred to as hydrogels.6,7 The co-existence of sol and gel components enables these materials to acquire unique physicochemical properties.1,8,9 Organic molecules such as amino acids, nucleic acids, polysaccharides, and lignocellulosic biomass, are being widely researched for the development of functional hydrogels.10–17Cellulose (C) and its derivatives are a fascinating class of polymers, owing to the long polysaccharide chains with reactive vicinal diols, providing a stable foundation for gelation.7,14,18–24 C-gels have been explored in the areas of actuators, soft robots, sensors, batteries, artificial wood, flame retardants, flexible electronics, drug delivery, and in the controlled release of pesticides and fertilizers.5,25–31 Different synthesis routes enable unique physicochemical properties and stimulus adaptability with respect to temperature, light, strain, pH, and electric and magnetic fields further expands the application space of C-hydrogels.6,32–38 On the flip side, the insolubility of C in water continues to be an obstacle to the development of hydrogels.6,26,39–43 Efforts have been made to address this limitation by surface functionalization using acetate, methyl or carboxymethyl groups: for example, carboxymethylcellulose (CMC) has emerged as an alternative gelator for the aforementioned applications.39,44–47 Further improvements to the gelation of CMC can be sought using (non)covalent cross-linking chemistry. Specifically, linkers based on ionic and hydrophobic interactions have been used in tandem to develop hydrogels, which showed enhanced viscoelastic properties compared to the traditional CMC-hydrogels.48 Depending on the gelator and cross-linker, chain tethering can be facilitated by both covalent and non-covalent (including hydrogen bonding, electrostatic, C–H–π and π–π stacking) interactions, and/or metal coordination.49–51 Although a mono-cross-linking strategy has been demonstrated to develop CMC hydrogels/thin-films with adjustable mechanical strength, pH responsivity and self-healing properties,49,52,53 multi-cross-linking approaches are seldom applied. Despite the promising possibilities enabled by cross-linking chemistry, molecular-level understanding of the local structure in the vicinity of cross-linkers and its connection to gelation properties and applications is still limited, calling attention for further investigation.
Here we present a dual-cross-linked gelation of CMC with citric acid (CA) and Al3+ ions as cross-linkers. A combination of covalent cross-linking and metal coordination is integrated into a facile approach to synthesize hydrogel films at a moderate temperature of ∼70 °C, followed by drying and immersion into an Al3+ ionic solution. A second synthesis procedure by adding both linkers at the beginning of gelation was also explored. Both methods resulted in stable film formation. Gels can be processed into thin films on glass substrates by using a programmable dip-coating technique. The ion-exchange properties of hydrogel thin films are examined for capturing alkali metals from aqueous solutions. The pH and thermal stability of mono- and dual-cross-linked hydrogels are examined and compared. Since these hydrogels contain heterogeneous compositions and structures that are difficult to characterize by long-range techniques such as X-ray diffraction, we used infrared spectroscopy and solid-state (ss)NMR spectroscopy to gain insights into the local structure. Specifically, we applied two-dimensional ssNMR spectroscopy at a high magnetic field of 28.2 T to characterize the local structures in xerogels. In this way, the gelation mechanism is corroborated by analyzing different intermolecular interactions, which help explain their enhanced stability and ion-exchange properties. These gels are open to a number of unexplored applications, such as drug delivery, coatings and ion-exchange membranes, upon further modification. The introduction of Al3+ cations into the hydrogel network (i) enhances the cross-linking ability by metal coordination and non-covalent hydrogen bonding interactions, and (ii) leads to the formation of Al-aqua complexes, which retain large amounts of water in the hydrogel matrix, enabling better flexibility of the gel. Therefore, Al3+ is preferred over other mono or divalent cations. It brings additional benefits, such as cost-effectiveness, low toxicity (in case gels are applied in a biomedical context), actuators, conductivity and ion-exchange properties.54–58 The facile gelation and multiscale characterization presented in this study is expected to pave the way towards understanding stability–property relationships in hydrogels.
Experimental
Materials
All materials were used as received. CMC was purchased from ThermoFisher Scientific (analytical grade, degree of substitution: 0.9, average molecular weight: ∼700 kDa). Anhydrous citric acid (CA), boric acid and aluminum sulfate octadecahydrate (Al2(SO4)3·18H2O) were procured from Sigma Aldrich (analytical grade). Millipore water was used to make the gels.Preparation of mono-cross-linked CMC–CA hydrogels
To obtain a 2 wt% homogeneous solution, CMC powder was added to distilled water and stirred at room temperature overnight. A variable quantity of CA (corresponding to CA:CMC anhydroglucose unit (AGU) molar ratios of 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.1:1, 0.5:1, 1:1 and 2:1) was added and stirred at room temperature for 1 h to ensure a uniform distribution. The mixture was stirred at 70 °C for 30 minutes for the cross-linking reaction to occur. It was degassed for 15 minutes to remove the bubbles and dried overnight at 50 °C to obtain CMC–CA transparent hydrogel films. These were hydrated with excess water to remove them from the beakers, dried at 50 °C overnight to obtain CMC–CA xerogels.
:1, 0.1:1, 0.5:1, 1:1 and 2:1) was added and stirred at room temperature for 1 h to ensure a uniform distribution. The mixture was stirred at 70 °C for 30 minutes for the cross-linking reaction to occur. It was degassed for 15 minutes to remove the bubbles and dried overnight at 50 °C to obtain CMC–CA transparent hydrogel films. These were hydrated with excess water to remove them from the beakers, dried at 50 °C overnight to obtain CMC–CA xerogels.
Preparation of CMC–CA–Al dual-cross-linked hydrogels
:Al ions = 10:1 molar equivalent) and CA (at a ratio of CMC AGU:CA = 1:1 molar equivalent) were dissolved in water to make a homogeneous solution. CMC powder (3 wt%) was slowly added to this solution with stirring. Complete dissolution was ensured by stirring overnight. Air bubbles were removed by degassing for 1 hour. An Ossila dip-coater was used to cast the CMC–CA–Al hydrogel film with a dip speed of 1.00 mm s−1 and withdrawal speed of 5.00 mm s−1 for 2 immersion cycles. The coated films were dried in an oven at 120 °C for 20 minutes. The gelation procedure is presented in Scheme 1.
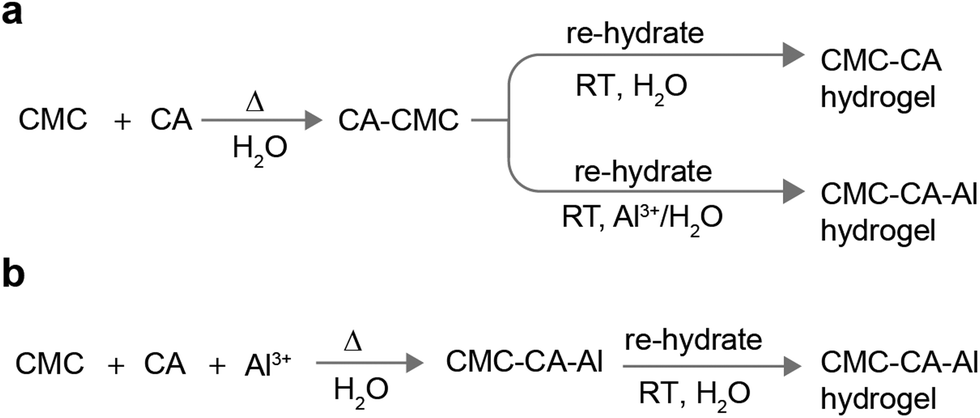 | ||
| Scheme 1 (a) Synthesis of mono-cross-linked CMC–CA hydrogel and dual-cross-linked CMC–CA–Al hydrogels obtained by the sequential addition of cross-linkers, i.e., CA is added first and then the gels are soaked in Al3+ solution. (b) Synthesis of dual-cross-linked CMC–CA–Al hydrogels obtained by the simultaneous addition of cross-linkers CA and Al3+ at the beginning of the gelation process. Both methods provided stable hydrogel films: see Fig. S1, ESI† for the chemical structures and photographs of the resulting hydrogels. | ||
Tap test
The stability of the formed gels was evaluated using both a tap test and vial inversion. The vials containing the gel matrix were gently tapped while held horizontally, and inverted to assess their clarity and self-standing integrity.pH-Responsive test
To evaluate the pH-responsive properties of the prepared hydrogel films, solutions of pH 4, 7 and 10 were used. CMC–CA–Al hydrogels of equal dimensions were immersed in the solutions and monitored for changes in swelling.Powder XRD characterization
The powder X-ray diffraction (PXRD) patterns of neat precursors and hydrogels were acquired using a Bruker D8 Advance A251 diffractometer equipped with Cu Kα radiation (λ = 1.5056 Å) at room temperature. Diffractograms were obtained from 2θ = 4° to 80° with a step width of 0.02° and a counting time of 0.5 s per step.Thermal analysis
Thermogravimetry (TGA) and differential scanning calorimetry (DSC) analysis of CMC and hydrogels were performed on approximately 10 mg of sample placed in an aluminum sample holder using a TA Q50 instrument. A temperature range of 25–600 °C was employed at a constant heating rate of 10 °C per minute. During the analysis, nitrogen gas was purged at a flow rate of 20 mL per minute to maintain an inert atmosphere.Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy
A PerkinElmer FTIR Spectrum Two UATR spectrometer was used to acquire the IR spectra of neat CMC, citric acid and the hydrogels. The spectra were acquired with 10 scans with a spectral resolution of 0.5 cm−1.Solid-state NMR spectroscopy
All materials for ssNMR analysis were used in powder form. 1D and 2D solid-state 1H and 27Al magic-angle spinning (MAS) NMR spectra were acquired on a 28.2 T Bruker Avance Neo spectrometer equipped with a 3.2-mm double-resonance H–X probe tuned to 1H and 27Al (Larmor frequencies are 1200.9 and 312.9 MHz, respectively). The MAS frequency was 20 kHz unless otherwise mentioned. The 1H 90° pulse duration was 2.3 μs. The 1H spectra of xerogels were acquired with 16 co-added transients using a recycle delay of 4 s. To acquire 1D 27Al spectra, a 90° pulse duration of 3.5 μs was used. Each spectrum was obtained by the co-addition of 512 transients with a recycle delay of 1 s. The 2D 27Al–1H correlation spectra of the hydrogel powders were acquired using a dipolar-mediated heteronuclear multiple-quantum coherence (D-HMQC) sequence with 27Al detection.59 The SR412 sequence60 was applied on the 1H channel to reintroduce the heteronuclear 27Al–1H dipolar couplings with a recoupling duration of τrcpl = 200 μs. The States-TPPI method was used for quadrature detection along the indirect 1H dimension. The 2D spectra resulted from 64 rotor-synchronized t1 increments, each with 32 co-added transients with a recycle delay of 1.5 s.For CA powder, all 1D 1H MAS and 2D 1H–1H correlation experiments were acquired with a 21.1 T Bruker Avance Neo spectrometer with a 1H Larmor frequency of 900 MHz. For 1D 1H the 90° pulse duration was 2 μs and the recycle delay was 50 s. All single-pulse 1H spectra of Cs adsorbed films were acquired with 4 co-added transients. The 2D 1H–1H spin-diffusion (SD) spectra were acquired using a three-pulse NOESY-like sequence with a mixing time τSD of 500 ms. The 2D spectra resulted from averaging 2 transients for each of 128 rotor-synchronized t1 increments. The spinning frequency was 50 kHz. The 2D 1H–1H double-quantum-single-quantum (DQ-SQ) spectrum was acquired using the back-to back (BaBa) sequence61,62 with one rotor period. For the indirect DQ dimension, 128 t1 increments were acquired each by coadding 16 transients using the States method to achieve sign discrimination. For CMC–CA and CMC–CA–Al, all 1D 23Na MAS NMR spectra were acquired on an 18.8 T Bruker Avance Neo spectrometer (Larmor frequencies: 1H = 800.1 MHz, 23Na = 211.7 MHz) equipped with a 1.3-mm double-resonance H–X probe tuned to 1H and 23Na. Each spectrum was obtained by the co-addition of 1024 transients with a recycle delay of 0.5 s. The 1H, 23Na, 27Al, 207Pb and 133Cs isotropic chemical shifts were calibrated using the 1H adamantane signal at 1.83 ppm as a secondary external reference, according to IUPAC nomenclature.63
Ion-exchange studies
To understand the Li+, Cs+, Sr2+ and Pb2+ adsorption properties, the CMC–CA–Al dip-coated films on glass substrates were immersed in aqueous solutions of metal ions for 1 hour. LiCl, CsCl and Pb(NO3)2 solutions were used (0.1, 0.5 and 20 wt%, respectively). The films were taken out from the solution and dried at room temperature overnight. The scratched films were powdered and used for analysis. 1D MAS NMR spectra were acquired on an 18.8 T Bruker Avance Neo spectrometer (Larmor frequencies: 1H = 800.1 MHz, 7Li = 311.0 MHz, 133Cs = 104.9 MHz and 207Pb = 167.4 MHz) equipped with a 1.3-mm double-resonance H–X probe tuned to 1H and X = 7Li or 133Cs or 207Pb. A spinning frequency of 50 kHz was employed. To acquire 1D 7Li MAS spectra, a 90° pulse duration of 1.4 μs was used with a relaxation delay of 0.5 s, by signal averaging with 1024 co-added transients. To collect 1D 133Cs NMR spectra, a 90° pulse duration of 1 μs and 1024 co-added transients were used. The adsorbed Cs+ ions on CMC–CA–Al films exhibited a shorter relaxation delay of 1 s, whereas a relaxation delay of 50 s was used for the CsCl salt. For 207Pb MAS NMR data collection, a 90° pulse duration of 1.25 μs was used with a relaxation delay of 0.5 s, using 20480 co-added transients.
Results and discussion
Fig. 1 presents the molecular structures of CMC and the cross-linkers used in this study, and compares a schematic of gelation between CMC and CA/Al3+ species together with the different steps involved. Although the chain entanglement of CMC in water leads to a highly viscous solution, it does not prevent the dissolution of CMC. As reported in the previous literature, CMC alone, i.e., without any covalent cross-linking agent being used, leads to poor gelation (ESI,† Fig. S2).64–68 Organic cross-linkers such as boric acid (BA) lead to poor gelation, and epichlorohydrin (ECH) leads to a stiff gel, but undergoes degradation over time (ESI,† Fig. S3 and S4). When a CMC:CA binary mixture in water (1:1, one CA equiv. per CMC anhydroglucose unit) was heated on a hot plate, covalent cross-linking between the OH of CMC and the COOH of CA occurs, yielding a stable hydrogel (Fig. 1b). In optimizing this process, we adjusted the CA concentration and temperature in the 70–150 °C range, but this did not show a substantial difference in gel setting, ESI,† Fig. S5, hence 70 °C was preferred. Gels made with CMC:CA [1:0.01] exhibited relatively low setting or started to flow downwards upon vial inversion. When [CA] was increased to 0.5 or 1, gelation was found to improve (ESI,† Fig. S6), indicating that CMC:CA [1:1] appears to yield better gel setting. However, further increasing [CA] to 2 or more resulted in reduced swelling and poor gelation. Once brought back to room temperature and allowed to settle, the CMC–CA [1:1] hydrogel leads to the formation of a thin layer at the gel/air interface, creating a barrier with the rest of the reaction mixture. Vial inversion, followed by a tap test during which the gel does not appear to run down, corroborates this observation (Fig. 1b, photographs in the inset), and hence the schematic presented in Fig. 1c. We reasoned that esterification occurs between –COOH (CA) and –OH (CMC). This reaction, depicted in the proposed mechanism (ESI,† Fig. S7a), leads to the formation of a thin layer at the gel/air interface. This esterification-mediated gel formation has been observed in similar systems in previous studies.64,69–75 It has been hypothesized that this reaction proceeds via the formation of a cyclic anhydride intermediate of CA in order to esterify the primary and secondary –OH groups of CMC,70 which may cause dehydration of excess water molecules as the cross-linking reaction of CMC evolves. Alternative mechanisms have also been proposed, as indicated in ESI,† Fig. S7b. Evaporation of excess of water results in a ‘nearly transparent’ layer at the bottom of the glass vial, necessitating a rehydration step to recover the hydrogel film. Consequently, these films are soaked in water to detach them from the vials to obtain stable hydrogel films. During this rehydration process, the Na+ ions present as counter cations in CMC are leached into the water. This was confirmed by acquiring and analyzing the 23Na MAS NMR spectra of the xerogels before and after soaking in water (ESI,† Fig. S8), where the low intensity of the 23Na peak in the washed gels confirms that the Na+ ions are weakly bound to the gel network. We were intrigued that the partial leaching of Na+ ions could provide opportunities to incorporate other multivalent cations via an ion-exchange process (vide infra), through which the gelation process could be adjusted.
 | ||
| Fig. 1 (a) Structures of gelator molecules and cross-linkers. (b) Gelation of CMC and CA together with photographs of the obtained gels with and without Al3+ linkers. The CMC–CA–Al gel exhibits excellent stability in water for over 180 days. (c) Schematic of cross-linked CMC–CA fibrils which are expected to lead to a hydrogel by cross-linking of CMC with CA and Al3+ ions, illustrating the use of both covalent and non-covalent interactions. | ||
The multivalent cations are expected to interact with the –COO− groups of CMC and CA, which could considerably improve the stability of the hydrogel thin films.76,77 Here we preferred Al3+ ions, owing to the ability of Al species to form coordinated aqua-complexes that can be brought into CMC–CA gels, enabling ionic cross-linking with the –COO− groups of CMC and CA moieties. In the subsequent stage, we aimed to obtain a dual-cross-linked CMC–CA–Al hydrogel, following the same synthesis step shown in Fig. 1b, except that the rehydration step involved an aqueous solution containing Al3+ ions. During this step, both Al3+ and their aqua complexes penetrated into the gel matrix and developed non-covalent interactions between the –COOH groups of CMC and CA. It is noteworthy that the addition of Al3+ to neither CMC nor CA alone causes stable film formation (ESI,† Fig. S9 and S10). Although the CMC–Al combination leads to the formation of hydrogels, the leaching of Al3+ ions into water upon soaking for 2 days leads to dissolution of the gel. This trend has been observed for similar systems with different metal ions used as cross-linkers.78–80 To this end, covalently and non-covalently dual-cross-linked CMC–CA–Al hydrogel films exhibit better stability and flexibility.
Understanding the gelation mechanism and the local structures of covalent/non-covalent linkages is paramount for identifying the stability–property relationships of hydrogel films, though it represents a characterization challenge. This is due, in part, to the compositional and structural heterogeneity associated with dual-cross-linked CMA–CA–Al films, which limits the use of long-range probes such as microscopy and X-ray scattering,81,82 or macroscopic property testing using rheology. Crystallinity and changes in long-range order upon gelation can be investigated by examining the X-ray diffraction patterns of these materials.83,84Fig. 2 compares the powder XRD patterns of CA and CMC with CMC–CA cross-linked hydrogel, where the well-resolved reflections of CA are due to high crystallinity that can be indexed based on the previously reported crystal structure.85,86 A broad PXRD feature centered at 20.4° for neat CMC suggests an amorphous nature, which is further reflected in the much broader PXRD patterns of CMC–CA and CMC–CA–Al xerogels (in comparison to CMC and CA), indicating a further reduction in the long-range order upon cross-linking reactions. Specifically, the absence of CA patterns in the hydrogel indicates that CA is dispersed into the CMC matrix at the molecular level due to cross-links and that at the given concentration CA does not phase separate to form any crystalline phase, a point that will be discussed again in the NMR analysis of local structures. These results are further supported by DSC (Fig. 2b) and TGA (Fig. 2c), which provide insights into the thermal stability, crystallization and melting properties. In the DSC plot of CMC (Fig. 2b), an endothermic peak at ∼49–51 °C is expected to occur due to the evaporation of physically bound water. For CMC–CA, two distinct exothermic peaks were observed in the ∼49–51 °C and 135–210 °C regions, which are attributable to the surface adsorbed water and the water molecules entrapped in the xerogel matrix, as reported previously.72,87 For CMC–CA–Al the exothermic peak at 195–260 °C indicates more stable water molecules entrapped in pores, which are closely associated with Al3+ species. In addition, these three compounds exhibit different melting behavior: CMC shows a relatively narrow feature (250–300 °C) compared to CMC–CA (250–300 °C) and CMC–CA–Al (with a low-intensity feature at 372 °C). TGA analysis corroborates these results, where different weight loss curves are observed for the xerogels of CMC powder, CMC–CA and CMC–CA–Al (Fig. 2c). Examining at ∼50% weight loss, a higher degree of thermal stability was observed for CMC–CA (∼350 °C) and CMC–CA–Al (∼370 °C) compared to neat CMC at ∼300 °C. In addition, a comparison of TGA analysis of CMC and cellulose is also presented in Fig. S11 (ESI†), in which the difference in thermal stability is due to the surface modification by carboxy methyl groups. Overall, these results indicate that the relatively high degree of thermal stability is due to the cross-linked CMC polymers.
 | ||
| Fig. 2 (a) Powder XRD patterns of CA, CMC, CMC–CA, and CMC–CA–Al. (b) DSC and (c) TGA plots of CMC, CMC–CA and CMC–CA–Al. In (c), the vertical dashed lines depict weight loss. | ||
To gain insights into the gelation process and the associated local chemical environments of CMC, CA and Al3+, we used attenuated total reflection Fourier transform infrared (FTIR) and ssNMR spectroscopy (Fig. 3). In the FTIR spectrum of CMC (Fig. 3a), stretching frequencies at ∼3490 cm−1 (νO–H), 2913 cm−1 (νC–H), 1616 cm−1 (antisymmetric νCOO), and 1412 cm−1 (symmetric νCOO) were detected. The vibrational frequencies of primary/secondary alcohol C–OH appear at 1106 cm−1, 1047 cm−1, 1019 cm−1 and 995 cm−1, and the β-1,4-glycosidic bonds between the glucose units exhibited a band at 898 cm−1. For CA, bands at 3279 cm−1 and at 1693 cm−1 are attributed to νO–H and hydrogen-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties, respectively. For CMC–CA, a new feature at 1715–1745 cm−1 suggests the formation of ester bonds between CMC(OH) and CA(COOH), and νO–H is at higher values 3500–3546 cm−1, which corroborates the formation of hydrogen bonds facilitating gelation. In CMC–CA–Al, νCO has shifted to a higher value, suggesting the formation of ionic/coordination links between Al3+ and COO− groups of CMC moieties. These results are further corroborated by magic-angle spinning (MAS) NMR spectroscopy that provides insights into the local structures and packing interactions in gels.88–92
O moieties, respectively. For CMC–CA, a new feature at 1715–1745 cm−1 suggests the formation of ester bonds between CMC(OH) and CA(COOH), and νO–H is at higher values 3500–3546 cm−1, which corroborates the formation of hydrogen bonds facilitating gelation. In CMC–CA–Al, νCO has shifted to a higher value, suggesting the formation of ionic/coordination links between Al3+ and COO− groups of CMC moieties. These results are further corroborated by magic-angle spinning (MAS) NMR spectroscopy that provides insights into the local structures and packing interactions in gels.88–92
 | ||
| Fig. 3 (a) Solid-state FTIR transmittance plots of CMC, CA, CMC–CA and CMC–CA–Al (1:0.5). (b) Solid-state 1D 1H MAS NMR spectra of CMC, CA, CMC + CA, CMC–CA, and CMC: CA–Al xerogels, and (c) 1D 27Al NMR spectra of CMC–CA–Al with different CMC:CA ratio as indicated. (d) Schematic of Al3+ complexes in close proximity to CMC and CA moieties. (e) Solid-state 2D 27Al–1H correlation NMR spectrum plotted along with 27Al and 1H skyline projections (inset) and the corresponding 1D spectra on the top and left-hand axes. The spectra presented in (b) were acquired at 21 T (1H = 900 MHz) with 50 kHz MAS, and in (c) and (e) were acquired at 28.2 T (1H = 1200.5 MHz and 27Al = 312.9 MHz) with 20 kHz MAS. | ||
In the 1D 1H MAS and 2D 1H–1H correlation NMR spectra of CMC (Fig. 3b and Fig. S12, ESI†), a broad feature at 2–5 ppm is due to overlapping contributions from anomeric protons, CH2 and OH groups. For CA, peaks at 2.4 and 3.0 ppm (CH2), at 5.5 ppm (OH), and at 10, 10.7, and 13.9 ppm (COOH) are identified (ESI,† Fig. S13 and S14). In contrast, the 1H NMR spectra of CMC–CA and CMC–CA–Al do not display the signals associated with –COOH groups of CA (10–14 ppm), due to the formation of covalent ester linkages, except when a high CA concentration of 5 equivalents was used (ESI,† Fig. S15). This observation is rationalized by a cross-linking reaction between COOH(CA) and the OH(CMC) groups. To test this, we acquired the 1H NMR spectrum of a physical mixture of CMC + CA (1 + 2), in which all of these peaks are retained, indicating that the cross-linking reactions between CA and CMC do not occur in the physical mixture. For CMC–CA and CMC–CA–Al, the absence of signals (10–14 ppm) could be an indication of the involvement of all COOH moieties of CA, unlike in the formation of a CA-cyclic intermediate that still holds a COOH group, which may occur through different mechanisms (ESI,† Fig. S7b). In the 1D 27Al NMR spectra of CMC–CA–Al (Fig. 3c and Fig. S16, ESI†), two distinct hexa-coordinated Al sites are detected: peaks at 0.5–2 ppm are attributed to a hexa-aqua [Al(H2O)6]3+ complex,93 where displacement and broadening of the 27Al peak is observed upon increasing the concentration of CA. An additional peak centered at 7 ppm, whose intensity increased as the concentration of CA cross-linker increased, is expected to originate from the secondary hexa-coordinated Al site binding, at least in part, to both CA and CMC moieties (Fig. 3d). Comparison of the 1D 27Al MAS NMR spectra of the xerogels and hydrogels (Fig. S17, ESI†) confirmed identical 27Al local coordination environments in both materials, as revealed by identical chemical shifts and peak intensities. Hence, for convenience we used xerogels for ssNMR spectroscopy analysis, including hydrogels prepared by the sequential and simultaneous addition of gelator and cross-linkers (Fig. S18, ESI†). Specifically, analysis of the 2D 27Al–1H correlation spectra (Fig. 3e and Fig. S19, ESI†) shed light on the local structures and through-space Al–H proximities in these species. While the 2D peak between 27Al (1.5 ppm) and 1H (4.7 ppm) is consistent with the incorporation of Al by means of [Al(H2O)6]3+ species, low-intensity peaks in 27Al (7.2–5.5 ppm) and 1H (3.8 ppm) confirm the spatial proximity between Al and the protons within the CMC–CA network (denoted by the doubled-headed arrows in Fig. 3d and red-shaded region in Fig. 3e).
A stability test of gels at different pH values is often required to assess their suitability for specific applications.94,95 The pH compatibility of CMC–CA and CMC–CA–Al was assessed by immersing them in buffer solutions (pH 4, 7, and 10) over a month (Fig. 4a and b). For both mono- and dual-cross-linked hydrogels, enhanced stability is observed at an acidic/neutral pH of 4–7. However, exposure to basic conditions (pH = 10) leads to swelling and partial distortion of the films. Overall, the CMC–CA–Al hydrogel films exhibited outstanding stability of over 150 days (ESI,† Fig. S20), in contrast to CMC–CA, owing to the ionic cross-linking through Al3+ ions. Yet long-term exposure to basic conditions may cause instability and the loss of structural integrity. Analysis of the FTIR spectra of gels before and after exposure to aqueous solutions with different pH values (Fig. 4c and d) corroborates these results, by means of changes in the vibrational bands associated with the carboxyl groups (vertical grey bands) and hydrogen-bonded protons (3000–3400 cm−1). At an acidic pH of 4, the IR peaks are nearly intact even after 90 days, suggesting the high stability of the films under acidic conditions. As the pH increases to 7, the broadening of the carboxylic stretching peaks is specifically observed, which could be attributed to weakening of ester linkages and enhanced swelling. At a basic pH of 10, the spectra are dominated by water peaks, suggesting dissolution of the gel matrix into the solution, owing to the loss of cross-links between polymer chains.
 | ||
| Fig. 4 Photographs of (a) CMC–CA (CMC:CA = 1:1) and (b) CMC–CA–Al (CMC:CA = 1:1 soaked in 0.1 M Al3+ solution) dual-cross-linked hydrogel films taken before and after immersion in aqueous solutions of pH = 4, 7 and 10 for up to 30 days. Solid-state attenuated FTIR transmittance plots of (c) CMC–CA and (d) CMC–CA–Al hydrogels acquired before and after exposure to pH 4, 7 and 10 for 90 days. | ||
In exploring the suitability of CMC–CA–Al gels as membranes for ion-exchange and environmental remediation applications, we examined the uptake of mono and divalent metals from aqueous solutions. Desalination involving hydrogels requires processing, such as extrusion, membrane formation and coating on the sorbate.96–98 Here we used dip-coated CMC–CA–Al on glass substrates, and the dried films were used to adsorb Li+, Cs+, Sr2+ and Pb2+ ions from aqueous solutions with variable concentrations of metal ions. The resulting materials are studied by analyzing the FTIR spectra before and after ion exchange (ESI,† Fig. S21) as well as the 7Li and 133Cs MAS NMR spectra (Fig. 5). The FTIR transmittance plots of ion-adsorbed hydrogel films (Fig. S21, ESI†) exhibited changes in the 1750–1600 cm−1 range (–CO stretching region), indicating interactions between carbonyl moieties and ions.
 | ||
| Fig. 5 (a) 1D 7Li MAS NMR spectra of Li+ adsorbed on hydrogel thin films together with the spectrum of Li2CO3, and (b) the normalized 7Li peak integral plotted as a function of concentration of Li2CO3 in water. (c) 1D 133Cs MAS NMR spectra of CsCl and Cs+-adsorbed Al–CA–CMC, and (d) photographs of Al–CA–CMC coated on glass substrates before and after Cs+ adsorption, and the 133Cs peak integral plotted as a function of concentration of CsCl in water. All spectra were acquired at 18.8 T (7Li = 311.0 MHz, and 133Cs = 104.9 MHz) with 50 kHz MAS and at room temperature. | ||
Fig. 5a presents the 7Li NMR spectra, in which the peaks in the vicinity of ∼0 ppm are characteristic of ionic Li+ species. The Li+ adsorption capacity of CMC–CA films (Fig. 5b) was estimated by analyzing the 7Li NMR peak integral with respect to a standard external calibration sample (powdered Li2CO3, in which Li = 18.8 at%). The mass of Li+ ions in Li2CO3 was used as a reference in order to estimate the amount of Li+ ions adsorbed on the hydrogel films, which was found to be ∼50 mg g−1 of sorbent.99,100 An identical protocol was used to study the Cs+ adsorption studies (Fig. 5c and d) using 133Cs NMR spectroscopy. When [Cs+] solution of less than 1 wt% was used, the Cs+ ions are entrapped in the CMC–CA–Al sorbent, which produces 133Cs NMR peaks in the 0–100 ppm range. Upon increasing the concentration of Cs+ ions in water, additional narrow features appeared in the 260–330 ppm range corresponding to phase-separated Cs+ ions resembling CsCl salt (photographs depicting the precipitate are shown in Fig. 5c). The concentration of Cs+ ions entrapped in the gel matrix was determined to be in the range 3–12.2 mg g−1 of sorbent, showing excellent efficiency (ESI,† Section S23).101,102 In an identical manner, the adsorption characteristics of Pb2+ ions by CMC–CA–Al gels were studied. 207Pb NMR peak at ∼3473 ppm (ESI,† Fig. S22) indicates the presence of Pb2+ or Pb(NO3)2 species. However, the poor sensitivity and resolution associated with 87Sr and 207Pb NMR and relatively longer acquisition times limit the study of adsorption isotherms. Nonetheless, 1H and 27Al ssNMR at 28.2 T is particularly suitable to characterize these xerogels.103,104
Conclusions
In summary, covalent and non-covalent interactions facilitate a stable gelation of CMC, CA and Al species, which can be processed into hydrogel thin films using a dip-coater. The resultant hydrogel films exhibit better thermal stability than CMC powder, indicating the covalent cross-linking of CMC polymers. The entanglement of CMC biopolymers by CA and Al3+ ions imparts distinct properties to the resulting hydrogel films, with greater flexibility, ion exchange and pH-responsive behavior. In doing so, we studied the gelation mechanism and local structures by FTIR and ssNMR spectroscopy. Hydrogel films exhibited excellent absorption/exchange properties with Li+, Na+, Cs+, Pb2+ and Al3+. These findings contribute valuable insights to hydrogel development, with implications for stimuli responsivity and processability. These gels are expected to open up opportunities for a number of unexplored applications. Among various applications, thin films dip coated on glass substrates are examined here for desalination processes. Further studies can be envisaged, such as the incorporation of dyes or drug molecules for biomedical applications, luminescent soft materials, environmental remediation, supercapacitors, 3D printing or additive manufacturing of hydrogels.Data availability
Data related to this work is available from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out under the EU H-2020 framework programme under Marie Skłodowska-Curie actions grant no. 795091. The Chevreul Institute is thanked for supporting CPER projects funded by the “Ministère de l’Enseignement Supérieur et de la Recherche”, the region “Hauts-de-France”, the ERDF program of the European Union and the “Métropole Européenne de Lille”. Financial support from the IR Infranalytics FR2054 for conducting the research is gratefully acknowledged. We thank Prof. Orlando Rojas for discussions.References
- L. Hu, P. L. Chee, S. Sugiarto, Y. Yu, C. Shi, R. Yan, Z. Yao, X. Shi, J. Zhi, D. Kai, H.-D. Yu and W. Huang, Adv. Mater., 2023, 35, 2205326 CrossRef.
- J. Y. C. Lim, S. S. Goh, S. S. Liow, K. Xue and X. J. Loh, J. Mater. Chem. A, 2019, 7, 18759–18791 RSC.
- K. Zhang, Q. Feng, Z. Fang, L. Gu and L. Bian, Chem. Rev., 2021, 121, 11149–11193 CrossRef.
- A. A. K. Das, J. Bovill, M. Ayesh, S. D. Stoyanov and V. N. Paunov, J. Mater. Chem. B, 2016, 4, 3685–3694 RSC.
- P. Sikdar, M. M. Uddin, T. M. Dip, S. Islam, M. S. Hoque, A. K. Dhar and S. Wu, Mater. Adv., 2021, 2, 4532–4573 RSC.
- S. H. Zainal, N. H. Mohd, N. Suhaili, F. H. Anuar, A. M. Lazim and R. Othaman, J. Mater. Res. Technol., 2021, 10, 935–952 CrossRef.
- S. K. Das, T. Parandhaman and M. D. Dey, Green Chem., 2021, 23, 629–669 RSC.
- H. Yuk, B. Lu and X. Zhao, Chem. Soc. Rev., 2019, 48, 1642–1667 RSC.
- H. Yuk, J. Wu and X. Zhao, Nat. Rev. Mater., 2022, 7, 935–952 CrossRef CAS.
- X.-Q. Dou and C.-L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef PubMed.
- Y. Zhang, L. Zhu, J. Tian, L. Zhu, X. Ma, X. He, K. Huang, F. Ren and W. Xu, Adv. Sci., 2021, 8, 2100216 CrossRef PubMed.
- J. Li, L. Mo, C.-H. Lu, T. Fu, H.-H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC.
- V. G. Muir and J. A. Burdick, Chem. Rev., 2021, 121, 10908–10949 CrossRef PubMed.
- X. Lin, P. Wang, R. Hong, X. Zhu, Y. Liu, X. Pan, X. Qiu and Y. Qin, Adv. Funct. Mater., 2022, 32, 2209262 CrossRef.
- D. M. Alshangiti, T. K. El-damhougy, A. Zaher, M. Madani and M. Mohamady ghobashy, RSC Adv., 2023, 13, 35251–35291 RSC.
- S. Mondal, S. Das and A. K. Nandi, Soft Matter, 2020, 16, 1404–1454 RSC.
- S. S. Das, D. Sharma, B. V. K. Rao, M. K. Arora, J. Ruokolainen, M. Dhanka, H. Singh and K. K. Kesari, Mater. Adv., 2023, 4, 6064–6091 RSC.
- L. Lewis, S. G. Hatzikiriakos, W. Y. Hamad and M. J. MacLachlan, ACS Macro Lett., 2019, 8, 486–491 Search PubMed.
- S. Bhaladhare and D. Das, J. Mater. Chem. B, 2022, 10, 1923–1945 Search PubMed.
- X. Shi, Y. Hu, K. Tu, L. Zhang, H. Wang, J. Xu, H. Zhang, J. Li, X. Wang and M. Xu, Soft Matter, 2013, 9, 10129–10134 Search PubMed.
- A. K. Tamo, J. Mater. Chem. B, 2024, 12, 7692–7759 Search PubMed.
- G. Dandegaonkar, A. Ahmed, L. Sun, B. Adak and S. Mukhopadhyay, Mater. Adv., 2022, 3, 3766–3783 RSC.
- M. A. Hossain, C. K. Roy, S. D. Sarkar, H. Roy, A. H. Howlader and S. H. Firoz, Mater. Adv., 2020, 1, 2107–2116 RSC.
- C. Palo-Nieto, A. Blasi-Romero, C. Sandström, D. Balgoma, M. Hedeland, M. Strømme and N. Ferraz, Mater. Adv., 2023, 4, 1555–1565 Search PubMed.
- D. Zhao, J. Huang, Y. Zhong, K. Li, L. Zhang and J. Cai, Adv. Funct. Mater., 2016, 26, 6279–6287 Search PubMed.
- S. M. F. Kabir, P. P. Sikdar, B. Haque, M. A. R. Bhuiyan, A. Ali and M. N. Islam, Prog. Biomater., 2018, 7, 153–174 Search PubMed.
- C. Zhou and Y. Wang, Sci. Technol. Adv. Mater., 2020, 21, 787–804 Search PubMed.
- S. Sugiarto, R. R. Pong, Y. C. Tan, Y. Leow, T. Sathasivam, Q. Zhu, X. J. Loh and D. Kai, Mater. Today Chem., 2022, 26, 101022 Search PubMed.
- Z. J. He, K. Chen, Z. H. Liu, B. Z. Li and Y. J. Yuan, J. Clean. Prod., 2023, 414, 137708 Search PubMed.
- Z. Hanif, D. Choi, M. Z. Tariq, M. La and S. J. Park, ACS Macro Lett., 2020, 9, 146–151 CrossRef CAS PubMed.
- A. Trubetskaya, J. Leppiniemi, S. Lipponen, S. Lombardo, W. Thielemans, T. Maloney, T. Pääkkönen, K. K. Kesari, J. Ruokolainen, V. P. Hytönen and E. Kontturi, Mater. Adv., 2024, 5, 570–583 RSC.
- P. K. Vemula and G. John, Acc. Chem. Res., 2008, 41, 769–782 CrossRef CAS.
- C. Chang, L. Zhang, J. Zhou, L. Zhang and J. F. Kennedy, Carbohydr. Polym., 2010, 82, 122–127 CrossRef CAS.
- X. Shen, J. L. Shamshina, P. Berton, G. Gurau and R. D. Rogers, Green Chem., 2015, 18, 53–75 RSC.
- Y. Dong, S. Zhao, W. Lu, N. Chen, D. Zhu and Y. Li, RSC Adv., 2021, 11, 10794–10803 RSC.
- Y. Chen, G. Cui, N. Dan, Y. Huang, Z. Bai, C. Yang and W. Dan, SN Appl. Sci, 2019, 1, 1–10 Search PubMed.
- S. Wang, L. Yu, S. Wang, L. Zhang, L. Chen, X. Xu, Z. Song, H. Liu and C. Chen, Nat. Commun., 2022, 13, 3408 CrossRef PubMed.
- J. Manasi Esther, R. Solanki, M. Dhanka, P. Thareja and D. Bhatia, Mater. Adv., 2024, 5, 5365–5393 RSC.
- X. He and Q. Lu, Carbohydr. Polym., 2023, 301, 120351 CrossRef.
- Y. Zhou, C. Wan, Y. Yang, H. Yang, S. Wang, Z. Dai, K. Ji, H. Jiang, X. Chen and Y. Long, Adv. Funct. Mater., 2019, 29, 1–8 Search PubMed.
- M. Hu Tu, M. Zhu, B. Duan and L. Zhang, Adv. Mater., 2021, 33, 200682 Search PubMed.
- C. Wu, J. Li, Y. Zhang, X. Li, S. Wang and D. Li, ChemSusChem, 2023, 16, e202300518 CrossRef PubMed.
- L. Zhang, B. Zhan, Y. He, Y. Deng, H. Ji, S. Peng and L. Yan, Green Chem., 2024, 26, 8794–8807 RSC.
- S. T. C. L. Ndruru, E. Pramono, D. Wahyuningrum, B. Bundjali and I. M. Arcana, Bull. Mater. Sci., 2021, 44, 761–778 Search PubMed.
- W. Zhang, Y. Liu, Y. Xuan and S. Zhang, Gels, 2022, 8, 529 CrossRef PubMed.
- P. Kaur, H. B. Bohidar, D. R. Nisbet, F. M. Pfeffer, A. Rifai, R. Williams and R. Agrawal, Cellulose, 2023, 30, 2713–2730 CrossRef.
- M. D. Islam, F. J. Uddin, T. U. Rashid and M. Shahruzzaman, Mater. Adv., 2023, 4, 4054–4102 RSC.
- H. Zhang, X. Wu, Z. Qin, X. Sun, H. Zhang, Q. Yu, M. Yao, S. He, X. Dong, F. Yao and J. Li, Cellulose, 2020, 27, 9975–9989 CrossRef.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Adv. Funct. Mater., 2020, 30, 1–22 CrossRef.
- C. Shao and J. Yang, in Self-Healing and Self-Recovering Hydrogels, ed. C. Creton and O. Okay, Springer International Publishing, Cham, 2020, pp. 319–354 Search PubMed.
- D. Massana Roquero, A. Othman, A. Melman and E. Katz, Mater. Adv., 2022, 3, 1849–1873 RSC.
- P. Sánchez-Cid, M. Jiménez-Rosado, A. Romero and V. Pérez-Puyana, Polymers, 2022, 14, 3023 CrossRef.
- Z. Li, F. Lu and Y. Liu, J. Agric. Food Chem., 2023, 71, 10238–10249 CrossRef.
- S. Anjum, P. Gurave, M. V. Badiger, A. Torris, N. Tiwari and B. Gupta, Polymer, 2017, 126, 196–205 CrossRef.
- H. Jiang, L. Fan, S. Yan, F. Li, H. Li and J. Tang, Nanoscale, 2019, 11, 2231–2237 RSC.
- H. Kumar, A. K. Gehlaut, H. Gupta, A. Gaur, S. Kamsonlian and D. Kumar, Polym. Compos., 2021, 42, 3899–3910 CrossRef.
- Y. Nakagawa, S. Ohta, M. Nakamura and T. Ito, RSC Adv., 2017, 7, 55571–55576 RSC.
- K. Ouyang, J. Zhuang, C. Chen, X. Wang, M. Xu and Z. Xu, Biomacromolecules, 2021, 22, 5033–5041 CrossRef PubMed.
- M. Taoufik, K. C. Szeto, N. Merle, I. Del Rosal, L. Maron, J. Trébosc, G. Tricot, R. M. Gauvin and L. Delevoye, Chem. – Eur. J., 2014, 20, 4038–4046 CrossRef PubMed.
- A. Brinkmann and A. P. M. Kentgens, J. Am. Chem. Soc., 2006, 128, 14758–14759 CrossRef PubMed.
- I. Schnell, A. Lupulescu, S. Hafner, D. E. Demco and H. W. Spiess, J. Magn. Reson., 1998, 133, 61–69 CrossRef.
- G. N. M. Reddy, M. Malon, A. Marsh, Y. Nishiyama and S. P. Brown, Anal. Chem., 2016, 88, 11412–11419 CrossRef PubMed.
- R. K. Harris, E. D. Becker, S. M. C. de Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef.
- M. Madaghiele, C. Demitri, I. Surano, A. Silvestri, M. Vitale, E. Panteca, Y. Zohar, M. Rescigno and A. Sannino, Sci. Rep., 2021, 11, 1–14 CrossRef.
- R. Nuisin, T. Siripongpreda, S. Watcharamul, K. Siralertmukul and S. Kiatkamjornwong, ChemistrySelect, 2022, 7, e202104598 CrossRef.
- M. F. Bósquez-Cáceres, L. De Lima, V. Morera Córdova, A. D. Delgado, J. Béjar, N. Arjona, L. Álvarez-Contreras and J. P. Tafur, Batteries, 2022, 8, 265 CrossRef.
- Z. Wang, W. Deng, J. Peng, L. Miao, Y. Chen and W. Chen, J. Electrochem. Soc., 2022, 169, 93501 CrossRef CAS.
- N. Antonova, Mater. Today Proc., 2021, 38, 1588–1591 CrossRef CAS.
- T. Nongnual, N. Butprom, S. Boonsang and S. Kaewpirom, Int. J. Biol. Macromol., 2024, 267, 131135 CrossRef.
- G. F. de Lima, A. G. de Souza and D. D. S. Rosa, Macromol. Symp., 2020, 394, 2000126 CrossRef.
- K. Wilpiszewska, A. K. Antosik, B. Schmidt, J. Janik and J. Rokicka, Polymers, 2020, 12, 1–16 CrossRef.
- K. Mali, S. Dhawale, R. Dias, N. Dhane and V. Ghorpade, Indian J. Pharm. Sci., 2018, 80, 657–667 Search PubMed.
- C. Demitri, R. Del Sole, F. Scalera, A. Sannino, G. Vasapollo, A. Maffezzoli, L. Ambrosio and L. Nicolais, J. Appl. Polym. Sci., 2008, 110, 2453–2460 CrossRef.
- S. Gorgieva, R. Vogrinčič and V. Kokol, J. Polym. Environ., 2019, 27, 318–332 CrossRef.
- V. S. Ghorpade, A. V. Yadav and R. J. Dias, Carbohydr. Polym., 2017, 164, 339–348 CrossRef.
- W. N. Sharratt, C. G. Lopez, M. Sarkis, G. Tyagi, R. O’connell, S. E. Rogers and J. T. Cabral, Gels, 2021, 7, 1–21 CrossRef PubMed.
- X. Li, M. Li, L. Tang, D. Shi, E. Lam and J. Bae, Mater. Chem. Front., 2023, 7, 5989–6034 RSC.
- W. Yang, Y. Ding, J. Liang, C. Li, H. Bian, H. Dai and C. Hu, Cellulose, 2023, 30, 3901–3913 CrossRef.
- S. Sun, L. Wang and A. Wang, J. Hazard. Mater., 2006, 136, 930–937 CrossRef PubMed.
- C. B. Godiya, X. Cheng, D. Li, Z. Chen and X. Lu, J. Hazard. Mater., 2019, 364, 28–38 CrossRef PubMed.
- V. S. Raghuwanshi and G. Garnier, Adv. Colloid Interface Sci., 2019, 274, 102044 CrossRef.
- H. Garcia, A. S. Barros, C. Gonçalves, F. M. Gama and A. M. Gil, Eur. Polym. J., 2008, 44, 2318–2329 CrossRef.
- K. S. Salem, N. K. Kasera, M. A. Rahman, H. Jameel, Y. Habibi, S. J. Eichhorn, A. D. French, L. Pal and L. A. Lucia, Chem. Soc. Rev., 2023, 52, 6417–6446 RSC.
- F. Lin, X. Lu, Z. Wang, Q. Lu, G. Lin, B. Huang and B. Lu, Cellulose, 2019, 26, 1825–1839 CrossRef.
- J. P. Glusker, J. A. Minkin and A. L. Patterson, Acta Crystallogr. B., 1969, 25, 1066–1072 CrossRef PubMed.
- G. Roelofsen and J. A. Kanters, Cryst. Struct. Commun., 1972, 1, 23 CAS.
- G. N. M. Reddy, D. S. Cook, D. Iuga, R. I. Walton, A. Marsh and S. P. Brown, Solid State Nucl. Magn. Reson., 2015, 65, 41–48 CrossRef CAS.
- M. Nonappa, B. Lahtinen, E. Behera, E. Kolehmainen and U. Maitra, Soft Matter, 2010, 6, 1748–1757 RSC.
- M. El Hariri El Nokab and P. C. A. van der Wel, Carbohydr. Polym., 2020, 240, 116276 CrossRef CAS PubMed.
- M. E. H. El Nokab, M. H. Habib, Y. A. Alassmy, M. M. Abduljawad, K. M. Alshamrani and K. O. Sebakhy, Polymers, 2022, 14, 1049 CrossRef.
- G. M. Peters, L. P. Skala, T. N. Plank, B. J. Hyman, G. N. M. Reddy, A. Marsh, S. P. Brown and J. T. Davis, J. Am. Chem. Soc., 2014, 136, 12596–12599 CrossRef CAS PubMed.
- G. N. M. Reddy, G. M. Peters, B. P. Tatman, T. S. Rajan, S. M. Kock, J. Zhang, B. G. Frenguelli, J. T. Davis, A. Marsh and S. P. Brown, Mater. Adv., 2020, 1, 2236–2247 RSC.
- M. Etou, T. Taketatsu, Y. Okaue, T. Inoue and T. Yokoyama, J. Solution Chem., 2023, 52, 1318–1328 CrossRef.
- A. Hendi, M. U. Hassan, M. Elsherif, B. Alqattan, S. Park, A. K. Yetisen and H. Butt, Int. J. Nanomedicine, 2020, 15, 3887–3901 CrossRef PubMed.
- S. R. Mane, A. Sathyan and R. Shunmugam, ACS Appl. Nano Mater., 2020, 3, 2104–2117 CrossRef.
- Y. Guo and G. Yu, Acc. Mater. Res., 2021, 2, 374–384 CrossRef.
- K. Zhang, X. Luo, L. Yang, Z. Chang and S. Luo, ACS ES T Water, 2021, 1, 1098–1116 CrossRef.
- J. Kaur, P. Sengupta and S. Mukhopadhyay, Ind. Eng. Chem. Res., 2022, 61, 1921–1954 CrossRef.
- L. Yang, Y. Tu, H. Li, W. Zhan, H. Hu, Y. Wei, C. Chen, K. Liu, P. Shao, M. Li, G. Yang and X. Luo, Angew. Chem., Int. Ed., 2023, 62, e202308702 CrossRef PubMed.
- S. H. Park, K. Kim, J. H. Lim and S. J. Lee, Sep. Purif. Technol., 2019, 212, 611–618 CrossRef.
- M. A. Olatunji, M. U. Khandaker, H. N. M. E. Mahmud and Y. M. Amin, RSC Adv., 2015, 5, 71658–71683 RSC.
- E. Cho, J. Kim, C. W. Park, K. W. Lee and T. S. Lee, J. Hazard. Mater., 2018, 360, 243–249 CrossRef PubMed.
- C. Welton, P. Raval, J. Trébosc and G. N. M. Reddy, Chem. Commun., 2022, 58, 11551–11554 RSC.
- N. Thomas, J. Dhainaut, A. Moissette, O. Lafon and G. N. M. Reddy, J. Phys. Chem. C, 2024, 25, 10248–10439 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details about materials and synthesis, reaction mechanism, photographs of gels and gelation with boric acid and epichlorohydrin are illustrated. Solid-state 1D 1H and 2D 1H–1H correlation NMR spectra of precursors, CMC–CA and CMC–CA gels are shown. For CMC–CA–Al gels, the pH stability, FTIR spectra of ion-exchanged gels, and 207Pb NMR spectra are presented. See DOI: https://doi.org/10.1039/d4ma00815d |
| This journal is © The Royal Society of Chemistry 2024 |