
Solvate ionic liquid-derived solid polymer electrolyte with lithium bis(oxalato) borate as a functional additive for solid-state lithium metal batteries†
Yan
Yuan
 *ab,
Xiuping
Peng
a,
Bin
Wang
a,
Kesi
Xue
a,
Zhengqian
Li
a,
Yitian
Ma
c,
Bin
Zheng
c,
Yonghui
Song
ab and
Hai
Lu
*c
*ab,
Xiuping
Peng
a,
Bin
Wang
a,
Kesi
Xue
a,
Zhengqian
Li
a,
Yitian
Ma
c,
Bin
Zheng
c,
Yonghui
Song
ab and
Hai
Lu
*c
aSchool of Metallurgical Engineering, Xi'an University of Architecture and Technology, Xi'an, 710055, China. E-mail: lingyi21@126.com; Tel: +86 29 82202923
bShaanxi Key Laboratory of Gold and Resources, Xi'an, 710055, China
cSchool of Materials Science and Engineering, Xi'an University of Science and Technology, Xi'an, 710054, China. E-mail: lhxust@126.com; Tel: +86 29 85587373
First published on 5th December 2022
Abstract
Solid-state lithium metal batteries have great application prospects due to their high theoretical energy density but are restricted by the low ionic conductivity of the solid electrolyte and unsatisfactory interface chemistry between the electrode and electrolyte. Herein, a novel solid polymer electrolyte (SPE) was obtained by incorporating a solvate ionic liquid (SIL) [Li(G4)1][TFSI] containing LiBOB as a functional additive into the polymer matrix. Profiting from the synergistic effect of the SIL and the Li+ conductive salt, the uniquely designed SPE has high ionic conductivity, wide electrochemical window, high Li+ transference number and excellent compatibility with metallic Li. Accordingly, it endows the solid-state lithium metal battery with superior long-term cyclability at room temperature. The addition of LiBOB not only improves the Li+ coordination and conductive environment, but also contributes to the rigid-flexible coupling interface chemistry to buffer the volume change in Li metal upon cycling and achieve homogeneous and dendrite-free Li deposition.
Introduction
Recently, Li metal has been attracting significant attention as a superior anode for next-generation secondary batteries due to its ultra-high theoretical capacity (3860 mA h g−1) and the lowest redox potential (−3.04 vs. SHE).1 However, an inhomogeneous and fragile solid electrolyte interphase (SEI) is usually formed on the Li surface, inducing the continuous consumption of liquid electrolytes and destabilizing the Li metal batteries (LMBs) with low coulombic efficiency.2 Moreover, uncontrolled Li dendrites gradually form during repeated Li plating/stripping, which can lead to cell failure and even safety issues such as an internal short circuit.3 In addition, the inclusion of liquid electrolytes is inevitably confronted with a series of risks such as leakage, combustion and gas generation. Therefore, it is essential to develop effective approaches to further improve both the cycle life and safety of LMBs.Replacing the flammable liquid electrolyte with a solid-state electrolyte is a highly desirable strategy to overcome the above challenges of the LMBs because of its inherent safety and the potential to prevent dendritic growth. As a typical representative, solid polymer electrolytes (SPEs)4 composed of lithium salt and polymer are particularly attractive owing to their excellent processability, desirable flexibility and good interfacial contact with the electrodes. Unfortunately, low ionic conductivity at room temperature (RT) caused by the high crystallinity of the polymer matrix impedes their application. Incorporating a liquid plasticizer into the polymer matrix to form a quasi-solid-state electrolyte5–7 can promote the segmental motion of polymer chains, increase the dissociation of the Li salt and reduce the interface contact resistance, but this usually results in poor mechanical strength. Alternatively, introducing inorganic fillers into the SPEs8–10 can also increase the proportion of amorphous regions to improve ionic mobility, as well as enhance the mechanical properties of the membrane; however, it is difficult to achieve the uniform distribution of the solid particles in the polymer matrix.
Solvate ionic liquid (SIL)11 is a particularly concentrated electrolyte in which ligand molecules strongly coordinate with the cations and/or anions of salts to form complex ions. The equimolar mixture of triglyme (G3) or tetraglyme (G4)-Li salt has the long-lived robust complex cation [Li(Gn)1]+ (n = 3 or 4), regarded as the prototype of the SILs.12 Because of the strong ion–dipole interactions between the glymes and Li+, the SILs based on the glyme-Li salt mixture indicate many similarities in the physical properties to common ILs, such as low melting point, low volatility and high thermal stability. In addition, they are intrinsically Li+-conductive with high Li+ transference numbers.13 More importantly, all of the lone pairs of the glyme oxygen atoms in the equimolar mixture are donated to the Li+ since the coordination number of Li+ is typically 4–5 (that is, theoretically without free or uncoordinated solvent),14 which greatly enhances the oxidation stability of the glyme and thus can afford the normal charge–discharge operation of the LMBs.15,16
Because of a series of merits offered by SIL, a few reports have attempted to employ the SIL [Li(Gn)1]X (X is counter anion) as both the lithium resource and liquid plasticizer to develop advanced SPEs with favorable ionic transport properties and high safety. For instance, Gao et al.17 constructed a gel polymer electrolyte by rapid UV-initiated polymerization with ethoxylated trimethylolpropane triacrylate (ETPTA) as a polymer monomer added into the SIL solution. The obtained SIL-based gel electrolyte exhibited enhanced thermal stability, robust mechanical strength and high ionic conductivity. Moreover, a close-contact electrode/electrolyte interface was formed by the in situ curing of the electrolyte on the electrode surface. Kido et al.18 investigated the effects of the polymer matrix (PEO, PMMA and PBA) on the thermal, ionic transport, and electrochemical properties of the SIL-based polymer electrolyte. However, these studies rarely referred to the application of the SPE towards 4V-class LMBs, and the latent relationships among the solvated structure of the SIL, physicochemical properties of the SPE, Li surface chemistry and battery performance remain to be investigated.
In this work, a novel SIL-based SPE was prepared by incorporating the [Li(G4)1][TFSI] into the PVDF-HFP matrix, with Li+ conductive salt LiBOB as a functional additive. The experimental studies combined with theoretical calculation were conducted to fundamentally understand the synergistic effect of the SIL and Li salt additive on the microscopic morphology, solvation structure and physicochemical properties of the SPE. Then, the optimized designed SPE was introduced in various LMBs to evaluate its application value. As a focus, a latent competition mechanism between different counteranions in interacting with Li+ was analyzed, and then a simple selection rule for the Li salt additive for the SIL-derived SPE was explored. The regulation principle of the LiBOB additive and the components of the SIL on the Li surface chemistry were intensively investigated and discussed.
Experimental section
Materials and preparation
PVDF-HFP (Mw ≈ 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Macklin), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, 99.95%, Sigma-Aldrich) and lithium bis(oxalato)borate (LiBOB, 99.0%, Aladdin) were directly used without further purification. G4 and THF solvents supplied by Aladdin were pretreated via activated molecular sieves prior to use.
000, Macklin), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, 99.95%, Sigma-Aldrich) and lithium bis(oxalato)borate (LiBOB, 99.0%, Aladdin) were directly used without further purification. G4 and THF solvents supplied by Aladdin were pretreated via activated molecular sieves prior to use.
[Li(G4)1][TFSI] (denoted as LG) was prepared by mixing LiTFSI with G4 in a molar ratio of 1:1. PVDF-HFP (30 wt%) and LG (70 wt%) were added to THF with continuous stirring at 60 °C, and then LiBOB was further introduced with a total mass of 1/3/5%. The obtained homogeneous solution was cast in a PTFE mold and dried in a vacuum oven at 60 °C for 12 h to remove THF. The obtained SPE containing LiBOB (referred to as PLGB-x, where x = 1, 3, 5) was cut into disks with a diameter of 20 mm and stored in an argon-filled glove box. SPE without LiBOB (denoted as PLG) was prepared based on a similar procedure. An all-solid-state electrolyte consisting of PVDF-HFP and LiTFSI was also fabricated for comparison.
Structural characterization
Raman spectra were recorded on a LabRAM HR evolution Raman spectrometer to analyze the coordination environment of the as-prepared liquid or solid electrolyte. Fourier transform infrared (FT-IR) spectra of the SPEs were collected on a TENSORII infrared spectrophotometer from 4000 cm−1 to 550 cm−1. The crystal structures of the SPEs were characterized by XRD (Smart Lab). The surface and cross-sectional morphologies of SPEs and electrodes were investigated by scanning electron microscopy (SEM, Hitachi S4800), accompanied by energy dispersive X-ray spectroscopy (EDS) for elemental analyses. XPS (Thermo Scientific K-Alpha+) with Al-Kα radiation was used for identifying the elemental species on the Li surface. Thermogravimetric analyses (TGA, TA SDT Q600) were measured from 30 °C to 800 °C at a heating rate of 20 °C min−1. The mechanical strengths of the SPEs were tested by controlling the elongation rate of 5 mm min−1 on the tensile tester (CMT6103).Electrochemical measurement
The SPE was clamped by two parallel stainless-steel plates (SS) in a CR2025-type coin cell. The electrochemical impedance spectroscopy (EIS) of the SS|SPE|SS cell was tested on an electrochemical workstation (DH7006) in the frequency range from 105 Hz to 0.1 Hz with the perturbation amplitude of 5 mV. The ionic conductivities (σ) of the SPE at different temperatures were calculated according to the following equation (eqn (1)): | (1) |
 | (2) |
The LiFePO4 (LFP) cathode was fabricated by uniformly dispersing 80 wt% LFP, 10 wt% super P, and 10 wt% PVDF in N-methyl-2-pyrrolidone (NMP). Then, the obtained slurry was coated on a piece of Al foil by a doctor blade and cut into discs with a diameter of 14 mm. The typical mass loading on the cathode was ∼2.5 mg cm−2. The LFP/Li coin cell was assembled in the glove box and cycled galvanostatically between 2.5 and 4.2 V on a Neware test system. Cyclic voltammetry (CV) was monitored on the electrochemical workstation at a scan rate of 0.1 mV s−1.
Simulation methods
Density functional theory (DFT) geometry optimization calculations were performed using the Dmol3 code.19 The exchange–correlation energy was defined by the B3LYP level of theory.20 All electron core treatments were used for all atoms, as was the double-numeric basis with polarization functions (DNP). The dispersion correction (Grimme) was adopted for describing the long-range interactions. The binding energy was obtained using eqn (3):| Eb = ELi+/A − ELi+ − EA | (3) |
The molecular dynamics simulations were conducted in the Forcite module in Materials Studio of Accelrys Inc. A condensed-phase optimized molecular potentials for atomistic simulation studies (COMPASS III) force field21 was chosen for the LiTFSI/G4 electrolyte, and a rule-based force field22 was used for the LiTFSI/G4/LiBOB electrolyte. The time step was fixed at 1 fs and the temperature was set at 298 K with a Nose thermostat. The simulation time was long enough to ensure that the equilibrium states of the electrolyte systems were reached. The coordination number was obtained by the integration,  ,23 where ρ0 is the average number density of the anion or solvent and rmin is the minimum position of g(r) after the first peak.
,23 where ρ0 is the average number density of the anion or solvent and rmin is the minimum position of g(r) after the first peak.
Results and discussion
Solvation environment
The MD simulation snapshots for the [Li(G4)1][TFSI] (LG) are shown in Fig. 1a. It is revealed that Li+ is closely surrounded by G4 molecules in the first solvation shell. Meanwhile, a considerable number of TFSI− anions enter the solvation sheath surrounding the central Li+. The RDF calculation in Fig. 1b confirms that the first solvation shell in the LG is dominated by the glyme solvent and counteranion, as indicated by the sharp peaks for Li–OG4 and Li–OTFSI at ∼2.0 Å. The resulting Li+ coordination number was calculated to be 2.56 O (G4) and 2.18 O (TFSI−). After adding LiBOB into the LG system (Fig. 1c, LGB), a new sharp peak for Li–OBOB appears at ∼2.1 Å, whereas the peak intensity for Li–OTFSI closest to the ion center slightly weakens, accompanied by a broadened peak around ∼4 Å. The coordination number from oxygen atoms of the BOB− reaches 0.77. These results suggest that the Li-TFSI distance increases to some extent after the introduction of LiBOB, and a part of TFSI− could be replaced by BOB− in the solvation structure of the SIL. | ||
| Fig. 1 (a) MD simulation snapshot of the [Li(G4)1][TFSI] and schematic of the first solvation shell around Li+. Radial distribution functions (RDFs) of the Li–O pairs in the (b) LG and (c) LGB. (d) Raman spectra and (e) species distribution of the LG and LGB. (f) Interactions of Li ions or solvated Li ions with two counteranions. (g) Raman spectra and (h) species distribution of the PLG and PLGB. | ||
To elucidate the effect of LiBOB on Li+ solvation chemistry, Raman spectroscopy was conducted on the two designed solutions. The strong absorption peak observed in the range of 730–760 cm−1 (Fig. 1d) is ascribed to the S–N symmetric stretching vibration coupled with the –CF3 bending arising from TFSI−,24 which can generally be divided into three bands due to the free anion (FA), loose ion pair (LIP) and intimate ion pair (IIP).25 The distribution of these solvate species is indicated in Fig. 1e and the detailed results are summarized in Table S1.† In the LG system, the FA scarcely exists and the LIP is the predominant species of TFSI− (considering that the chelating effect from G4 separates the coordination of TFSI− towards Li+), similar to the characteristics of those high-concentration electrolytes.26 The addition of LiBOB to the system leads to the decrease in LIP and the increase in FA, implying the substitution of BOB− for TFSI− in the solvation structure, consistent with the above RDF results.
Since the [Li(glyme)]X with weak Lewis basic anions (such as X = TFSI−) can form long-lived complex cations, the Li(G4)+ can be considered to be an integral unit for interacting with the counteranions, which should mainly contribute to the solvent-shared LIP in the SIL. The DFT calculation in Fig. 1f reveals that Li(G4)+-BOB− has a lower binding energy than Li(G4)+-TFSI−. In other words, solvated Li+ due to the chelating effect of the G4 will preferentially coordinate with BOB− rather than TFSI−. This explains why a part of TFSI− in the LIP can be replaced by BOB−. However, the slightly increased IIP proportion suggests that it is difficult for the additive in the liquid solution to dissociate those close-contact ion pairs (one anion coordinating with two or more Li+, e.g., aggregates),27 which is probably due to strong Li-TFSI interactions. This can be confirmed by the interaction difference of two counter anions with bare Li+, as the binding energy of Li+-TFSI− is lower than that of Li+-BOB−. Moreover, it is likely that part of TFSI− from dissociated LIP recombined with Li+ to generate a compact Li-TFSI complex, resulting in the increase of IIP in the LGB.
The solvation structure and cation–anion interaction in the SPE were also investigated by Raman technology. The results in Fig. 1g and h and Table S1† indicate a notable decrease in LIP and IIP if incorporating the LG into the PVDF-HFP matrix (PLG, in contrast to the original LG); meanwhile, a certain amount of the FA species appear. It should be noted that an additional influencing factor from the polymer matrix arises in the SPE, which is different from the above pure liquid environment. On one hand, the interaction between G4 and PVDF-HFP (so-called “plasticizing effect”) shares a part of the glyme solvent for the polymer. Thus, insufficient G4 impels the conversion of the solution species from LIP to FA and/or IIP. On the other hand, it is likely that the latent interaction between cation/anion and polar functional group in the polymer chain weakens the Li+-TFSI− association. Consequently, BOB− could easily attack the close-contact ion pairs28 and then release the free TFSI−, which also leads to the increase in FA from the conversion of IIP. Therefore, the FA is inevitably produced in the PLG.
Afterwards, the introduction of LiBOB in the SPE give rise to a similar effect on Li+ coordination as the liquid electrolyte, that is, the decrease of LIP followed by the increase of both FA and IIP, owing to the competition mechanism of the added BOB− with TFSI− in interacting with solvated Li+. The resulting solvation structure, consisting of rich LIP and moderate amounts of FA and IIP brought by the synergistic effect of the SIL, LiBOB and polymer matrix, forms a unique ion transport model. Based on the above analyses, the typical characteristics of the LiBOB employed in this study can be concluded according to the order of binding energy (E), that is, ELi-BOB > ELi-TFSI, while ELi(solvent)-BOB < ELi(solvent)-TFSI. A simple classification rule of the Li salt additive for the SIL-derived SPE is proposed in Fig. S1.†
Electrolyte structure and properties
The surface and cross-sectional SEM images of the as-prepared SPEs are shown in Fig. 2a, b and S2c.† The PLG and PLGB all present dense, uniform and single-phase structures with low porosity (thickness ∼130 μm), regardless of the addition of LiBOB. The EDS mapping in Fig. 2c1–c4 reveals the uniform distribution of various components (LiTFSI, LiBOB, G4, PVDF-HFP) in the PLGB without visible aggregation, which reflects the excellent compatibility of the SIL and the Li salt additive with the polymer matrix. In contrast, the all-solid-state electrolyte merely consisting of LiTFSI and PVDF-HFP delivers a rough surface with severe phase separation, probably due to the insufficient dissolution of LiTFSI in the polymer (as shown in Fig. S2a and b†), which will cause disordered ion transport in the SPE, poor interface contact with the electrodes and inferior mechanical properties of the membrane. The stress–strain curves provided in Fig. S3† demonstrate that the integration of G4 from SIL dramatically improves the stretchability of the resulting membrane with higher elongation at break when comparing PLG with PVDF-HFP/LiTFSI at the same thickness. Therefore, the polar solvent G4 is regarded as a key factor in the structural stability and homogeneity of the SIL-derived SPE. Moreover, the introduction of LiBOB further increases the tensile strength and modulus from 3.45/8.83 MPa in PLG to 4.98/12.86 MPa in PLGB; hence, it is capable of withstanding repeated stretching or curling operation without structure deformation. As exhibited in Fig. 2d, the self-standing PLGB has outstanding flexibility since it can maintain the original state after being folded several times. | ||
| Fig. 2 SEM images of (a) PLG and (b) PLGB; (c1–c4) EDS mapping of PLGB. (d) Photographs of the PLGB membrane undergoing bending tests. (e) FTIR spectra and (f) XRD patterns of the as-prepared SPEs and their components. | ||
FT-IR analyses of the as-prepared SPEs and their components are provided in Fig. 2e. Two strong characteristic peaks at 2870 cm−1 and 1100 cm−1 correspond to the stretching vibration of the CH3 group saturated at the end group and the stretching vibration of the C–O bond in the G4, respectively.29 On dissolving equimolar LiTFSI in the G4, the clear position shift and intensity fading of the above peaks can be observed, reflecting the significant interaction of G4 with LiTFSI and the formation of the solvated structure [Li(G4)1][TFSI]. The FT-IR spectrum of PLG illustrates the characteristic absorptions assigned to PVDF-HFP (e.g., 1400 cm−1, 870 cm−1 and 793 cm−1),30 and that of PLGB verifies the presence of LiBOB (new bands centered at 1810 cm−1 and 1660 cm−1 are characterized to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the carbonyl group in the LiBOB).31 The inconspicuous effect of incorporating LiBOB on the FT-IR response of the SPE would be attributed to a limited additive amount. However, the divided FT-IR spectrum at 720–755 cm−1 (as shown in Fig. S4 and Table S2†) reflects the same conclusion as the above Raman analyses, that is, the existence of LiBOB in the SPE leads to more free TFSI− and reduced LIP.
O bond of the carbonyl group in the LiBOB).31 The inconspicuous effect of incorporating LiBOB on the FT-IR response of the SPE would be attributed to a limited additive amount. However, the divided FT-IR spectrum at 720–755 cm−1 (as shown in Fig. S4 and Table S2†) reflects the same conclusion as the above Raman analyses, that is, the existence of LiBOB in the SPE leads to more free TFSI− and reduced LIP.
The XRD patterns of the as-prepared SPEs are compared in Fig. 2f. The characteristic peaks located at 18.2°, 20.2°, 26.8°, and 40° reflect the crystalline α-phase of PVDF-HFP.32 With the incorporation of the SIL, the absorption peaks at 18.2° and 26.8° nearly disappear and the one at 40° becomes significantly less intense, indicating that the interaction between SIL and PVDF-HFP disrupts the regular arrangement of the polymer chain and accordingly increases the fraction of the amorphous phase, which is convenient for chain movement and ion migration in the SPE. The addition of LiBOB further causes a slight decline in the overall crystallinity of the SPE. The TGA analyses of the two SPEs in Fig. S5† present a continuous weight loss beginning at around 30 °C until 800 °C, which can be ascribed to the evaporation of G4 from the solid phase in the initial stage and subsequent decomposition of the Li salts, yet the thermal stability of the SPE is still suitable for battery operation under normal temperatures.
The effects of the SIL and LiBOB on the ionic conductivity of SPE were investigated. The bulk resistance extracted from the EIS plots in Fig. 3a was used to calculate the ionic conductivity according to eqn (1). The room temperature ionic conductivity of the all-solid-state polymer electrolyte PVDF-HFP/LiTFSI is only 1.58 × 10−6 S cm−1, whereas it reaches 7.57 × 10−4 S cm−1 at 25 °C for the PLG (Fig. 3b), revealing a preferable ionic transport path along the polymer matrix assisted by the SIL. The addition of LiBOB further enhances the ionic conductivity of the SPE, which mainly benefited from the regulative solvation environment and decreased crystallinity of the polymer matrix. When the mass ratio is up to 3 wt%, the ionic conductivity of PLGB-3 reaches as high as 2.18 × 10−3 S cm−1 at 25 °C, superior to previously reported SPEs containing ionic liquids or other organic solvents (as shown in Table S3†). However, a higher LiBOB content is less significant to the improvement of the ionic conductivity. The dependence of ionic conductivity on temperature follows the Arrhenius behavior. The LSV curves in Fig. 3c indicate that the PLGB-group SPEs all have better oxidation stability relative to the counterpart PLG. Moreover, the increased additive amount brings about a wider stable potential window of the electrolyte. This is because the glyme oxygen atoms also need to be donated to the Li+ in the LiBOB, and then the solvent reactivity in the higher-concentration electrolyte brought by the Li salt additive would be further weakened.
 | ||
| Fig. 3 Electrochemical characterizations of PLG and PLGB-x (x = 1, 3, 5): (a) Nyquist plots at RT; (b) Arrhenius plots of the conductivities. (c) The LSV measurement of SS|SPE|Li cells; (d) DC polarization profiles of the symmetrical Li|PLGB-3|Li cell (the inset shows the impedance spectra before and after polarization). | ||
The tLi+ of the SPE was evaluated via the chronoamperometry method in a symmetrical Li|SPE|Li cell with a constant voltage of 10 mV. The tLi+ of PLGB-3 (Fig. 3d) was calculated to be ∼0.86, which is much higher than that of PLG (∼0.77, as exhibited in Fig. S6†) and PVDF-HFP/LiTFSI reported in the literature.33 The anion contribution to the total ion migration number will be greatly limited by the scarce FA in the SIL. Even though the moderate introduction of LiBOB releases a part of the free TFSI− as well as provides additional BOB−, it would be preferable for the Li+ migration owing to the reformative ion–solvent, ion–polymer and anion–cation interactions; therefore, increased tLi+ in the PLGB-3 is still obtained. The SPE with high tLi+ is conductive to low polarization during cell operation and uniform Li deposition, as discussed below.
To probe the interfacial stability of the SPE against Li metal, Li plating/stripping cycling in the symmetric Li/SPE/Li cells was performed. The formation of Li dendrites is usually the result of multi-phase Li+ deposition caused by uneven surface current density during the plating/stripping process and side reactions at the interface. As presented in Fig. 4a, the Li|PLG|Li cell has a low overpotential (<30 mV) but a short circuit occurs after only 500 h of testing. The PLGB-type SPEs deliver a higher initial overpotential than PLG but tend to stabilize upon prolonged cycling, which may be related to the building of the SEI layer.34 Among them, the PLGB-3 favors the stable cycle up to 2000 h with a relatively lower polarization voltage of ≈150 mV in contrast to other LiBOB-containing SPEs at a current density of 0.1 mA cm−2 (Fig. 4b and c). The symmetrical Li|PLGB-3|Li cell can even cycle for more than 4500 h at 0.05 mA cm−2 (Fig. S7†). Also, it exhibits acceptable plating/stripping cyclability and reversibility at higher current density (e.g., 0.2 and 0.5 mA cm−2) with a controlled cycle number (as shown in Fig. S8–S10†). The Li deposition morphology from the cycled Li|PLGB-3|Li cell after 2000 h is given in Fig. S11.† A smooth and integrated surface without any detectable dendrites is observed, indicating that a suitable amount of LiBOB effectively regulates the Li deposition and hence achieves excellent compatibility of the SPE with metallic Li. Associated with the highest redox peak exhibited in the CV profile of the LFP|PLGB-3|Li cell (Fig. S12†), the optimum LiBOB content is selected as 3 wt% for subsequent investigations.
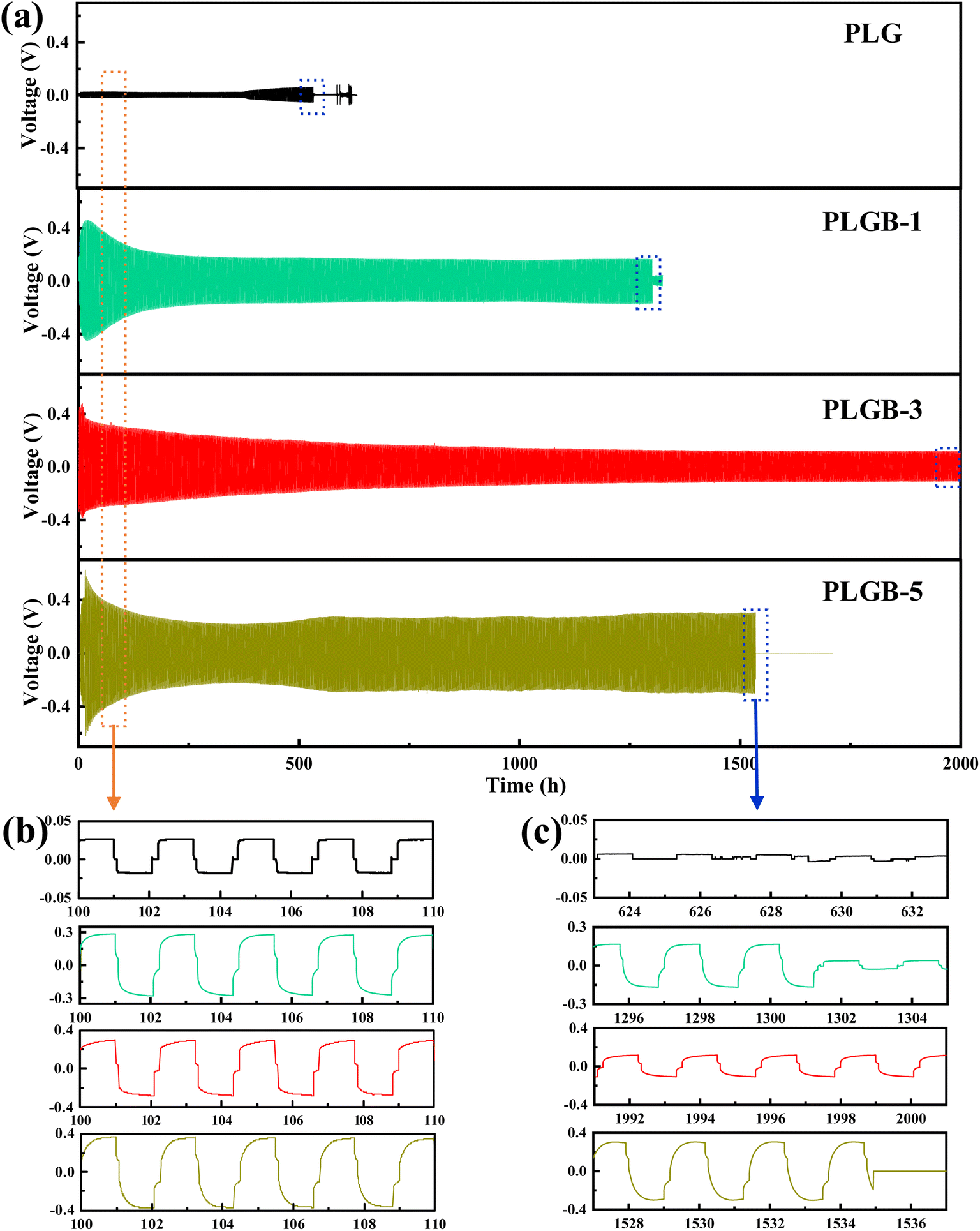 | ||
| Fig. 4 (a) Galvanostatic cycling profiles of the symmetrical Li|SPE|Li cells with various SPEs at the current density of 0.1 mA cm−2 at RT. Enlarged voltage profiles for various SPEs during (b) the initial 100–110 h and (c) the last 10 h. | ||
Electrochemical performance
The optimized design of PLGB-3 with favorable overall properties was assembled in the LFP|Li cells. The solid-state battery demonstrates excellent rate capability ranging from 0.1C to 2C at RT (Fig. 5a and b), which profited from the good ionic conductivity of the SPE. A high specific capacity of 159.9 mA h g−1 with well-defined voltage plateaus of ∼3.3 V is achieved at 0.1C, and the capacity retention reaches 63.0% when the rate is increased by 20 times to 2C (100.8 mA h g−1). When the discharge current is back to 0.1C, the specific capacity almost recovers to its initial value. The CV curves in Fig. 5c further demonstrate the excellent redox reversibility of the cell, and the DCD profiles in Fig. 5d reflect the acceptable charge/discharge stability. In particular, the room-temperature capacity retention of the LFP|PLGB-3|Li cell after 500 cycles at 0.5C (Fig. 5e) is as high as 95.9% with average coulombic efficiency of 99.96%, which is comparable and even superior to those reported SPE systems, as listed in Table S3.† In contrast, the LFP|PLG|Li cell sustains acceptable cycling, exceeding 30 cycles at 0.1C (as confirmed in Fig. S13†) since low current density corresponds to a longer Li nucleation time, and consequently restrains the dendrite growth controlled by the Li+ transport step. However, the dendrite formation is accelerated and hence continual capacity decay is observed in the cell at a higher current density of 0.5C (dropped to 43.1 mA h g−1 at only 200 cycles). The PLGB-3 also favors the stable cycling of the as-assembled LiNi0.5Co0.2Mn0.3O2 (NCM523)/Li cell and LiCoO2 (LCO)/Li cell at the discharge current of 0.5C at RT, as indicated in Fig. S14.† The addition of LiBOB plays a key role in improving the cyclability of these Li metal batteries, e.g., enabling high interfacial stability of the SPE with the metallic Li. | ||
| Fig. 5 The electrochemical performance of the LFP|PLGB-3|Li cell at RT: (a) rate performance; (b) DCD profiles at different C-rates; (c) CV curves; (d) DCD curves at different cycle stages. (e) A comparison of the long-term cycle performance between LFP|PLG|Li and LFP|PLGB-3|Li cells at 0.5C. | ||
Interface chemistry
The electrodes disassembled from the cycled solid-state batteries were characterized by SEM. The cycled cathode in the LFP|PLG|Li cell (Fig. 6b) presents a rough morphology with a large number of irregular aggregates, whereas that in the LFP|PLGB|Li cell (Fig. 6c) remains intact and flat, close to its pristine state (Fig. 6a). The more significant difference in surface morphology appears at the Li anode side (Fig. 6d–f). It is found that the cycled Li metal from the LFP|PLG|Li cell (Fig. 6e) has been deeply corroded with clear holes/cracks and massive deposits originating from undesired side reactions. In contrast, the anode surface remains quite smooth and dense after the LFP|PLGB|Li cell was cycled (Fig. 6f). It is considered that the added LiBOB would contribute to a successful surface modification on both electrodes, especially the Li anode. | ||
| Fig. 6 SEM images of LFP cathodes and Li anodes: (a) the pristine cathode; harvested cathode from the (b) LFP|PLG|Li cell and (c) LFP|PLGB-3|Li cell after 500 cycles; (d) fresh Li metal; harvested Li metal from the (e) LFP|PLG|Li cell and (f) LFP|PLGB-3|Li cell after 500 cycles. | ||
To elucidate the interfacial chemistry differences induced by different SPEs, the SEI composition on the cycled Li anodes were investigated by XPS. The high-resolution XPS spectra of C, O, F and B elements are presented in Fig. 7a–d and the surface atomic percentages are compared in Fig. 7e. The C 1s spectra can be divided into five fitted peaks, in which C–C, C–O and CO peaks always represent those organic degradation products.35 The strong Li2CO3/–CF2 signal in the PLG case is related to the decomposition of electrolyte components (salt, solvent and polymer)36 but it disappears in the cycled PLGB cell; instead, a prominent –CF3 group is detected, which is mainly derived from TFSI−. As evidenced by previous results, free TFSI− arises in the PLGB, enabling it or its decomposition product to be easily incorporated into the SEI layer (more sulfur species found in Fig. 7e can be deemed as another signal from TFSI−). Besides, a small amount of Li2O/LiOH responsible for the Li–O band in the O 1s spectra is identified after adding LiBOB.
 | ||
| Fig. 7 High-resolution XPS patterns of (a) C 1s, (b) O 1s, (c) F 1s, and (d) B 1s on the surface of cycled Li metal from the LFP|PLG|Li cell (upper layer) and LFP|PLGB-3|Li cell (bottom layer). (e) Comparison of the atomic percent on the surface; (f) HOMO and LUMO energy levels of the Li salts and the solvent by DFT calculation. (g) An illustration of the mechanism for regulating the SEI constitution to achieve a dendrite-free Li interface by PLGB. | ||
The F 1s spectrum demonstrates that more LiF is produced in the PLG. Its accumulation in the SEI could result in continuously increased interfacial resistance and hinder the electrochemical reaction.37,38 In comparison, the relatively low intensity of LiF and limited Li content in the PLGB reveals that lithium fluoride is not the main constituent of the SEI. Instead, other fluoride species containing –CFx (e.g., –CF3 from TFSI−) dominate the Li surface. In particular, an additional peak relating to the B–F/O–B–F compound appears, which can also be observed in the B 1s spectra, suggesting that the LiBOB participates in the SEI construction. The molecular orbital energy calculation via DFT in Fig. 7f confirms that the LiBOB has a lower LUMO energy level (−2.805 eV) than LiTFSI (−1.173 eV) and G4 so that it can be more preferentially reduced on the surface of the Li anode.
The above XPS results are helpful for the clear recognition of the role of LiBOB in the interface chemistry on the Li anode. A credible working mechanism of the additive in regulating SEI is illustrated in Fig. 7g. The Li surface without LiBOB mainly consists of large amounts of organic species followed by lithium carbonate and lithium fluoride produced by the electrolyte decomposition. The organic–inorganic hybrid SEI has low interfacial energy for Li, which suppresses the lateral Li+ flux along Li/SEI39 but promotes the vertical dendrite growth and uncontrollable side-reaction between the electrolyte and fresh Li. After introducing LiBOB as a functional additive, it is firstly reduced on the Li anode, building a B-rich surface with abundant B–F/O–B–F based polymer species (such as (CO2BF2)2 dimers)40 and moderate content of Li2O/LiOH. This makes the SEI more elastic41,42 and can buffer the volume evolution of Li metal during cycling. Afterwards, the released TFSI− due to the substitution of LiBOB fabricates a stiff top layer rich in –CFx, conducive to preventing the dendrite growth and realizing homogeneous Li deposition. Therefore, these distinctive components in the PLGB case are more likely to act synergistically for building a rigid-flexible coupling SEI on the Li surface with good elasticity, adequate mechanical strength and high Li+ conductivity.
Conclusion
In summary, a novel PVDF-HFP-based SPE was successfully developed by introducing [Li(G4)1][TFSI] and LiBOB into the polymer matrix. The SIL with a unique solvated structure modifies the membrane morphology and provides favorable ionic transport kinetics along the polymer matrix. The additional LiBOB further regulates the Li+ coordination environment by competing with TFSI−. Combined with the two advantages of the SIL and the additive, the optimized PLGB-3 exhibits high ionic conductivity (2.18 × 10−3 S cm−1) at RT, a high lithium-ion transfer number (0.86) and superior electrochemical stability (up to 5.7 V vs. Li/Li+). Moreover, the excellent Li compatibility of the PLGB-3 favors the stable Li plating/stripping cycle of the symmetrical cell beyond 2000 h without detectable dendrites. The SEM and XPS investigations further confirm that the LiBOB participates in interface construction and then contributes to a favorable SEI on the Li anode, together with released TFSI anions. Consequently, the assembled solid-state LFP|Li cell delivers a high capacity of 143.2 mA h g−1 and capacity retention as high as 95.9% after 500 cycles at a rate of 0.5C at RT. This study provides a convenient and scalable solid electrolyte strategy (without rare raw material) for the application of next-generation LMBs with high energy density, long lifespan and good safety.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 51704222), the Key Project of Natural Science Basic Research Plan of Shaanxi Province (No. 2022JZ-25) and Scientific Research Program of Shaanxi Provincial Education Department (22JC049).References
- J. Lang, L. Qi, Y. Luo and H. Wu, Energy Storage Mater., 2017, 7, 115 CrossRef.
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 1 CrossRef.
- P. Jaumaux, J. Wu, D. Shanmukaraj, Y. Wang, D. Zhou, B. Sun, F. Kang, B. Li, M. Armand and G. Wang, Adv. Funct. Mater., 2020, 31, 2008644 CrossRef.
- X. Yang, K. R. Adair, X. Gao and X. Sun, Energy Environ. Sci., 2021, 14, 643 RSC.
- J. Hwang, K. Matsumoto, C.-Y. Chen and R. Hagiwara, Energy Environ. Sci., 2021, 14, 5834 RSC.
- S. Xu, R. Xu, T. Yu, K. Chen, C. Sun, G. Hu, S. Bai, H.-M. Cheng, Z. Sun and F. Li, Energy Environ. Sci., 2022, 15, 3379–3387 RSC.
- Y. Yuan, Z. Li, X. Peng, K. Xue, M. Liu, Z. Fang, L. Wang, H. Du and H. Lu, Electrochim. Acta, 2022, 410, 140052 CrossRef CAS.
- R. Fang, B. Xu, N. S. Grundish, Y. Xia, Y. Li, C. Lu, Y. Liu, N. Wu and J. B. Goodenough, Angew. Chem., Int. Ed., 2021, 60, 17842 CrossRef.
- Z. Sun, Y. Li, S. Zhang, L. Shi, H. Wu, H. Bu and S. Ding, J. Mater. Chem. A, 2019, 7, 11069 RSC.
- W. Zhang, J. Nie, F. Li, Z. L. Wang and C. Sun, Nano Energy, 2018, 45, 413 CrossRef CAS.
- T. Mandai, K. Yoshida, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 8761 RSC.
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323 CrossRef CAS PubMed.
- S. Zhang, A. Ikoma, Z. Li, K. Ueno, X. Ma, K. Dokko and M. Watanabe, ACS Appl. Mater. Interfaces, 2016, 8, 27803 CrossRef CAS PubMed.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121 CrossRef CAS PubMed.
- A. Orita, K. Kamijima, M. Yoshida, K. Dokko and M. Watanabe, J. Power Sources, 2011, 196, 3874 CrossRef CAS.
- K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Electrochem. Soc., 2012, 159, A1005 CrossRef CAS.
- X. Gao, W. Yuan, Y. Yang, Y. Wu, C. Wang, X. Wu, X. Zhang, Y. Yuan, Y. Tang, Y. Chen, C. Yang and B. Zhao, ACS Appl. Mater. Interfaces, 2022, 14, 43397 CrossRef CAS PubMed.
- R. Kido, K. Ueno, K. Iwata, Y. Kitazawa, S. Imaizumi, T. Mandai, K. Dokko and M. Watanabe, Electrochim. Acta, 2015, 175, 5 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 508 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- H. Sun, Z. Jin, C. Yang, R. L. Akkermans, S. H. Robertson, N. A. Spenley, S. Miller and S. M. Todd, J. Mol. Model., 2016, 22, 47 CrossRef PubMed.
- N. Yao, X. Chen, Z. H. Fu and Q. Zhang, Chem. Rev., 2022, 122, 10970 CrossRef CAS PubMed.
- S. C. Kim, X. Kong, R. A. Vila, W. Huang, Y. Chen, D. T. Boyle, Z. Yu, H. Wang, Z. Bao, J. Qin and Y. Cui, J. Am. Chem. Soc., 2021, 143, 10301 CrossRef CAS PubMed.
- H. Lu, Y. Zhu, B. Zheng, H. Du, X. Zheng, C. Liu, Y. Yuan, J. Fang and K. Zhang, New J. Chem., 2020, 44, 361 RSC.
- X. He, Y. Ni, Y. Hou, Y. Lu, S. Jin, H. Li, Z. Yan, K. Zhang and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 22672 CrossRef CAS PubMed.
- W. Liu, C. Yi, L. Li, S. Liu, Q. Gui, D. Ba, Y. Li, D. Peng and J. Liu, Angew. Chem., Int. Ed., 2021, 60, 12931 CrossRef CAS PubMed.
- P. Zeng, Y. Han, X. Duan, G. Jia, L. Huang and Y. Chen, Mater. Res. Bull., 2017, 95, 61 CrossRef CAS.
- H. Moon, T. Mandai, R. Tatara, K. Ueno, A. Yamazaki, K. Yoshida, S. Seki, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2015, 119, 3957 CrossRef CAS.
- Y. Shang, N. Chen, Y. Li, S. Chen, J. Lai, Y. Huang, W. Qu, F. Wu and R. Chen, Adv. Mater., 2020, 32, 2004017 CrossRef CAS PubMed.
- H. Jiang, Y. Wu, J. Ma, Y. Liu, L. Wang, X. Yao and H. Xiang, J. Membr. Sci., 2021, 640, 119840 CrossRef CAS.
- Q. Zhao, P. Chen, S. Li, X. Liu and L. A. Archer, J. Mater. Chem. A, 2019, 7, 7823 RSC.
- H. Lu, R. Feng, P. Wang, Y. Yuan, Z. Zhang, H. Du and X. Li, J. Mater. Sci.: Mater. Electron., 2022, 33, 16828 CrossRef CAS.
- W. Xiong, T. Huang, Y. Feng, X. Ye, X. Li, J. Liang, S. Ye, X. Ren, Y. Li, Q. Zhang and J. Liu, J. Mater. Chem. A, 2021, 9, 18338 RSC.
- F. Xu, S. Deng, Q. Guo, D. Zhou and X. Yao, Small Methods, 2021, 5, 2100262 CrossRef CAS PubMed.
- H. Xiang, P. Shi, P. Bhattacharya, X. Chen, D. Mei, M. E. Bowden, J. Zheng, J.-G. Zhang and W. Xu, J. Power Sources, 2016, 318, 170 CrossRef CAS.
- J. Yu, X. Lin, J. Liu, J. T. T. Yu, M. J. Robson, G. Zhou, H. M. Law, H. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2021, 12, 2102932 CrossRef.
- L. Yu, S. Guo, Y. Lu, Y. Li, X. Lan, D. Wu, R. Li, S. Wu and X. Hu, Adv. Energy Mater., 2019, 9, 1900257 CrossRef.
- L. Xia, S. Lee, Y. Jiang, Y. Xia, G. Z. Chen and Z. Liu, ACS Omega, 2017, 2, 8741 CrossRef CAS PubMed.
- T. Deng, L. Cao, X. He, A.-M. Li, D. Li, J. Xu, S. Liu, P. Bai, T. Jin, L. Ma, M. A. Schroeder, X. Fan and C. Wang, Chem, 2021, 7, 3052 CAS.
- P. Bai, X. Ji, J. Zhang, W. Zhang, S. Hou, H. Su, M. Li, T. Deng, L. Cao, S. Liu, X. He, Y. Xu and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202202731 CAS.
- Z. Cao, X. Zheng, Q. Qu, Y. Huang and H. Zheng, Adv. Mater., 2021, 33, 2103178 CrossRef CAS PubMed.
- Y. Qin, D. Wang, M. Liu, C. Shen, Y. Hu, Y. Liu and B. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 49445 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07393e |
| This journal is © The Royal Society of Chemistry 2023 |